

Abstract
Blood brain barrier (BBB) disruption occurring within the first few hours of ischemic stroke onset is closely associated with hemorrhagic transformation following thrombolytic therapy. However, the mechanism of this acute BBB disruption remains unclear. In the neurovascular unit, neurons do not have direct contact with the endothelial barrier, however they are highly sensitive and vulnerable to ischemic injury, and may act as the initiator for disrupting BBB when cerebral ischemia occurs. Herein we employed oxygen-glucose deprivation (OGD) and an in vitro BBB system consisting of brain microvascular cells and astrocytes to test this hypothesis. Neurons (CATH.a cells) were exposed to OGD for 3-hours before co-culturing with endothelial monolayer (bEnd 3 cells), or endothelial cells plus astrocytes (C8-D1A cells). Incubation of OGD-treated neurons with endothelial monolayer alone did not increase endothelial permeability. However, when astrocytes were present, the endothelial permeability was significantly increased, which was accompanied by loss of occludin and claudin-5 proteins as well as increased VEGF secretion into the conditioned medium. Importantly, all these changes were abolished when VEGF was knocked down in astrocytes by siRNA. Our findings suggest that ischemic neurons activate astrocytes to increase VEGF production, which in turn induces endothelial barrier disruption.
Keywords: blood brain barrier, oxygen glucose deprivation, neuron, astrocyte, VEGF
Introduction
Acute blood brain barrier (BBB) disruption occurring within the first few hours of ischemic stroke onset has been increasingly appreciated as it negatively impacts the safety and efficacy profiles of thrombolytic therapy for ischemic stroke. In an early clinical magnetic resonance imaging (MRI) study, Warach and Latour (Warach & Latour 2004) reported that 8 out of 9 stroke patients who had early ischemic BBB damage underwent cerebral bleeding following thrombolytic therapy and showed worse outcome. Several recent animal and human studies have shown that brain regions with a compromised BBB at early stroke stages often undergo hemorrhagic transformation at later times during thrombolytic reperfusion (Hjort et al. 2008, Hom et al. 2011, Sun et al. 2010, Jin et al. 2012). Thus, protecting BBB integrity during the acute phase of cerebral ischemia is crucial to reducing thrombolysis-associated hemorrhagic complications. However, the mechanism underlying acute ischemic BBB damage remains elusive.
The BBB, also known as the neurovascular unit, is formed by the endothelium of brain capillaries, surrounded by extracellular matrix components and several associated cell types including astrocytes, pericytes and neurons (Zlokovic 2008, Yang & Rosenberg 2011a, Sa-Pereira et al. 2012). Ischemic damage to non-neuronal cells and the extracellular matrix has been extensively studied in cellular and animal stroke models, in an attempt to understand the molecular mechanisms underlying BBB disruption (Liu et al. 2012b, Borlongan et al. 2012, Ballabh et al. 2004). However, how ischemic neurons contribute to BBB damage has been largely neglected, probably due to the fact that the neuronal cells are not in direct contact with brain capillaries. In general, neurons are very sensitive and vulnerable to ischemic insult due to their high demand on oxygen and nutrition (Bickler & Donohoe 2002), which may render them the first cells in the brain to respond to ischemia. Previous studies showed that damaged neurons could produce molecules that activate astrocytes, such as FGF-1 (Pehar et al. 2005), CXC and CC chemokines (de Haas et al. 2007). During homeostasis, astrocytes interact with capillary endothelial cells to maintain the BBB integrity. Under pathological conditions, activated astrocytes may produce many active factors such as VEGF and cytokines to disrupt the BBB (Haseloff et al. 2005, Argaw et al. 2012, Chaitanya et al. 2011, Heinemann et al. 2012). In this context, we speculated that ischemic neurons, though not being in direct contact with the BBB, may be an early contributor to ischemic BBB damage through interaction with astrocytes.
In the present study, we tested this hypothesis using an in vitro BBB system consisting of brain microvascular cells and astrocytes. The results showed that incubation of astrocytes with ischemic neurons for 24 hours and then with endothelial monolayer for another 24 hours increased endothelial monolayer permeability, compared with controls. Moreover, increased VEGF production from ischemic neuron-primed astrocytes was the key mediator of this endothelial permeability increase.
Materials and methods
Cell culture
Neuronal cell CATH.a, astrocytic cell C8-D1A and brain microvascular endothelial cell bEnd3 were obtained from American Type Culture Collection (hereafter referred to as neurons, astrocytes, and endothelial cells or ECs, respectively). Neurons were cultured in RPMI 1640 medium containing 2 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM HEPES, 1.0 mM sodium pyruvate, 8% horse serum, 4% fetal bovine serum and antibiotics (penicillin, 100 U/mL and streptomycin, 100 μg/mL). Astrocytes and ECs were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics. All of the cells were grown in a humidified atmosphere of 5% CO2/95% air at 37 °C.
In vitro BBB model of endothelial monolayer
An endothelial monolayer grown on a cell culture insert was widely used for in vitro BBB models. Nunc cell culture inserts (for 24 well plates) with 0.02 μm Anapore membranes (Nunc Inc., Naperville, IL) were coated with collagen by incubating with 70 μg/mL type I collagen (BD Biosciences, San Jose, CA) in 20 mM acetic acid for 1 hour at room temperature. Inserts were then washed with serum free medium to remove excess protein before adding complete medium to the inserts and the wells to equilibrate the membrane for 3 hours at 37°C in a cell culture incubator. ECs were trypsinized from tissue culture flasks, washed 3 times with complete medium, and seeded on inserts at 20,000 cells/cm2. The seeded cells were allowed to grow for 3–4 days at 37°C in 5% CO2/95% air to achieve full confluence, which was confirmed under a phase contrast microscopy.
Exposure of neurons to OGD treatment
To mimic ischemic condition in vitro, neurons were exposed to oxygen and glucose deprivation (OGD) as we described previously (Furuichi et al. 2005). In brief, neurons were seeded on Nunc cell culture inserts at 20,000 cells/cm2. When they reached 60–70% confluency, the inserts with cells were subjected to ischemic injury by transferring the cultures to glucose free medium (DMEM without glucose) pre-equilibrated with 95% N2 and 5% CO2. Cells were then incubated in a humidified airtight chamber (Billups-Rothberg Inc.) equipped with an air lock and flushed with 5% CO2/95% N2 for 15 minutes. The chamber was then sealed and kept at 37°C for 3 hours. The oxygen concentration was below 0.2% as monitored by an oxygen analyzer (Sable Systems). Control cultures were incubated with normal DMEM medium without FBS at 37°C in 5% CO2/95% air for 3 hours. OGD was terminated by removing cells from the hypoxic chamber, followed by changing back to complete culture medium. Trypan blue staining shown that all of the neurons were alive after 3 hours OGD treatment.
Co-culture of endothelial monolayer with astrocytes and ischemic neurons
To determine whether ischemic neurons impact BBB permeability, we applied a co-culture system consisting of endothelial monolayer grown on Nunc cell culture inserts, with astrocytes grown on 24 well plates and neurons grown on the inserts pre-exposed or not to 3 hours OGD as described above. The inserts with ischemic or normal neurons were transferred to 24 well plates with astrocytes already grown on the bottom of the wells (with the 20,000 cells/cm2 of initial seeded cells and reach 70% confluence before use) and co-cultured for 24 hours. Then, the neuronal inserts were replaced with endothelial inserts having 100% confluent ECs grown on them. The endothelial inserts were co-cultured with astrocytes for another 24 hours before measuring the endothelial monolayer permeability as described below.
Endothelial monolayer permeability assay
Permeability of the EC monolayer was measured as described previously (Vincent et al. 2008). In brief, at the end of 24 hours incubation, 750 μL assay medium (RPMI-1640 supplemented with 10 mM HEPES, antibiotics and 0.5% BSA) was added to each lower chamber (i.e. the well containing the insert) and 150 μL assay medium containing 100 μg/mL FITC-BSA (Invitrogen, Carlsbad, CA) was added to each insert (upper chamber). These volumes ensured similar hydrostatic pressure on both sides of the bEnd3 monolayer. Incubation continued for 1 hour at 37°C in 5% CO2/95% air. Afterwards the inserts were carefully removed, and the medium in each lower chamber was thoroughly mixed. Aliquots of 150 μL conditioned media was collected from the lower chambers, FITC-BSA assay medium (total fluorescence added to inserts), and the assay medium itself (background) was used for measuring fluorescence intensity on a fluorescence plate reader (Fluorescence Concentration Analyzer, IDEXX Laboratories, Westbrook, ME). Endothelial monolayer permeability was quantitated as clearance of FITC-BSA from the upper chamber to the lower chamber after subtracting background fluorescence intensity using the following equation: Clearance (%) = (Fluorescence in lower chamber/Total fluorescence added in upper chamber) × 100. After permeability assessment, the endothelial inserts were examined under phase-contrast microscopy to check the intactness of each endothelial monolayer. Data obtained from inserts with disrupted monolayers were excluded.
Knockdown of VEGF with siRNA
VEGF siRNA pools and transfection reagents were purchased from Santa Cruz Biotechnology (Santa Cruz Biotechnology, Santa Cruz, CA). Astrocytes grown to 60–80% confluence on 24 well plates were used for siRNA knockdown. The following solutions were prepared, solution A: for each transfection, 3 μL of siRNA duplex (sc-36815 or sc-37007) was diluted into 50 μL siRNA transfection medium (sc-36868); solution B: for each transfection, 3 μL of siRNA transfection reagent (sc-29528) was diluted into 50 μL siRNA transfection medium. Then, solution A was gently mixed with solution B and incubated for 30 minutes at room temperature. For each transfection, 0.4 mL siRNA transfection medium was added to 0.1 mL siRNA transfection reagent mixture (solution A + solution B) and mixed gently. Then the mixture was overlaid onto astrocytes that were pre-rinsed with antibiotics free DMEM medium. After incubation at 37° C in a CO2 incubator for 6 hours, 0.5 mL of DMEM containing 20% FBS, 200 U/mL penicillin, and 200 μg/mL streptomycin was added without removing the transfection mixture. The cells were incubated for an additional 18 hours before replacing the medium with fresh normal growth medium. After another 48 hours, the astrocytes were used for indicated experiments. Specific silencing of VEGF expression was confirmed by western blot analysis.
Western Blot analysis
Cell lysate (30 μg of protein) was resolved on an 8–10% SDS-polyacrylamide gel and transferred onto nitrocellulose membrane (Bio-Rad, Hercules, CA), and incubated for 1 hour in TBST (10 mM Tris, pH 8.0, 150 mM NaCl and 0.1% Tween 20) containing 5% non-fat milk at room temperature. The membrane was then incubated overnight at 4 °C with primary antibodies against occludin (Invitrogen, 1:1000 dilution), claudin-5 (Invitrogen, 1:1000 dilution) and VEGF (Abcam, Cambridge, MA, 1:1000 dilution). After washing with TBST, the membrane was incubated for 1 hour with horseradish peroxidase conjugated secondary antibodies (Santa Cruz Biotech., 1:2000 dilution). Protein bands on membranes were detected on a Kodak image station 4000MM after incubating with the SuperSignal West Pico chemiluminescent reagents (Pierce, Rockford, IL). To control for sample loading and protein transfer, the membrane was stripped and re-probed to detect β-actin (from Santa Cruz Biotech., 1:500 dilution). Protein band intensities were quantified using Kodak molecular imaging software version 4.0.
ELISA assay for VEGF
VEGF contents in cell culture supernatants were assessed using an ELISA kit from Invitrogen. Briefly, cell culture supernatants were diluted 5 times in standard diluent buffer. Aliquots of 100 μL of diluted supernatants were added to the appropriate microtiter wells provided by the kit. For the standard curve, 100 μL of different concentration VEGF standards and the blank standard were added to the appropriate microtiter wells. The plate was then incubated for 1 hour at room temperature. After discarding the liquid of each well and washing 4 times with the wash buffer, 100 μL of biotinylated anti-mouse VEGF solution was added into each well except the chromogen blanks and incubated for 1 hour at room temperature. After washing, 100 μL streptavidin-HRP working solution was added to each well, except the chromogen blanks, and incubated for 30 minutes at room temperature. Lastly, 100 μL of stabilized chromogen was added to each well, and incubated for 30 minutes at room temperature in the dark before the addition of 100 μL of stop solution. The absorbance at 450 nm was recorded on a plate reader. A standard curve was generated and the concentrations for unknown samples were obtained from the standard curve.
Data analysis
Data were presented as means ± S.D. Statistical analysis was performed using ANOVA. A value of P< 0.05 was considered statistically significant.
Results
Ischemic neurons contribute to endothelial barrier disruption
To demonstrate an important role of ischemic neurons in contributing to BBB damage, we exposed neurons to OGD for 3 hours before co-culturing them with endothelial monolayer in the presence or absence of astrocytes. The integrity of the endothelial barrier was assessed by measuring the permeability of the endothelial monolayer to FITC-BSA. As shown in Figure 1, under normal condition (Control), the permeability of the endothelial cell monolayer to FITC-BSA was quite low. Co-culturing the endothelial monolayer with astrocytes slightly decreased its permeability to FITC-BSA, supporting an important role of astrocytes in maintaining BBB integrity (Ronaldson & Davis 2012). However, when OGD-treated neurons were included in the experimental system, the integrity of the endothelial monolayer was disrupted, as reflected by a two-fold increase of FITC-BSA permeability when compared to the control group (Figure 1). As expected, incubation of normal neurons with astrocytes and the endothelial monolayer had no effect on endothelial permeability (Figure 1).
Fig. 1.

OGD-treated neurons interact with astrocytes to increase endothelial monolayer permeability. Neuronal cells (CATH.a cells) grown on inserts were exposed to OGD for 3 hours and then co-cultured with astrocytes (C8-D1A cells) for 24 hours. After that, the neurons were removed and the astrocytes were incubated with endothelial monolayer (bEnd.3 cells) for another 24 hours. Endothelial monolayer permeability was assessed by quantitating the clearance of FITC-BSA from luminal compartment to abluminal compartment. The co-culture of OGD-treated neurons with astrocytes significantly increased endothelial barrier permeability. Data are expressed as mean ± SD, n = 3. *P < 0.05 vs Control. Control: endothelial monolayer alone; W/Astro: co-culturing endothelial monolayer with astrocytes; W/Astro/N-Neu: astrocytes were first co-cultured with normal neurons and then with endothelial monolayer; W/Astro/OGD-Neu: astrocytes were first co-cultured with OGD-treated neurons and then with endothelial monolayer
To uncover the mechanism by which ischemic neurons contribute to BBB damage, we assessed two important tight junction proteins occludin and claudin-5 using western blots. As shown in Figure 2A, control endothelial cells expressed high levels of occludin and claudin-5, and both proteins were significantly reduced after incubation with astrocytes that were pre-incubated with OGD-treated neurons for 24 hours (Figure 2B and 2C). As expected, co-culture of the endothelial monolayer with normal astrocytes or astrocytes pre-incubated with untreated neurons (i.e., not exposed to OGD) did not induce significant changes to these proteins (Figure 2A, 2B and 2C). These results were in good agreement with endothelial permeability changes observed above (Figure 1), suggesting that OGD-treated neurons may contribute to endothelial barrier disruption via interfering with tight junction proteins.
Fig. 2.
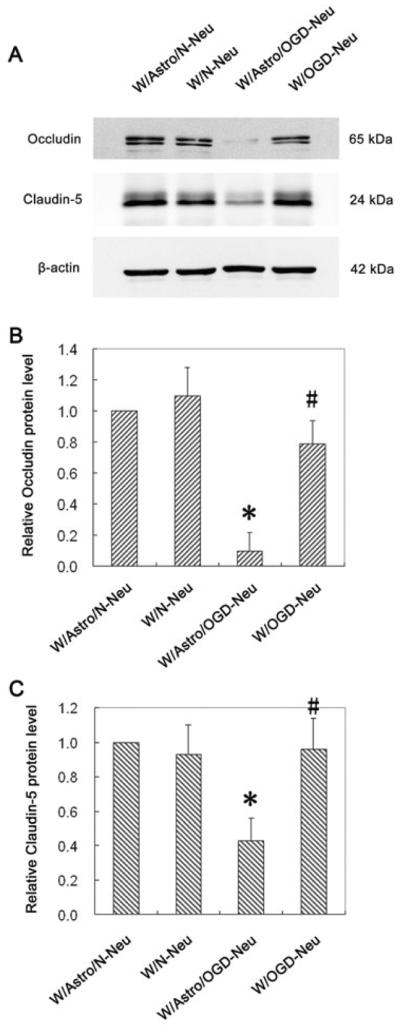
OGD-treated neurons interact with astrocytes to reduce occludin and claudin-5 protein levels in endothelial monolayer. Neuronal cells (CATH.a) grown on inserts were exposed to OGD for 3 hours and then co-cultured with astrocytes (C8-D1A) for 24 hours. After that, the neurons were removed and the astrocytes were incubated with endothelial monolayer (bEnd.3 cells) for another 24 hours before assessing occludin and claudin-5 protein levels by western blot. (A) Representative blots for occludin, claudin-5 and β-actin. The relative quantity of occludin protein (B) or claudin-5 protein (C) was calculated after normalization to β-actin. Co-culturing OGD-treated neurons with astrocytes significantly decreased occludin and claudin-5 protein levels in endothelial cells. Data represent the mean ± S.D, n = 3. *P < 0.05 vs Control. Control: endothelial monolayer alone; W/Astro: co-culturing endothelial monolayer with astrocytes; W/Astro/N-Neu: astrocytes were first co-cultured with normal neurons and then with endothelial monolayer; W/Astro/OGD-Neu: astrocytes were first co-cultured with OGD-treated neurons and then with endothelial monolayer
Astrocytes are required for ischemic neurons to exert damaging effects on the endothelial barrier
The above results showed that pre-incubation of astrocytes with OGD-treated neurons, but not control neurons, led to endothelial monolayer permeability increase, however it was not clear whether OGD-treated neurons acted directly on the endothelial monolayer or acted first on astrocytes to induce this change. To answer this question, we first assessed the impact of OGD-treated neurons on endothelial barrier integrity in the presence or absence of astrocytes. As shown in Figure 3, normal neurons did not induce any significant changes to endothelial barrier integrity no matter regardless of astrocyte presence. To our surprise, when astrocytes were removed from the ischemic model system, the effects of OGD-treated neurons on endothelial permeability completely disappeared (Figure 3), suggesting an important role of astrocytes in BBB damage from ischemic neurons.
Fig. 3.

Astrocytes are required for ischemic neurons to exert its damaging effect on endothelial barrier. Neuronal cells (CATH.a cells) grown on inserts were exposed to OGD for 3 hours and then cultured for another 24 hours with or without astrocytes (C8-D1A cells). After that, the neurons were removed, and the astrocytes were incubated with endothelial monolayer for another 24 hours before assessing endothelial monolayer permeability to FITC-BSA. Data are expressed as mean ± SD, n = 3. *P < 0.05 vs W/Astro/N-Neu, #P < 0.05 vs W/Astro/OGD-Neu. W/Astro/N-Neu: astrocytes were first incubated with normal neurons and then with endothelial monolayer; W/N-Neu: Conditioned media collected from normal neurons and then with endothelial monolayer; W/Astro/OGD-Neu: astrocytes were first incubated with OGD-treated neurons and then with endothelial monolayer; W/OGD-Neu: incubating endothelial monolayer with the conditioned media collected from OGD-treated neurons
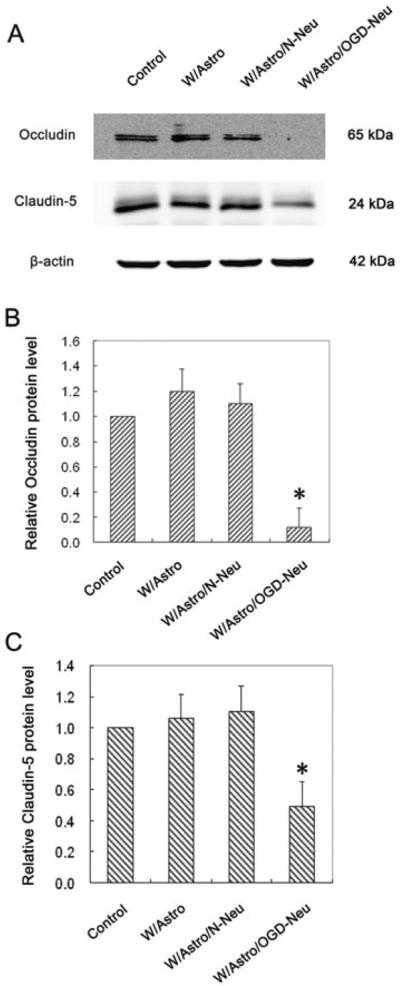
Next, we evaluated the impact of OGD-treated neurons on occludin and claudin-5 protein levels in endothelial cells in the presence or absence of astrocytes. Consistent with endothelial permeability trends observed above, we found that OGD-treated neurons induced dramatic decrease in occludin and claudin-5 proteins in endothelial cells, which was abolished when the astrocytes were removed from the experimental system (Figure 4). These results suggest that ischemic neurons require the presence of astrocytes to exert its damaging effect on the BBB.
Fig. 4.

Removal of astrocytes abolishes the impact of OGD-treated neurons on occludin and claudin-5 protein levels in endothelial cells. Neuronal cells (CATH.a cells) grown on inserts were exposed or not to OGD for 3 hours and then co-cultured with astrocytes (C8-D1A cells) /or not for 24 hours. After that, the neurons were replaced by endothelial monolayer and incubated for another 24 hours before assessing occludin and claudin-5 protein levels by western blot. (A) Representative blots for occludin, claudin-5 and β-actin. The relative quantity of occludin protein (B) or claudin-5 protein (C) was calculated after normalization to β-actin. Data are expressed as mean ± SD, n = 3. *P < 0.05 vs W/Astro/N-Neu, #P < 0.05 vs W/Astro/OGD-Neu. W/Astro/N-Neu: astrocytes were first co-cultured with normal neurons and then with endothelial monolayer; W/N-Neu: incubating with normal neuron-conditioned media; W/Astro/OGD-Neu: astrocytes were first co-cultured with OGD-treated neurons and then with endothelial monolayer; W/OGD-Neu: incubating with conditioned media collected from OGD-treated neurons
VEGF from astrocytes by OGD-treated neurons is a potential predominant inducer of endothelial barrier disruption
We next asked the question of how astrocytes mediated ischemic neuron-induced BBB damage. In our model system, astrocytes did not have direct contact with endothelial cells, it is therefore reasonable to speculate that the incubation of astrocytes with OGD-treated neurons resulted in the secretion of certain molecules by astrocytes that subsequently acted on endothelial cell monolayer. VEGF is the most important angiogenic factor and is also a potent inducer of BBB disruption following cerebral ischemia. It has also been demonstrated to be an important upstream molecule for down-regulation of both occludin and claudin-5 (Argaw et al. 2009). So we detected VEGF production and secretion by astrocytes after incubation with OGD-treated neurons. Indeed, OGD-treated neurons, but not normal neurons, greatly stimulated VEGF production in astrocytes, as reflected by a 1.5-fold increase in astrocytic extracts (Figure 5A) and an approximate 60-fold increase in VEGF secretion compared to basal level of VEGF production (Figure 5B). Co-culturing astrocytes with normal neurons did not increase VEGF production (Figure 5). These results demonstrate that OGD-treated neurons strongly stimulate VEGF production and secretion in astrocytes. In order to demonstrate that VEGF alone is enough to induce the disruption of endothelial barrier, recombinant VEGF was added to the culture medium of endothelial monolayer to evaluate its effect on the endothelial barrier. As shown in Figure 6, VEGF increased the endothelial barrier permeability and decreased the occludin and claudin-5 proteins in a dose-dependent manner, supporting the key role of VEGF on disruption of endothelial barrier as well as the loss of tight junction proteins. Together, these results indicate that VEGF from astrocytes by OGD-treated neurons is a potential predominant inducer of endothelial barrier disruption.
Fig. 5.

OGD-treated neurons interact with astrocytes to increase VEGF production and secretion in astrocytes. The neuronal cells (CATH.a) were exposed or not to OGD for 3 hours before co-culturing with astrocytes (C8-D1A) for 24 hours. After co-culture, VEGF production and secretion were assessed by measuring its content in astrocytic extracts and conditioned media using western blot and ELISA, respectively. (A) OGD-treated neurons (OGD-Neuron), but not normal neurons (N-Neuron) induced a slight but significant increase in VEGF protein in astrocytic extract. Upper panel: representative blots for VEGF and β-actin. Lower panel: The relative quantity of VEGF protein was calculated after normalization to β-actin. Data are expressed as mean ± SD, n = 3, *P <0.05 vs Astrocyte (astrocytic culture alone). (B) VEGF secreted into the conditioned media was assessed using an ELISA kit. Data are expressed as mean ± SD, n = 3, *P <0.05 vs Astrocyte (astrocytic culture alone)
Fig. 6.

VEGF induces the disruption of endothelial barrier and the loss of occludin and claudin-5 proteins. Different concentrations of VEGF were added to the culture medium of the endothelial monolayer (bEnd.3 cells) with full confluence. After 24 hours incubation, the endothelial barrier permeability was assayed as described in “materials and methods” (A), and the protein levels of occludin and claudin-5 were assayed by western blot (B): the upper panel shows representative blots for occludin, claudin-5 and β-actin; the lower panel shows the relative quantity of occludin and claudin-5 protein after normalization to β-actin. *P < 0.05 vs Control (0 ng/mL group), n = 3.
Knockdown of VEGF in astrocytes abolishes the effect of OGD-treated neurons on endothelial barrier integrity
To establish a causal link between astrocyte-derived VEGF production and the ability of OGD-treatment to cause neuronally-induced endothelial barrier disruption, we applied siRNA approach to knock down VEGF expression. Specific silencing was confirmed by western blot analysis, which showed a greater than 80% reduction in VEGF protein levels in the astrocytes transfected with VEGF siRNA (Figure 7A). Most importantly, knockdown of VEGF significantly reduced VEGF secretion, occludin and claudin-5 reduction, and endothelial monolayer permeability increase induced by OGD-treated neurons when compared to control siRNA (Figure 7B, 7C and 7D). These results suggest that OGD-treated neurons may activate astrocytes to increase VEGF production and secretion, which in turn decreases occludin and claudin-5 proteins, consequently leading to endothelial barrier disruption.
Fig. 7.
VEGF mediates induced by co-culturing OGD-treated neurons with astrocytes. Astrocytes (C8-D1A) were transfected with control or VEGF siRNA for 72 hours before co-culturing them with OGD-treated neurons (CATH.a) and endothelial monolayer. (A) Transfection of astrcocytes with VEGF siRNA for 72 hours significantly reduced (~80% reduction) VEGF protein levels. The upper panel shows representative blots for VEGF and β-actin; the lower panel shows the relative quantity of VEGF protein after normalization to β-actin. *P < 0.05 vs Control siRNA. Knockdown of VEGF in astrocytes significantly blocked the impact of OGD-treated neurons on astrocytes and endothelial cells, as reflected by the findings that VEGF secretion was significantly reduced in astrocytes (B), occludin and claudin-5 protein loss was reversed in endothelial monolayer (C), and endothelial barrier permeability increase was significantly reduced (D). Controls are astrocytes alone (B), endothelial monolayer alone (C and D), respectively. Data are expressed as mean ± SD, *P < 0.05 vs Control, #P < 0.05 vs Control siRNA (ConsiRNA), n = 3.
Discussion
Emerging evidence suggests that acute BBB disruption predisposes ischemic brain tissue to hemorrhaging following thrombolytic therapy for ischemic stroke. However, the contribution of brain cells to early ischemic BBB damage and the underlying mechanisms remain unclear. In the present study, an in vitro BBB system consisting of brain microvascular endothelial cells and astrocytes was employed to assess the impact of ischemic neurons on BBB permeability. We found that OGD-treated neurons triggered a cascade inducing BBB breakdown, which required the activation of astrocytes and increased VEGF secretion from astrocytes, leading to disruption of occludin and claudin-5 proteins in endothelial cells. Notably, all these changes were abolished when VEGF expression in astrocytes was knocked down by siRNA. Our results suggest that, despite no direct contact with brain microvascular endothelial cells, ischemic neurons can contribute to BBB damage via stimulation of VEGF expression in astrocytes.
The BBB consists of the endothelial cells lining brain capillaries, surrounded by a basal membrane with several associated cell types including astrocytes, pericytes and neurons (Sa-Pereira et al. 2012, Yang & Rosenberg 2011a, Zlokovic 2008). Endothelial cells are polarized into luminal (blood side) and abluminal (brain side) plasma membrane domains, and are connected by extensive tight junctions that restrict movement of substances between endothelial cells (Liu et al. 2012c, Mark & Davis 2002, Luissint et al. 2012). At the abluminal side, the endothelial cells are covered by pericytes and astrocytes throughout the brain capillaries (Sa-Pereira et al. 2012, Dore-Duffy & Cleary 2011, Rajkowska et al. 2013). Pericytes communicate with other cells of the neurovascular unit by direct contact or through signaling pathways and regulate several important microcirculatory functions including the maintenance of BBB integrity and the distribution of the capillary blood flow to match the local metabolic need of the nearby cells (Vandenhaute et al. 2011, Winkler et al. 2011, Armulik et al. 2010, Quaegebeur et al. 2010). Astrocytes are the most abundant cell type in the brain, and some of them directly contact with the brain capillaries via their end-feet to help maintain the endothelial barrier's integrity (Ronaldson & Davis 2012, Ezan et al. 2012). However, when activated, astrocytes are also an important source of matrix metalloproteinases and inflammatory cytokines (Wiese et al. 2012), and thus contribute to BBB disruption under various pathological CNS conditions (Liu et al. 2012a, Yang et al. 2013).
At the neurovascular unit, the neurons are the only cellular component that does not have direct contact with brain capillaries (Zlokovic 2008). This may be why the neurons have been the least-studied neural cell type of the neurovascular unit in respect to their impact on BBB integrity. Given that neurons have a high demand for oxygen and nutrition (Bickler & Donohoe 2002) and are very sensitive and vulnerable to ischemia or hypoxia, it is conceivable that neurons may be the earliest cell type to respond to cerebral ischemia and so communicates with other cell types at the neurovascular unit to contribute to ischemic BBB damage. Indeed, using the in vitro BBB model (co-culture of endothelial cells with astrocytes), we found that OGD-treated neurons significantly increased BBB permeability (Figure 1). In our model system, the ischemic (OGD-treated) neurons did not have direct contact with the endothelial cells. The neurons could only influence the endothelial barrier through either secretion of active molecules which directly acted on endothelial cells, or indirectly regulating BBB permeability by activating astrocytes. Our results showed that removal of astrocytes almost completely abolished the increase of BBB permeability induced by OGD-treated neurons. The conditioned medium from OGD-treated neurons had marginal impact on BBB permeability (Figure 3). These observations indicate that the ischemic neurons communicate with astrocytes to exert its damaging effect on the BBB.
The tight junctions are key BBB structural components that seal the gaps between adjacent endothelial cells (Liu et al. 2012c, Mark & Davis 2002, Luissint et al. 2012). Occludin and claudins are transmembrane tight junction proteins that interact with adaptor proteins to form the tight junctions (Mark & Davis 2002, Ballabh et al. 2004). Disruption of occludin or claudin-5 is frequently reported to contribute to BBB damage in various CNS pathological conditions (Yang & Rosenberg 2011b, Liu et al. 2012a, Argaw et al. 2009, Xu et al. 2012, Schubert-Unkmeir et al. 2010, Lochhead et al. 2010). Therefore, we speculated that ischemic neurons may also contribute to endothelial barrier disruption through interfering with these two proteins. Indeed, we observed a significant reduction in occludin and claudin-5 protein levels in an endothelial monolayer co-cultured with OGD-treated neurons and astrocytes (Figure 2). Similar to the changes in endothelial permeability, when astrocytes were excluded from our experimental system, loss of tight junction proteins were abolished (Figure 4), suggesting an essential role for astrocytes in mediating BBB disruption induced by ischemic neurons.
In our co-culture system, astrocytes did not have direct contact with endothelial cells. Therefore, astrocytes could only exert their influence on the endothelial monolayer via secreted soluble molecules. VEGF is the most potent BBB “opener” produced by astrocytes (Argaw et al. 2012). Previous studies have demonstrated that VEGF increases BBB permeability by disrupting claudin-5 protein (Argaw et al. 2009). Here, we show that co-culture of ischemic neurons and astrocytes significantly increased VEGF protein production and in particular its secretion (~ 60 times of increase) (Figure 5), and this level of VEGF is sufficient to induce the disruption of BBB and loss of tight junction proteins (Figure 6). Moreover, knockdown of VEGF in astrocytes almost completely blocked the reduction in occludin and claudin-5 proteins as well as the increase of BBB permeability (Figure 7). These results demonstrate that VEGF secreted by activated astrocytes is the key molecule mediating the BBB disruption induced by ischemic neurons. Although our data clearly indicate that the activated astrocytes are the main source of VEGF, there is a possibility that astrocytes trigger OGD-treated neurons to secret VEGF because there is evidence that neurons express VEGF under certain condition (Sondell et al. 1999). Our data that neither untreated neurons nor OGD-treated neurons alone induced BBB disruption and knockdown of VEGF in astrocytes with siRNA inhibited BBB disruption suggest that neurons may only produce a small amount of VEGF, if any, under our experimental conditions. In this study, we did not further explore how ischemic neurons activate astrocytes. Future studies are warranted to answer this important question.
In summary, our results define an important role of neurons in mediating ischemic BBB damage. We propose that ischemic neurons activate astrocytes to increase VEGF secretion, which eventually leads to disruption of tight junction proteins and a BBB permeability increase. These findings may expand our understanding of the molecular events that are associated with early ischemic BBB injury.
Acknowledgments
This work was supported by grants from National Institutes of Health (P30GM103400 and R01AG031725), American Heart Association (10BGIA3190010), National Natural Science Foundation of China (81100892, 81171242 and 81200928), and The Second Affiliated Hospital of Medical School, Xi'an Jiaotong University (YJ (ZD) 201005).
Abbreviations
- BBB
blood brain barrier
- OGD
oxygen-glucose deprivation
- VEGF
vascular endothelial growth factor
- MRI
magnetic resonance imaging
- FGF-1
fibroblast growth factor-1
- FITC-BSA
fluorescein isothiocyanate labelled bovine serum albumin
Footnotes
Conflicts of interest The authors declare no conflict of interest.
References
- Argaw AT, Asp L, Zhang J, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Mae M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Donohoe PH. Adaptive responses of vertebrate neurons to hypoxia. J Exp Biol. 2002;205:3579–3586. doi: 10.1242/jeb.205.23.3579. [DOI] [PubMed] [Google Scholar]
- Borlongan CV, Rodrigues AA, Jr., Oliveira MC. Breaking the barrier in stroke: what should we know? A mini-review. Curr Pharm Des. 2012;18:3615–3623. doi: 10.2174/138161212802002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitanya GV, Cromer WE, Wells SR, et al. Gliovascular and cytokine interactions modulate brain endothelial barrier in vitro. J Neuroinflammation. 2011;8:162. doi: 10.1186/1742-2094-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haas AH, van Weering HR, de Jong EK, Boddeke HW, Biber KP. Neuronal chemokines: versatile messengers in central nervous system cell interaction. Mol Neurobiol. 2007;36:137–151. doi: 10.1007/s12035-007-0036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore-Duffy P, Cleary K. Morphology and properties of pericytes. Methods Mol Biol. 2011;686:49–68. doi: 10.1007/978-1-60761-938-3_2. [DOI] [PubMed] [Google Scholar]
- Ezan P, Andre P, Cisternino S, et al. Deletion of astroglial connexins weakens the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1457–1467. doi: 10.1038/jcbfm.2012.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi T, Liu W, Shi H, Miyake M, Liu KJ. Generation of hydrogen peroxide during brief oxygen-glucose deprivation induces preconditioning neuronal protection in primary cultured neurons. J Neurosci Res. 2005;79:816–824. doi: 10.1002/jnr.20402. [DOI] [PubMed] [Google Scholar]
- Haseloff RF, Blasig IE, Bauer HC, Bauer H. In search of the astrocytic factor(s) modulating blood-brain barrier functions in brain capillary endothelial cells in vitro. Cell Mol Neurobiol. 2005;25:25–39. doi: 10.1007/s10571-004-1375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Kaufer D, Friedman A. Blood-brain barrier dysfunction, TGFbeta signaling, and astrocyte dysfunction in epilepsy. Glia. 2012;60:1251–1257. doi: 10.1002/glia.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjort N, Wu O, Ashkanian M, Solling C, Mouridsen K, Christensen S, Gyldensted C, Andersen G, Ostergaard L. MRI detection of early blood-brain barrier disruption: parenchymal enhancement predicts focal hemorrhagic transformation after thrombolysis. Stroke. 2008;39:1025–1028. doi: 10.1161/STROKEAHA.107.497719. [DOI] [PubMed] [Google Scholar]
- Hom J, Dankbaar JW, Soares BP, et al. Blood-brain barrier permeability assessed by perfusion CT predicts symptomatic hemorrhagic transformation and malignant edema in acute ischemic stroke. AJNR Am J Neuroradiol. 2011;32:41–48. doi: 10.3174/ajnr.A2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Liu J, Yang Y, Liu KJ, Liu W. Spatiotemporal evolution of blood brain barrier damage and tissue infarction within the first 3h after ischemia onset. Neurobiol Dis. 2012;48:309–316. doi: 10.1016/j.nbd.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Liu J, Jin X, Liu KJ, Liu W. Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J Neurosci. 2012a;32:3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Agalliu D, Yu C, Fisher M. The role of pericytes in blood-brain barrier function and stroke. Curr Pharm Des. 2012b;18:3653–3662. doi: 10.2174/138161212802002706. [DOI] [PubMed] [Google Scholar]
- Liu WY, Wang ZB, Zhang LC, Wei X, Li L. Tight junction in blood-brain barrier: an overview of structure, regulation, and regulator substances. CNS Neurosci Ther. 2012c;18:609–615. doi: 10.1111/j.1755-5949.2012.00340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, Davis TP. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. 2012;9:23. doi: 10.1186/2045-8118-9-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark KS, Davis TP. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am J Physiol Heart Circ Physiol. 2002;282:H1485–1494. doi: 10.1152/ajpheart.00645.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, Vargas MR, Cassina P, Barbeito AG, Beckman JS, Barbeito L. Complexity of astrocyte-motor neuron interactions in amyotrophic lateral sclerosis. Neurodegener Dis. 2005;2:139–146. doi: 10.1159/000089619. [DOI] [PubMed] [Google Scholar]
- Quaegebeur A, Segura I, Carmeliet P. Pericytes: blood-brain barrier safeguards against neurodegeneration? Neuron. 2010;68:321–323. doi: 10.1016/j.neuron.2010.10.024. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Hughes J, Stockmeier CA, Javier Miguel-Hidalgo J, Maciag D. Coverage of blood vessels by astrocytic endfeet is reduced in major depressive disorder. Biol Psychiatry. 2013;73:613–621. doi: 10.1016/j.biopsych.2012.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Davis TP. Blood-brain barrier integrity and glial support: mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr Pharm Des. 2012;18:3624–3644. doi: 10.2174/138161212802002625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa-Pereira I, Brites D, Brito MA. Neurovascular unit: a focus on pericytes. Mol Neurobiol. 2012;45:327–347. doi: 10.1007/s12035-012-8244-2. [DOI] [PubMed] [Google Scholar]
- Schubert-Unkmeir A, Konrad C, Slanina H, Czapek F, Hebling S, Frosch M. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog. 2010;6:e1000874. doi: 10.1371/journal.ppat.1000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci. 1999;19:5731–5740. doi: 10.1523/JNEUROSCI.19-14-05731.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Zhou W, Mueller C, Sommer C, Heiland S, Bauer AT, Marti HH, Veltkamp R. Oxygen therapy reduces secondary hemorrhage after thrombolysis in thromboembolic cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:1651–1660. doi: 10.1038/jcbfm.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenhaute E, Dehouck L, Boucau MC, et al. Modelling the neurovascular unit and the blood-brain barrier with the unique function of pericytes. Curr Neurovasc Res. 2011;8:258–269. doi: 10.2174/156720211798121016. [DOI] [PubMed] [Google Scholar]
- Vincent T, Saikali P, Cayrol R, Roth AD, Bar-Or A, Prat A, Antel JP. Functional consequences of neuromyelitis optica-IgG astrocyte interactions on blood-brain barrier permeability and granulocyte recruitment. J Immunol. 2008;181:5730–5737. doi: 10.4049/jimmunol.181.8.5730. [DOI] [PubMed] [Google Scholar]
- Warach S, Latour LL. Evidence of reperfusion injury, exacerbated by thrombolytic therapy, in human focal brain ischemia using a novel imaging marker of early blood-brain barrier disruption. Stroke. 2004;35:2659–2661. doi: 10.1161/01.STR.0000144051.32131.09. [DOI] [PubMed] [Google Scholar]
- Wiese S, Karus M, Faissner A. Astrocytes as a source for extracellular matrix molecules and cytokines. Front Pharmacol. 2012;3:120. doi: 10.3389/fphar.2012.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14:1398–1405. doi: 10.1038/nn.2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Feng X, Xie X, Zhang J, Wu D, Xu L. HIV-1 Tat protein increases the permeability of brain endothelial cells by both inhibiting occludin expression and cleaving occludin via matrix metalloproteinase-9. Brain Res. 2012;1436:13–19. doi: 10.1016/j.brainres.2011.11.052. [DOI] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011a;42:3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA. MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol Biol. 2011b;762:333–345. doi: 10.1007/978-1-61779-185-7_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Thompson JF, Taheri S, Salayandia VM, McAvoy TA, Hill JW, Estrada EY, Rosenberg GA. Early inhibition of MMP activity in ischemic rat brain promotes expression of tight junction proteins and angiogenesis during recovery. J Cereb Blood Flow Metab. 2013 doi: 10.1038/jcbfm.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]