

Abstract
Methicillin-resistant S. aureus (MRSA) has emerged as a leading contributor to mortality during recent influenza pandemics. The mechanism for this influenza-induced susceptibility to secondary S. aureus infection is poorly understood. Here we show that innate antibacterial immunity was significantly suppressed during the recovery stage of influenza infection, despite the fact that MRSA super-infection had no significant effect on viral burdens. Compared to mice infected with bacteria alone, post-influenza MRSA infected mice exhibited impaired bacterial clearance, which was not due to defective phagocyte recruitment, but rather coincided with reduced intracellular reactive oxygen species (ROS) levels in alveolar macrophages and neutrophils. NADPH oxidase is responsible for ROS production during phagocytic bacterial killing, a process also known as oxidative burst. We found that gp91phox-containing NADPH oxidase activity in macrophages and neutrophils was essential for optimal bacterial clearance during respiratory MRSA infections. In contrast to WT animals, gp91phox−/− mice exhibited similar defects in MRSA clearance before and after influenza infection. Using gp91phox+/− mosaic mice, we further demonstrate that influenza infection inhibits a cell-intrinsic contribution of NADPH oxidase to phagocyte bactericidal activity. Together, our results establish that influenza infection suppresses NADPH oxidase-dependent bacterial clearance and leads to susceptibility to secondary MRSA infection.
Introduction
Viral influenza is often a seasonal infection that can lead to primary influenza pneumonia. However, secondary bacterial infections, commonly associated with Streptococcus pneumoniae, Staphylococcus aureus and Haemophilus influenzae, are known to be more frequent causes of severe morbidity and mortality (1). Among these, methicillin-resistant S. aureus (MRSA) has emerged as the leading contributor to mortality during recent influenza pandemics and epidemics (2–7). A dysregulated immune defense against either influenza virus or S. aureus has been proposed as a contributor to co-infection pathogenesis (8–11); however, an incomplete understanding of co-infection pathophysiology has slowed the development of effective treatments (12–14).
NADPH oxidase produces superoxide, which can spontaneously form hydrogen peroxide that will undergo further reactions to generate reactive oxygen species (ROS). During influenza virus infection alone, gp91phox-containing NADPH oxidase has been shown to exacerbate lung inflammation (15–18), even though there is no evidence linking its detrimental effect to enhanced enzymatic activity. In contrast, during S. aureus lung infection alone, phagocyte ROS generation is essential for antibacterial immunity (19, 20). However, the outcome of these conflicting contributions of NADPH oxidase to immune defense against influenza and S. aureus co-infection has not been addressed in any reported studies. Instead, several reports have demonstrated that synergistic influenza and S. aureus co-infection is associated with dysregulation of antibacterial immunity (8–11). Therefore, we sought to determine the regulatory effect of influenza infection on ROS-dependent antibacterial activity and its contribution to susceptibility to subsequent S. aureus infection. In our model, we found that innate antibacterial immunity was significantly suppressed following influenza infection. In addition, mice were more susceptible to respiratory MRSA infection in the absence of NADPH oxidase activity. Importantly, we show that influenza infection inhibited the cell-intrinsic contribution of NADPH oxidase to phagocytic bacterial killing, which led to susceptibility to secondary MRSA infection.
Materials and Methods
Murine model of viral and bacterial infection
Specific pathogen-free, C57BL/6 WT, gp91phox-/y, gp91phox−/−, p47phox−/−, NOS2−/− and Mafia mice were initially purchased from the Jackson Laboratory (Bar Harbor, ME) and bred at Albany Medical College following IACUC guidelines.
Viral challenge was performed with A/PR/8 or H1N1 CA4 administered i.n. to anesthetized, sex and age-matched adult mice in 50 μl of sterile PBS. Unless otherwise specified, bacterial co-infection was performed at 7 days after influenza infection. To induce bacterial pneumonia, anesthetized mice were inoculated i.n. with 50 μl of PBS containing 107- 2×108 CFU of community-acquired MRSA strain BAA-1695 (an isolate from patient sputum), or MRSA BAA-1692 (an isolate from human sinus) obtained from ATCC. Titers of virus stocks and viral levels in the bronchoalveolar lavage fluids (BALF) and lungs of infected mice were determined by plaque assays on MDCK cell monolayers (21). Bacterial burdens in the lungs and BALF were measured by sacrificing infected mice at the indicated time points, and plating serial 10-fold dilutions of each sample onto blood agar plates.
Bronchoalveolar lavage (BAL) cell analysis
BAL was collected by making an incision in the trachea and gavaging the lung twice with 0.8 ml PBS, pH 7.4. The BAL cells were incubated with 2.4G2 mAb against FcγRII/III, and stained with PE- or APC-conjugated anti-CD11c (Caltag Laboratories, Burlingame, CA), FITC- or PE-Cy7-conjugated anti-CD11b (BD Biosciences), FITC- or PE-conjugated anti-Ly6G mAb (Clone 1A8, BD Biosciences), and APC-Cy7-conjugated anti-Ly6C mAb (BD Biosciences). The stained cells were analyzed on a BD FACSCanto using BD FACSDiva software.
Detection of gp91phox WT and deficient neutrophils
Gp91phox expression in neutrophils was assessed with an anti-mouse gp91phox mAb (BD Biosciences) (22). Briefly, after staining for neutrophils with FITC-conjugated anti-mouse Ly6G mAb and PE-Cy7-conjugated anti-mouse CD11b mAb, cells in suspension were fixed with 2% paraformaldehyde for 20 min. Cells were then washed with PBS and permeabilized with BD Perm Buffer IV for 20 min. Cells were washed twice with 2% BSA in PBS and then incubated with purified mouse anti-gp91phox mAb for 30 min. Cells were washed and incubated with PE-conjugated rat anti-mouse IgG1 Ab (BD Biosciences) prior to analysis by flow cytometry.
Flow cytometry analysis of oxidative stress in BALF cells
Seven days after i.n. inoculation of 50 PFU PR8 influenza virus, mice were infected i.n. with 107 CFU MRSA and 24 h later, BALF cells were collected and incubated with CellROX Deep Red reagent (Invitrogen) at a final concentration of 5 mM in complete RPMI medium for 30min at 37°C, washed three times with PBS, and then fixed with 3.5% formaldehyde for 15 min before staining for surface marker expression (Supplemental Fig. 1). BALF cells incubated with RPMI medium alone were used for background staining of individual cell subsets.
Neutrophil depletion during MRSA infection
Anti-Gr-1 mAb was purified from culture supernatants of the RB6-8C5 hybridoma using anti-rat IgG agarose columns (Sigma, St. Louis, MO). For neutrophil depletion, mice were injected i.p. 48 and 24 h before bacterial infection with 0.1 mg of RB6-8C5 anti-Gr-1 mAb or with rat IgG as a control. The efficiency and specificity of neutrophil depletion in BALF of MRSA-infected mice was confirmed by flow cytometry analysis (Supplemental Fig. 2).
Alveolar macrophage depletion during MRSA infection
“Mafia” transgenic mice have an inducible Fas suicide/apoptotic system driven by the mouse colony stimulating factor-1 receptor promoter (23). The transgene insert contains a mutant human FK506 binding protein 1A, which preferentially binds the dimerization drug, AP20187. Administration of AP20187 induces apoptosis specifically in macrophages and dendritic cells.
AP20187 was a gift from Ariad Pharmaceuticals. Lyophilized AP20187 was dissolved in 100% ethanol at a concentration of 13.75 mg/ml. A working solution for peritoneal injections was prepared from the stock diluted to 0.55 mg/ml in 4% ethanol, 10% PEG-400, and 1.7% Tween. Mafia mice were injected i.p. for 5 consecutive days with 300 μl of AP20187 or diluent control. The mice were then rested for another 6 days before i.n. infection with S. aureus. The efficiency of pulmonary macrophage depletion was confirmed by flow cytometry (24).
Statistical analyses
Results are expressed as means ± s.d. Significant differences between experimental groups were determined using a Student t-test (to compare two samples), or an ANOVA analysis followed by Tukey’s multiple comparisons test (to compare multiple samples) in GraphPad Prism 6 (La Jolla, CA). Survival analyses were performed using the Kaplan-Meier log rank test. For all analyses, a P value <0.05 was considered to be significant.
Results
Influenza infection impairs innate defense against a subsequent S. aureus respiratory challenge
To establish a mouse model that mimics the clinical observation that influenza infection predisposes individuals to S. aureus infection, we followed an experimental protocol similar to our previous secondary pneumococcal infection studies (25, 26). Specifically, C57BL/6 mice were infected with a sublethal dose of H1N1 CA04 influenza virus, and followed by i.n. challenge with 107 CFU MRSA BAA-1692 at days 5, 7 or 9 after viral infection. Co-infected mice were sacrificed 24 h after MRSA infection (i.e. days 6, 8 and 10 post-influenza infection) for determination of lung viral and bacterial burdens. Compared to animals infected with MRSA only, influenza and MRSA co-infected mice exhibited decreased ability to clear bacteria from their lungs (Fig. 1A). However, the relative defect in bacterial clearance was significantly exacerbated in mice superinfected with MRSA at day 7 post-influenza as compared to day 5 superinfected animals (Fig. 1B), indicating greater susceptibility to secondary S. aureus infection at the beginning of the viral recovery phase (Fig. 1C).
Figure 1. Influenza enhances susceptibility to MRSA infection during the recovery stage of viral infection.
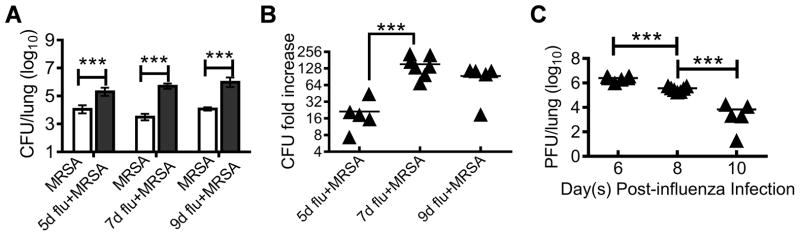
(A) Bacterial burdens, (B) influenza-induced bacterial outgrowth, and (C) viral burdens in the lungs at various days after 3×103 PFU H1N1 CA04 influenza virus infection of B6 WT mice and 24 h after infection with 107 CFU/mouse of MRSA BAA-1692 (5–7 mice/group). In (B), the relative increases of bacterial burdens in influenza-infected mice 24 h after MRSA infection are represented as fold increases relative to mean lung bacterial CFUs in mice infected with MRSA alone (A) included at each time point. In (A&C) P<0.001, ANOVA, in (B), P<0.01, ANOVA; ***P< 0.001, Tukey’s multiple comparisons test.
To determine the possible strain-specific effect of influenza virus or S. aureus on co-infection outcomes, mice were next infected with PR8 virus and superinfected with the community-acquired MRSA strain BAA-1695 on day 7 post-influenza. Consistent with the observations above, bacterial burdens were significantly increased in PR8-infected mice (Fig. 2A). Interestingly, MRSA super-infection did not appear to affect viral loads in the lung (Fig. 2B). Accordingly, co-infection was associated a high mortality rate (>90%), after an increased (2×108 CFU) MRSA challenge dose, which was not observed with either infectious agent alone (Fig. 2C). Collectively, these results indicate that anti-MRSA immunity is profoundly inhibited at the recovery stage of influenza infection, which is correlated with a lethal outcome following influenza and S. aureus co-infection.
Figure 2. Influenza infection impairs innate immunity against respiratory MRSA challenge.
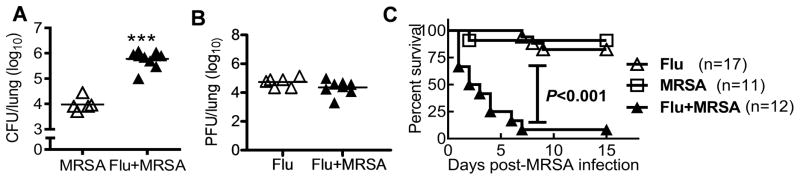
(A) Bacterial and (B) viral burdens in the lungs 24 h after inoculation of naïve (MRSA) or day 7 postinfluenza infected (Flu+MRSA) C57BL/6 mice with 107 CFU MRSA BAA-1695 (Flu+MRSA) or PBS (Flu). ***P< 0.001, compared to mice not infected with influenza virus, unpaired t test. (C) Survival of C57BL/6 mice after infection with 2×108 CFU of MRSA BAA-1695 alone (MRSA) or on day 7 after PR8 infection (Flu + MRSA). Also shown, survival of mice infected with 50 PFU PR8 and inoculated with PBS control, instead of MRSA, 7 days later (Flu).
Influenza infection inhibits phagocyte antibacterial function in the airways
To determine how influenza infection leads to defective bacterial clearance, we next examined airway phagocyte recruitment after influenza and/or MRSA infection. Based on the published reports (27, 28), alveolar macrophages were defined as CD11c+CD11blow, neutrophils as CD11b+Ly6G+, and inflammatory monocytes as CD11b+Ly6G− (Supplemental Fig. 1). Pulmonary S. aureus infection induced significant neutrophil (CD11b+Ly6G+) recruitment into the respiratory tract (Fig. 3A). However, comparable numbers of neutrophils were detected in MRSA-infected and co-infected mice, which were higher than neutrophil counts in mice infected with influenza alone. In fact, there were increased total numbers of phagocytes in co-infected mice due to the recruitment of inflammatory monocytes (CD11b+Ly6ChiLy6G−) following influenza infection. Based on this observation, we hypothesized that the antibacterial function of these phagocytes is impaired, which then leads to defective bacterial clearance in co-infected mice.
Figure 3. Influenza infection regulates the antibacterial ability of airway phagocytes.
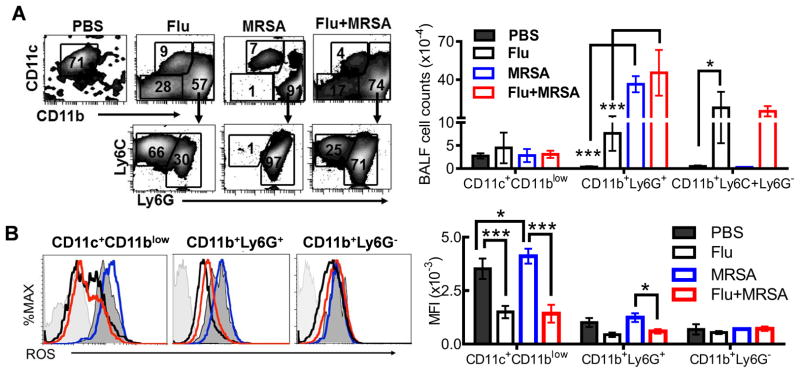
(A) Airway phagocyte profiles (3 mice/group), and (B) intracellular ROS levels (3 mice/group) at 24 h after infection with 107 CFU of MRSA BAA-1695 alone (MRSA) or on day 7 after PR8 infection (Flu + MRSA). In (A&B), P<0.001, ANOVA; *P< 0.05, ***P< 0.001, Tukey’s multiple comparisons test. Control mice were inoculated with only PBS (PBS) or infected with 50 PFU PR8 and inoculated with PBS (Flu) 7 days after PR8 infection. The data are representative of two independent experiments.
Given the critical role of NADPH oxidase in innate defense against S. aureus lung infection, we next determined whether phagocyte oxidative burst was impaired following influenza infection. Significantly decreased ROS levels were observed in alveolar macrophages (CD11c+CD11blow) and neutrophils (CD11b+Ly6G+) isolated from influenza or co-infected mice compared with animals infected with MRSA alone (Fig. 3B and Supplemental Fig. 1). It is noteworthy that alveolar macrophages from normal mice exhibited increased ROS mean fluorescence intensity (MFI) compared to neutrophils, which was not necessarily due to their greater ROS-generating capacity, but rather, their higher autofluorescence (29). This result suggests that influenza infection impairs the ability of alveolar macrophages and neutrophils to mediate ROS-dependent bactericidal function.
Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial killing
We next assessed the possible innate immune components involved in pulmonary S. aureus killing without influenza infection, since influenza-induced susceptibility to MRSA infection is essentially a result of the negative regulation of these antibacterial factors. NADPH oxidase is responsible for ROS production during phagocytic killing of S. aureus (30) and both gp91phox and p47phox are critical subunits for enzyme activity. Consistent with findings from other in vivo S. aureus infection studies (19, 20), we showed that NADPH oxidase was necessary for optimal killing of S. aureus using in the current model, as demonstrated by significantly increased lung bacterial burdens in gp91phox−/− and p47phox−/−, but not NOS2−/− mice (Fig. 4A).
Figure 4. Macrophage oxidative burst is essential for lung S. aureus clearance.

Numbers of bacteria remaining in the lungs 24 h after MRSA BAA-1695 infection of C57BL/6 (A) WT, NOS2−/−, gp91phox−/−, and p47phox−/− mice, (B) α-PMN antibody-treated WT and gp91phox−/− mice, and (C) Mafia mice. In (B), mice were treated with RB6-8C5 mAb for neutrophil depletion. In (C), Mafia mice were depleted of macrophages using AP20187 dissolved in ethanol. Control WT mice were also treated with AP20187. (D) Survival of α-PMN antibody-treated WT mice after i.n. infection with 108 CFU of MRSA. Control WT mice were treated with Rat IgG. (E) Survival of α-PMN antibody-treated WT and gp91phox−/− mice after i.n. infection with 107 CFU of MRSA. In (A, B&C), P<0.001, ANOVA; ***P< 0.001, Tukey’s multiple comparisons test.
On the other hand, previous reports suggest that innate clearance of S. aureus only requires neutrophils but not macrophages (20, 31, 32). Gp91phox is the specific catalytic subunit of NADPH oxidase in phagocytes, including both macrophages and neutrophils. Our results above demonstrated that influenza infection reduces ROS levels in alveolar macrophages. To provide direct evidence that macrophages and their oxidative burst are essential for pulmonary clearance of S. aureus, we compared bacterial burdens in neutrophil-depleted WT and gp91phox−/− mice. Pulmonary MRSA burdens were significantly increased after neutrophil depletion (Fig. 4B). Interestingly, there was a significant difference in bacterial burdens between neutrophil-depleted gp91phox−/− and neutrophil-depleted WT mice (Fig. 4B), suggesting an important contribution of macrophages to NADPH oxidase-dependent control of bacterial outgrowth. Consistent with this, increased bacterial burdens were found in macrophage-depleted “Mafia” mice (Fig. 4C). The essential role of neutrophils in host defense against pulmonary MRSA infection was revealed by increased mortality rates in neutrophil-depleted mice (Fig. 4D). Prolonged macrophage depletion compromises the health of Mafia mice (23); therefore, it was not feasible to directly evaluate the contribution of macrophages to survival from MRSA infection. Nonetheless, the highly elevated bacterial counts in neutrophil-depleted gp91phox−/− mice were accompanied by increased mortality after a relatively low dose (1×107 CFU) MRSA infection (Fig. 4E). Taken together, these results indicate that neutrophils, macrophages and their associated NADPH oxidase are essential for efficient pulmonary MRSA containment.
To determine the possible effect of influenza infection on ROS-dependent bacterial clearance, we next examined anti-MRSA immunity in influenza-infected gp91phox−/− mice. We hypothesized that if influenza infection impaired anti-MRSA immune mechanisms other than NADPH oxidase-mediated bacterial killing, co-infected gp91phox−/− mice should have further increased bacterial burdens as compared to MRSA-infected gp91phox−/− animals. On the contrary, we found that gp91phox−/− mice exhibited similar defects in MRSA clearance both before and after influenza infection, while WT mice were defective only after influenza infection (Fig. 5A). This indicates that influenza infection predominantly inhibits NADPH oxidase-dependent MRSA clearance. In addition, BALF cytology revealed an increase in neutrophil counts in the co-infected gp91phox−/− mice relative to co-infected WT mice (Fig. 5B). However, in the absence of downstream effector NADPH oxidase, this enhanced antibacterial immune response failed to facilitate bacterial clearance in co-infected gp91phox−/− mice (Fig. 5A). These results further established that susceptibility to post-influenza MRSA is due to defective phagocyte antibacterial function. Interestingly, influenza infection resulted in defective bacterial clearance in NOS2−/− mice in a pattern similar to WT mice (data not shown), despite the fact that NOS2 also participates in phagocyte oxidative burst.
Figure 5. Influenza infection inhibits NADPH oxidase-dependent bacterial killing.
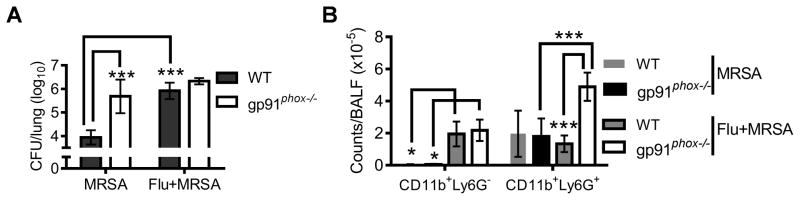
(A) Numbers of bacteria (4–8 mice/group) in the lungs, and (B) airway inflammatory cell accumulation at 24 h after infection WT and gp91phox−/− mice (n=4) with 107 CFU of MRSA BAA-1695 alone (MRSA) or on day 7 after PR8 infection (Flu + MRSA). In (A&B), P<0.001, ANOVA; *P< 0.05, ***P< 0.001, Tukey’s multiple comparisons test.
Despite of its detrimental effect on lung inflammation (15–18), an overall impact of NADPH oxidase on animal mortality was not evident after a lethal dose of influenza infection alone (Fig. 6A). Conversely, both p47phox−/− and gp91phox−/− mice exhibited increased susceptibility to S. aureus infection alone compared to WT mice (Fig. 6B). Furthermore, gp91phox−/− mice were highly susceptible to post-influenza bacterial infection, as revealed by rapid mortality kinetics and decreased survival rates compared with WT animals (Fig. 6C and D, respectively). These data suggest that NADPH oxidase-dependent anti-MRSA immunity is impaired, though not absolutely absent, in co-infected WT mice.
Figure 6. Influenza infection inhibits NADPH oxidase-dependent immune defense against S. aureus.

(A) Survival of WT and gp91phox−/− mice after infection with 103 PFU of PR8 virus alone. (B) Survival of WT and gp91phox−/− mice after infection with 1×108 CFU of MRSA BAA-1695 alone. (C&D) Survival of WT and gp91phox−/− mice after infection with (C) 2×108 CFU and (D) 1×108 CFU of MRSA BAA-1695 on day 7 after PR8 infection.
The cell-intrinsic contribution of NADPH oxidase to the immune defense against post-influenza MRSA infection
The data above demonstrate that influenza infection inhibits NADPH oxidase-dependent bacterial clearance and predisposes mice to secondary MRSA infection. Conversely, it has been reported that gp91phox-containing NADPH oxidase promotes influenza infection-associated immunopathology to surrounding tissues, due to the nonspecific toxic effect of extracellular ROS (15–18). To differentiate these concomitant but conflicting contributions of NADPH oxidase, we next sought to determine whether influenza infection impairs the cell-intrinsic contribution of NADPH oxidase to phagocytic bacterial killing. The gp91phox subunit is an X-chromosome-linked gene. Female gp91phox+/− (mosaic) mice have both gp91phox WT and deficient circulating neutrophils due to random inactivation of the X-chromosome in individual cells (Fig. 7A). Therefore, the potential cell-intrinsic NADPH oxidase-dependent phagocytic killing could be determined in such mosaic animals. Influenza infection induced an increase in the percentage of blood neutrophils in gp91phox mosaic mice (Fig. 7A&B). However, a similar ratio of gp91phox WT and deficient blood neutrophils was detected in mosaic mice before and after influenza infection, which was also comparable to that in airways after influenza infection (Fig. 7B). These observations suggest that gp91phox WT and deficient neutrophil subpopulations in mosaic mice perform similarly in terms of differentiation and migration. Conversely, despite the abundance of gp91phox WT neutrophils in mosaic mice, their lung bacterial burdens were greatly increased compared with WT mice after MRSA infection (Fig. 7C). Moreover, similar to gp91phox−/− mice, mosaic mice exhibited defective MRSA clearance both before and after influenza infection (Fig. 7C). Together, these results further demonstrated that influenza infection suppresses cell-intrinsic contributions of NADPH oxidase to phagocytic bacterial killing.
Figure 7. Influenza infection inhibits cell-intrinsic contribution of NADPH oxidase to phagocytic bacterial killing.

(A) Flow cytometry analysis of gp91phox expression in blood neutrophils (CD11b+Ly6G+) from female gp91phox WT, knockout (KO) and mosaic mice (n=5). (B) Composition of gp91phox WT and deficient neutrophils in blood and airways of gp91phox+/− mosaic mice (n=5) at day 7 after influenza infection. (C) Numbers of bacteria (5 mice/group) in the lungs 24 h after challenge of naïve (MRSA) or day 7 postinfluenza infected (Flu+MRSA) female WT and gp91phox+/− mosaic mice with 107 CFU MRSA BAA-1695. In (C), P<0.01, ANOVA; **P< 0.01, ***P< 0.001, Tukey’s multiple comparisons test.
Discussion
In the present study, we demonstrated that influenza and MRSA co-infection is associated with defective antibacterial immunity. Compared to mice challenged with bacteria alone, post-influenza MRSA-infected animals were characterized by significantly increased bacterial burdens, increased numbers of inflammatory monocytes, reduced ROS levels in alveolar macrophages and neutrophils in the airways. At the same time, no significant effect of MRSA super-infection on antiviral immunity was observed. Furthermore, gp91phox−/− phagocytes were defective in their antibacterial function. This cell-intrinsic contribution of NADPH oxidase activity to phagocytic bacterial killing was established utilizing mosaic mice containing both NADPH oxidase WT and deficient phagocyte subpopulations. Importantly, similar to gp91phox−/− mice, these mosaic mice exhibited similar defective MRSA clearance both before and after influenza infection. Collectively, the results presented in this study demonstrate that influenza virus infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and leads to susceptibility to secondary MRSA infection.
Multiple mechanisms have been implicated in the adverse outcome of influenza and bacterial co-infection, namely: 1) direct viral cytotoxicity associated with increased viral burden (8, 10); 2) cytotoxicity as a result of bacterial outgrowth (9, 10, 33–37); and 3) inflammatory tissue damage due to excessive production of proinflammatory mediators and defective resolution/tissue repair responses (38–40). The observed differences in the relative contributions of virus, bacteria and host inflammation to the pathogenesis of co-infection primarily reflect variation in the timing of bacterial inoculation in these experimental models. When mice were infected with MRSA at the beginning of the viral recovery phase, we found that co-infection-induced high mortality rates that were associated with impaired antibacterial immunity and excessive inflammatory infiltrates but not exacerbated viral infection. These effects were observed with both PR8 and H1N1 CA04 influenza virus infection (25, 26).
In recent studies, impaired antibacterial immunity has been shown to be associated with dysregulated pulmonary cytokine responses following influenza infection. Small et al. reported that influenza infection impairs TNF-α production and causes increased susceptibility to subsequent S. aureus infection (9). It has also been reported that influenza A inhibits IL-1β and Th17-mediated host defense against S. aureus pneumonia in mice, although the molecular mechanisms involved remain elusive (11, 33). Whereas those studies described an association between suppression of proinflammatory responses and defective bacterial clearance during influenza infection, how decreased cytokine responses lead to defective antibacterial immunity remains unclear. Nonetheless, it has been show that TNF-α and IL-1β prime the respiratory burst in phagocytes (41, 42), their reduced production in influenza-infected mice might be responsible for decreased ROS. Our previous work with secondary pneumococcal infection demonstrated that IFN-γ is responsible for inhibition of innate bacterial clearance (25). The dynamics of susceptibility to post-influenza MRSA infection resemble those of post-influenza pneumococcal infection. Moreover, an in vitro study suggests that IFN-γ can inhibit macrophage-mediated phagocytosis of S. aureus (43). Conversely, it is shown that T cell-derived IFN-γ can actually perpetuate infection in a S. aureus wound infection model by increasing neutrophil recruitment, cells that can serve a reservoir for bacterial growth (44). Therefore, IFN-γ might have a different effect on phagocyte-mediated clearance of S. aureus versus pneumococci. This differential regulatory role may be related to differences in the effector mechanisms that are needed to eliminate these two pathogens. For example, MARCO is critical for alveolar macrophage-mediated clearance of pneumococci but not S. aureus clearance (unpublished data). Conversely, NADPH oxidase activity, which is likely to overcome the antioxidant action of endogenous staphylococcal catalase, is essential for pulmonary S. aureus clearance but is dispensable for pneumococcal eradiation (24).
There is controversy surrounding the relative contribution of macrophages to pulmonary clearance of S. aureus (20, 31, 32). However, we found that the immunological killing of S. aureus requires both neutrophils and macrophages. NADPH oxidase has also been found to be important for antibacterial immunity (20). Consistent with this, we observed increased bacterial loads and mortality after MRSA infection of NADPH oxidase-deficient mice. Since 98% of leukocytes in the airways of MRSA-infected mice were composed of alveolar macrophages and neutrophils (Fig. 3A and Supplemental Fig. 1), the lack of control of MRSA infection observed in gp91phox−/− mice is likely due to defective oxidase-dependent bacterial killing by these phagocytes. In post-influenza MRSA-infected mice, analysis of myeloid cell profiles in the airway revealed accumulation of neutrophils and inflammatory monocytes, suggesting that the defective antibacterial immunity in these mice was not associated with total numbers of phagocytes but rather their reduced antimicrobial ability. It is noteworthy that in contrast to our observations in animal models, in vitro co-culture of IAV/S. pneumonia with neutrophils increased the respiratory burst activity of these host cells (45), probably because pneumolysin released by S. pneumonia is a potent activator of intracellular oxygen radical production (46). Nonetheless, unlike S. aureus, NADPH oxidase is not essential for phagocytic killing of S. pneumonia (24, 47). In fact, gp91phox−/− mice demonstrate increased resistance to pneumococcal infection compared to WT controls (48). Nonetheless, we found that influenza infection impaired ROS generation in both alveolar macrophages and neutrophils, which, together with the observation that similar defective bacterial clearance was detected in gp91phox deficient and mosaic mice with or without influenza infection, suggests that NADPH oxidase-dependent bacterial killing is the principal pathway by which influenza infection regulates innate antibacterial immunity.
Although phagocyte NADPH oxidase is critical for host antibacterial immunity, it has also been shown to promote detrimental inflammatory responses during influenza infection (15–18). As such, alleviated lung pathologies and reduced accumulation of airway inflammatory cells were detected in NADPH oxidase-deficient mice in response to inactivated H5N1 avian influenza virus (18) or early after low pathogenicity H3N2 influenza infection (15). Therefore, the negative regulation of phagocyte ROS generation at the recovery stage of influenza infection, as revealed in the current study, has likely evolved to minimize influenza infection-associated lung damage. In agreement with our findings, it has been shown that both basal and phorbol dibutyrate-stimulated superoxide production in airway inflammatory cells are decreased from day 3 to day 7 after influenza infection, which is associated with a suppressive effect of Nox1 oxidase in tissue cells (49). Considering that gp91phox-dependent generation of superoxide is not absent, but rather reduced in influenza-infected WT mice, it is not surprising that gp91phox−/− mice exhibit attenuated lung inflammatory damage compared to WT controls and inhibition of NADPH oxidase reduces disease severity during influenza infection alone, i.e. in the absence of secondary S. aureus infection (15–17).
The present studies were conducted using a mouse infection model which is well-accepted for studying influenza and influenza-compromised bacterial infection (50). However, mice are not only highly resistant to S. aureus infection but also exhibit very rapid mortality kinetics after a lethal inoculum. In order to minimize complications from compensatory or secondary inflammatory responses due to lethal bacterial burdens, we infected mice with a relatively low dose of MRSA to examine the influence of influenza infection on protective antibacterial immunity, and then confirmed these findings with higher inoculums for survival studies. The results of this study establish, for the first time, that influenza infection suppresses cell-intrinsic contribution of NADPH oxidase to phagocytic bacterial killing, and predisposes hosts to secondary S. aureus infection.
Supplementary Material
Acknowledgments
This work was supported by a Pfizer Young Investigator Award and National Institutes of Health/National Heart, Lung, and Blood Institute R01 HL118408 to K.S. and by National Institutes of Health/National Institute of Allergy and Infectious Diseases grants R01 AI41517 and RO1 AI75312 to D.W.M.
We thank Drs. Tammy Kielian and Victor Huber for critical review of the manuscript and Ms. Kari Nelson for editorial comments.
References
- 1.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louria DB, Blumenfeld HL, Ellis JT, Kilbourne ED, Rogers DE. Studies on influenza in the pandemic of 1957–1958. II. Pulmonary complications of influenza. J Clin Invest. 1959;38:213–265. doi: 10.1172/JCI103791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease, C., and Prevention. Severe methicillin-resistant Staphylococcus aureus community-acquired pneumonia associated with influenza--Louisiana and Georgia, December 2006–January 2007. MMWR Morb Mortal Wkly Rep. 2007;56:325–329. [PubMed] [Google Scholar]
- 4.Finelli L, Fiore A, Dhara R, Brammer L, Shay DK, Kamimoto L, Fry A, Hageman J, Gorwitz R, Bresee J, Uyeki T. Influenza-associated pediatric mortality in the United States: increase of Staphylococcus aureus coinfection. Pediatrics. 2008;122:805–811. doi: 10.1542/peds.2008-1336. [DOI] [PubMed] [Google Scholar]
- 5.Murray RJ, Robinson JO, White JN, Hughes F, Coombs GW, Pearson JC, Tan HL, Chidlow G, Williams S, Christiansen KJ, Smith DW. Community-acquired pneumonia due to pandemic A(H1N1)2009 influenzavirus and methicillin resistant Staphylococcus aureus co-infection. PloS one. 2010;5:e8705. doi: 10.1371/journal.pone.0008705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randolph AG, Vaughn F, Sullivan R, Rubinson L, Thompson BT, Yoon G, Smoot E, Rice TW, Loftis LL, Helfaer M, Doctor A, Paden M, Flori H, Babbitt C, Graciano AL, Gedeit R, Sanders RC, Giuliano JS, Zimmerman J, Uyeki TM I Pediatric Acute Lung, N. Sepsis Investigator’s, L. the National Heart, and A. C. T. N. Blood Institute. Critically ill children during the 2009–2010 influenza pandemic in the United States. Pediatrics. 2011;128:e1450–1458. doi: 10.1542/peds.2011-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Centers for Disease, C., and Prevention. Severe Coinfection with Seasonal Influenza A (H3N2) Virus and Staphylococcus aureus - Maryland, February-March 2012. MMWR Morb Mortal Wkly Rep. 2012;61:289–291. [PubMed] [Google Scholar]
- 8.Tashiro M, Ciborowski P, Klenk HD, Pulverer G, Rott R. Role of Staphylococcus protease in the development of influenza pneumonia. Nature. 1987;325:536–537. doi: 10.1038/325536a0. [DOI] [PubMed] [Google Scholar]
- 9.Small CL, Shaler CR, McCormick S, Jeyanathan M, Damjanovic D, Brown EG, Arck P, Jordana M, Kaushic C, Ashkar AA, Xing Z. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. Journal of immunology. 2010;184:2048–2056. doi: 10.4049/jimmunol.0902772. [DOI] [PubMed] [Google Scholar]
- 10.Iverson AR, Boyd KL, McAuley JL, Plano LR, Hart ME, McCullers JA. Influenza virus primes mice for pneumonia from Staphylococcus aureus. J Infect Dis. 2011;203:880–888. doi: 10.1093/infdis/jiq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, Alcorn JF. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. Journal of immunology. 2013;191:5153–5159. doi: 10.4049/jimmunol.1301237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vardakas KZ, Matthaiou DK, Falagas ME. Incidence, characteristics and outcomes of patients with severe community acquired-MRSA pneumonia. Eur Respir J. 2009;34:1148–1158. doi: 10.1183/09031936.00041009. [DOI] [PubMed] [Google Scholar]
- 13.Blyth CC, Webb SA, Kok J, Dwyer DE, van Hal SJ, Foo H, Ginn AN, Kesson AM, Seppelt I, Iredell JR A. I. I. on behalf of the, and C. M. Investigators. The impact of bacterial and viral co-infection in severe influenza. Influenza Other Respi Viruses. 2012 doi: 10.1111/j.1750-2659.2012.00360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Metersky ML, Masterton RG, Lode H, File TM, Jr, Babinchak T. Epidemiology, microbiology, and treatment considerations for bacterial pneumonia complicating influenza. Int J Infect Dis. 2012;16:e321–331. doi: 10.1016/j.ijid.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Vlahos R, Stambas J, Bozinovski S, Broughton BR, Drummond GR, Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS pathogens. 2011;7:e1001271. doi: 10.1371/journal.ppat.1001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akaike T, Noguchi Y, Ijiri S, Setoguchi K, Suga M, Zheng YM, Dietzschold B, Maeda H. Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:2448–2453. doi: 10.1073/pnas.93.6.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snelgrove RJ, Edwards L, Rae AJ, Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur J Immunol. 2006;36:1364–1373. doi: 10.1002/eji.200635977. [DOI] [PubMed] [Google Scholar]
- 18.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133:235–249. doi: 10.1016/j.cell.2008.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jann NJ, Schmaler M, Ferracin F, Landmann R. TLR2 enhances NADPH oxidase activity and killing of Staphylococcus aureus by PMN. Immunol Lett. 2011;135:17–23. doi: 10.1016/j.imlet.2010.09.007. [DOI] [PubMed] [Google Scholar]
- 20.Kohler J, Breitbach K, Renner C, Heitsch AK, Bast A, van Rooijen N, Vogelgesang S, Steinmetz I. NADPH-oxidase but not inducible nitric oxide synthase contributes to resistance in a murine Staphylococcus aureus Newman pneumonia model. Microbes Infect. 2011;13:914–922. doi: 10.1016/j.micinf.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 21.Gaush CR, Smith TF. Replication and plaque assay of influenza virus in an established line of canine kidney cells. Applied microbiology. 1968;16:588–594. doi: 10.1128/am.16.4.588-594.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandra R, Federici S, Nemeth ZH, Horvath B, Pacher P, Hasko G, Deitch EA, Spolarics Z. Female X-chromosome mosaicism for NOX2 deficiency presents unique inflammatory phenotype and improves outcome in polymicrobial sepsis. Journal of immunology. 2011;186:6465–6473. doi: 10.4049/jimmunol.1100205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chinnery HR, Carlson EC, Sun Y, Lin M, Burnett SH, Perez VL, McMenamin PG, Pearlman E. Bone marrow chimeras and c-fms conditional ablation (Mafia) mice reveal an essential role for resident myeloid cells in lipopolysaccharide/TLR4-induced corneal inflammation. Journal of immunology. 2009;182:2738–2744. doi: 10.4049/jimmunol.0803505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun K, Gan Y, Metzger DW. Analysis of murine genetic predisposition to pneumococcal infection reveals a critical role of alveolar macrophages in maintaining the sterility of the lower respiratory tract. Infection and immunity. 2011;79:1842–1847. doi: 10.1128/IAI.01143-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. 2008;14:558–564. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- 26.Sun K, Ye J, Perez DR, Metzger DW. Seasonal FluMist vaccination induces cross-reactive T cell immunity against H1N1 (2009) influenza and secondary bacterial infections. Journal of immunology. 2011;186:987–993. doi: 10.4049/jimmunol.1002664. [DOI] [PubMed] [Google Scholar]
- 27.Seo SU, Kwon HJ, Ko HJ, Byun YH, Seong BL, Uematsu S, Akira S, Kweon MN. Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS pathogens. 2011;7:e1001304. doi: 10.1371/journal.ppat.1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. Journal of immunology. 2008;180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 29.Duan M, Li WC, Vlahos R, Maxwell MJ, Anderson GP, Hibbs ML. Distinct macrophage subpopulations characterize acute infection and chronic inflammatory lung disease. Journal of immunology. 2012;189:946–955. doi: 10.4049/jimmunol.1200660. [DOI] [PubMed] [Google Scholar]
- 30.Hampton MB, Kettle AJ, Winterbourn CC. Involvement of superoxide and myeloperoxidase in oxygen-dependent killing of Staphylococcus aureus by neutrophils. Infection and immunity. 1996;64:3512–3517. doi: 10.1128/iai.64.9.3512-3517.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robertson CM, Perrone EE, McConnell KW, Dunne WM, Boody B, Brahmbhatt T, Diacovo MJ, Van Rooijen N, Hogue LA, Cannon CL, Buchman TG, Hotchkiss RS, Coopersmith CM. Neutrophil depletion causes a fatal defect in murine pulmonary Staphylococcus aureus clearance. J Surg Res. 2008;150:278–285. doi: 10.1016/j.jss.2008.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rigby KM, DeLeo FR. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol. 2012;34:237–259. doi: 10.1007/s00281-011-0295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, Khader SA, Dubin PJ, Enelow RI, Kolls JK, Alcorn JF. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. Journal of immunology. 2011;186:1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee MH, Arrecubieta C, Martin FJ, Prince A, Borczuk AC, Lowy FD. A postinfluenza model of Staphylococcus aureus pneumonia. J Infect Dis. 2010;201:508–515. doi: 10.1086/650204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, Deng JC. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. 2009;119:1910–1920. doi: 10.1172/JCI35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, van Rijt LS, Lambrecht BN, Sirard JC, Hussell T. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205:323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Navarini AA, Recher M, Lang KS, Georgiev P, Meury S, Bergthaler A, Flatz L, Bille J, Landmann R, Odermatt B, Hengartner H, Zinkernagel RM. Increased susceptibility to bacterial superinfection as a consequence of innate antiviral responses. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:15535–15539. doi: 10.1073/pnas.0607325103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kash JC, Walters KA, Davis AS, Sandouk A, Schwartzman LM, Jagger BW, Chertow DS, Li Q, Kuestner RE, Ozinsky A, Taubenberger JK. Lethal synergism of 2009 pandemic H1N1 influenza virus and Streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses. MBio. 2011;2 doi: 10.1128/mBio.00172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karlstrom A, Heston SM, Boyd KL, Tuomanen EI, McCullers JA. Toll-like receptor 2 mediates fatal immunopathology in mice during treatment of secondary pneumococcal pneumonia following influenza. J Infect Dis. 2011;204:1358–1366. doi: 10.1093/infdis/jir522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karlstrom A, Boyd KL, English BK, McCullers JA. Treatment with protein synthesis inhibitors improves outcomes of secondary bacterial pneumonia after influenza. J Infect Dis. 2009;199:311–319. doi: 10.1086/596051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, DeLeo FR, Quinn MT. Role of NF-kappaB in transcriptional regulation of the phagocyte NADPH oxidase by tumor necrosis factor-alpha. Journal of leukocyte biology. 2007;82:729–741. doi: 10.1189/jlb.1206735. [DOI] [PubMed] [Google Scholar]
- 42.El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol. 2008;30:279–289. doi: 10.1007/s00281-008-0118-3. [DOI] [PubMed] [Google Scholar]
- 43.Hang do TT, Choi EJ, Song JY, Kim SE, Kwak J, Shin YK. Differential effect of prior influenza infection on alveolar macrophage phagocytosis of Staphylococcus aureus and Escherichia coli: involvement of interferon-gamma production. Microbiol Immunol. 2011;55:751–759. doi: 10.1111/j.1348-0421.2011.00383.x. [DOI] [PubMed] [Google Scholar]
- 44.McLoughlin RM, Lee JC, Kasper DL, Tzianabos AO. IFN-gamma regulated chemokine production determines the outcome of Staphylococcus aureus infection. Journal of immunology. 2008;181:1323–1332. doi: 10.4049/jimmunol.181.2.1323. [DOI] [PubMed] [Google Scholar]
- 45.Engelich G, White M, Hartshorn KL. Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. Journal of leukocyte biology. 2001;69:50–56. [PubMed] [Google Scholar]
- 46.Martner A, Dahlgren C, Paton JC, Wold AE. Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infection and immunity. 2008;76:4079–4087. doi: 10.1128/IAI.01747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marriott HM, Hellewell PG, Whyte MK, Dockrell DH. Contrasting roles for reactive oxygen species and nitric oxide in the innate response to pulmonary infection with Streptococcus pneumoniae. Vaccine. 2007;25:2485–2490. doi: 10.1016/j.vaccine.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marriott HM, Jackson LE, Wilkinson TS, Simpson AJ, Mitchell TJ, Buttle DJ, Cross SS, Ince PG, Hellewell PG, Whyte MK, Dockrell DH. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. American journal of respiratory and critical care medicine. 2008;177:887–895. doi: 10.1164/rccm.200707-990OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Selemidis S, Seow HJ, Broughton BR, Vinh A, Bozinovski S, Sobey CG, Drummond GR, Vlahos R. Nox1 oxidase suppresses influenza a virus-induced lung inflammation and oxidative stress. PloS one. 2013;8:e60792. doi: 10.1371/journal.pone.0060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. Journal of immunology. 2013;191:2047–2052. doi: 10.4049/jimmunol.1301152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.