

Abstract
The judicious use of antibiotics to combat infections in children relies upon appropriate selection of an agent, dose and duration to maximise efficacy and to minimise toxicity. Critical to dose optimisation is an understanding of the pharmacokinetics and pharmacodynamics of available drugs. Optimal dosing strategies may take advantage of pharmacokinetic/pharmacodynamic (PK/PD) principles so that antibiotic dosing can be individualised to assure effective bacterial killing in patients who have altered pharmacokinetics or who have infections with less susceptible or resistant organisms. This review will outline the fundamentals of antimicrobial pharmacokinetics/pharmacodynamics through discussion of antibacterial agents most often used in children. We aim to highlight the importance of dose optimisation in paediatrics and describe non-conventional dosing strategies that can take advantage of PK/PD principles at the bedside.
Keywords: Dose optimisation, Pharmacokinetics, Pharmacodynamics, PK/PD, Antibiotics
1. Introduction
Antibiotics are among the most commonly administered medications in children. In ambulatory settings in the USA, antibiotics are prescribed during as many as one in five paediatric visits [1]. Cross-sectional point prevalence studies have shown that more than 35% and 40% of hospitalised children are receiving antimicrobials at any given time in European and non-European countries, respectively [2].
Whilst the judicious use of antibiotics is clearly needed in all paediatric settings, appropriate drug selection and dose optimisation are also important to improve the management of bacterial infections. An understanding of pharmacokinetic/pharmacodynamic (PK/PD) principles may allow for the selection of the best drug to treat a specific bacterial pathogen while utilising the ideal dosing regimen to eradicate the infection, limit toxicity and reduce development of bacterial resistance.
As Dr Abraham Jacobi formally acknowledged more than 100 years ago, children are not little adults. Responses to medications differ depending on organ function, size and maturation of the child [3]. Age-related differences in absorption, distribution, metabolism and elimination of drugs preclude the use of a one-dose-fits-all approach for prescribing medications in children [4]. In addition, there are special populations within paediatrics—individuals with critical illness, cystic fibrosis, morbid obesity, immune compromise, and others—for whom ‘standard’ age-based dosing regimens may be inadequate. These groups often demand optimal antibiotic exposure to assure eradication of infection and limit the development of resistant pathogens. Therefore, it is crucial to understand and apply PK/PD principles when using antimicrobials and to appreciate how dosing regimens can be individualised to optimise therapy in these vulnerable patients. The goal of this review is to provide a general overview of PK/PD principles and to discuss how strategies can be employed in clinical practice to optimise antibiotic therapy in paediatric patients. Particular attention will be given to special populations to highlight the importance of dose optimisation.
2. Pharmacokinetics/pharmacodynamics of antibiotics
Pharmacokinetics refers to the drug concentration in serum, tissues and other body fluids over time and is dependent on the absorption, metabolism, distribution and elimination of the drug. Pharmacodynamics denotes the pharmacological effects of the drug for a given exposure. Whilst pharmacokinetics is dependent on patient factors, antimicrobial pharmacodynamics also involves the pathogen. The susceptibility of a bacterium to antibiotic killing can vary widely based on the particular organism and the agent of interest. Traditionally, an in vitro measure of the minimum concentration of drug needed to inhibit bacterial growth, called the minimum inhibitory concentration (MIC), is the PD parameter most frequently utilised clinically to describe antibiotic activity against a pathogen. Knowledge of the MIC alone cannot predict antibiotic success because the MIC does not account for the dynamic in vivo processes that influence antibacterial killing over time. Integration of pharmacokinetics (exposure) and pharmacodynamics (antimicrobial activity) at the site of infection predicts the agent’s efficacy against any given pathogen.
Antimicrobial effects are generally described as being either concentration-dependent or time-dependent [5,6]. Concentration-dependent drugs maximally kill bacteria with increasing concentrations, whereas the effect of increasing concentrations of time-dependent drugs plateaus; the latter are most effective when exposure is prolonged. Some drugs also have persistent or post-antibiotic effects (PAE), referring to the continued suppression of bacterial growth following the removal of drug after exposure, varying based upon the mechanism of drug action, the pathogen and how the drug is administered [7,8]. Thus, based on optimal bactericidal conditions, antibiotics can be divided into three categories (Table 1): concentration-dependent killing with moderate-to-persistent PAEs; time-dependent killing with minimal-to-no persistent bactericidal effects; and time-dependent killing with prolonged persistent effects [10]. The PK/PD indices correlating with clinical efficacy differ depending on the mechanism of antimicrobial activity. Understanding which category best describes an antibiotic’s mechanism allows for rationale dose adjustments to maximise activity.
Table 1.
Pattern of activity of different antibacterial drugs and their associated pharmacokinetic/pharmacodynamic (PK/PD) targets a
| Mechanism of bactericidal effects based on in vitro data | Antibiotic class | PK/PD parameter(s) associated with efficacy | Goal of dosing regimen |
|---|---|---|---|
| Concentration- dependent killing with moderate-to-persistent bactericidal effects | Aminoglycosides Fluoroquinolones Metronidazole Daptomycin Ketolides |
Cmax/MIC AUC0–24/MIC |
Maximise concentration: increase dose |
| Time-dependent killing with minimal-to-no persistent bactericidal effects | β-Lactams: Penicillins Cephalosporins Carbapenems Aztreonam Erythromycin |
T>MIC | Maximise the duration of exposure: increase duration of infusion or frequency of administration |
| Time-dependent killing with moderate-to-prolonged persistent bactericidal effects | Macrolides Tetracyclines Glycopeptides Clindamycin Linezolid b |
AUC0–24/MIC | Maximise drug exposure: increase dose, frequency of administration or duration of infusion |
Cmax, maximum serum concentration; MIC, minimum inhibitory concentration; AUC0–24, area under the concentration–time curve over a 24-h period; T>MIC, percentage of the dosing interval above the MIC.
Adapted with permission from Taylor and Francis Group LLC Books [10].
T>MIC has also been reported to be an appropriate PK/PD target for linezolid [9].
Historically, PK/PD relationships have been determined from in vitro or animal infection models [11] and confirmed in trials of adult patients [10]. Studies corroborating PK/PD endpoints in paediatric populations are often lacking. Nevertheless, the mechanism of action and PK/PD parameters that correlate with efficacy for the treatment of specific infections in adults should theoretically be similar for children. There are three primary PK/PD parameters that are most often described as the clinical targets for antibiotic exposure because they have been shown to correlate with clinical efficacy for different antibiotic classes (Fig. 1): the maximum serum concentration (Cmax) to MIC ratio (Cmax/MIC) for concentration-dependent drugs; the percentage of the dosing interval above the MIC (T>MIC) for time-dependent drugs with minimal-to-no persistent effects; and the ratio of the area under the concentration–time curve over a 24-h period (AUC0–24) to MIC ratio (AUC0–24/MIC) for time-dependent drugs with moderate-to-prolonged persistent effects. It is important to note that drug molecules bound to plasma proteins are not free to act upon bacteria, thus PD parameters often relate to the free fraction of drug in serum [12]. In the next sections, we will discuss each of the categories of antimicrobial effects in more detail, using specific examples to demonstrate how PK/PD information can influence dose optimisation in paediatric clinical practice.
Fig. 1.
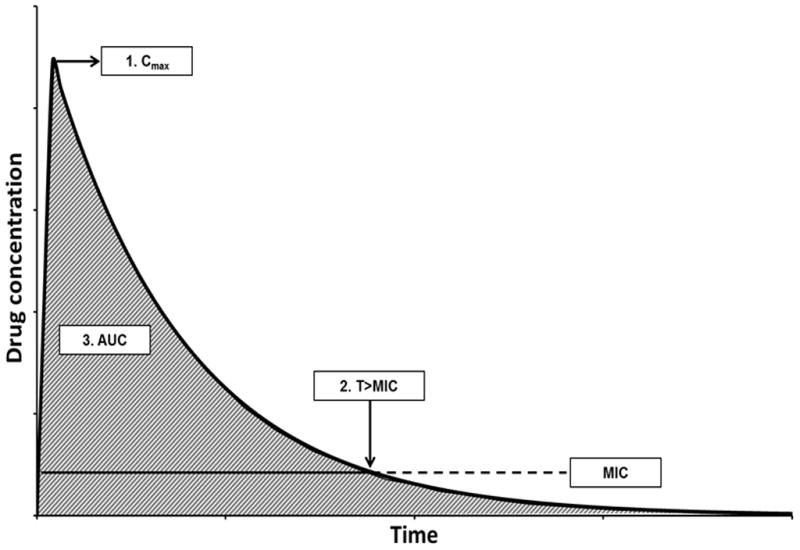
Pharmacokinetic/pharmacodynamic (PK/PD) relationships between antibiotic concentrations relative to the minimum inhibitory concentration (MIC) over time. Three principle PK/PD parameters most often correlate with the clinical efficacy of different antibiotic classes: (1) maximum concentration (Cmax)/MIC ratio for concentration-dependent drugs; (2) percentage of the dosing interval above the MIC (T>MIC) for time-dependent drugs with minimal-to-no persistent effects; and (3) area under the concentration–time curve (AUC, shaded area)/MIC ratio for time-dependent drugs with moderate-to-prolonged persistent effects.
Before proceeding, two important points should be conveyed. First, PK/PD parameters are highly inter-related: dose adjustments influence multiple PK/PD parameters simultaneously. For instance, a higher dose will increase the Cmax and AUC0–24 in relation to the MIC, as well as T>MIC. Therefore, multiple PK/PD parameters may correlate with efficacy, depending on the mechanism of action of the antibiotic. The pharmacokinetics of the drug will impact the extent to which dosing alterations affect each PK/PD parameter.
Second, it is critical to recognise that an organism’s MIC strongly influences the likelihood of PK/PD target attainment in clinical practice. Small increases in MIC dramatically impact the Cmax/MIC and AUC0–24/MIC ratios and T>MIC for any given exposure. For highly susceptible organisms, even suboptimal dosing may be adequate. However, in organisms with decreased susceptibility or resistance, appropriate antibiotic dosing is paramount to successful treatment.
3. Antibiotics with concentration-dependent killing with moderate-to-persistent bactericidal effects
There are two major classes of antibiotics used in paediatrics that demonstrate concentration-dependent killing of bacteria, namely aminoglycosides and fluoroquinolones. Other drugs, such as ketolides, daptomycin and metronidazole, also act in this manner [10]. Concentration-dependent drugs exhibit maximum bactericidal activity at highest concentrations and a PAE, suppressing bacterial growth after concentrations drop below the MIC of the organism. These properties allow for dosing regimens that aim to maximise the concentration (large doses) while taking advantage of the prolonged effects of the drugs (extended-interval dosing). Optimising concentration-dependent antibiotics in this manner not only promotes maximum efficacy but may prevent the development of resistance and adverse reactions. As aminoglycosides are the most widely used concentration-dependent antibiotics in paediatric patients, optimised dosing of this class will be discussed.
3.1. Aminoglycosides
Derived from Streptomyces spp., aminoglycosides have been used in paediatric patients for more than 60 years. A wealth of literature regarding their clinical utility, mechanisms of action, pharmacokinetics and pharmacodynamics, and toxicity exists [13–15]. The best described aminoglycoside in the paediatric population is gentamicin, although it represents only a small part of the large, structurally heterogeneous group. Aminoglycosides elicit their effects by binding irreversibly to the 16S ribosomal RNA of the 30S bacterial ribosomal subunit, thereby inhibiting protein synthesis. Peak serum concentrations consistently occur at the end of intravenous (i.v.) infusions, which allows for direct measurement of Cmax following infusion and the distribution phase, where drug diffuses from the plasma into the tissues. These medications demonstrate a broad spectrum of Gram-negative bacterial coverage as well as synergistic effects against select Gram-positive organisms.
Moore et al. first described the association between improved patient outcomes and higher Cmax/MIC ratios for aminoglycosides [16], demonstrating improved response among adults with Gram-negative infections when the Cmax/MIC ratio was 8–10. This PK/PD target has been reproduced in several other studies in adults [17,18] and is often utilised when deriving individualised PK dosing regimens in children. Although traditional dosing of aminoglycosides involved multiple daily administrations, optimal dosing strategies include high doses administered once daily [19,20]. Such extended-interval dosing takes advantage of the concentration-dependent bactericidal activity of the drugs and is clinically aided by the substantial PAE and leukocyte enhancement, minimising bacterial re-growth after the serum concentration has fallen below the bacterial MIC [14]. Extended-interval dosing also addresses the PD process known as adaptive resistance, wherein bacteria develop reversible impedance to the bactericidal action of the drugs. It allows for complete clearance of the drugs, giving adequate time for the targeted bacteria to return to a susceptible stage and minimising potential complications caused by developing resistance.
Concerns surrounding nephrotoxicity and ototoxicity limit administration of aminoglycosides to use in clinically significant infections, particularly of multidrug-resistant (MDR) organisms. Nephrotoxicity is linked to drug accumulation, which occurs due to binding of the medication to the brush borders of renal cells [21]. Low sustained concentrations result in more effective uptake in these tissues than when exposed to high intermittent levels [20]. Ototoxicity is due to production of free radicals that damage cochlear and vestibular hair cells [22]. Ototoxicity is often irreversible, and nephrotoxicity, while reversible in the majority of cases, may lead to prolonged hospitalisation and increased hospital costs in children and adults [23]. Individualised PK dosing has been shown to reduce the incidence of nephrotoxicity [24,25].
The extensive documentation of differences in PK/PD parameters for aminoglycosides in children allows for ready application of dose optimisation strategies. For example, dosing strategies in premature infants are driven predominantly by PK considerations. The lack of mature renal function in this population slows medication clearance, prolonging the expected half-life from 2–3 h to 8–12 h. This could result in higher trough concentrations, increasing the risk for toxicity if no dosing interval modifications are made. Premature infants also exhibit lower peak concentrations owing to their relatively larger volume of distribution (Vd) [26], and PK models support the administration of higher doses at lower frequency in this population, such as every 24–48 h [19].
In the case of gentamicin administration for neonatal sepsis, goal peak serum concentrations should be >8–10 μg/mL when treating organisms with an MIC of ≤1 μg/mL, and goal trough concentrations should be <0.5–1 μg/mL prior to re-dosing [19,20]. This represents a prime example for the utility of individualised dose optimisation, as personalised adaptation improves outcomes by reducing toxicity and decreases healthcare costs [27]. Clinicians often obtain these concentrations following the third to fourth dose, waiting for the patient’s levels to approximate steady-state concentrations. However, this practice may delay necessary dose modifications when treating documented infections. Use of appropriate population PK models and Bayesian adaptive control can individualise dosing schedules as soon as the clinical need for a prolonged antibiotic course is determined, maximising clinical efficacy of the drug and minimising potential toxicity [28].
Patients with cystic fibrosis (CF) also benefit from extended-interval dosing, although for different reasons than those in neonates. The role of aminoglycoside therapy in patients with CF is to combat Gram-negative organisms, in particular Pseudomonas aeruginosa. Higher doses are needed in patients with CF for three reasons: (i) bacteria carried by patients with CF often demonstrate decreased susceptibility to antibacterial agents owing to frequent antibiotic exposure over time; (ii) patients with CF have higher total body clearance and larger Vd than other patients and higher doses are needed to achieve the same serum concentrations; and (iii) high concentrations are more difficult to achieve in pulmonary infections than serum [29,30]. Extended-interval dosing of aminoglycosides allows clinicians to administer large doses, achieve high peak concentrations, and maximise efficacy against more resistant organisms [31]. Extended-interval dosing also allows sufficient time for complete clearance of the drug prior to re-administration, thereby reducing the likelihood of nephrotoxicity. Studies have shown that once-daily dosing is the ideal dosing strategy in patients with CF [32] and this is now endorsed by the Cystic Fibrosis Foundation as the preferred dosing strategy [33]. Although the Cmax/MIC ratio has been described as the optimal PK/PD target for clinical efficacy in the treatment of bronchopneumonia in CF [34], no specific peak to MIC ratio has been clearly established.
4. Antibiotics with time-dependent bactericidal activity with little-to-no persistent effects
Time-dependent antibiotics exhibit bactericidal activity by maintaining serum concentrations above the MIC of the organism. Some time-dependent drugs have little-to-no persistent effects, and re-growth of most organisms occurs soon after the serum levels decrease below the MIC. For antibiotics in this class, killing is not improved at higher concentrations, and the PD parameter associated with efficacy is T>MIC, with the goal to maximise the duration of drug exposure in order to optimise bacterial killing [6]. β-Lactams are such antibiotics.
4.1. β-Lactam antibiotics
Fleming discovered penicillin G in 1928 and its first use against Staphylococcus spp. was in 1941 [35]. Since that time, over 50 β-lactam derivatives, including cephalosporins and carbapenems, have been produced, in part because of the many possible chemical combinations and their effectiveness in killing a variety of organisms. Cephalosporins, which have a slow rate of killing, require a T>MIC of 60–70% of the dosing interval for maximum efficacy; less time is considered acceptable for penicillins (50%) and carbapenems (40%), which have faster rates of killing and longer PAEs [6,12]. β-Lactams are often the drugs of choice in paediatrics owing to their relatively high safety profile and they are used both in common and severe paediatric infections. As an example of how the PK/PD properties of β-lactam antibiotics can be optimised depending on the site of infection, we will contrast their use in acute otitis media and bacteraemia.
The most common pathogens causing acute otitis media in infants and children include Streptococcus pneumoniae, Haemophilus influenzae and Moraxella catarrhalis. Resistance to β-lactams in S. pneumoniae is due to alterations in penicillin-binding proteins and can be overcome in a dose-dependent manner. Owing to the increasing prevalence of resistant S. pneumoniae, larger doses of penicillins are often required. Susceptible S. pneumoniae isolates have an MIC of <2 μg/mL for i.v. penicillin, <0.06 μg/mL for oral penicillin and ≤1.0 μg/mL for i.v. cephalosporins for non-meningitis infections [36]. On average, the likelihood of bacteriological cure is 80–85% with a T>MIC of 40–50% and is near 100% with a T>MIC of 60–70% [37]. Fortunately, the middle ear has a rich capillary bed and a small volume of interstitial fluid that allows antibiotics to readily cross. This leads to slower responses to changes in serum concentrations resulting in blunted peaks and high trough values [37]. Higher doses of amoxicillin given twice daily can achieve a saturation of antibiotic in the middle ear and overcome resistance; thus, the recommended dosing regimen to treat penicillin-resistant S. pneumoniae has become 75–90 mg/kg/day [38,39]. A single 50 mg/kg intramuscular dose of ceftriaxone will achieve middle ear fluid concentrations of 9.5 μg/mL at 72 h and 4.8 μg/mL at 96 h, achieving a T>MIC of 100% for several days even for resistant S. pneumoniae [37,40].
When treating bacteraemia, or other more serious infections, there is no third space available to saturate with antibiotics and clinical efficacy is dependent on free drug concentrations in serum. In critically ill or immunocompromised patients, where it may be essential to maximise killing throughout the dosing regimen, continuous or extended infusions may offer benefit over traditional intermittent dosing. Extended or continuous i.v. infusion of β-lactam antibiotics will increase the time the drug concentration is above the MIC of the organism (Fig. 2) and may be superior to intermittent administration when treating pathogens with high MICs. A systematic review of adult randomised clinical trials conducted by Kasiakou et al. corroborated this hypothesis [41]. In addition, Monte Carlo simulations of children aged 2 years and 12 years receiving a variety of cefepime, ceftazidime, imipenem/cilastatin, meropenem and piperacillin/tazobactam regimens against P. aeruginosa with various MICs suggest target attainment is more difficult to achieve at higher MICs with intermittent dosing versus a prolonged or continuous infusion [42]. Extended-infusion β-lactams have shown benefit in decreased mortality, length of hospital stay and hospital costs in adults [43,44]; however, studies demonstrating clinical superiority are lacking in children. Although a recent systematic review failed to support routine use in paediatric patients [45], future, well designed prospective clinical trials will be needed to evaluate ways in which extended or continuous infusions improve outcomes in paediatric patients in addition to optimising PK/PD target attainment.
Fig. 2.
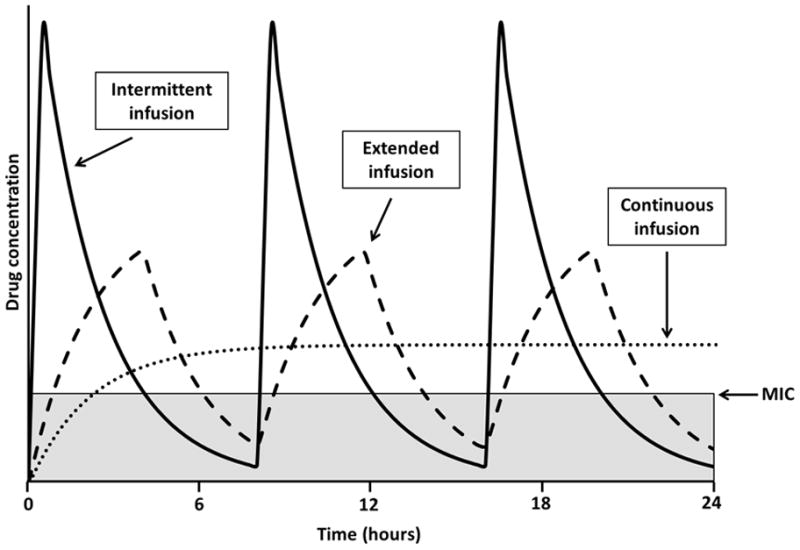
Time above the minimum inhibitory concentration (MIC) for intermittent, extended and continuous infusion of time-dependent drugs. Extended or continuous infusion of time-dependent drugs can improve the percentage of the dosing interval above the MIC (T>MIC), particularly when treating infections with elevated MICs. Intermittent infusion (solid line), extended infusion (dashed line) and continuous infusion (dotted line) are compared with the time that each dosing regimen is below the MIC, highlighted by the grey area. Curves were generated using the same total daily dose and consistent pharmacokinetic parameters for each regimen.
5. Antibiotics with time-dependent bactericidal activity with prolonged persistent effects
Whilst β-lactams need to maintain free concentrations above the MIC of the organism, other time-dependent antimicrobials have additional antibacterial activity after serum concentrations have dropped below the MIC. Antimicrobials that fall into this category include the macrolides, tetracyclines, clindamycin, linezolid and vancomycin. As an example, the PAE of vancomycin is dependent upon organism and ranges from 0.7 h to 2.6 h for Staphylococcus aureus and from 4.3 h to 6.5 h for Staphylococcus epidermidis [46]. Because of the moderate-to-persistent suppressive effects, the PK/PD parameter most closely associated with efficacy for this category of antibiotics is the AUC0–24/MIC ratio [47,48]. As examples of these types of antibiotics we will discuss vancomycin and linezolid and will contrast their use in drug-resistant Gram-positive infections.
5.1. Vancomycin
Vancomycin is a glycopeptide that has been commercially available for use since 1958 [49]. Clinical use was prevalent in the 1950s with the emergence of penicillinase-producing staphylococci; it is now back in frequent use due to meticillin-resistant S. aureus (MRSA). In adults with MRSA infections, an AUC0–24/MIC of >400 has been associated with improved clinical outcomes [50,51]. Current clinical practice guidelines for the treatment of MRSA infections from the Infectious Diseases Society of America (IDSA) recommend dosing regimens that utilise this PK/PD target [52,53]. In children with serious or invasive MRSA infections, initiating i.v. vancomycin 15 mg/kg/dose every 6 h is recommended as a means of reaching that goal for isolates with an MIC of ≤1 μg/mL. However, data in children linking improved outcomes with attainment of an AUC0–24/MIC >400 are lacking.
The ability to achieve an AUC0–24/MIC >400 has been evaluated in multiple paediatric population PK models. Frymoyer et al. studied children aged 2–12 years and predicted the AUC0–24/MIC for vancomycin daily doses of 40 mg/kg/day and 60 mg/kg/day [54]. These authors found that doses of 40 mg/kg/day were unlikely to achieve an AUC0–24/MIC >400 with an MIC ≥ 1 μg/mL, whilst this was more easily achieved with doses of 60 mg/kg/day [54]. A Monte Carlo simulation reiterated this point, with 58–66% of children predicted to achieve AUC0–24/MIC >400 with doses of 40 mg/kg/day, whilst doses >60 mg/kg/day achieved this target in 88–98% of children when the MIC was ≤1 μg/mL [55]. This may be less achievable in clinical practice: Chhim et al. found that only 40% of 96 children receiving 60 mg/kg/day vancomycin achieved an AUC0–24/MIC >400 [56].
The current IDSA guidelines suggest that trough values of 15–20 μg/mL should be targeted in adult patients with invasive MRSA infection, as troughs in this range correlate with AUC0–24/MIC >400 for isolates with an MIC ≤ 1 μg/mL [52]. Troughs of 15–20 μg/mL are often chosen in paediatric patients as well, although there are limited data to support this practice. Frymoyer et al. and Le et al. suggest that lower troughs may be adequate in children [57,58]. Via population PK modelling and simulation, Frymoyer et al. found that troughs of 7–10 μg/mL achieved an AUC0–24/MIC >400 in >90% of children receiving 15 mg/kg/dose every 6 h when the MIC was 1 μg/mL [57]. On the other hand, Le et al. utilised population-based PK modelling and Monte Carlo simulation to show that 60–70 mg/kg/day divided every 6 h achieved the AUC0–24/MIC >400 target in 75% of virtual patients over a range of MICs; an AUC0–24/MIC >400 corresponded with troughs of 8–9 μg/mL [58]. At Cincinnati Children’s Hospital Medical Center (Cincinnati, OH), only 67% of children achieved an AUC0–24 >400 when trough concentrations were 8–10 μg/mL, suggesting that lower troughs may be appropriate for MICs < 1 μg/mL but inadequate when MICs are ≥1 μg/mL [59].
Although targeting trough levels is much simpler for clinicians, it is not clear that the trough goals established in adult patients are ideal in children. And whilst an AUC0–24/MIC >400 has been associated with improved outcomes in adult patients with MRSA, paediatric studies are lacking and prospective studies are needed to validate the currently accepted PK/PD target. In addition, some children may demonstrate rapid clearance of vancomycin leading to use of excessive doses and potentially unnecessary frequent dose adjustments. Whilst the above studies bring up important topics for discussion, based on vancomycin PK and AUC0–24 data, we would not recommend a change in clinical practice that simply sought lower vancomycin serum trough values. Instead, personalising medical treatment and using information about the AUC and MIC for each person requiring vancomycin for invasive staphylococcal disease could provide the best outcomes for paediatric patients [59].
5.2. Linezolid
Resistant Gram-positive organisms such as MRSA and vancomycin-resistant enterococci (VRE) are becoming increasingly prevalent in children [60,61]. Infections with MRSA with high vancomycin MICs (vancomycin-intermediate S. aureus) and VRE are problematic because they are hard to treat and often carry greater morbidity and mortality than do those due to vancomycin-susceptible isolates [62]. Therapeutic options for these pathogens are limited; linezolid, an oxazolidinone antibiotic, is the most extensively utilised in paediatric patients. This agent, which inhibits bacterial protein synthesis by preventing formation of the bacterial 70S ribosomal subunit, is highly bioavailable and has excellent tissue penetration, making it an appealing alternative to vancomycin [63–65]. Although studies have shown linezolid to be as effective as vancomycin in treating serious Gram-positive infections [66,67], its use should be limited to preserve activity against drug-resistant organisms.
Linezolid exhibits bacteriostatic activity against Gram-positive organisms such as staphylococci and enterococci and has time-dependent killing with moderate PAEs, which are increased at higher concentrations relative to the MIC of the organism [68]. A study by Rayner et al. found that an AUC0–24/MIC of 80–120 and T>MIC of >85% of the dosing interval were associated with successful treatment of bacteraemia, lower respiratory tract infections, and skin and soft-tissue infections in debilitated adult patients [69]. The authors also found that despite the in vitro bacteriostatic activity of linezolid against staphylococci and enterococci, eradication of bacteraemia was improved and more rapid when the AUC0–24/MIC was >105 and T>MIC was >82.1%, results that suggest bactericidal activity clinically. The PK/PD parameters of T>MIC and AUC0–24/MIC have been corroborated by a number of other studies [70,71].
As with many drugs, the pharmacokinetics of linezolid are dependent on organ maturation and allometric increases in weight, with faster clearance, smaller AUC and a shorter elimination half-life in children compared with adults [72,73]. Studies have determined that more frequent doses (every 8 h) are required for children <12 years of age to achieve appropriate exposure. In septic patients, Vd and clearance are often increased resulting in significant interindividual variation in PK parameters for many drugs, including linezolid [74,75]. This is particularly problematic in young children with sepsis as the use of standard dosing regimens may lead to inadequate linezolid exposure and subtherapeutic drug concentrations. Use of continuous infusion of linezolid could promote better targeted drug levels and improve the likelihood of PK/PD target attainment, whether using an AUC0–24/MIC of 80–120 or T>MIC of >85% [74]. Alternative linezolid dosing strategies in septic or critically ill children have not been formally studied.
Again, children with CF also have altered pharmacokinetics compared with other children [29,30,76]. Clearance when normalised for body weight is often increased, and individuals with CF typically require increased mg/kg doses for most antibiotics. Santos et al. verified this for linezolid and found that standard linezolid dosing (10 mg/kg/dose i.v. every 8 h) was inadequate to achieve an AUC0–24/MIC >80 for S. aureus [77]. As MRSA colonisation and lower respiratory tract infections in paediatric patients with CF increase, exploration of alternative dosing strategies in this patient population may be needed.
Importantly, linezolid also demonstrates dose-dependent haematological toxicity, with higher serum AUC and elevated troughs being associated both with thrombocytopenia and anaemia [78–80]. Thus, appropriate exposure is paramount both for maximising efficacy and limiting toxicity. Continuous infusion, which can allow for lower total daily doses compared with higher or more frequent dosing schedules, may be a reasonable approach for treatment of resistant organisms, in patients with altered PK profiles, or in those at higher risk for toxicity. Again, more studies in paediatric patients are needed.
6. A future of drug resistance
To view the approaching horizon of antimicrobial resistance in children, one may look to patterns emerging in the adult population. In 2009, the IDSA reported a list of pathogens that could be expected to bring upon the post-antibiotic era [81]. A US Centers for Disease Control and Prevention (CDC) report, ‘Antibiotic resistance threats in the United States, 2013’, details the pathogens that pose the highest threat due to emerging drug resistance against currently available antibiotics [82]. With the decreased number of newly developed antibiotics, there is an increased need to optimise dosing of the existing armamentarium. The Gram-positive organisms (VRE and MRSA) can be treated using several antibiotics when susceptible. Few antibiotics can be used to treat the more resistant members of these species, including daptomycin, linezolid, quinupristin/dalfopristin, tigecycline, and fifth-generation cephalosporins. Of these, few PK/PD data are published in the paediatric population.
The MDR Gram-negative organisms (Klebsiella spp., Escherichia coli, Acinetobacter baumannii, P. aeruginosa and Enterobacter spp.) have fewer therapeutic options, leading to the use of formerly abandoned antibiotics such as colistimethate. Most of these organisms have shown clinical resistance to almost all antibiotic classes through the production of extended-spectrum β-lactamases, carbapenemases, efflux pumps, or alterations in the binding affinity of antimicrobial targets. Although several new therapies for these organisms are in the pipeline, a delay in the study of paediatric PK/PD data will be expected, heightening the importance of studying and applying the optimisation of current antibiotics to increase their efficacy and to prevent resistance.
Ultimately, knowledge of how to use an antibiotic appropriately is as important as knowing which antibiotic to choose. In the majority of clinical situations in paediatrics, choosing the right drug is sufficient. However, in other circumstances such as sepsis (neonatal or otherwise), infections in patients on renal replacement therapy, bronchopneumonia in CF patients, or when combating MDR organisms in immunocompromised children, correct dosing of the drug is as important as the antibiotic chosen. Utilisation of PK/PD principles may preserve the efficacy of existing drugs in the battle against resistant organisms. In the years to come, the importance of dose optimisation will become even more evident.
7. Individualised dose optimisation: a practical approach to pharmacokinetics/pharmacodynamics
Whilst PK studies typically describe the average behaviour of drugs in populations, they do not necessarily offer specific guidance on optimal dosing for individual patients. As with most drugs, antibiotics display significant interindividual variability in drug exposure that is dependent upon the underlying condition of the patient. Empirical dosing based on PK/PD principles will maximise the likelihood of achieving therapeutic targets. However, therapeutic drug management (TDM) is paramount for identifying the need for dose adjustments and assuring optimal doses throughout an antibiotic course.
At most institutions, TDM is available for only a minority of antibiotics, such as aminoglycosides and vancomycin. Despite extensive knowledge of the pharmacokinetics of many antibiotics, all antibiotics can be better prescribed with the direct measurement of drug concentrations. Frequently, TDM is utilised to minimise toxicity, whilst approximating efficacy, owing to practical and cost-related concerns. Yet direct measurement of (free) drug concentrations can be used to more accurately estimate attainment of PK/PD targets and to inform rational dose adjustments as needed. Dosing software can implement non-linear regression or other mathematical techniques such as Bayesian algorithms to estimate individual PK parameters (AUC, Cmax) that can be used to achieve desired targets for a given drug.
Population models can be also be helpful to optimise individualised therapy. These models provide a summary of PK parameter estimates in a specific population and can be employed to develop a dosing regimen that is most likely to achieve a desired target based on the clinical characteristics of the patient [83]. When drug concentration measurements are not available, clinical and demographic information is incorporated to provide empirical dosing regimens. Using drug concentrations as feedback to inform the model of how the patient is handling the drug will increase the accuracy of PK parameter estimation and improve target attainment. A variety of software programs exist to assist practitioners in identifying ideal drug regimens that maximise the probability of PK/PD target attainment in specific patients. These packages can assist practitioners in taking PK/PD dosing principles to the bedside.
8. Conclusions
Knowledge of PK/PD principles is crucial when prescribing antibiotics in children, particularly when treating serious infections and in special populations of children who have altered pharmacokinetics. Although standard dosing regimens may be adequate for most children, recognition of those patients who will benefit most from non-traditional dosing strategies is important. In addition, as paediatricians become engaged in the battle against MDR organisms once reserved for adult patients, dosing of antibiotics based on PK/PD principles may allow for continued use of common antibiotics and preservation of newer agents for those that truly lack other therapeutic options. We believe that PK/PD dose optimisation is an important component of judicious antibiotic prescribing, which is the foundation of antimicrobial stewardship.
Acknowledgments
Funding: Eunice Kennedy Shriver National Institute of Child Health and Human Development under a fellowship training grant [NIH 5 T32 HD069054] to KJD, AH and JW, and The Joint Commission and Pfizer Independent Grants for Learning & Change [grant 8116513] to JC.
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health (NIH).
Competing interests: None declared.
Ethical approval: Not required.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hersh AL, Shapiro DJ, Pavia AT, Shah SS. Antibiotic prescribing in ambulatory pediatrics in the United States. Pediatrics. 2011;128:1053–61. doi: 10.1542/peds.2011-1337. [DOI] [PubMed] [Google Scholar]
- 2.Versporten A, Sharland M, Bielicki J, Drapier N, Vankerckhoven V, Goossens H ARPEC Project Group Members. The Antibiotic Resistance and Prescribing in European Children Project: a neonatal and pediatric antimicrobial Web-based point prevalence survey in 73 hospitals worldwide. Pediatr Infect Dis J. 2013;32:e242–53. doi: 10.1097/INF.0b013e318286c612. [DOI] [PubMed] [Google Scholar]
- 3.Anderson BJ, Holford NH. Tips and traps analyzing pediatric PK data. Paediatr Anaesth. 2011;21:222–37. doi: 10.1111/j.1460-9592.2011.03536.x. [DOI] [PubMed] [Google Scholar]
- 4.Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349:1157–67. doi: 10.1056/NEJMra035092. [DOI] [PubMed] [Google Scholar]
- 5.Drusano GL. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’. Nat Rev Microbiol. 2004;2:289–300. doi: 10.1038/nrmicro862. [DOI] [PubMed] [Google Scholar]
- 6.Craig WA. Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis. 1998;26:1–10. doi: 10.1086/516284. quiz 11–2. [DOI] [PubMed] [Google Scholar]
- 7.Bundtzen RW, Gerber AU, Cohn DL, Craig WA. Postantibiotic suppression of bacterial growth. Rev Infect Dis. 1981;3:28–37. doi: 10.1093/clinids/3.1.28. [DOI] [PubMed] [Google Scholar]
- 8.Craig WA, Ebert SC. Killing and regrowth of bacteria in vitro: a review. Scand J Infect Dis Suppl. 1990;74:63–70. [PubMed] [Google Scholar]
- 9.Di Paolo A, Malacarne P, Guidotti E, Danesi R, Del Tacca M. Pharmacological issues of linezolid: an updated critical review. Clin Pharmacokinet. 2010;49:439–47. doi: 10.2165/11319960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 10.Craig WA. Pharmacodynamics of antimicrobials: general concepts and applications. In: Nightingale CH, Ambrose PG, Drusano GL, Murakawa T, editors. Antimicrobial pharmacodynamics in theory and clinical practice. 2. New York, NY: CRC Press; 2007. pp. 1–19. [Google Scholar]
- 11.Eagle H, Fleischman R, Levy M. ‘Continuous’ vs‘discontinuous’ therapy with penicillin; the effect of the interval between injections on therapeutic efficacy. N Engl J Med. 1953;248:481–8. doi: 10.1056/NEJM195303192481201. [DOI] [PubMed] [Google Scholar]
- 12.Drusano GL. Prevention of resistance: a goal for dose selection for antimicrobial agents. Clin Infect Dis. 2003;36(Suppl 1):S42–50. doi: 10.1086/344653. [DOI] [PubMed] [Google Scholar]
- 13.Jones EM, Howard WL. Streptomycin in the treatment of tuberculosis in children. Dis Chest. 1949;16:744–60. doi: 10.1378/chest.16.6.744. [DOI] [PubMed] [Google Scholar]
- 14.Pagkalis S, Mantadakis E, Mavros MN, Ammari C, Falagas ME. Pharmacological considerations for the proper clinical use of aminoglycosides. Drugs. 2011;71:2277–94. doi: 10.2165/11597020-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 15.Touw DJ, Westerman EM, Sprij AJ. Therapeutic drug monitoring of aminoglycosides in neonates. Clin Pharmacokinet. 2009;48:71–88. doi: 10.2165/00003088-200948020-00001. [DOI] [PubMed] [Google Scholar]
- 16.Moore RD, Lietman PS, Smith CR. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimal inhibitory concentration. J Infect Dis. 1987;155:93–9. doi: 10.1093/infdis/155.1.93. [DOI] [PubMed] [Google Scholar]
- 17.Zelenitsky SA, Harding GK, Sun S, Ubhi K, Ariano RE. Treatment and outcome of Pseudomonas aeruginosa bacteraemia: an antibiotic pharmacodynamic analysis. J Antimicrob Chemother. 2003;52:668–74. doi: 10.1093/jac/dkg403. [DOI] [PubMed] [Google Scholar]
- 18.Kashuba AD, Nafziger AN, Drusano GL, Bertino JS. Optimizing aminoglycoside therapy for nosocomial pneumonia caused by Gram-negative bacteria. Antimicrob Agents Chemother. 1999;43:623–9. doi: 10.1128/aac.43.3.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mohamed AF, Nielsen EI, Cars O, Friberg LE. Pharmacokinetic–pharmacodynamic model for gentamicin and its adaptive resistance with predictions of dosing schedules in newborn infants. Antimicrob Agents Chemother. 2012;56:179–88. doi: 10.1128/AAC.00694-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rao SC, Srinivasjois R, Hagan R, Ahmed M. One dose per day compared to multiple doses per day of gentamicin for treatment of suspected or proven sepsis in neonates. Cochrane Database Syst Rev. 2011;(11):CD0058091. doi: 10.1002/14651858.CD005091.pub3. [DOI] [PubMed] [Google Scholar]
- 21.Rougier F, Claude D, Maurin M, Maire P. Aminoglycoside nephrotoxicity. Curr Drug Targets Infect Disord. 2004;4:153–62. doi: 10.2174/1568005043340858. [DOI] [PubMed] [Google Scholar]
- 22.Beaubien AR, Desjardins S, Ormsby E, Bayne A, Carrier K, Cauchy MJ, et al. Incidence of amikacin ototoxicity: a sigmoid function of total drug exposure independent of plasma levels. Am J Otolaryngol. 1989;10:234–43. doi: 10.1016/0196-0709(89)90002-1. [DOI] [PubMed] [Google Scholar]
- 23.Zappitelli M, Moffett BS, Hyder A, Goldstein SL. Acute kidney injury in noncritically ill children treated with aminoglycoside antibiotics in a tertiary healthcare centre: a retrospective cohort study. Nephrol Dial Transplant. 2011;26:144–50. doi: 10.1093/ndt/gfq375. [DOI] [PubMed] [Google Scholar]
- 24.Bartal C, Danon A, Schlaeffer F, Reisenberg K, Alkan M, Smoliakov R, et al. Pharmacokinetic dosing of aminoglycosides: a controlled trial. Am J Med. 2003;114:194–8. doi: 10.1016/s0002-9343(02)01476-6. [DOI] [PubMed] [Google Scholar]
- 25.van Lent-Evers NA, Mathot RA, Geus WP, van Hout BA, Vinks AA. Impact of goal-oriented and model-based clinical pharmacokinetic dosing of aminoglycosides on clinical outcome: a cost-effectiveness analysis. Ther Drug Monit. 1999;21:63–73. doi: 10.1097/00007691-199902000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Nielsen MEI, Sandström M, Honoré PH, Ewald U, Friberg LE. Developmental pharmacokinetics of gentamicin in preterm and term neonates: population modelling of a prospective study. Clin Pharmacokinet. 2009;48:253–63. doi: 10.2165/00003088-200948040-00003. [DOI] [PubMed] [Google Scholar]
- 27.Streetman DS, Nafziger AN, Destache CJ, Bertino JS. Individualized pharmacokinetic monitoring results in less aminoglycoside-associated nephrotoxicity and fewer associated costs. Pharmacotherapy. 2001;21:443–51. doi: 10.1592/phco.21.5.443.34490. [DOI] [PubMed] [Google Scholar]
- 28.Neely M, Jelliffe R. Practical, individualized dosing: 21st century therapeutics and the clinical pharmacometrician. J Clin Pharmacol. 2010;50:842–7. doi: 10.1177/0091270009356572. [DOI] [PubMed] [Google Scholar]
- 29.Rey E, Tréluyer JM, Pons G. Drug disposition in cystic fibrosis. Clin Pharmacokinet. 1998;35:313–29. doi: 10.2165/00003088-199835040-00004. [DOI] [PubMed] [Google Scholar]
- 30.Touw DJ, Vinks AA, Mouton JW, Horrevorts AM. Pharmacokinetic optimisation of antibacterial treatment in patients with cystic fibrosis. Current practice and suggestions for future directions. Clin Pharmacokinet. 1998;35:437–59. doi: 10.2165/00003088-199835060-00003. [DOI] [PubMed] [Google Scholar]
- 31.Lam W, Tjon J, Seto W, Dekker A, Wong C, Atenafu E, et al. Pharmacokinetic modelling of a once-daily dosing regimen for intravenous tobramycin in paediatric cystic fibrosis patients. J Antimicrob Chemother. 2007;59:1135–40. doi: 10.1093/jac/dkm097. [DOI] [PubMed] [Google Scholar]
- 32.Smyth AR, Bhatt J. Once-daily versus multiple-daily dosing with intravenous aminoglycosides for cystic fibrosis. Cochrane Database Syst Rev. 2010;(1):CD002009. doi: 10.1002/14651858.CD002009.pub3. [DOI] [PubMed] [Google Scholar]
- 33.Flume PA, Mogayzel PJ, Jr, Robinson KA, Goss CH, Rosenblatt RL, Kuhn RJ, et al. Cystic fibrosis pulmonary guidelines: treatment of pulmonary exacerbations. Am J Respir Crit Care Med. 2009;180:802–8. doi: 10.1164/rccm.200812-1845PP. [DOI] [PubMed] [Google Scholar]
- 34.Burkhardt O, Lehmann C, Madabushi R, Kumar V, Derendorf H, Welte T. Once-daily tobramycin in cystic fibrosis: better for clinical outcome than thrice-daily tobramycin but more resistance development? J Antimicrob Chemother. 2006;58:822–9. doi: 10.1093/jac/dkl328. [DOI] [PubMed] [Google Scholar]
- 35.Tozuka Z, Murakawa T. β-Lactam pharmacodynamics. In: Nightingale CH, Ambrose PG, Drusano GL, Murakawa T, editors. Antimicrobial pharmacodynamics in theory and clinical practice. 2. New York, NY: CRC Press; 2007. pp. 129–46. [Google Scholar]
- 36.Ampofo K, Byington CL. Streptococcus pneumoniae. In: Long SS, Pickering LK, Prober CG, editors. Principles and practice of pediatric infectious disease. 4. Churchill Livingstone; 2012. pp. 721–8. [Google Scholar]
- 37.Craig WA, Andes D. Pharmacokinetics and pharmacodynamics of antibiotics in otitis media. Pediatr Infect Dis J. 1996;15:255–9. doi: 10.1097/00006454-199603000-00015. [DOI] [PubMed] [Google Scholar]
- 38.Canafax DM, Yuan Z, Chonmaitree T, Deka K, Russlie HQ, Giebink GS. Amoxicillin middle ear fluid penetration and pharmacokinetics in children with acute otitis media. Pediatr Infect Dis J. 1998;17:149–56. doi: 10.1097/00006454-199802000-00014. [DOI] [PubMed] [Google Scholar]
- 39.Seikel K, Shelton S, McCracken GH., Jr Middle ear fluid concentrations of amoxicillin after large dosages in children with acute otitis media. Pediatr Infect Dis J. 1998;17:969–70. doi: 10.1097/00006454-199810000-00042. [DOI] [PubMed] [Google Scholar]
- 40.Gudnason T, Gudbrandsson F, Barsanti F, Kristinsson KG. Penetration of ceftriaxone into the middle ear fluid of children. Pediatr Infect Dis J. 1998;17:258–60. doi: 10.1097/00006454-199803000-00022. [DOI] [PubMed] [Google Scholar]
- 41.Kasiakou SK, Lawrence KR, Choulis N, Falagas ME. Continuous versus intermittent intravenous administration of antibacterials with time-dependent action: a systematic review of pharmacokinetic and pharmacodynamic parameters. Drugs. 2005;65:2499–511. doi: 10.2165/00003495-200565170-00006. [DOI] [PubMed] [Google Scholar]
- 42.Courter JD, Kuti JL, Girotto JE, Nicolau DP. Optimizing bactericidal exposure for β-lactams using prolonged and continuous infusions in the pediatric population. Pediatr Blood Cancer. 2009;53:379–85. doi: 10.1002/pbc.22051. [DOI] [PubMed] [Google Scholar]
- 43.Mouton JW, Vinks AA. Continuous infusion of β-lactams. Curr Opin Crit Care. 2007;13:598–606. doi: 10.1097/MCC.0b013e3282e2a98f. [DOI] [PubMed] [Google Scholar]
- 44.Bauer KA, West JE, O’Brien JM, Goff DA. Extended-infusion cefepime reduces mortality in patients with Pseudomonas aeruginosa infections. Antimicrob Agents Chemother. 2013;57:2907–12. doi: 10.1128/AAC.02365-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker MC, Lam WM, Manasco KB. Continuous and extended infusions of β-lactam antibiotics in the pediatric population. Ann Pharmacother. 2012;46:1537–46. doi: 10.1345/aph.1R216. [DOI] [PubMed] [Google Scholar]
- 46.Lowdin E, Odenholt I, Cars O. In vitro studies of pharmacodynamic properties of vancomycin against Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob Agents Chemother. 1998;42:2739–44. doi: 10.1128/aac.42.10.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rybak MJ. The pharmacokinetic and pharmacodynamic properties of vancomycin. Clin Infect Dis. 2006;42(Suppl 1):S35–9. doi: 10.1086/491712. [DOI] [PubMed] [Google Scholar]
- 48.Knudsen JD, Fuursted K, Raber S, Espersen F, Frimodt-Moller N. Pharmacodynamics of glycopeptides in the mouse peritonitis model of Streptococcus pneumoniae or Staphylococcus aureus infection. Antimicrob Agents Chemother. 2000;44:1247–54. doi: 10.1128/aac.44.5.1247-1254.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hermsen ED, Ross GH, Rotschafer JC. Glycopeptide pharmacodynamics. In: Nightingale CH, Ambrose PG, Drusano GL, Murakawa T, editors. Antimicrobial Pharmacodynamics in theory and clinical practice. 2. New York, NY: CRC Press; 2007. pp. 189–215. [Google Scholar]
- 50.Kullar R, Davis SL, Levine DP, Rybak MJ. Impact of vancomycin exposure on outcomes in patients with methicillin-resistant Staphylococcus aureus bacteremia: support for consensus guidelines suggested targets. Clin Infect Dis. 2011;52:975–81. doi: 10.1093/cid/cir124. [DOI] [PubMed] [Google Scholar]
- 51.Moise-Broder PA, Forrest A, Birmingham MC, Schentag JJ. Pharmacodynamics of vancomycin and other antimicrobials in patients with Staphylococcus aureus lower respiratory tract infections. Clin Pharmacokinet. 2004;43:925–42. doi: 10.2165/00003088-200443130-00005. [DOI] [PubMed] [Google Scholar]
- 52.Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52:285–92. doi: 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- 53.Rybak M, Lomaestro B, Rotschafer JC, Moellering R, Jr, Craig W, Billeter M, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm. 2009;66:82–98. doi: 10.2146/ajhp080434. [DOI] [PubMed] [Google Scholar]
- 54.Frymoyer A, Hersh AL, Benet LZ, Guglielmo BJ. Current recommended dosing of vancomycin for children with invasive methicillin-resistant Staphylococcus aureus infections is inadequate. Pediatr Infect Dis J. 2009;28:398–402. doi: 10.1097/INF.0b013e3181906e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frymoyer A, Hersh AL, Coralic Z, Benet LZ, Joseph Guglielmo B. Prediction of vancomycin pharmacodynamics in children with invasive methicillin-resistant Staphylococcus aureus infections: a Monte Carlo simulation. Clin Ther. 2010;32:534–42. doi: 10.1016/j.clinthera.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chhim RF, Arnold SR, Lee KR. Vancomycin dosing practices, trough concentrations, and predicted area under the curve in children with suspected invasive staphylococcal infections. J Pediatr Infect Dis Soc. 2013;2:259–62. doi: 10.1093/jpids/pis083. [DOI] [PubMed] [Google Scholar]
- 57.Frymoyer A, Guglielmo BJ, Hersh AL. Desired vancomycin trough serum concentration for treating invasive methicillin-resistant staphylococcal infections. Pediatr Infect Dis J. 2013;32:1077–9. doi: 10.1097/INF.0b013e318299f75c. [DOI] [PubMed] [Google Scholar]
- 58.Le J, Bradley JS, Murray W, Romanowski GL, Tran TT, Nguyen N, et al. Improved vancomycin dosing in children using area under the curve exposure. Pediatr Infect Dis J. 2013;32:e155–63. doi: 10.1097/INF.0b013e318286378e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hahn A, Vinks AA. Lower vancomycin serum concentrations might not be the answer. Pediatr Infect Dis J. 2013;32:1403–4. doi: 10.1097/INF.0000000000000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Creech CB, Kernodle DS, Alsentzer A, Wilson C, Edwards KM. Increasing rates of nasal carriage of methicillin-resistant Staphylococcus aureus in healthy children. Pediatr Infect Dis J. 2005;24:617–21. doi: 10.1097/01.inf.0000168746.62226.a4. [DOI] [PubMed] [Google Scholar]
- 61.McCracken M, Wong A, Mitchell R, Gravel D, Conly J, Embil J, et al. Molecular epidemiology of vancomycin-resistant enterococcal bacteraemia: results from the Canadian Nosocomial Infection Surveillance Program, 1999–2009. J Antimicrob Chemother. 2013;68:1505–9. doi: 10.1093/jac/dkt054. [DOI] [PubMed] [Google Scholar]
- 62.DiazGranados CA, Zimmer SM, Klein M, Jernigan JA. Comparison of mortality associated with vancomycin-resistant and vancomycin-susceptible enterococcal bloodstream infections: a meta-analysis. Clin Infect Dis. 2005;41:327–33. doi: 10.1086/430909. [DOI] [PubMed] [Google Scholar]
- 63.Dehghanyar P, Bürger C, Zeitlinger M, Islinger F, Kovar F, Müller M, et al. Penetration of linezolid into soft tissues of healthy volunteers after single and multiple doses. Antimicrob Agents Chemother. 2005;49:2367–71. doi: 10.1128/AAC.49.6.2367-2371.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Traunmüller F, Schintler MV, Spendel S, Popovic M, Mauric O, Scharnagl E, et al. Linezolid concentrations in infected soft tissue and bone following repetitive doses in diabetic patients with bacterial foot infections. Int J Antimicrob Agents. 2010;36:84–6. doi: 10.1016/j.ijantimicag.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 65.Gee T, Ellis R, Marshall G, Andrews J, Ashby J, Wise R. Pharmacokinetics and tissue penetration of linezolid following multiple oral doses. Antimicrob Agents Chemother. 2001;45:1843–6. doi: 10.1128/AAC.45.6.1843-1846.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaplan SL, Deville JG, Yogev R, Morfin MR, Wu E, Adler S, et al. Linezolid versus vancomycin for treatment of resistant Gram-positive infections in children. Pediatr Infect Dis J. 2003;22:677–86. doi: 10.1097/01.inf.0000078160.29072.42. [DOI] [PubMed] [Google Scholar]
- 67.Chiappini E, Conti C, Galli L, de Martino M. Clinical efficacy and tolerability of linezolid in pediatric patients: a systematic review. Clin Ther. 2010;32:66–88. doi: 10.1016/j.clinthera.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 68.MacGowan AP. Pharmacokinetic and pharmacodynamic profile of linezolid in healthy volunteers and patients with Gram-positive infections. J Antimicrob Chemother. 2003;51(Suppl 2):ii17–25. doi: 10.1093/jac/dkg248. [DOI] [PubMed] [Google Scholar]
- 69.Rayner CR, Forrest A, Meagher AK, Birmingham MC, Schentag JJ. Clinical pharmacodynamics of linezolid in seriously ill patients treated in a compassionate use programme. Clin Pharmacokinet. 2003;42:1411–23. doi: 10.2165/00003088-200342150-00007. [DOI] [PubMed] [Google Scholar]
- 70.Dailey CF, Dileto-Fang CL, Buchanan LV, Oramas-Shirey MP, Batts DH, Ford CW, et al. Efficacy of linezolid in treatment of experimental endocarditis caused by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2001;45:2304–8. doi: 10.1128/AAC.45.8.2304-2308.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oramas-Shirey MP, Buchanan LV, Dileto-Fang CL, Dailey CF, Ford CW, Batts DH, et al. Efficacy of linezolid in a staphylococcal endocarditis rabbit model. J Antimicrob Chemother. 2001;47:349–52. doi: 10.1093/jac/47.3.349. [DOI] [PubMed] [Google Scholar]
- 72.Jungbluth GL, Welshman IR, Hopkins NK. Linezolid pharmacokinetics in pediatric patients: an overview. Pediatr Infect Dis J. 2003;22(9 Suppl):S153–7. doi: 10.1097/01.inf.0000086954.43010.63. [DOI] [PubMed] [Google Scholar]
- 73.Kearns GL, Abdel-Rahman SM, Blumer JL, Reed MD, James LP, Jacobs RF, et al. Single dose pharmacokinetics of linezolid in infants and children. Pediatr Infect Dis J. 2000;19:1178–84. doi: 10.1097/00006454-200012000-00012. [DOI] [PubMed] [Google Scholar]
- 74.Adembri C, Fallani S, Cassetta MI, Arrigucci S, Ottaviano A, Pecile P, et al. Linezolid pharmacokinetic/pharmacodynamic profile in critically ill septic patients: intermittent versus continuous infusion. Int J Antimicrob Agents. 2008;31:122–9. doi: 10.1016/j.ijantimicag.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 75.Dong H, Wang X, Dong Y, Lei J, Li H, You H, et al. Clinical pharmacokinetic/pharmacodynamic profile of linezolid in severely ill intensive care unit patients. Int J Antimicrob Agents. 2011;38:296–300. doi: 10.1016/j.ijantimicag.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 76.Touw DJ. Clinical pharmacokinetics of antimicrobial drugs in cystic fibrosis. Pharm World Sci. 1998;20:149–60. doi: 10.1023/a:1008634911114. [DOI] [PubMed] [Google Scholar]
- 77.Santos RP, Prestidge CB, Brown ME, Urbancyzk B, Murphey DK, Salvatore CM, et al. Pharmacokinetics and pharmacodynamics of linezolid in children with cystic fibrosis. Pediatr Pulmonol. 2009;44:148–54. doi: 10.1002/ppul.20966. [DOI] [PubMed] [Google Scholar]
- 78.Sasaki T, Takane H, Ogawa K, Isagawa S, Hirota T, Higuchi S, et al. Population pharmacokinetic and pharmacodynamic analysis of linezolid and a hematologic side effect, thrombocytopenia, in Japanese patients. Antimicrob Agents Chemother. 2011;55:1867–73. doi: 10.1128/AAC.01185-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hiraki Y, Tsuji Y, Hiraike M, Misumi N, Matsumoto K, Morita K, et al. Correlation between serum linezolid concentration and the development of thrombocytopenia. Scand J Infect Dis. 2012;44:60–4. doi: 10.3109/00365548.2011.608712. [DOI] [PubMed] [Google Scholar]
- 80.Tsuji Y, Hiraki Y, Matsumoto K, Mizoguchi A, Kobayashi T, Sadoh S, et al. Thrombocytopenia and anemia caused by a persistent high linezolid concentration in patients with renal dysfunction. J Infect Chemother. 2011;17:70–5. doi: 10.1007/s10156-010-0080-6. [DOI] [PubMed] [Google Scholar]
- 81.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 82.US Centers for Disease Control and Prevention (CDC) Antibiotic resistance threats in the United States, 2013. Washington, DC: US Department of Health and Human Services; 2013. [Google Scholar]
- 83.Vinks AA. The application of population pharmacokinetic modeling to individualized antibiotic therapy. Int J Antimicrob Agents. 2002;19:313–22. doi: 10.1016/s0924-8579(02)00023-7. [DOI] [PubMed] [Google Scholar]