

Abstract
Vibrio nigripulchritudo is an emerging pathogen of farmed shrimp in New Caledonia and other regions in the Indo-Pacific. The molecular determinants of V. nigripulchritudo pathogenicity are unknown; however, molecular epidemiological studies have suggested that pathogenicity is linked to particular lineages. Here, we performed high-throughput sequencing-based comparative genome analysis of 16 V. nigripulchritudo strains to explore the genomic diversity and evolutionary history of pathogen-containing lineages and to identify pathogen-specific genetic elements. Our phylogenetic analysis revealed three pathogen-containing V. nigripulchritudo clades, including two clades previously identified from New Caledonia and one novel clade comprising putatively pathogenic isolates from septicemic shrimp in Madagascar. The similar genetic distance between the three clades indicates that they have diverged from an ancestral population roughly at the same time and recombination analysis indicates that these genomes have, in the past, shared a common gene pool and exchanged genes. As each contemporary lineage is comprised of nearly identical strains, comparative genomics allowed differentiation of genetic elements specific to shrimp pathogenesis of varying severity. Notably, only a large plasmid present in all highly pathogenic (HP) strains encodes a toxin. Although less/non-pathogenic strains contain related plasmids, these are differentiated by a putative toxin locus. Expression of this gene by a non-pathogenic V. nigripulchritudo strain resulted in production of toxic culture supernatant, normally an exclusive feature of HP strains. Thus, this protein, here termed ‘nigritoxin', is implicated to an extent that remains to be precisely determined in the toxicity of V. nigripulchritudo.
Keywords: horizontal gene transfer, nigritoxin, phylogeny, shrimp, virulence, vibrio
Introduction
The family Vibrionaceae comprises a diverse group of bacteria that are widespread within marine environments (Thompson et al., 2004). It encompasses the well-studied human pathogen Vibrio cholerae and some less thoroughly characterized species that are responsible for infections in aquatic animals (Austin, 2010). The diseases due to these organisms have serious environmental and economic consequences. It is now widely accepted that intensive aquaculture practices are accompanied by considerable risks both for the amplification of existing pathogenic strains and for the spread of novel virulence determinants from these pathogens into existing commensal bacteria (Waterfield et al., 2004).
V. nigripulchritudo was first isolated in New Caledonia in 1995 from diseased shrimp (Litopenaeus stylirostris) affected by a winter vibriosis called Syndrome 93 (Goarant et al., 2006b). Initially, it was only reported in two adjoining farms and hence considered as a geographically restricted phenomenon. However, in 1997, V. nigripulchritudo was isolated from moribund shrimp collected from a farm some 50 km away from the original location, albeit this time during summer. As these conditions did not fit the Syndrome 93 epidemiology, which typically occurs when water temperature is below 25 °C, this new disease was named ‘Summer Syndrome' (Goarant et al., 2006a). Mass mortalities of other penaeid shrimp (Marsupenaeus japonicus and Penaeus monodon) ascribed to V. nigripulchritudo infections have also been reported in Japan (Sakai et al., 2007) and Madagascar (E Chung personal communication), suggesting that these bacterial pathogens may affect wider areas in the Indo-Pacific.
A collection of V. nigripulchritudo isolates obtained from a variety of sites in New Caledonia over an 8-year period was previously characterized by both multilocus sequence typing and experimental infections of L. stylirostris (Goarant et al., 2006a,b). Although these environmental isolates were genotypically diverse, all strains isolated from shrimp affected by Summer Syndrome and Syndrome 93 grouped into two distinct clades (A and B, respectively). The virulence of representative strains was assessed using three complementary experimental infection models (Le Roux et al., 2011). First, when shrimp were intramuscularly injected with live bacteria, strains could be operationally classified into highly pathogenic (HP), moderately pathogenic (MP) and non-pathogenic (NP) variants based on >80%, 20–80% and <20% observed host mortality, respectively. Second, when shrimp were transiently immersed into water inoculated with different isolates, only HP strains were able to induce mortality. Finally, when shrimp were intramuscularly injected with culture supernatants, those from HP strains but not MP strains caused death (exotoxicity). Strains from clade A were found to be either MP or HP (Reynaud et al., 2008), while most strains assigned to clade B were HP (Goarant et al., 2006b). Non-pathogenic isolates were genetically diverse. Hence, these results suggested that virulence is specific to clades A and B.
To gain better insight into the genetic and genomic underpinnings of the disease, we developed an HP strain from Summer Syndrome clade A (designated SFn1) as a model. This strain carries two plasmids, pASFn1 and pBSFn1 (formerly pA1066 and pB1067), of 250 and 11 kb, respectively (Reynaud et al., 2008; Le Roux et al., 2011). By infecting shrimp with derivatives of SFn1 from which pASFn1 and/or pBSFn1 had been lost, we found that both plasmids are necessary for full virulence. In the immersion model, only the wild-type strain containing both pASFn1 and pBSFn1 was virulent. In bacterial injection assays, SFn1 was more virulent than a mutant containing pASFn1 alone, while a mutant containing only pBSFn1 was avirulent. When supernatants were injected, toxicity was dependent upon the presence of pASFn1 only and attributed to a large, heat-sensitive factor. It is likely that differences between results obtained with the three challenge protocols reflect the different host-imposed barriers that are encountered by the bacteria. They may also indicate that there are multiple pathways by which the bacterium impairs shrimp viability.
Here, we further explore the genomic diversity and evolutionary history of pathogen-containing lineages of V. nigripulchritudo. We ask what types of genetic elements (i) differentiate each lineage and (ii) are shared among the HP strains within and across lineages. To address these questions, we performed high-throughput sequencing-based comparative genome analysis. Our phylogenetic analyses suggest that recent differential adaptation of lineages was followed by horizontal acquisition of genetic material, which distinguishes pathotypes within clades. A unique region carried by a large plasmid is specific to all HP strains and contains a gene encoding a putative toxin. Expression of this gene in a NP strain results in production of toxic culture supernatant.
Materials and methods
Bacterial strains, plasmids and culture conditions
The V. nigripulchritudo strains used for the genomic analyses are described in Table 1. Other bacterial strains and plasmids are described in Supplementary Tables S1 and S2. V. nigripulchritudo strains were grown in Luria-Bertani (LB) or LB-agar (LBA)+NaCl 0.5 M, marine broth or marine agar at 30 °C. Escherichia coli strains were grown in LB or on LBA at 37 °C. Spectinomycin (100 μg ml−1), thymidine (0.3 mM) and diaminopimelate (0.3 mM) were added as supplements when necessary. Induction of the PBAD promoter was achieved by the addition of 0.2% L-arabinose to the growth media, and conversely, was repressed by the addition of 1% D-glucose.
Table 1. Vibrio nigripulchritudo strains included in the study.
| Strains | Origin | Pathogenicity | Clade | Chr (Mb) | Plasmid (kb) | CDSs chr | CDSs pA_ | CDSs pB_ | Accession numbers |
|---|---|---|---|---|---|---|---|---|---|
| SFn1 | Summer syndrome, moribund L. stylirostris, March 2000, New Caledonia | HP | A | 4,11+2,21 | 250+11 | 5653 (3618+2035) | 189 | 9 | FO203526 VIBNI_A (Chr1), FO203527 VIBNI_B (Chr2), NC_015156 (pA), NC_010733 (pB) |
| SFn27 | Summer syndrome, sediment pore water, diseased pond, December 2000, New Caledonia | HP | A | 6.22 | 250+11 | 5655 | 191 | 9 | CAOB01000001-CAOB01000122 |
| SFn135 | Summer syndrome, growout pond water, diseased pond, December 2002, New caledonia | HP | A | 6.22 | 250+11 | 5672 | 191 | 9 | CAOD01000001-CAOD01000125 |
| Wn13 | Moribund L. stylirostris hemoculture, November 2002, New Caledonia | MP | A | 6.18 | 260 | 5611 | 197 | — | CAOG01000001-CAOG01000119 |
| ENn2 | Moribund L. stylirostris hemoculture, June 2000, New Caledonia | MP | A | 6.2 | 260 | 5631 | 199 | — | CANV01000001-CANV01000117 |
| BLFn1 | Moribund L. stylirostris hemoculture, March 2001, New Caledonia | MP | A | 6.16 | 260 | 5578 | 196 | — | CANU01000001-CANU01000120 |
| FTn2 | Healthy L. stylirostris, healthy pond, healthy farm, March 2003, New Caledonia | HP | B | 6.43 | 200 | 6080 | 147 | — | CANW01000001-CANW01000168 |
| SO65 | Syndrome 93, moribund L. stylirostris hemoculture, May 1995, New Caledonia | HP | B | 6.17 | 200 | 5768 | 151 | — | CAOE01000001-CAOE01000181 |
| AM115 | Syndrome 93, moribund L. stylirostris hemoculture, May 1995, New Caledonia | HP | B | 6.17 | 200 | 5927 | 149 | — | CANT01000001-CANT01000208 |
| POn4 | Healthy L. stylirostris hemoculture, healthy pond, healthy farm, April 2002, New Caledonia | NP | B | 6.07 | — | 5652 | — | — | CAOA01000001-CAOA01000149 |
| SOn1 | Moribund L. stylirostris hemoculture, July 2001, New Caledonia | NP | 6.44 | approx190 | 6117 | 231 | — | CAOF01000001-CAOF01000202 | |
| SFn118 | Growout pond water, diseased pond, April 2002, New Caledonia | NP | 6.11 | — | 5752 | — | — | CAOC01000001-CAOC01000235 | |
| ATCC 27043T | Seawater, chitin enrichment, USA | NP | 6.11 | — | 5770 | — | — | NZ-AFWJ00000000 | |
| Mada3021 | Mud crab, September 2007, Madagascar | Nd | M | 6.14 | 160 | 5775 | 127 | — | CANX01000001-CANX01000136 |
| Mada3020 | Moribund P. monodon hemoculture, April 2007, Madagascar | Nd | M | 6.15 | 160 | 5758 | 127 | — | CANY01000001-CANY01000140 |
| Mada3029 | Moribund P. monodon hemoculture, Pond B11, September 2007, Madagascar | Nd | M | 6.15 | 160 | 5768 | 127 | — | CANZ01000001-CANZ01000131 |
Abbreviations: CDSs, coding DNA sequences; Chr, chromosomes; HP, highly pathogenic; MP, moderately pathogenic; ND, Not determined; NP, non-pathogenic.
Genome sequencing, assembly and annotation
The complete genome sequence of SFn1 strain was obtained from (1) a Sanger library sequencing leading to a fourfold coverage; (2) a 454-single read library sequencing leading to a 16-fold coverage (Supplementary Methods). The 14 other V. nigripulchritudo strains were sequenced using the Illumina HiSeq2000 technology (Illumina, San Diego, CA, USA) with ∼50-fold coverage. Contigs were assembled de novo using Velvet (Zerbino and Birney, 2008) and genome assembly was improved by contig mapping against the SFn1 reference genome. Computational prediction of coding DNA sequences (CDSs) together with functional assignments was performed using the automated annotation pipeline implemented in the MicroScope platform (Vallenet et al., 2009). Antismash and NapDoS softwares were used to identify and annotate secondary metabolite biosynthesis gene clusters in the V. nigripulchritudo genomes (Medema et al., 2011, Ziemert et al., 2012). As the partial genome of V. nigripulchritudo type strain (ATCC 27043T) is available in Genbank (Hoffmann et al., 2012), it was also included in this study.
In silico analyses
To investigate the core and flexible genomes, an all-versus-all BlastP search was performed using genomic sequences of 134 organisms: 118 Vibrionaceae (21 completes, 97 whole-genome shotguns, including V. nigripulchritudo ATCC 27043T) and 1 Shewanella baltica (strain OS155, complete genome) available on Genbank and the 15 V. nigripulchritudo sequenced in the present study (Table 1). A dedicated precomputing repository (marshaling) was created to perform comparative genomic and phylogenomic analyses. Orthologous proteins were defined as reciprocal best hit proteins with 80% MaxLrap and a minimum of 30% and 60% identity cutoff was used for intra- and inter-species analysis, respectively (Daubin et al., 2002). The nucleic acid sequences were aligned using Muscle (Edgar, 2004) and filtered by Gblocks (Castresana, 2000). Phylogenetic trees were built using the parallel version of PhyML applied to Maximum-likelihood algorithm and General Time Reversible (GTR) model as parameters (NNIs, γ4, invariant site) (Guindon et al., 2010). Reliability was assessed by the bootstrap method with 100 replicates. Tree topologies were analyzed using the ETE2 Python library (Huerta-Cepas et al., 2010).
Vector construction and mutagenesis
Alleles carrying an internal deletion were generated in vitro using a two-step PCR construction method (Supplementary Methods and Supplementary Table S3), and cloned into an R6K γ-ori-based suicide vector that encodes the ccdB toxin gene under the control of an arabinose-inducible and glucose-repressible promoter, PBAD (Le Roux et al., 2007). Matings between E. coli and V. nigripulchritudo were performed at 30 °C as described previously (Le Roux et al., 2011). Selection of the plasmid-borne drug marker (SpecR) resulted in integration of the entire plasmid in the chromosome by a single crossover. Elimination of the plasmid backbone resulting from a second recombination step was selected by arabinose induction of the ccdB toxin gene. Mutants were screened by PCR.
The gfp and nigritoxin genes were PCR amplified and cloned under the control of a PBAD promoter in a P15A-ori-based replicative vector then transferred from E. coli to SFn118 by conjugation (Supplementary Methods). The same replicative vector was used to clone and express the reb genes cluster in E. coli.
Transmission electron microscopy
Transmission electron microscopy was used to explore R-body production by V. nigripulchritudo SFn1 and E. coli carrying the reb gene cluster expression vector (GV691). Expression of recombinant R-bodies was carried out by incubating the GV691 culture at 37 °C until OD600 nm of 0.4, then adding arabinose and shifting the culture to 25 °C for another 18 h of incubation. The bacteria were fixed, post fixed, dehydrated, embedded and ultracut as described previously (Schrallhammer et al., 2012).
Experimental challenges
Bacterial supernatants were prepared from bacterial cultures grown overnight in marine broth +/− glucose or arabinose. Specimens of L. stylirostris were injected with either 100 μl of filtered (0.22 μm) supernatant or sterile culture medium as a control following a previously described procedure (Le Roux et al., 2011). Experiments were conducted in triplicate (5 shrimp per tank, n=15). Survival was monitored for 24 h, preliminary trials having established that no significant mortality occurred after this time. The experiment was repeated three times.
Results
General features of the V. nigripulchritudo genomes
The genome of strain SFn1 was closed and annotated manually. It consists of two circular chromosomes of 4.1 (chromosome 1) and 2.2 (chromosome 2) Mb with an average percent G+C content of 45.9 and 45.5, respectively (Table 1). As described previously, SFn1 also carries two plasmids of 250 (pASFn1) and 11 (pBSFn1) kb (Le Roux et al., 2011). There is a total of 5653 predicted CDSs, 3618 being located on chromosome 1, 2035 on chromosome 2, 189 on pASFn1 and 9 on pBSFn1. Chromosomes 1 and 2 contain 8 and 0 rRNA, and 97 and 6 tRNA genes, respectively. The genome sequences of the other 14 strains were partially assembled, with contigs number per strain ranging from 123 to 208. The approximate genome sizes ranged from 6.1 to 6.4 Mb (Table 1).
Comparative genomics revealed pathotype consistent distribution of two plasmids. Like SFn1, the two other HP strains belonging to clade A (AHP) carry two plasmids of 250 and 11 kb, while the three MP strains belonging to clade A (AMP) carry only a large plasmid of 260 kb. Within clade B, the three HP strains (BHP) carry a large plasmid of 200 kb that is not present in the non-pathogenic BNP strain. A plasmid was also evident in the NP strain SOn1; however, it appears only distantly related to the other plasmids, and due to higher sequence divergence, it was not possible to complete its assembly using a PCR strategy based on the fully assembled genome SFn1. The NP strains SFn118 and ATCC 27043T do not carry any plasmid. Finally, the three strains isolated in Madagascar carry a plasmid of 160 kb. The pathogenicity of these strains to the native shrimp P. monodon has not yet been assessed, due to the lack of experimental bio-secure facilities in Madagascar. However, the culture supernatant of Mada3020 was demonstrated to be toxic for L. stylirostris (unpublished results).
Genes differentiating V. nigripulchritudo from other Vibrionaceae
Phylogenetic analysis of concatenated nucleic-acid sequences derived from 122-shared proteins from 133 Vibrionaceae genome sequences including V. nigripulchritudo, confirmed the cohesive genotypic structure of V. nigripulchritudo with relatively little diversity among genomes and only distant relationship to other currently characterized Vibrio species (Supplementary Figure S1).
Intraspecific genomic comparisons revealed that 4421 proteins are shared by all sequenced V. nigripulchritudo strains. Among these proteins, 620 were unique to V. nigripulchritudo genomes (317 on chromosome 1 and 303 on chromosome 2), whereas 486 (242 on chromosome 1 and 244 on chromosome 2) are shared between 1 and 5 strains of other Vibrionaceae species (Supplementary Figure S2). Similarly to other vibrios, chromosome 1 carries a higher proportion of the core genes, whereas chromosome 2 mainly carries accessory genes (Reen et al., 2006).
A phenotypic characteristic of all V. nigripulchritudo strains is the formation of brown-pigmented colonies. We identified two genes encoding a laccase (VIBNI_B0280) and a phosphotyrosinase (VIBNI_B1404), which might be involved in melanin production (Nosanchuk and Casadevall, 2003). The deletion of each gene in the SFn1 strain decreased the colony pigmentation (Figure 1). However, mutants from the two genes reverted to full pigmentation over time, suggesting possible functional redundancy and physiological readjustments. Several attempts to create a double mutant were unsuccessful, suggesting that the presence of these two genes may be essential for SFn1. It is noteworthy that the lack of a second usable resistance marker prevents demonstration that the double mutant can only be constructed when one of the two genes is provided in trans. We are currently exploring a larger panel of antibiotic resistance genes to allow the development of such strategy in the future.
Figure 1.
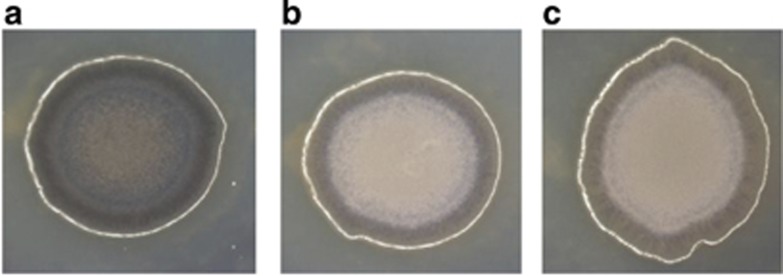
Colony pigmentation of V. nigripulchritudo strains SFn1 (a), Δ VIBNI_B0280 (laccase) (b) and Δ VIBNI_B1404 (phosphotyrosinase) (c) derivatives grown on marine agar supplemented with 1% glycerol.
Polyketide synthases (PKS-type I) have also been implicated in melanin production (Kroken et al., 2003). Among the putative PKS-non ribosomal peptide synthetase identified in SFn1 (Supplementary Table S4), only the plasmid gene VIBNI_pA0055 was annotated as a PKS-type I. However, curing pASFn1 plasmid from SFn1 did not result in a decrease of colony pigmentation (Le Roux et al., 2011). In addition, strains SFn118 and ATCC 27043T do not contain the large plasmid but still produce a brown pigment. Taken together, these data exclude a role of this gene in melanin production.
A cluster containing four reb genes (VIBNI_A3101, 3105, 3106 and 3107), potentially involved in the formation of refractile bodies (R-bodies) was identified in all V. nigripulchritudo strains (Supplementary Figure S3). R-bodies are coiled proteinaceous ribbons produced by Paramecium endosymbionts to which they confer a killer phenotype (Pond et al., 1989). A similar syntenic group was also found in the genome sequences of V. coralliilyticus (ATCC_BAA_450) and Marinomonas mediterranea (MMB-1). Within the cluster, six other genes encoding proteins of unknown function are also conserved between these bacteria. R-bodies were not observed by electron microscopy in cultured strains of V. nigripulchritudo and V. coralliilyticus (not shown). However, when this cluster of genes (VIBNI_A3108 to 3099) was cloned under the arabinose-inducible promoter PBAD in E. coli, transmission electron microscopy observations revealed the presence of R-body-like structures only in the induced cells (Figure 2), and resemble closely R-bodies of Caedibacter taeniospiralis and M. mediterranea (Schrallhammer et al., 2012).
Figure 2.
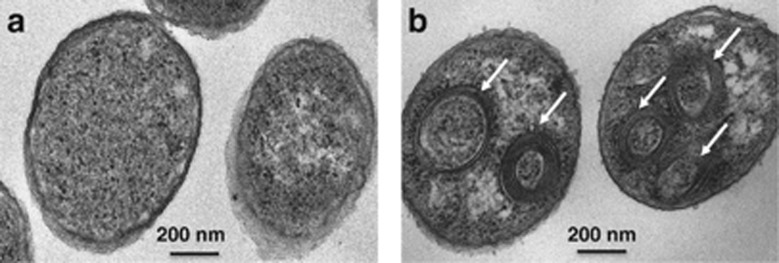
Transmission electron micrographs of bacterial cells. (a) Not induced. (b) Induced expression of the reb gene cluster (VIBNI_A3108 to 3099) cloned under the arabinose-inducible promoter PBAD in E. coli (GV691) and producing several recombinant R-bodies (white arrows).
Recent emergence of three lineages within V. nigripulchritudo
The phylogenetic relationship based on the core genome of strains included in this study, was investigated (Figure 3). The main outcome of this analysis was the grouping of all HP isolates, along with some MP and NP strains, into three clades with very little intra-clade diversity. Clades A and B contain all the HP strains along with some MP and NP isolates confirming previous multilocus sequence typing analysis (Goarant et al., 2006b) while all isolates from Madagascar form a distinct clade (termed M). This clade is a sister to clade A while clade B isolates are also more closely related to the non-pathogenic isolates SOn1 and SFn118 (Figure 3).
Figure 3.

Phylogenetic analysis based on concatenated alignments of nucleic acid sequences of 4421 core genes from 16 V. nigripulchritudo strains (ATCC 27043T as outgroup). Trees were built by the Maximum-Likelihood method based on sequences aligned using Muscle. Branch lengths are drawn to scale and are proportional to the number of nucleotide changes. Numbers at each node represent the percentage value given by bootstrap analysis of 100 replicates. The pathotype of each strain, when tested, is indicated in parentheses.
To investigate evidence for past recombination among the clades, we compared the phylogenetic relationships of each core genes (Supplementary Figure S4). This confirmed the placement of isolates into the three clades for 81% of the genes with >75% bootstrap values; however, the relationship among the three clades remains unresolved in 44% of the cases, while in 19, 17 and 20% clade affiliation switches between AM, BM or AB, suggesting that recombination has shuffled genes between clades.
As phylogenetic analysis indicated that the core genome of each clade contains very little diversity (Figure 3), we further investigated the percent nucleotide identities of each gene among all strains within each clade. Within clades A, B and M, the extent of gene identities ranges from 99.5 to 100, 98.6 to 100, 99.8 to 100%. Overall, this suggests that the clades are of near clonal and relatively recent origin as neither mutation nor recombination has introduced much diversity into their core genomes since the emergence from a common ancestral population.
A common, highly recombinogenic plasmid
We identified variants of the pASFn1 plasmid in AMP, BHP, M strains and SOn1, raising the possibility that these plasmids are extensively circulating among the V. nigripulchritudo strains and convey some sort of selective advantage to their hosts. The sequence comparison of these replicons revealed modules that differentiate the plasmids by geography, clade and pathogenicity (Figure 4). These plasmids contain a conserved core, consisting of 40 genes, that are localized within 14 modules (C1–14, Figure 4). Core genes encode a replication/segregation system (C1), a polyketide synthase cluster (C3) and a conjugation machinery (C6). A phylogenetic tree based on a concatenated alignment of all 40 nucleic acid sequences split V. nigripulchritudo plasmids into five clades with 100% bootstrap support (Supplementary Figure S5). Plasmid-containing hosts, which were grouped into clade A by chromosomal markers, split into two clades termed AHP and AMP, recapitulating different levels of pathogenicity (that is, highly and moderatly). In the plasmid-based comparison, these two clades are a sister to clade BHP and more distantly related to clade M (Supplementary Figure S5).
Figure 4.

Plasmid diversity among the different pathotypes. Modules are defined by gene(s) found in all plasmids (core C1 to 14 in gray); genes that are specific to strains isolated in New Caledonia (NC1 to 5, in green) or in Madagascar (M1 to 4, in turquoise); genes that are specific to clade A (A1 to 7, in pink), clade B (B in blue) or shared by clades M and B (M/B in black); genes that are specific to MP (MP1 to 3, in yellow) or specific to all HP (HP1 to 4, in red). The SFn1 strain-specific gene labels are VIBNI_: C1 [pA0002–pA0019], C2 [pA0028–pA0045], C3 [pA0051–pA0066], C4 [pA0088–pA0089], C5 [pA0113], C6 [pA0118–pA0134], C7 [pA0150], C8 [pA0169–pA0170], C9 [pA0178–pA0180], C10 [pA0198–pA0205], C11 [pA0207–pA0208], C12 [pA0210–pA0214], C13 [pA0222], C14 [pA0225], NC1 [pA0020–pA0027], NC2 [pA0047–pA0049], NC3 [pA0115–pA0117], NC4 [pA0135–pA0140], NC5 [pA0167–pA0168], A1 [pA0068–pA0087], A2 [pA0090–pA0112], A3 [pA0206], A4 [pA0209], A5 [pA0215–pA0221], A6 [pA0223–pA0224], A7 [pA0226–pA0239], HP1 [pA0141–pA0149], HP2 [pA0151–pA0166], HP3 [pA0171–pA0177], HP4 [pA0181–pA0196]. The BLFn1 strain-specific gene labels are: VIBNIBLFn1_: MP1 [p0128–p0145], MP2 [p0147–pA0154], MP1 [p0164–p0188]. The strain AM115 gene labels were VIBNIAM115_: B [p0058–p0066], M/B [p0155–p0165]. The strain Mada3020 gene labels were: VIBNIMada3020_: M1 [p0052–p0057], M2 [p0061–p0069], M3 [p0088–p0089], M4 [p0091–p0099].
To determine whether individual gene histories are consistent with the concatenated phylogeny, analyses were performed for each of the 40 gene sequences of the core plasmid using SOn1 as an outgroup (not shown). A total of 18/40 trees split the strains in three clades with >75% bootstrap values; 22% show a trifurcation of A, M and B clades, whereas in 0%, 17% and 22% of the trees AM, BM or AB were sister clades, respectively. In addition, three topologies (17, 11 and 11%) cluster all HP strains (AHP and BHP). Overall, these results suggest that this plasmid is a highly recombinogenic evolutionary mosaic.
Horizontal gene acquisition by HP strains within each lineage
Although all HP strains belong to near clonal lineages, none of these lineages exclusively consist of HP strains. We, therefore, performed comparative genomic analyses to identify HP-specific genes by first identifying genes present in all AHP but not AMP genomes, and in all BHP but not BNP genomes. These data were then used to define a set of genes common to all HP strains across lineages where the plasmids were major contributors to the HP-specific gene pool.
As noted above (Table 1), the three AHP strains carry two plasmids of 250 and 11 kb, previously demonstrated to be necessary for virulence in SFn1 (Le Roux et al., 2011), while the three AMP strains contain a replicon of 260 kb closely related to but distinct from the 250 kb plasmid. The plasmid in AMP strains lacks four modules that are specific to HP strains, (HP1-4), and instead carries three modules that are not present in the AHP large plasmid (MP1-3) (Figure 4). AMP-specific genes encode several ABC transporters, metabolic proteins and several proteins of unknown function. Four modules are AHP-specific and encode a galactose utilization operon (HP1), a siderophore ABC transporter, a peptidase (HP2) and genes involved in purine metabolism (HP3) (Supplementary Figure S6). In module HP4, the gene VIBNI_pA0182 encodes a protein annotated as a ‘putative toxin' (described in detail below). Additionally, 25 AHP-specific genes were located within three chromosomal islands (Figure 5). The first and second islands originated from the integration of a phage within the chromosomal thymidine synthase gene (thyA) (Figure 5a) and a tRNA gene (Figure 5b), respectively. These two islands encode common phage-related proteins (for example, integrase, helicase, relaxase and restriction endonuclease system) (Figure 5 CDSs in blue), as well as other proteins of unknown function. The third island is an operon encoding an ABC transporter and a probable hemolysin translocator, but does not contain a hemolytic toxin hlyA gene (Figure 5c).
Figure 5.

Structure of chromosomal genomic islands present in all AHP but absent in all AMP strains ((a) [VIBNI_A0333–0352], (b) [VIBNI_B1043–1063] and (c) [VIBNI_B1464–1471]), or present in all BHP and absent in the BNP strain ((d) [VIBNIAM115_840001–84152], (e) [VIBNIAM115_1620066–1620139]). CDSs in blue indicate a phage origin (a, b and d). CDSs in brown indicate a putative Hly translocator complex (c). CDSs in white encode proteins with unknown function. CDSs in black indicate the mobile genetic element boundaries, labeled ‘hypo' when encoding a hypothetical protein. ‘End' indicates the end of a contig. Within the ICE, recombination hot spot (HS) genes are indicated in pink (HS5), turquoise (HS1), orange (HS2), green (HS4), and yellow (HS3). The HS contents are shared by one or two other BHP strains (subscript). Regulator genes are indicated by a star.
We next identified genes shared by all BHP genomes but absent from the only BNP genome (POn4). This analysis yielded more genes than the AHP vs AMP comparison, in part because the large (200 kb) plasmid present in all BHP strains is absent in POn4 (Table 1). The four AHP modules described above are also found in the BHP plasmids and one module is specific to BHP plasmids (Figure 4, module B). In addition, 130 chromosomal genes are present in all BHP strains but absent from POn4. Over 75% of these genes are located within two large genomic islands (Figure 5d). The first island, integrated at a tRNA gene, appears to correspond to a prophage as it contains Cro regulators and phage E structural genes (Figure 5d). The second island has the typical structure of an integrative conjugative element (ICE) (Figure 5e). The three BHP strains share 52 core genes required for ICE function and differ at several recombinational hot spots (Figure 5e, HS1 to 5) (Wozniak and Waldor, 2010).
Finally, we determined which genes are present in all HP strains (AHP and BHP), but absent from the AMP and BNP strains. This showed that only the four plasmid modules (HP1 to 4) described above are present in and specific to all HP strains (Figure 4 and Supplementary Figure S6). Interestingly, the module HP3 and a part of HP4 were also identified in the plasmid carried by clade M isolates (Figure 4) and the culture supernatant of Mada3020 was demonstrated to be toxic for L. stylirostris (unpublished results). These results raise the possibility that the HP3 and/or HP4 modules might be particularly important in the emergence of pathogenic strains.
Nigritoxin, a new toxin sufficient to kill shrimp
As noted above, the HP4 module contains a gene (VIBNI_pA0182) annotated as a putative toxin. This gene (2274 bp) encodes a 757 amino-acid polypeptide corresponding to a putative protein with a theoretical molecular mass of 82.9 kDa. Using InterProScan, PFAM and Figfam we were unable to identify any particular functional domain within the putative protein. However, BlastP analysis revealed that amino acids 28 to 373 of the VIBNI_pA0182 gene product shares about 30% identity with a region of a putative toxin named Afp18 described in Serratia entomophila (amino acids 978–1329) (Hurst et al., 2004) and Yersinia ruckeri (amino acids 937–1286) (Supplementary Figure S7).
Numerous attempts to disrupt specific genes, including but not limited to VIBNI_pA0182, within the large plasmid of SFn1 were unsuccessful. Previous work suggests that genetic tools that have been successfully used to mutagenize V. nigripulchritudo chromosomal and pBSFn1 genes (R6K-based suicide vector) interfere in an unknown fashion with pASFn1 (Le Roux et al., 2011). Therefore, to assess whether the putative toxin is linked to V. nigripulchritudo-associated exotoxicity, a plasmid containing VIBNI_pA0182 under the control of a PBAD promoter was introduced in the avirulent SFn118 strain and the supernatant toxicity was assayed by injection into shrimp. Supernatant from SFn118 containing a control plasmid, which carries a gene encoding green fluorescent protein (GFP), was also tested. For this assay, strains were cultivated in the presence of 1% glucose or 0.2% arabinose in order to either repress or activate the PBAD promoter.
Strikingly, 1 day post-injection, 100% mortality occurred in shrimp injected with supernatant prepared from PBAD-VIBNI_pA0182 transconjuguants cultivated in the presence of arabinose, whereas no significant mortality was observed when animals were challenged with supernatant prepared from transconjuguants grown in the presence of glucose (Figure 6). No significant mortality was observed in shrimp injected with PBAD-GFP transconjuguant supernatants cultivated in arabinose or in glucose. Additionally, no mortality was observed following injection of supernatants prepared from E. coli carrying the same plasmids (PBAD-VIBNI_pA0182 or PBAD- gfp) in the presence of arabinose or glucose and 100% mortality occurred in shrimp injected with supernatant prepared from the HP strain SFn1. These data indicate that VIBNI_pA0182 contributes to the supernatant toxicity associated with HP strains. Culture supernatants were analyzed using SDS-polyacrylamide gel electrophoresis in an attempt to confirm that supernatant toxicity correlates with the amount of the overproduced protein. However, the complexity of the protein profiles prevented the identification of a single protein (data not shown).
Figure 6.

Shrimp mortality in response to injection of bacterial culture supernatants. Shrimp, L. stylirostris (n=5), were injected with 100 μl of bacterial supernatants prepared from overnight cultures. Survival was assessed after 24 h. Experiments were conducted three times. Shrimp were intramuscularly injected with: supernatants prepared from the V. nigripulchritudo HP strain SFn1 and avirulent strain SFn118; from the PBAD-VIBNI_pA0182 transconjuguant of SFn118 cultivated in the presence of arabinose or glucose; from the PBAD-GFP transconjuguant of SFn118 cultivated in the presence of arabinose or glucose; from E. coli carrying the PBAD-VIBNI_pA0182 plasmid cultivated in the presence of arabinose or glucose; from E. coli carrying the PBAD-GFP plasmid cultivated in the presence of arabinose or glucose; with medium Zobell or LB as a control.
Discussion
We performed high-throughput sequencing-based comparative genome analysis of 16 V. nigripulchritudo strains to explore the genomic diversity and evolutionary history of pathogen-containing lineages within the species and investigate virulence determinants. Phylogenetic analysis based on the V. nigripulchritudo core genome suggests a recent emergence of three lineages (A, B and M) containing all disease-associated HP strains but not exclusively so. HP-specific genes acquired by HGT were evidenced in comparative genomic analyses. These genes include a toxin homolog, VIBNI_pA0182, which is encoded by the large plasmid present in all AHP, BHP and M strains. Heterologous expression of VIBNI_pA0182 conferred a toxic phenotype following bacterial supernatant injection to shrimp, demonstrating its contribution to the supernatant toxicity associated with V. nigripulchritudo, a specific feature of HP strains.
Among genes unique to V. nigripulchritudo genomes or shared with a few other Vibrio species, two genes encoding a laccase and a phosphotyrosinase could be involved in melanin production (Nosanchuk and Casadevall, 2003). Melanins have been associated with enhanced virulence in many microorganisms including (i) possession of antioxidant properties that can attenuate macrophage superoxide production, (ii) interference with the action of endogenous antimicrobial peptides and (iii) facilitation of ferrous iron uptake through a specific transport system. Whether melanin production has a role in V. nigripulchritudo virulence and/or is essential for this bacterium are interesting issues that require a complete inactivation of the pigment synthesis, presently hampered by the lack of available resistance markers to allow multiple gene knock outs in these multiresistant strains.
Another feature of V. nigripulchritudo genomes, shared with V. coralliilyticus, is the presence of a cluster of reb genes potentially involved in the formation of ‘R-bodies' (Pond et al., 1989). These cytoplasmic inclusions were first described in Caedibacter taeniospirilis, an obligate symbiont of paramecia to which they confer a killer phenotype. This enables them to kill symbiont-free competitors by an unknown mechanism. Recently a study demonstrated that Reb homologs are widely distributed in members of proteobacteria (Raymann et al., 2013). In the present study, although we did not observe R-body structures in cultured strains of V. nigripulchritudo or V. coralliilyticus, the induction of the expression of V. nigripulchritudo reb genes in E. coli was sufficient to produce R-bodies, suggesting that specific conditions are required for reb gene expression in Vibrio. In the future, it will be interesting to perform grazing tests to investigate R-bodies in V. nigripulchritudo or V. coralliilyticus and test their killing potential in protists.
Several genes related to antibiotic synthesis, particularly PKS-non ribosomal peptide synthetase, are shared by all sequenced V. nigripulchritudo and specific to this species. These enzymes produce polyketides, a large class of secondary metabolites with a wealth of pharmacologically important activities, including antimicrobial (Staunton and Weissman, 2001). PKS have recently been implicated in mediation of social interactions among Vibrio populations where the antibiotics are produced in a highly specific manner by few members of the population while all others are resistant, giving rise to the idea that they may have a role in interference competition between populations (Cordero et al., 2012).
In New Caledonia, V. nigripulchritudo strains have been isolated from different sources (water, sediment, crustaceans) and were previously shown by multilocus sequence typing to be genetically diverse (Goarant et al., 2006b). However, disease-associated isolates were grouped into two fairly monomorphic clades, termed A and B. In the present study, a phylogenetic analysis based on the concatenated V. nigripulchritudo core genome showed that isolates from Madagascar form a new clade (termed M), which is a sister to clade A and more distantly related to clade B. However, this phylogeny varies when individual genes are analyzed, suggesting that past recombination has shaped the diversity within these clades. Within clades A, B and M the strains seem nearly clonal, suggesting that the expansion of these lineages is relatively recent and independent from each other as neither mutation nor recombination has introduced much diversity into the core genome differentiating the contemporary strains. Importantly, because clades A and B also contain NP and MP strains, it was possible to identify genes and genetic elements acquired in the evolution of pathogenesis.
We previously demonstrated that it is possible to discriminate AHP from AMP strains based on the pASFn1 plasmid genotype (Le Roux et al., 2011). Here, we show that a plasmid sharing a core set of genes with pASFn1 occurs in all pathogenic strains. These plasmids also contain distinct modules that permit plasmid classification based on geography, clade and pathogenicity. However, analysis of all genes individually shows, like in the case of the chromosomes, discordant phylogeny, suggesting this plasmid is a highly recombinogenic evolutionary mosaic. Interestingly, a few genes cluster AHP and BHP, suggesting recombination events between plasmids carried by strains from different lineages. Further evidence of such genetic exchanges is the presence of four modules found only in AHP and BHP.
Within module HP4, a putative toxin-encoding gene, VIBNI_pA0182, was identified. This gene encodes a protein with a theoretical MW of 82.9 kDa, which is consistent with the molecular mass of the toxic proteinaceous compounds previously detected in SFn1 culture supernatant and shown to be dependent upon the presence of pASFn1 (Le Roux et al., 2011). In the present study, we demonstrated that the expression of VIBNI_pA0182 is sufficient to induce supernatant toxicity in the NP strain SFn118. We propose to name the product of VIBNI_pA0182 ‘nigritoxin'. Although we do not yet know the pathotype of disease-associated strains from Madagascar, the fact that (i) the supernatant of Mada3020 was demonstrated to be toxic for L. stylirostris and (ii) the nigritoxin gene was also present in strains belonging to clade M strongly suggest that disease-associated isolates from Madagascar are HP strains. Altogether, our results demonstrate that the nigritoxin is a key factor of V. nigripulchritudo shrimp pathogenesis shared by different lineages.
The nigritoxin is similar to a putative toxin described in Serratia entomophila (Afp18), which causes amber disease in the grass grub Costelytra zealandica (Coleoptera: Scarabaeidae) (Hurst et al., 2004). Larval disease symptoms include cessation of feeding, clearance of the gut, amber coloration and eventual death. A 155-kb plasmid, pADAP, is essential for production of amber disease symptoms. Within this plasmid, a defective prophage (afp) carries the antifeeding genes (Hurst et al., 2004). It has been postulated that the first 16 CDSs of the afp cluster comprise the carriage region that forms an R-type pyocin structure, which functions as the delivery system for the putative toxin Afp18. As the expression of Afp18 in E. coli failed to cause the antifeeding pathotype, the authors suggested that the R-type pyocin is mandatory for the transport of the toxin to the target site (Hurst et al., 2007). Here, aside from the nigritoxin gene, afp encoding phage tail-like structures could not be identified in the HP genomes. Expression of the nigritoxin in E. coli failed to confer a supernatant toxicity but was sufficient to induce a toxic phenotype in SFn118. As SFn118 does not contain any of the plasmids, ICE or prophage described as HP-specific traits, the secretion of the nigritoxin may be regulated by V. nigripulchritudo core genes, but the partners of this process are still unknown. On the other hand, we cannot rule out the possibility that the recombinant nigritoxin may accumulate within E. coli cells as insoluble inclusion bodies, known to be devoid of biological activities. In the future, mutagenesis of the nigritoxin gene will allow identification of the domain(s) of the protein required for processing, export and toxic effects.
As microbial pathogenesis is often multifactorial (Finlay and Falkow, 1997), it is likely that other genes than the nigritoxin are involved in the V. nigripulchritudo virulence process. We previously demonstrated that pBSFn1 is necessary for the expression of full virulence in SFn1 (Le Roux et al., 2011). Other mobile genetic elements discriminate the pathotypes within each lineage. In addition to pBSFn1, three chromosomal genomic islands are specific to AHP strains. One island encodes an ABC transporter and a probable hemolysin translocator. In E. coli, hemolysin translocator has been demonstrated to be essential for the secretion of the Repeats-in-Toxin (RTX) hemolytic toxin HlyA (Pimenta et al., 2005). Several putative Repeats-in-Toxin proteins have been identified in the V. nigripulchritudo genomes but were not restricted to HP strains (not shown). In clade B, two large genomic islands, a prophage and an ICE, are specific to BHP strains. As no CDSs annotated in these mobile genetic elements can be assigned clearly to a virulence factor, it remains to be seen whether these HP-specific genetic elements are required for virulence.
Overall, it appears that three lineages containing shrimp pathogens have evolved independently and fairly recently within V. nigripulchritudo. As these lineages contain strains with varying pathogenicity, as well as non-pathogens, it is possible to reconstruct at least partially the genomic changes that have accompanied evolution of the HP pathotype. A key factor appears to have been the acquisition of the nigritoxin, which is located within a large, mobile plasmid, suggesting a plausible avenue for rapid and parallel evolution from harmless to pathogenic.
Acknowledgments
We acknowledge Dr Eliane Chung and Iony Manitra Razanajatovo for providing the strains isolated in Madagascar, Sophie Le Panse for the transmission electron microscopy analysis, the staff of the station Ifremer Nouvelle Calédonie, the ABIMS and LABGeM plateform for technical support, Dr Simonetta Gribaldo, Otto Cordero and Dr Cyrille Goarant for fruitful discussions and Dr Brigid Davis for critical reading of the manuscript. The present study has been supported by the Genoscope (appel d'offre 2007 and 2011) Region Bretagne (SAD Vibrigen 6633), the Institut Pasteur, the CNRS (UMR3525), the ANR blanc (11-BSV7-023-01 ‘VIBRIOGEN') and Ifremer (DG post doctoral funding).
Footnotes
Supplementary Information accompanies this paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Austin B. Vibrios as causal agents of zoonoses. Vet Microbiol. 2010;140:310–317. doi: 10.1016/j.vetmic.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Cordero OX, Wildschutte H, Kirkup B, Proehl S, Ngo L, Hussain F, et al. Ecological populations of bacteria act as socially cohesive units of antibiotic production and resistance. Science. 2012;337:1228–1231. doi: 10.1126/science.1219385. [DOI] [PubMed] [Google Scholar]
- Daubin V, Gouy M, Perriere G. A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history. Genome Res. 2002;12:1080–1090. doi: 10.1101/gr.187002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay BB, Falkow S. Common themes in microbial pathogenicity revisited. Microbiol Mol Biol Rev. 1997;61:136–169. doi: 10.1128/mmbr.61.2.136-169.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goarant C, Ansquer D, Herlin J, Domalain F, Imbert F, de Decker S. ‘Summer syndrome' in litopenaeus stylirostris in New Caledonia: pathology and epidemiology of the etiological agent, Vibrio nigripulchritudo. Aquaculture. 2006a;253:105–113. [Google Scholar]
- Goarant C, Reynaud Y, Ansquer D, de Decker S, Saulnier D, le Roux F. Molecular epidemiology of Vibrio nigripulchritudo, a pathogen of cultured penaeid shrimp (Litopenaeus stylirostris) in New Caledonia. Syst Appl Microbiol. 2006b;29:570–580. doi: 10.1016/j.syapm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, Monday SR, Allard MW, Strain EA, Whittaker P, Naum M, et al. Vibrio caribbeanicus sp. nov., isolated from the marine sponge Scleritoderma cyanea. Int J Syst Evol Microbiol. 2012;62:1736–1743. doi: 10.1099/ijs.0.032375-0. [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J, Dopazo J, Gabaldon T. ETE: a python environment for tree exploration. BMC Bioinformatics. 2010;11:24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst MR, Beard SS, Jackson TA, Jones SM. Isolation and characterization of the Serratia entomophila antifeeding prophage. FEMS Microbiol Lett. 2007;270:42–48. doi: 10.1111/j.1574-6968.2007.00645.x. [DOI] [PubMed] [Google Scholar]
- Hurst MR, Glare TR, Jackson TA. Cloning Serratia entomophila antifeeding genes—a putative defective prophage active against the grass grub Costelytra zealandica. J Bacteriol. 2004;186:5116–5128. doi: 10.1128/JB.186.15.5116-5128.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroken S, Glass NL, Taylor JW, Yoder OC, Turgeon BG. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad Sci USA. 2003;100:15670–15675. doi: 10.1073/pnas.2532165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux F, Binesse J, Saulnier D, Mazel D. Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl Environ Microbiol. 2007;73:777–784. doi: 10.1128/AEM.02147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux F, Labreuche Y, Davis BM, Iqbal N, Mangenot S, Goarant C, et al. Virulence of an emerging pathogenic lineage of Vibrio nigripulchritudo is dependent on two plasmids. Environ Microbiol. 2011;13:296–306. doi: 10.1111/j.1462-2920.2010.02329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, et al. AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic acids Res. 2011;39:W339–W346. doi: 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosanchuk JD, Casadevall A. The contribution of melanin to microbial pathogenesis. Cell Microbiol. 2003;5:203–223. doi: 10.1046/j.1462-5814.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- Pimenta AL, Racher K, Jamieson L, Blight MA, Holland IB. Mutations in HlyD, part of the type 1 translocator for hemolysin secretion, affect the folding of the secreted toxin. J Bacteriol. 2005;187:7471–7480. doi: 10.1128/JB.187.21.7471-7480.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond FR, Gibson I, Lalucat J, Quackenbush RL. R-body-producing bacteria. Microbiol Rev. 1989;53:25–67. doi: 10.1128/mr.53.1.25-67.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymann K, Bobay LM, Doak TG, Lynch M, Gribaldo S. A genomic survey of reb homologs suggests widespread occurrence of R-bodies in proteobacteria. G3 (Bethesda) 2013;3:505–516. doi: 10.1534/g3.112.005231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reen FJ, Almagro-Moreno S, Ussery D, Boyd EF. The genomic code: inferring Vibrionaceae niche specialization. Nat Rev Microbiol. 2006;4:697–704. doi: 10.1038/nrmicro1476. [DOI] [PubMed] [Google Scholar]
- Reynaud Y, Saulnier D, Mazel D, Goarant C, Le Roux F. Correlation between detection of a plasmid and high-level virulence of Vibrio nigripulchritudo, a pathogen of the shrimp Litopenaeus stylirostris. Appl Environ Microbiol. 2008;74:3038–3047. doi: 10.1128/AEM.02680-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai T, Hirae T, Yuasa K, Kamaishi T, Matsuyama T, Miwa S, et al. Mass mortality of cultured kuruma prawn Peneaus japonicus caused by Vibrio nigripulchritudo. Fish Pathol. 2007;42:141–147. [Google Scholar]
- Schrallhammer M, Galati S, Altenbuchner J, Schweikert M, Gortz HD, Petroni G. Tracing the role of R-bodies in the killer trait: absence of toxicity of R-body producing recombinant E. coli on paramecia. Eur J Protistol. 2012;48:290–296. doi: 10.1016/j.ejop.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- Thompson FL, Iida T, Swings J.2004Biodiversity of vibrios Microbiol Mol Biol Rev 68403–431.(table of contents). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallenet D, Engelen S, Mornico D, Cruveiller S, Fleury L, Lajus A, et al. MicroScope: a platform for microbial genome annotation and comparative genomics. Database (Oxford) 2009;2009:bap021. doi: 10.1093/database/bap021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterfield NR, Wren BW, Ffrench-Constant RH. Invertebrates as a source of emerging human pathogens. Nat Rev Microbiol. 2004;2:833–841. doi: 10.1038/nrmicro1008. [DOI] [PubMed] [Google Scholar]
- Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Microbiol. 2010;8:552–563. doi: 10.1038/nrmicro2382. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR. The natural product domain seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS One. 2012;7:e34064. doi: 10.1371/journal.pone.0034064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.