

Abstract
Pin1 is a prolyl isomerase that recognizes phosphorylated Ser/Thr-Pro sites and phosphatase inhibitor-2 (I-2) is phosphorylated during mitosis at a PSpTP site that is expected to be a Pin1 substrate. However, we previously discovered I-2, but not phospho-I-2, bound to Pin1 as an allosteric modifier of Pin1 substrate specificity (Li et al., Biochemistry 47: 292, 2008). Here, we used binding assays and NMR spectroscopy to map the interactions on Pin1 and I-2 to elucidate the organization of this complex. Despite only ~ 50% sequence identity, human, Xenopus and Drosophila I-2 proteins all exhibited identical, saturable binding to GST-Pin1 with K0.5 of 0.3 μM. The [1H-15N] Heteronuclear Single Quantum Coherence spectra for both the WW domain and isomerase domain of Pin1 showed distinctive shifts upon addition of I-2. Conversely, in NMR spectroscopy specific regions of I-2 were affected by addition of Pin1. A single residue substitution I68A in I-2 reduced binding to Pin1 by half and essentially eliminated binding to the isolated WW domain. On the other hand truncation of I-2 to residue 152 had minimal effect on binding to WW domain but eliminated binding to the isomerase domain. Size exclusion chromatography revealed that wild type I-2 plus Pin1 formed a large (> 300 kDa) complex, whereas I-2(I68A) formed a complex of half the size that we propose are a heterotetramer and a heterodimer, respectively. Pin1 and I-2 are conserved among eukaryotes from yeast to human, and we propose they are an ancient partnership that provides a means to regulate Pin1 specificity and function.
Pin1 is the most widely studied member of the parvulin family of peptidyl-prolyl cis-trans isomerase proteins, with the unique feature of selectively recognizing the phosphorylated Ser/Thr-Pro motif in protein (1, 2). First characterized in 1996 as a regulator of the mitotic kinase NIMA in the fungus Aspergillus nidulans (3), Pin1 is homologous to the previously reported essential gene in Saccharomyces cerevisiae (Ess1)(4) and to the Drosophila gene dodo (5). The Pin1 protein comprises 163 amino acids in two distinct domains, an N-terminal WW domain (Pin1ww; residues 1–39) and a C-terminal catalytic domain (Pin1cat: residues 45–163), and three dimensional structures have been determined by crystallography (6) and NMR spectroscopy (7). Whereas the crystal structure shows the domains compressed together in a single, overall globular shape the solution structure by NMR spectroscopy depicts the domains separated from one another in a more extended configuration. Isomerization of phosphoproteins by Pin1 is considered to modulate many biological processes especially those involving key proteins such as cyclin D1 (8), c-jun (9), c-Myc (10), p53 (11, 12), tau (13–15). The over expression of Pin1 in various human tumors (9, 16–20) and recognition that its depletion from cells induces mitotic arrest has made Pin1 an attractive therapeutic target for drug development (21–23). A first Pin1 inhibitor is juglone, which covalently modifies the Cys in the active site of the isomerase domain (24), but as a reactive compound lacks selectivity. Other Pin1 inhibitors have been described (25, 26), but as far as we know have not yet entered clinical trials. Because both WW and isomerase domains can bind phosphosites it has been proposed that Pin1 uses simultaneous interaction with two different phosphosites in a particular substrate (27). However, Ser16 in the WW is phosphorylated by PKA (28), which then occupies the site where a sulfate ion binds in the 3D structure (6). Moreover, dynamic measurements by NMR spectroscopy indicate that dually phosphorylated peptides tend to interact only with the isomerase domain in Pin1 (29). The presumption has been that Pin1 acts as a monomer to isomerize phosphorylated sites in many different proteins, yet the basis for Pin1 substrate specificity is poorly understood (29).
Inhibitor-2 (I-2) was discovered in 1976 (30) as a thermostable protein that inhibited protein phosphatase activity, and later was used to distinguish type-1 (I-2 sensitive) from type-2 (I-2 insensitive) protein Ser/Thr phosphatases (31). I-2 is the most ancient of the more than 200 PP1 binding proteins, with recognized homologues in yeast (Glc8), Drosophila, Xenopus, and all mammals (32). The most conserved feature of eukaryotic I-2 proteins is a Pro-X-Thr-Pro (PXTP) sequence motif. The heterodimer of I-2 with PP1 was studied as an “MgATP-dependent phosphatase”, wherein the phosphorylation and dephosphorylation of Thr73 in the PXTP motif causes conformational activation of the bound PP1 (33). GSK3, MAPK and CDK kinases phosphorylate this motif in biochemical assays (34–37). We discovered >25-fold increase in PXTP phosphorylation during mitosis (38), catalyzed by CDK1:cyclinB1 (36) in a reaction enhanced by Suc1 (32), making this site a potential Pin1 substrate. However, T73 phosphorylated I-2 was not a substrate and did not bind to Pin1, but the negative controls in this assay, using unphosphorylated or T73A I-2, showed formation of a Pin1-I-2 complex (39). In the presence of I-2, compared to serum albumin as a control, GST-Pin1 binding to a panel of known mitotic phosphoprotein substrates was allosterically modified, with evidence for both enhanced and restricted binding (39). These results showed that Pin1 association with I-2 does not occlude its phosphopeptide binding sites, but does alter substrate binding specificity. Our hypothesis is that functions of Pin1 and I-2 are interdependent.
Recent results have discovered that I-2 acts as a critical regulator of cellular events related to mitosis. The protein is localized at centrosomes and serves to activate Nek2 kinase by inhibition of associated PP1 (40). Centrosomes lie at the base of the primary cilium, and I-2 is concentrated in the cilium, as seen by immunofluorescent microscopy (41). Knockdown of I-2 prevents formation of the cilium and reduces acetylation of tubulin in the cilium (41). I-2 is a maternal gene in Drosophila, the I-2 protein is concentrated in oocytes, and is required for the proper execution of early mitosis in the syncytial embryo (42). Hypomorphic embryos show abnormal mitotic spindles and lagging chromosomes, with lethal consequences. The defects are rescued by dose-dependent transgenic expression of D-I-2 (42). Likewise, in human epithelial cells knockdown of I-2 caused defects in mitosis, with appearance of lagging chromosomes and failed cytokinesis, resulting in multinucleated cells, phenotypes that were rescued by co-expression of I-2 (43). Direct binding of I-2 that activates Aurora A kinase (44) reduces the threshold for CDK activation at entry into mitosis (45). The biological functions of I-2 can in part be attributed to inhibition of PP1, but that alone seems inadequate to account for these phenotypes.
In this study, we have explored the binding of I-2 with Pin1 at atomic resolution. We found that the binding of I-2 with Pin 1 is conserved among species, despite only ~50% sequence identity between the proteins. We used NMR spectroscopy and reciprocal labeling to titrate and map the interactions on the surfaces of these two proteins. When we found that a single residue substitution in I-2 gave 50% binding at saturation compared to wild type we realized that multimers must be involved, and used size exclusion chromatography to demonstrate that Pin1 and I-2 form a tetramer in solution. We used truncated and mutated proteins in binding assays to assign which regions of I-2 interact with the WW and isomerase domains in Pin1 to generate a model for this complex. We propose that Pin1 and I-2 functions are, like the proteins themselves, inextricably intertwined, and this opens new directions for future research.
MATERIALS AND METHODS
Restriction enzymes and reagents for polymerase chain reaction were purchased from New England BioLabs. Oligonucleotides were synthesized by Integrated DNA Technologies. Affinity purified sheep anti-HI-2, anti-GST, anti-X-I-2 and anti-D-I-2 antibodies have been described previously (42). Buffers and chemicals were purchased from Thermo Fisher Scientific. Phospho-S16Pin1 antibodies were from Cell Signaling Technologies. Alexa Fluor® 680 goat anti-rabbit IgG were purchased from Invitrogen, and donkey anti-rabbit IRDye 800CW was purchased from LI-COR Biosciences (Lincoln, NE).
Cloning and Bacterial Expression of Proteins
Cloning of all Inhibitor 2 proteins used in this study are performed according to the methods described previously (32). Human Pin1 and WW domain (residues 1–44) and isomerase domain (residues 46–163) were cloned in pGEX-4T-1 (Amersham Pharmacia Biotech, Freiburg, Germany). In vitro phosphorylation of purified recombinant GST-Pin1 and GST-WW domain was performed using pure PKA catalytic subunit by previously described method (46).
E. coli strain BL21-CodonPlus (DE3)-RIPL (Agilent Technologies) was transformed with pET-I-2 vectors, or pGEX-4T-1(Pin1) bacterial expression vectors and grown overnight at 37 °C in 10 mL of TB medium (1.2% tryptone, 2.4 % yeast extract, 2%glucose, 0.017 M KH2PO4, 0.072 M K2HPO4), 30 μg/mL of kanamycin (for pET vectors), 30 μg/mL of Ampicilin (for pGEX vectors) and 20 μg/mL of chloramphenicol. The culture was inoculated into 1.0 L of TB medium including 30 μg/mL of either Kanamycin or Ampicilin and 20 mL of ethanol. The transformed cells were grown to A600nm of 0.6 at 37 °C, and the expression of I-2 proteins was induced by addition of isopropyl-1-thio-β-D-galactopyranoside into the culture at a final concentration of 1 mM for 12–16 h at 18 °C. The bacteria were collected by centrifugation at 5000 g for 10 min, and the cell pellet was washed by suspending in ice-cold PBS, centrifuged at 5000 g for 10 min, and cell pellets frozen at −80 °C.
Purification of His6-tagged I-2 proteins
Pellet was suspended by a buffer containing 50 mM MOPS-NaOH (pH 7.4), 300 mM NaCl, 10 mM imidazole. 10% glycerol 1 mM DDT, 1 tablet Complete EDTA-free tablet (Roche Applied Science) per 30 ml buffer, 1.0 mg/mL of lysozyme, and frozen at −80 °C. After complete freezing, the pellet was thawed for 10 min in a 37 °C bath, and sonicated for 30 sec pulses eight times for lysis of the cells. The homogenates were centrifuged at 20,000 g for 1 h at 4°C, and the supernatant heated 10 min in boiling water and centrifuged for 20 min at 20,000 g. The supernatant was transferred into a 50 mL tube containing 3 ml prewashed TALON® His-Tag resin (Clontech) to adsorb protein for 1 h at 4°C. The beads were packed into a column. The column was washed extensively with lysis buffer and bound proteins eluted with buffer containing 150 mM imidazole/HCl. The eluted proteins were dialyzed against 10 mM Tris-HCl (pH 7.4) and Complete EDTA-free protease inhibitor tablet was added to the dialyzed proteins that were stored at −80 °C.
Purification of GST and GST-Tagged proteins
Pelleted bacteria were suspended in a buffer containing 100 mM Tris-HCl (pH 8.0), 200 mM NaCl, 10% glycerol, plus 1 tablet Complete EDTA-free tablet per 30 ml buffer, 1.0 mg/mL of lysozyme, and frozen at −80 °C. The frozen pellet was thawed for 10 min in a 37 °C bath, and sonicated for 30 sec eight times for lysis of the cells. The homogenate was centrifuged at 20,000 g for 1 h at 4°C, and the supernatant transferred into a 50 mL tube containing 3 ml prewashed Glutathione Sepharose™ 4B resin (GE Healthcare Life Sciences and Laboratories). After 1 h at 4°C the resin was packed into a column, washed with lysis buffer and the bound proteins eluted using 10 mM reduced glutathione (GSH, Sigma Aldrich) in the same buffer. Eluted proteins were dialyzed against 10 mM Tris-HCl (pH 7.4) and Complete EDTA-free tablet was added to the dialyzed proteins that were stored at −80 °C.
Site Directed Mutagenesis
Site directed mutagenesis of HI-2 was performed using the Phusion® Site directed Mutagenesis Kit (New England Biolabs) according to the manufacturer’s protocol. Mutagenic primers were designed using Primer X software available online. The mutations were all verified by automated DNA sequencing in the core facility, University of Virginia School of Medicine.
Protein-protein Binding Assays
The binding of purified I-2 to GST-Pin1 was performed using pull-down assays, quantified by fluorescent immunoblotting. A key feature to reduce background was saturation of beads, accomplished by incubating 10 μl pre-washed glutathione Sepharose beads with excess recombinant GST-Pin1 in binding buffer containing 50 mM HEPES (pH 7.4), 100 mM NaCl, 10 mM MgCl2, 0.5 mM MnCl2, 5 mM EGTA, 1 mg/ml BSA, 0.5 mM DDT and 1 mM Pefabloc (Roche Applied Sciences). The GST-Pin1 beads were pelleted by centrifugation and washed once with the same buffer. Various concentrations of recombinant I-2 proteins were added to the GST-Pin1 beads in a total volume of 1 ml with binding buffer and incubated for 1 h at room temperature. The beads were pelleted and washed 3X by centrifugation with 1 ml of the same buffer, without BSA. Bound proteins were eluted with 2X SDS sample buffer and subjected to 12% SDS-PAGE and Western blotting as described previously (47).
Expression and purification of Pin1 for NMR spectroscopy experiments
The BL21(DE3) E. coli strain was transformed with the pET15b plasmid (Merck) carrying pin1. The recombinant strain was grown at 37°C in a LB medium until A600nm reached about 0.8, then the induction phase was started by addition of 0.5 mM IPTG, for 3 h at 31°C. The cells were harvested by centrifugation and the pellet was resuspended in a lysis buffer [50 mM Na2HPO4 / NaH2PO4 pH 8.0, 300 mM NaCl (Buffer A)], with 10 mM imidazole, 1 mM DTT, 0.1 % NP40 and a protease inhibitor cocktail (Roche)). Cell lysis was performed by incubation with 10 mg lysozyme and 0.5 mg DNase I per liter of culture, followed by a brief sonication, and an extract prepared by centrifugation. The soluble extract was loaded on a nickel affinity column (Chelating Sepharose Fast Flow 5 ml, GE Healthcare). Unbound proteins were washed away with 20 mM imidazole in buffer A and the protein of interest was eluted by 250 mM imidazole in buffer A. The same protocol was performed for the production of 15N-labeled Pin1, with the use of M9 minimal medium complemented with 1 g 15NH4Cl (Cambridge Isotope Laboratories, Cambridge, MA) as nitrogen source, 4 g/L glucose, 1 mM MgSO4, 100 mg/L ampicillin and MEM vitamin cocktail (Sigma). Homogeneous fractions were buffer-exchanged on a PD10 G-25 column (GE Healthcare) equilibrated in the NMR spectroscopy sample buffer (50 mM Tris-d11 pH 6.4, 25 mM NaCl, 1 mM DTT, 1 mM EDTA).
Expression and purification of I-2 for NMR spectroscopy experiments
The BL21(DE3) E. coli strain was transformed with the pET28a plasmid (Merck) encoding HI-2 and grown at 37°C in a LB medium until A600nm reached about 0.8, then 0.5 mM IPTG was added for 16 h at 20°C. The cells were harvested by centrifugation and the pellet was resuspended in a lysis buffer [50 mM Na2HPO4 / NaH2PO4 pH 8.0, 300 mM NaCl (Buffer A)], with added 10 mM imidazole, 1 mM DTT, 0.1 % NP40 and a protease inhibitor cocktail (Roche, Switzerland)). Cell lysis was performed by incubation with 5 mg lysozyme per liter of culture followed by sonication and centrifugation. The soluble extract was incubated at 75°C for 15 min and centrifuged at 15,000 g. The protein in the supernatant was purified on a 1 ml column of Chelating Sepharose Fast Flow (GE Healthcare) following the same procedure as for Pin1. The same protocol was performed for the production of 15N- or 15N/13C-labeled I-2 with the use of M9 minimal medium complemented with 1 g 15NH4Cl (Cambridge Isotope Laboratories, Cambridge, MA) as nitrogen source, 4 g/L glucose or 2 g [13C6]-glucose, respectively, 1 mM MgSO4, 20 mg/L kanamycin and MEM vitamin cocktail (Sigma). The same 1H-15N HSQC spectrum was obtained for I-2, whether the heating step of the soluble extract at 75°C was performed or not during the purification, indicating that heating did not modify the natively unfolded conformation of the I-2 protein. The eluted fractions containing most of the I-2 were pooled and purified by gel filtration (HiLoad 16/60 Superdex75 prep grade, GE Healthcare) in 50 mM ammonium bicarbonate. Fractions were checked on a 12% SDS polyacrylamide gel, pooled and lyophilized before being redissolved in the NMR spectroscopy sample buffer (50 mM Tris-d11 pH 6.4, 25 mM NaCl, 1 mM DTT, 1 mM EDTA) or directly in Pin1 solution for titration.
NMR spectroscopy
Interactions between I-2 and Pin1 were mapped either with 15N-labeled I-2 at 100 μM and unlabeled Pin1 added to reach 0.5, 1, 2 or 12 molar equivalent of Pin1, or 15N-labeled Pin1 at 62.5 μM and unlabeled I-2 as lyophilized aliquots to reach 0.5 or 1 molar equivalent of I-2. Spectra were acquired at 293K or 277K on a 600 or 900 MHz spectrometer equipped with a cryogenic probehead. All 1H spectra were calibrated with 1 mM sodium 3-trimethylsilyl-3,3′,2,2′-d4-propionate as a reference. 1H-15N HSQC spectra were recorded with at least 2048 points in the proton and 256 points in the nitrogen dimensions. The chemical shift perturbations of individual amide resonances in I-2 protein were calculated with the equation (1), taking into account the relative dispersion of the proton and nitrogen chemical shifts (1 ppm and 20 ppm respectively). Instead, a correction coefficient of 0.2 was applied to the nitrogen dimension for the mapping of Pin1 resonances.
| Equation (1) |
Superose 12 Size Exclusion Chromatography
Chromatography on a Superose 12 HR 10/300 column was performed to study Pin1 and I-2 complex formation. The GST tag was cleaved from Pin1 by on-column thrombin digestion using the previously described method (5). Pin1 was concentrated on a centrifugal ultrafilter (10 kDa MWCO, Millipore), and mixed with final concentration of 1.25 μM HI-2 and incubated for 1 h at room temperature. An aliquot (200 μl) was applied to the column and eluted with 50 mM HEPES (pH 7.4), 100 mM NaCl, 10 mM MgCl2, 0.5 mM MnCl2, 5 mM EGTA, 1 mg/ml BSA, 0.5 mM dithiothreitol and 1 mM Pefabloc at a flow rate of 0.6 ml/min. Fractions of 1 min (0.6 ml) were collected and elution profiles developed by dot-blot immunoblotting. Samples (0.2 ml) of individual fractions were adsorbed on nitrocellulose using a 96 well vacuum manifold and developed as immunoblots for I-2 and Pin1. The staining intensities were quantified by fluorescent scanning and plotted into an elution profile.
RESULTS
Binding of inhibitor-2 proteins with Pin1
To define the structural requirements for interaction we compared binding of I-2 proteins from different species: human (HI-2), Xenopus (XI-2) and Drosophila (DI-2), with human GST-Pin1 in a pull-down assay. The recombinant proteins were expressed in bacteria and extensively purified by affinity chromatography, and the final products analyzed by Coomassie staining following SDS-PAGE. (Figure 1A). The GST and GST-Pin1 were detected as single bands by anti-GST immunoblotting (Figure 1B). The binding assay used a range of concentration of added I-2 proteins (0.1 – 5 μM) mixed with GST-Pin1 that was bound to glutathione Sepharose beads. The bound proteins were quantified by fluorescent immunoblotting with species-specific anti-I-2 antibodies. Because the I-2 sequences are so divergent no single anti-I-2 antibody reacts with these three proteins, therefore we prepared and affinity purified species-specific anti-HI-2, anti-XI-2 and anti-DI-2 antibodies for immunoblotting detection (Figure 1C). Staining intensities on immunoblots were quantified and calibrated over a range of concentrations, using recombinant I-2 standards with the absolute amount of the proteins determined by A280nm. The binding of HI-2, X-I-2 and D-I-2 to GST-Pin1 was indistinguishable, with a K0.5 of ~ 0.3 μM, and all showed saturation at 1.25 to 5 μM (Figure 1D), which is well below the estimated cellular concentration of I-2 (43). The total amount of each I-2 protein bound to GST-Pin1 at saturation was identical (Fig. 1E). GST was used as the negative control, and the assay was optimized so there was essentially no I-2 binding to GST beads. This showed that the I-2 proteins were binding to the Pin1 portion of the fusion protein, not to GST itself. These results demonstrated that stable, saturable association with Pin1 is a conserved function of the I-2 family of proteins. Multiple sequence alignment generated for these I-2 proteins revealed the location and clustering of identical residues (boxed in Figure 1F) that are the most probable regions for interaction with human Pin1.
Figure 1. Binding of Human, Xenopus and Drosophila I-2 proteins to GST-Pin1.

(A) Coomassie staining of purified proteins used in binding assays after SDS-PAGE. Left panel: lane 1, molecular size standard proteins; lane 2, human I-2 (HI-2); lane 3, Xenopus I-2 (XI-2); and lane 4, Drosophila I-2 (DI-2). Right panel: lane 1, molecular size standards; lane 2, GST control protein and lane 3, GST-Pin1. (B) Immunoblotting of GST protein and GST-Pin1 using anti-GST antibody. (C) Immunoblotting different I-2 using species-specific antibodies for assay of direct binding of recombinant His6-I-2 proteins to GST-Pin1 in a pull-down assay, as described in Materials and Methods. (D) Concentration dependence for I-2 from different species. Symbols: diamonds, HI-2; squares, XI-2; triangles, DI-2. (E) Quantification of the amount of different I-2 proteins bound to GST-Pin1, relative to recombinant protein standards. (F) Sequence alignment for human I-2 (HI-2), Xenopus I-2 (XI-2) and Drosophila I-2 (DI-2) using ClustalW 2.1. Conserved residues are marked with an asterisk and a box drawn around them. Polyacidic region shown in boldface and underline. HI-2 is 60% identical to XI-2 and 40% identical to DI-2.
Association of HI-2 with phospho-Pin1 and separate Pin1 domains
Binding of HI-2 to Pin1 used proteins expressed in bacteria, therefore they were both devoid of phosphorylations that occur in live animal cells, such as phosphorylation of Ser16 in the Pin1 WW domain (28). We reacted full-length GST-Pin1 with purified PKA and showed phosphorylation of Ser16 by immunoblotting with a phosphosite-specific antibody (Figure 2A). Binding of HI-2 to the PKA phosphorylated GST-Pin1 was decreased only slightly compared to the binding of HI-2 with non-phosphorylated GST-Pin1 (Figure 2A, D). These results show that phosphorylation of Pin1 by PKA, involving at least Ser16, did not affect the formation of a protein-protein complex with HI-2.
Figure 2. Comparison of I-2 binding to Pin1, phospho-Pin1, and separate WW and isomerase domains.
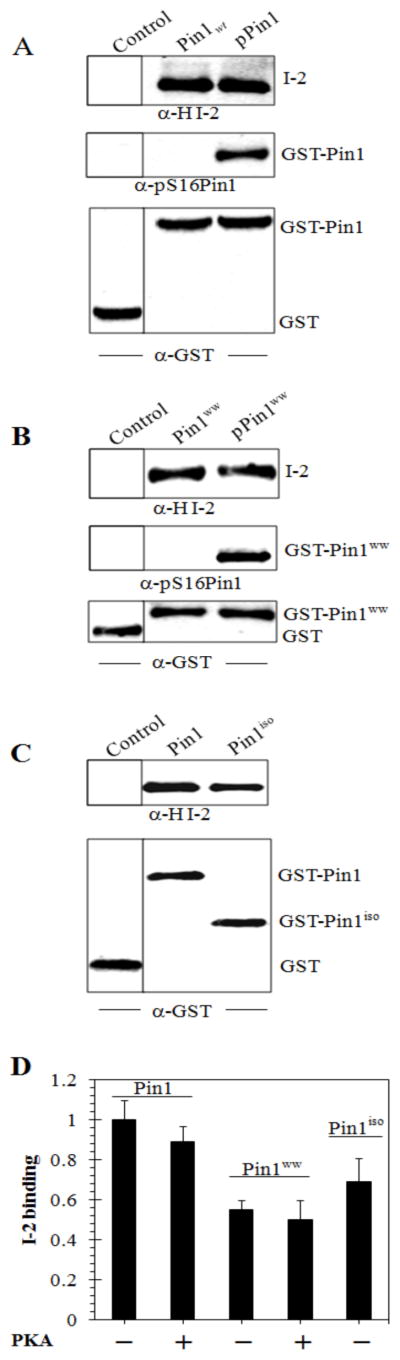
GST-Pin and GST-Pinww and their PKA phosphorylated forms, and GST-Pin1iso were prepared as described in Materials & Methods. (A) Glutathione Sepharose beads were saturated with GST (Control), wild-type GST-Pin1 (Pin1wt), or phosphorylated GST-Pin1 (pPin1) and pull down assay with human I-2 protein (1.25 μM) done as described in Materials & Methods. Proteins were eluted and immunoblotted with anti-human I-2 (top panel), anti-phosphoS16Pin1 (center panel) and anti-GST (lower panel) antibodies. (B) GST (Control), GST-Pin1ww (Pin1ww), and PKA phosphorylated GST-Pin1ww (pPin1ww) were incubated for 1 h in with HI-2 protein and processed as in (A). (C) Direct binding of H I-2 to GST (Control, left lane), GST-Pin1 (center lane) and GST-Pin1 isomerase domain (right lane). Upper panel immunoblots for bound H I-2 and lower panel immunoblots for GST as loading control. (D) Quantification of pull down assays depicted in A–C, showing average values and SD for three independent experiments.
We expressed the N-terminal WW domain of Pin1 (Pin1ww) and C terminal isomerase catalytic domain (Pin1iso) fused to GST and used them in pull down assays with purified HI-2. The amount of HI-2 bound to the Pin1ww was about half the level that bound to Pin1 (Figure 2B, D). Phosphorylation of the Pin1ww by PKA was demonstrated by immunoblotting with the pSer16 phosphosite antibody (Figure 2B). Phosphorylation of the Pin1ww domain resulted in no difference in binding of HI-2 in the pull down assay, reinforcing the results with full length Pin1. These results showed that the WW domain of Pin1 alone was sufficient for interaction with HI-2, albeit at about half the level relative to the full length Pin1. Assays with the isomerase domain of Pin1 (Piniso) revealed that the amount of HI-2 bound was ~30% lower compared to full length Pin1 (Figure 2C, D), indicating that the isomerase domain also was sufficient for association with I-2. The results indicate that I-2 independently associates with both the WW domain and the isomerase domain of Pin1.
NMR spectroscopy analysis of I-2 association with Pin1
Pin1 and I-2 proteins were separately 15N-labelled by expression in bacteria, purified, and their interaction in solution investigated by NMR spectroscopy. The 2D [15N-1H] Heteronuclear Single Quantum Coherence (HSQC) spectra of 100 uM 15N-labelled Pin1 alone and mixed with purified unlabeled HI-2 were recorded and super imposed. The spectrum of Pin1 alone (black) and after the addition of 0.5 (red) and 1.0 (blue) molar equivalents of HI-2 showed weak but reproducible shifts of selected resonances in Pin1 (Figure 3A), indicative of physical contacts with HI-2. To delineate the HI-2 binding sites on Pin1, the residues with induced chemical shift perturbations were mapped on the Pin1 3D structure from PDB (PDID: 1PIN), depicted front and back in Figure 3B. Cross-peak movements that correspond to residues with combined chemical shift variations greater than 0.04 ppm (shown in red), in the 0.02–0.04 ppm range (orange) and in the 0.015–0.02 ppm range (yellow) mapped residues in contiguous areas on the surfaces of both the WW domain and catalytic domains of Pin1. The structure is rotated 180 degrees to view the opposite side, where relatively fewer interactions appeared. These images support our results in binding assays that I-2 interacts with the separate, isolated Pin1ww and Pin1iso domains. The contacts of I-2 with the WW domain involved R17, S18 and S32, plus several hydrophobic side chains, including V22, F25 and W34. On the other hand, interactions with the isomerase catalytic domain were dominated by charged side chains, K77, R68, D153 and K132. The I-2 contact surface on Pin1 catalytic domain includes the putative active site residue R68, but does not appear to occlude the active site so as to interfere with substrate binding. This is consistent with our previous report that I-2 allosterically modifies Pin1 specificity.
Figure 3. NMR spectroscopy mapping of surface residues in Pin1 that contact I-2.

(A) The 1H-15N HSQC spectrum of 15N-labeled Pin1 alone (black) overlaid with spectra after addition of 0.5 molar equivalent (red) and 1.0 molar equivalent (blue) of unlabelled I-2 showing the shifts of resonances, marked with single letter code and residue number. Zoom-in areas are enlarged to the left side for closer examination. (B) Mapping of the I-2 interaction sites on Pin1 structure (PDB ID: 1PIN). The isomerase domain (Pin1CAT) is to the upper left and the WW domain (Pin1ww) to the lower right side. The structure on the right is rotated 180° to show the reverse side. Residues with combined chemical shift variations (calculated as described in Methods) greater than 0.04 ppm are colored red, those in the 0.02 – 0.04 ppm range colored orange, and those in the 0.015 – 0.02 ppm range colored yellow.
Reciprocal labeling experiments involved the titration of purified 15N-labelled HI-2 with unlabeled Pin1, which were done under different conditions (both 4°C and 20°C) and different ratios of the proteins (from 0.5 to 12 mol/mol). The HSQC spectra of HI-2 (Figure 4A) in the presence of increasing amounts of Pin1 were superimposed over the spectrum of HI-2 alone (black) to show significant perturbations that correspond to residues in HI-2 in physical contact with Pin1. HI-2 is known to be an extended polypeptide in solution and other than one short helical region is devoid of secondary or tertiary structure, so the results could not be mapped on a 3D structure as for Pin1 (see above) and instead we plotted shifts as a function of sequence position in HI-2 (Figure 4B). This analysis reveals multiple regions in I-2 appear to be in contact with Pin1. These include the N and C termini, residues 85–95 and 105–125. The residue 60–85 region was so broadened by addition of excess Pin1 the delta could not be calculated. These relatively small variations were reproducible under different conditions, and our interpretation was that the extreme flexibility of I-2 did not allow for larger changes. These NMR spectroscopy results with labeled I-2 show binding with Pin1 involved multiple sites of protein-protein interaction.
Figure 4. NMR spectroscopy mapping of I-2 residues that contact Pin1.

(A) The complete (upper panel) and an enlarged zoomed area (lower panel) of the 1H-15N HSQC spectrum of 15N-labeled I-2 alone (black) overlaid with spectra recorded after addition of 0.5 (red), 1.0 (green), 2.0 (blue) and 12 (magenta) molar equivalents of unlabeled Pin1, recorded at 4°C showing the shifts of specific residues. (B) Graphical representation of the chemical shift variations in HDSQ spectrum of I-2 resonances (Δδ) upon addition of excess Pin1, as done in (A).
Defining regions in HI-2 involved in binding to Pin1
To further dissect the structure to function relationship for I-2 we produced truncated and mutated forms of HI-2 and assayed binding to GST-Pin1 (Figure 5). The HI-2 variants used were: full length wild type I-2(WT); C-terminal truncated I-2(1–197); substitution of a conserved aliphatic side chain, I-2(I68A); deletion of both N- and C-terminal regions, I-2 (14–197); and larger deletions of C terminus, I-2(1–152) and I-2(1–119). Recombinant I-2 proteins were affinity purified and assayed at a final concentration of 1.25 μM. The results (Figure 5A) showed no significant difference between the binding of I-2(205) and I-2(197), showing that the extreme C terminal region was not required for HI-2 association with GST-Pin1. In contrast, truncation of the N terminal 13 residues that includes the SILK motif common to many PP1 binding proteins reduced by half the binding of I-2 to Pin1. Further truncation of I-2 from the C terminus to residue 152, or residue 119, resulted in a significant 75% loss of binding to GST-Pin1. These results reinforced the NMR spectroscopy assignment of a site in I-2 encompassing residues 170–200 as strongly interacting with Pin1. We suspect involvement of an uninterrupted stretch of acidic residues, which is a conserved feature among I-2 proteins that does not align exactly in the same residue positions. It is important to note here that 1–152 was tested because its inhibitory potency for PP1C is identical to full length I-2 (48).
Figure 5. Binding of I-2 variants to GST-Pin1.
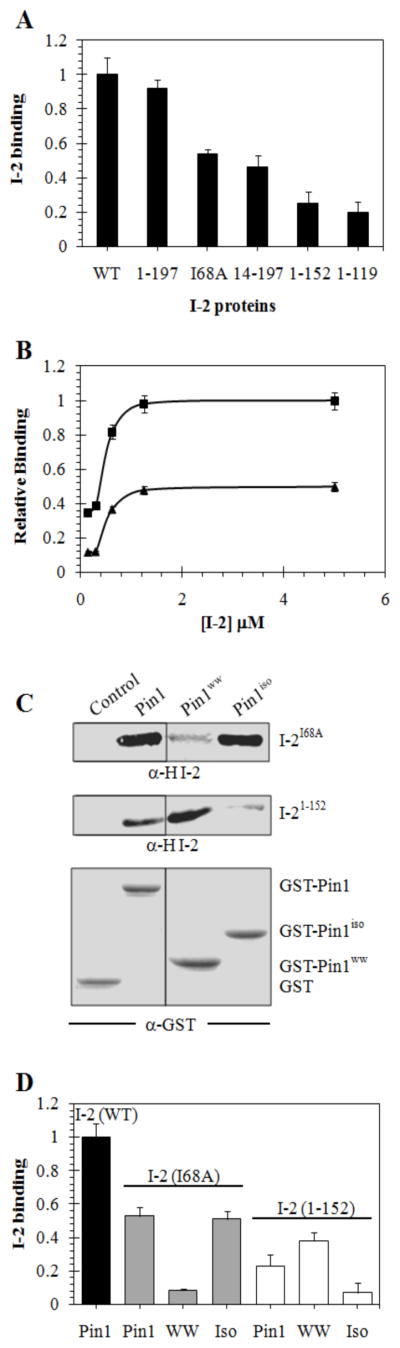
(A) Binding of wild type (WT) and truncated and substituted HI-2 to GST-Pin1 in pull down assay as described in Materials & Methods. The recovery of bound I-2 was quantitated using Odyssey software V1.2 by immunoblotting, with correction for amount of GST-Pin1 by simultaneous dual wavelength scanning of two different secondary antibodies on the same filter (Odyssey, Li-Cor Industries). Values from three independent experiments were normalized to WT, averaged, and plotted with S.D. (B) Dose response for binding of wt HI-2 (squares) and I-2(I68A) (triangles) to GST-Pin1 in pull down assay (n=3). Immunoblotting analyzed as described in (A) and data plotted with S.D. using Sigma plot 10.0 software. (C and D) Pull down assays for binding of 1.25 μM either HI-2(I68A) [I-2I68A] (upper panels in C, grey bars in D) or HI-2(1–152) [I-21–152] (center panels in C, open bars in D) with GST, GST-Pin1 [Pin1], GST-Pin1ww [Pin1ww, WW], or GST-Pin1iso [Pin1iso, Iso]. Immunoblotting for GST and fusion proteins is shown in C, lower panel. For control and normalization of other samples HI-2 wild type (WT) was pulled down with GST-Pin1. Data (n=3) analyzed as described in (A).
We had reported association of I-2 with Pin1 was exceedingly sensitive to disruption by detergents (39), presumably due to hydrophobic interaction between the proteins. The highly conserved 60–85 region of I-2 is likely engaged with Pin1 based on results of NMR spectroscopy. Within this region most of the conserved residues are charged, with the exception of Ile68, which is a strictly conserved, aliphatic residue that we suspected could be involved in a hydrophobic protein-protein interface. Therefore, we produced an I68A substituted form of I-2 and found its binding with Pin1 was reduced to half that of wild type HI-2 when both proteins were compared at 1.25 μM (Figure 5A). We assayed for binding to Pin1 over a range of concentrations and found a reduced the slope in the initial portion (< 1 μM) of the saturation curve (Figure 5B). However, more provocative was the observation that when binding of I-2(I68A) reached saturation at about the same concentration as wild type I-2, there was only 50% of the amount of I-2 protein bound. This suggested to us that there were two binding sites for wild type I-2 binding to Pin1, and I-2(I68A) bound to only one of them.
We compared binding of I-2, I-2(I68A) and I-2(152) to full length Pin1 and Pin1ww and Pin1iso in pull down assays (Figure 5C,D). Mutation of the single residue I68A resulted in essentially a complete loss of binding to the WW domain, whereas the binding to the Pin1 isomerase domain was the same as binding to full length Pin1. Our conclusion was that I68 was critical for binding to the WW domain, but was not required for and did not much affect I-2 binding to the isomerase domain of Pin1. On the other hand, truncation of I-2 to 152 residues essentially eliminated binding to the isomerase domain, while I-2(152) bound to full length Pin1 and Pin1ww. Our hypothesis was that each domain of Pin1 bound separate regions of I-2, and Pin1 bound to two I-2 molecules, one to each domain.
Analysis of HI-2–Pin1 complexes by size exclusion chromatography
We tested our hypothesis using Superose 12 column chromatography to analyze the complexes formed between His6-HI-2 and Pin1 (Figure 6). This was a homogeneous solution assay that involved simple mixing of soluble components, compared to the heterogeneous assay that used a GST fusion protein immobilized on beads. Pin1 protein was recovered free of GST by on-column thrombin cleavage of the GST-Pin1 fusion protein purified from bacteria (see inset, Figure 6D). This preparation of Pin1 eluted as a symmetrical peak centered at 15.6 ml (dashed line, Figure 6D), corresponding to monomeric Pin1 (Figure 6A–D). Purified I-2 eluted from Superose 12 as a peak centered at 13.2 ml (dotted line, Figure 6A–D). Due to its extended and unstructured conformation I-2 has been known to behave as a larger molecule in solution and indeed I-2 eluted before ovalbumin (43 kDa, 13.8 ml, Figure 6E) in size exclusion chromatography. When recombinant I-2 was mixed with Pin1 and the sample resolved on Superose 12, immunoblotting of the fractions for I-2 revealed two prominent peaks: a major symmetrical peak centered at 10.2 ml that contained about 60% of the total I-2 protein, and a second peak at 11.4 ml, corresponding to unbound monomeric I-2 (Figure 6A). Immunoblotting of the fractions for Pin1 showed exact co-elution of Pin1 with I-2 in the peak at 10.2 ml and also a second peak at 15.6 ml, corresponding to free monomeric Pin1. These results showed the formation of a stable, large complex between purified Pin1 and I-2 that eluted at a size larger than catalase (260 kDa, 10.8 ml, Figure 6E). On the other hand, when the I68A mutant of I-2 was mixed with Pin1 the complex that formed eluted at 11.4 ml (Figure 6B). This complex is smaller than catalase, but still larger than immunoglobulin (150 kDa, 12.0 ml, Figure 6E) and corresponds to about half the relative size of the wild type I-2:Pin1 complex. The distribution of I-2 protein between the Pin1 complexes and the free monomer was about the same for I-2(I68A) and for wild type I-2 (Figure 6A and 6B), and our interpretation was that these different I-2 proteins had about the same affinity for Pin1. Analysis of I-2(152) binding to Pin1 showed a very low yield of complex between the proteins in solution, with most of the proteins eluted separately as monomers (Figure 6C). What little complex was formed eluted at 11.4 ml, not 10.2 ml. The excluded volume (V0) of this column is 9 ml thus the complexes formed were discrete molecular species and not simply polymers. Because the complexes eluting at 10.2 ml and 11.4 ml both contained equivalent amounts of I-2 and Pin1, we concluded that these peaks represented tetrameric (Pin1)2(I-2)2 and dimeric (Pin1)(I-2) complexes, respectively.
Figure 6. Size Exclusion chromatography analysis of Pin1 and I-2 complexes.


Recombinant GST-Pin1 was cleaved by thrombin, and the Pin1 protein concentrated, incubated with recombinant HI-2 and applied to a Superose 12 HR 10/300 column, as described under Materials & Methods. Aliquots of the eluted fractions were immunoblotted with anti-Pin1 (dashed lines) and anti-human I-2 (dotted lines) antibodies using dot blot technique with a 96 well vacuum manifold. (A) Elution profile for mixture of Pin1 and wild type HI-2. (B) Elution profile for mixture of Pin1 and I68A substituted form of HI-2. (C) Elution profile for mixture of Pin1 and truncated form of HI-2(1–152). (D) Superimposition of separate elution profiles for Pin1 and wild type HI-2. Inset is image of Coomassie stained SDS-PAGE of lane 1, molecular size standards; lane 2, the purified Pin1; and lane 3 purified HI-2 protein used in these experiments. (E) Superimposition of separate elution profiles for purified proteins used as molecular size standards: catalase (260 kDa, 10.8 ml), immunoglobulin (150 kDa, 12.0 ml), and ovalbumin (43 kDa, 13.8 ml).
DISCUSSION
The results of this study reveal molecular interactions between two ancient and essential proteins, Pin1 and I-2. Purified recombinant I-2 proteins from divergent species exhibited saturable binding to Pin1 with sub-micromolar affinity. Estimates for the intracellular concentrations of Pin1 and I-2 are micromolar (43, 49), which would predict that the proteins are complexed together in living cells and tissues. Our results argue that binding to Pin1 is an evolutionarily conserved function and our hypothesis was that interactions involved regions of the I-2 protein identical in sequence among the different species. Indeed, NMR spectroscopy of reciprocally labeled proteins dynamically interacting in solution showed that multiple conserved sequence regions in I-2 were in contact with surface sites on Pin1, in both the WW and isomerase domains. One key conclusion from the NMR spectroscopy is that the I-2 interactions mapped on the surfaces of Pin1 do no occlude the active site. This supports the concept that I-2 does not interfere with substrate binding but acts as an allosteric modulator of Pin1 substrate specificity, as we previously demonstrated using a panel of mitotic phosphoproteins (39).
Mapping the sites of I-2 interaction on Pin1 by NMR spectroscopy revealed surface regions on the WW domain and on the isomerase domain. Residues in the WW domain that contacted I-2 did not include Ser16, consistent with unimpeded I-2 binding to PKA phosphorylated Pin1. Phosphorylation by PKA involves at least Ser16 and this phosphoryl group is thought to preclude the binding of phosphorylated substrates to the WW domain (28). Binding of I–2 was insensitive to PKA phosphorylation of either the full-length Pin1 protein or the WW domain, so the phosphoryl group at Ser16 did not exert steric or electrostatic constraints on association with I-2. These results are important in envisioning a model for the complex between Pin1 and I-2 and potentially will influence understanding how signaling events regulate Pin1 function. The NMR spectroscopy analysis also identified several hydrophobic residues in the Pin1 WW domain that contacted I-2. Pin1 was already known to elaborate a hydrophobic surface on the WW domain that faces the isomerase domain. This forms a hydrophobic cleft that binds polyethylene glycol during crystallization, as visualized in the 3-D structure determined by X-ray crystallography (6). There are relatively few conserved hydrophobic residues in I-2 that would be available for generating an aliphatic protein-protein interface with Pin1. Searching among the conserved sequence regions in I-2 that NMR spectroscopy identified as Pin1 contact sites, we selected Ile68 as a likely candidate. The I68A substitution was a relatively minor modification in the entire 205 residue protein, but had profound effects on binding to Pin1. Binding was reduced about 50% but more important, the amount of I-2(I68A) bound at saturating concentrations was only half as much as wild type. This was unexpected, and forced us to consider there were two binding sites for I-2 on Pin1, only one of which bound I-2(I68A).
This idea was borne out by assay of I-2 binding to Pin1 and the separate WW and isomerase domains. The I68A mutation essentially eliminated I-2 binding to the isolated WW domain, but did not equally impair binding to the Pin1 isomerase domain. Based on this we assign the I68 and surrounding 45–80 region of I-2 as the part of the protein engaged with the WW domain of Pin1. On the other hand, truncation of I-2 to residue 152 eliminates the Pin1 interacting site at residue 170 seen by NMR spectroscopy, which we suspect is a long uninterrupted stretch of acidic residues that appears as a conserved feature among I-2 from different species (boldface and underlined in Figure 1). The I-2(1–152) had reduced binding to Pin1, however, the level of binding to Pin1 and its isolated WW domain was about the same, whereas there was almost no binding to the isomerase domain. Our interpretation of these results is that the polyacidic site in I-2 probably interacts with the multiple adjacent basic residues across the surface of the isomerase domain in Pin1. Assays with mutated I-2 and Pin1 separate domains were consistent with the results of mapping with NMR spectroscopy, and allowed us to assign interactions of different I-2 regions to individual Pin1 domains.
The results led us to consider the idea that if two molecules of I-2 were bound to a single Pin1, at different sites, in the two domains, then it is possible that a second molecule of Pin1 could contact these same two I-2 proteins to form a sandwich, with the Pin1 proteins positioned opposite one another bridged together by two molecules of I-2 that are predominantly unstructured and extended (Figure 7). The [Pin1]2[I-2]2 heterotetramer is proposed as the native active form of Pin1. Bringing two molecules of Pin1 together positions two phosphoSer/Thr binding sites in proximity, and the involvement of I-2 in the complex generates a composite protein surface surrounding the Pin1 isomerase domain, which is predicted to dictate the substrate specificity. This tetramer represents quite a different target for drug development compared to monomeric Pin1.
Figure 7. Model for Pin1 and I-2 complex.
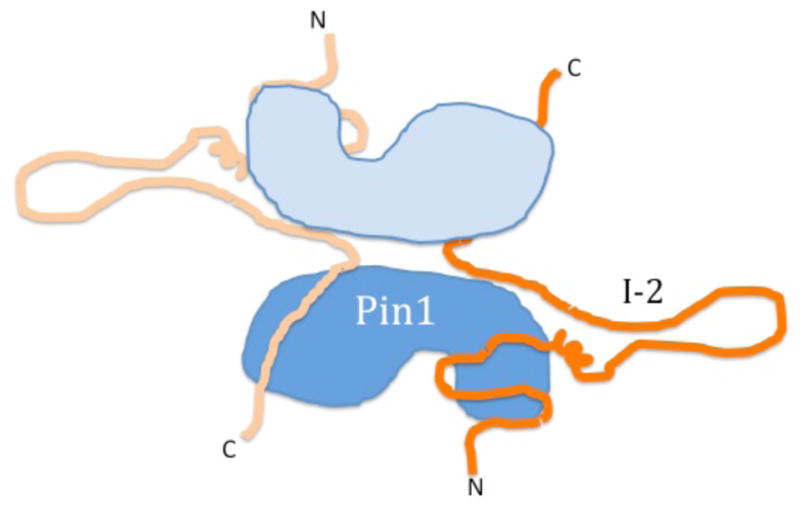
A cartoon to show arrangement of two molecules of Pin1 with two molecules of I-2 in a complex formed in solution. Pin1 is depicted in light and dark blue as two lobes, composed of a smaller WW domain and a larger isomerase catalytic domain. I-2 is depicted in orange as an unstructured polypeptide with N and C termini labeled, using multiple regions for binding to separate domains of Pin1.
Furthermore, our model accommodates phosphorylation as a means of Pin1 regulation. While PKA phosphorylation of S16 in Pin1 does not affect association with I-2, this phosphorylation of two sites on the surface of the [Pin1]2[I-2]2 complex could serve to alter binding to different substrates. Phosphorylation of Thr72 in I-2, which is catalyzed by CDK/cyclinB during mitosis (36), prevents I-2 binding to Pin1 (39). Therefore, we predict upon entry into mitosis phosphorylation of I-2 in the [Pin1]2[I-2]2 complex will cause dissociation into separate components, releasing monomeric Pin1 to interact with mitotic phosphoproteins. Mitotic exit will involve dephosphorylation of I-2 and reassembly with Pin1 into the heterotetramer. This model implies dynamic regulation of Pin1 complexes that would affect substrate specificity at different stages of mitosis.
We note that I-2 residues 95–105 and 120–135 are engaged by Pin1. For I-2 bound to PP1C the 95–105 segment is looped out and does not form contacts with PP1C, and NMR spectroscopy shows some tendency for this region to be in an alpha-helix (50). In a recent publication it was speculated that this putative helix could possibly contact another I-2 partner, such as Aurora A or Pin1 (51). This would fit with our results. The 120–135 region we see affected by Pin1 only partly overlaps with the 130–142 region of I-2 that is 70% populated in a helix conformation, as observed by NMR spectroscopy (50). The 130–142 segment of I-2 was visualized as a helix bound across the PP1C active site in the co-crystal structure, with only 25% of the I-2 showing discernable electron density (52). Even when in complex with its primary partner PP1C, most of I-2 is thought to remain as flexible as it is when alone in solution. Overall, we are impressed that our NMR spectroscopy results and those from Peti and collaborators map different I-2 residues interacting with Pin1 compared to those that contact PP1C monomer, or PP1C complexed to neurabin (51). Although different regions of I-2 are used in these various complexes, we do not imagine this implies assembly of supercomplexes with multiple partners, but rather assembly of I-2 into multiple complexes with different partners. Our previous research has revealed that, in addition to PP1C, I-2 forms complexes with neurabin (53), KPI-2 (54) (a.k.a. LMKT2, BREK), Aurora A (44) and, of course, Pin1 (39). This emphasizes the possibilities available to an unstructured protein to employ various sequence segments to engage different protein partners. The ancient and conserved function of I-2 involves a web of protein-protein interactions and we speculate binding to Pin1 may be as ancient, and fundamentally important, as binding to PP1, the property for which I-2 was originally named.
Acknowledgments
This work was supported by NIH grant GM56362 (to DLB) and by CNRS (to GL). The NMR spectroscopy facility was supported by the TGE RMN THC (FR-3050, France).
We thank Drs. Jean-Michel Wieruszesk and Bernd Fritzinge for their support in recording the NMR spectra. We dedicate the article to the memory of Professor Emanuel Margoliash, a pioneer in the use of homologous proteins from various species to provide insights into structure and function.
Abbreviations
- GST
glutathione S-transferase
- HI-2
human phosphatase inhibitor-2
- XI-2
Xenopus phosphatase inhibitor-2
- DI-2
Drosophila phosphatase inhibitor-2
- Pin1ww
Human Pin1 WW domain (residues 1–44)
- Pin1iso or Pin1cat
Pin1 isomerase domain (residues 46–163)
- HSQC
Heteronuclear Single Quantum Coherence
References
- 1.Shen M, Stukenberg PT, Kirschner MW, Lu KP. The essential mitotic peptidyl-prolyl isomerase Pin1 binds and regulates mitosis-specific phosphoproteins. Genes Dev. 1998;12:706–720. doi: 10.1101/gad.12.5.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schutkowski M, Bernhardt A, Zhou XZ, Shen M, Reimer U, Rahfeld JU, Lu KP, Fischer G. Role of phosphorylation in determining the backbone dynamics of the serine/threonine-proline motif and Pin1 substrate recognition. Biochemistry. 1998;37:5566–5575. doi: 10.1021/bi973060z. [DOI] [PubMed] [Google Scholar]
- 3.Lu KP, Hanes SD, Hunter T. A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature. 1996;380:544–547. doi: 10.1038/380544a0. [DOI] [PubMed] [Google Scholar]
- 4.Hanes SD, Shank PR, Bostian KA. Sequence and mutational analysis of ESS1, a gene essential for growth in Saccharomyces cerevisiae. Yeast. 1989;5:55–72. doi: 10.1002/yea.320050108. [DOI] [PubMed] [Google Scholar]
- 5.Maleszka R, Lupas A, Hanes SD, Miklos GL. The dodo gene family encodes a novel protein involved in signal transduction and protein folding. Gene. 1997;203:89–93. doi: 10.1016/s0378-1119(97)00522-2. [DOI] [PubMed] [Google Scholar]
- 6.Ranganathan R, Lu KP, Hunter T, Noel JP. Structural and functional analysis of the mitotic rotamase Pin1 suggests substrate recognition is phosphorylation dependent. Cell. 1997;89:875–886. doi: 10.1016/s0092-8674(00)80273-1. [DOI] [PubMed] [Google Scholar]
- 7.Bayer E, Goettsch S, Mueller JW, Griewel B, Guiberman E, Mayr LM, Bayer P. Structural analysis of the mitotic regulator hPin1 in solution: insights into domain architecture and substrate binding. J Biol Chem. 2003;278:26183–26193. doi: 10.1074/jbc.M300721200. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Wang S, Zhu T, Zhou J, Xu Q, Lu Y, Ma D. Pin1 contributes to cervical tumorigenesis by regulating cyclin D1 expression. Oncol Rep. 2006;16:491–496. [PubMed] [Google Scholar]
- 9.Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, Lu KP. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. Embo J. 2001;20:3459–3472. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, Hahn WC, Stukenberg PT, Shenolikar S, Uchida T, Counter CM, Nevins JR, Means AR, Sears R. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–318. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 11.Wulf GM, Liou YC, Ryo A, Lee SW, Lu KP. Role of Pin1 in the regulation of p53 stability and p21 transactivation, and cell cycle checkpoints in response to DNA damage. J Biol Chem. 2002;277:47976–47979. doi: 10.1074/jbc.C200538200. [DOI] [PubMed] [Google Scholar]
- 12.Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G, Gu L, Tang X, Lu KP, Xiao ZX. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature. 2002;419:849–853. doi: 10.1038/nature01116. [DOI] [PubMed] [Google Scholar]
- 13.Hamdane M, Dourlen P, Bretteville A, Sambo AV, Ferreira S, Ando K, Kerdraon O, Begard S, Geay L, Lippens G, Sergeant N, Delacourte A, Maurage CA, Galas MC, Buee L. Pin1 allows for differential Tau dephosphorylation in neuronal cells. Mol Cell Neurosci. 2006;32:155–160. doi: 10.1016/j.mcn.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Lim J, Lu KP. Pinning down phosphorylated tau and tauopathies. Biochim Biophys Acta. 2005;1739:311–322. doi: 10.1016/j.bbadis.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, Stoller G, Kullertz G, Stark M, Fischer G, Lu KP. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Mol Cell. 2000;6:873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 16.Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, Wheeler TM, Lu KP, Bao L. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244–6251. [PubMed] [Google Scholar]
- 17.Wulf G, Ryo A, Liou YC, Lu KP. The prolyl isomerase Pin1 in breast development and cancer. Breast Cancer Res. 2003;5:76–82. doi: 10.1186/bcr572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–1737. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim CJ, Cho YG, Park YG, Nam SW, Kim SY, Lee SH, Yoo NJ, Lee JY, Park WS. Pin1 overexpression in colorectal cancer and its correlation with aberrant beta-catenin expression. World J Gastroenterol. 2005;11:5006–5009. doi: 10.3748/wjg.v11.i32.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou CX, Gao Y. Aberrant expression of beta-catenin, Pin1 and cylin D1 in salivary adenoid cystic carcinoma: relation to tumor proliferation and metastasis. Oncol Rep. 2006;16:505–511. [PubMed] [Google Scholar]
- 21.Ryo A, Liou YC, Lu KP, Wulf G. Prolyl isomerase Pin1: a catalyst for oncogenesis and a potential therapeutic target in cancer. J Cell Sci. 2003;116:773–783. doi: 10.1242/jcs.00276. [DOI] [PubMed] [Google Scholar]
- 22.Lu KP. Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell. 2003;4:175–180. doi: 10.1016/s1535-6108(03)00218-6. [DOI] [PubMed] [Google Scholar]
- 23.Wildemann D, Erdmann F, Alvarez BH, Stoller G, Zhou XZ, Fanghanel J, Schutkowski M, Lu KP, Fischer G. Nanomolar inhibitors of the peptidyl prolyl cis/trans isomerase Pin1 from combinatorial peptide libraries. J Med Chem. 2006;49:2147–2150. doi: 10.1021/jm060036n. [DOI] [PubMed] [Google Scholar]
- 24.Hennig L, Christner C, Kipping M, Schelbert B, Rucknagel KP, Grabley S, Kullertz G, Fischer G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry. 1998;37:5953–5960. doi: 10.1021/bi973162p. [DOI] [PubMed] [Google Scholar]
- 25.Xu GG, Etzkorn FA. Pin1 as an anticancer drug target. Drug news & perspectives. 2009;22:399–407. doi: 10.1358/dnp.2009.22.7.1381751. [DOI] [PubMed] [Google Scholar]
- 26.Zhao S, Etzkorn FA. A phosphorylated prodrug for the inhibition of Pin1. Bioorganic & medicinal chemistry letters. 2007;17:6615–6618. doi: 10.1016/j.bmcl.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science (New York, N Y. 1997;278:1957–1960. doi: 10.1126/science.278.5345.1957. [DOI] [PubMed] [Google Scholar]
- 28.Lu PJ, Zhou XZ, Liou YC, Noel JP, Lu KP. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. J Biol Chem. 2002;277:2381–2384. doi: 10.1074/jbc.C100228200. [DOI] [PubMed] [Google Scholar]
- 29.Lippens G, Landrieu I, Smet C. Molecular mechanisms of the phospho-dependent prolyl cis/trans isomerase Pin1. Febs J. 2007;274:5211–5222. doi: 10.1111/j.1742-4658.2007.06057.x. [DOI] [PubMed] [Google Scholar]
- 30.Huang FL, Glinsmann WH. Separation and characterization of two phosphorylase phosphatase inhibitors from rabbit skeletal muscle. Euro J Biochem. 1976;70:419–426. doi: 10.1111/j.1432-1033.1976.tb11032.x. [DOI] [PubMed] [Google Scholar]
- 31.Ingebritsen TW, Cohen P. The protein phosphatases involved in cellular regulation. 1. Classification and substrate specificities. Eur J Biochem. 1983;132:255–261. doi: 10.1111/j.1432-1033.1983.tb07357.x. [DOI] [PubMed] [Google Scholar]
- 32.Li M, Satinover DL, Brautigan DL. Phosphorylation and functions of inhibitor-2 family of proteins. Biochemistry. 2007;46:2380–2389. doi: 10.1021/bi602369m. [DOI] [PubMed] [Google Scholar]
- 33.Ballou LM, Fischer EH. Phosphoprotein Phosphatases. In: Krebs PDBaEG., editor. The Enzymes. Academic Press, Inc; Orlando: 1986. pp. 311–361. [Google Scholar]
- 34.Hemmings BA, Resink TJ, Cohen P. Reconstitution of a Mg-ATP-dependent protein phosphatase and its activation through a phosphorylation mechanism. FEBS Lett. 1982;150:319–324. doi: 10.1016/0014-5793(82)80760-6. [DOI] [PubMed] [Google Scholar]
- 35.Puntoni F, Villa-Moruzzi E. Phosphorylation of the inhibitor-2 of protein phosphatase-1 by cdc2-cyclin B and GSK3. Biochem & Biophys Res Commun. 1995;207:732–739. doi: 10.1006/bbrc.1995.1248. [DOI] [PubMed] [Google Scholar]
- 36.Li M, Stefansson B, Wang W, Schaefer EM, Brautigan DL. Phosphorylation of the Pro-X-Thr-Pro site in phosphatase inhibitor-2 by cyclin-dependent protein kinase during M-phase of the cell cycle. Cell Signal. 2006;18:1318–1326. doi: 10.1016/j.cellsig.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 37.Wang QM, Guan KL, Roach PJ, DePaoli-Roach AA. Phosphorylation and activation of the ATP-Mg-dependent protein phosphatase by the mitogen-activated protein kinase. J Biol Chem. 1995;270:18352–18358. doi: 10.1074/jbc.270.31.18352. [DOI] [PubMed] [Google Scholar]
- 38.Leach C, Shenolikar S, Brautigan DL. Phosphorylation of phosphatase inhibitor-2 at centrosomes during mitosis. J Biol Chem. 2003;278:26015–26020. doi: 10.1074/jbc.M300782200. [DOI] [PubMed] [Google Scholar]
- 39.Li M, Stukenberg PT, Brautigan DL. Binding of phosphatase inhibitor-2 to prolyl isomerase Pin1 modifies specificity for mitotic phosphoproteins. Biochemistry. 2008;47:292–300. doi: 10.1021/bi701819k. [DOI] [PubMed] [Google Scholar]
- 40.Eto M, Elliott E, Prickett TD, Brautigan DL. Inhibitor-2 regulates protein phosphatase-1 complexed with NimA-related kinase to induce centrosome separation. J Biol Chem. 2002;277:44013–44020. doi: 10.1074/jbc.M208035200. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, Brautigan DL. Phosphatase inhibitor 2 promotes acetylation of tubulin in the primary cilium of human retinal epithelial cells. BMC Cell Biol. 2008;9:62. doi: 10.1186/1471-2121-9-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang W, Cronmiller C, Brautigan DL. Maternal phosphatase inhibitor-2 is required for proper chromosome segregation and mitotic synchrony during Drosophila embryogenesis. Genetics. 2008;179:1823–1833. doi: 10.1534/genetics.108.091959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang W, Stukenberg PT, Brautigan DL. Phosphatase inhibitor-2 balances protein phosphatase 1 and aurora B kinase for chromosome segregation and cytokinesis in human retinal epithelial cells. Mol Biol Cell. 2008;19:4852–4862. doi: 10.1091/mbc.E08-05-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Satinover DL, Leach CA, Stukenberg PT, Brautigan DL. Activation of Aurora-A kinase by protein phosphatase inhibitor-2, a bifunctional signaling protein. Proc Natl Acad Sci U S A. 2004;101:8625–8630. doi: 10.1073/pnas.0402966101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Satinover DL, Brautigan DL, Stukenberg PT. Aurora-A Kinase and Inhibitor-2 Regulate the Cyclin Threshold for Mitotic Entry in Xenopus Early Embryonic Cell Cycles. Cell Cycle. 2006;5 doi: 10.4161/cc.5.19.3316. [DOI] [PubMed] [Google Scholar]
- 46.Liu J, Wu J, Oliver C, Shenolikar S, Brautigan DL. Mutations of the serine phosphorylated in the protein phosphatase-1-binding motif in the skeletal muscle glycogen-targeting subunit. Biochemical Journal. 2000;346:77–82. [PMC free article] [PubMed] [Google Scholar]
- 47.Hall EH, Daugherty AE, Choi CK, Horwitz AF, Brautigan DL. Tensin1 requires protein phosphatase-1alpha in addition to RhoGAP DLC-1 to control cell polarization, migration, and invasion. J Biol Chem. 2009;284:34713–34722. doi: 10.1074/jbc.M109.059592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang HB, Horiuchi A, Watanabe T, Shih SR, Tsay HJ, Li HC, Greengard P, Nairn AC. Characterization of the inhibition of protein phosphatase-1 by DARPP-32 and inhibitor-2. J Biol Chem. 1999;274:7870–7878. doi: 10.1074/jbc.274.12.7870. [DOI] [PubMed] [Google Scholar]
- 49.Winkler KE, Swenson KI, Kornbluth S, Means AR. Requirement of the prolyl isomerase Pin1 for the replication checkpoint. Science (New York, N Y. 2000;287:1644–1647. doi: 10.1126/science.287.5458.1644. [DOI] [PubMed] [Google Scholar]
- 50.Dancheck B, Nairn AC, Peti W. Detailed structural characterization of unbound protein phosphatase 1 inhibitors. Biochemistry. 2008;47:12346–12356. doi: 10.1021/bi801308y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dancheck B, Ragusa MJ, Allaire M, Nairn AC, Page R, Peti W. Molecular investigations of the structure and function of the protein phosphatase 1-spinophilin-inhibitor 2 heterotrimeric complex. Biochemistry. 2011;50:1238–1246. doi: 10.1021/bi101774g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hurley TD, Yang J, Zhang L, Goodwin KD, Zou Q, Cortese M, Dunker AK, DePaoli-Roach AA. Structural basis for regulation of protein phosphatase 1 by inhibitor-2. J Biol Chem. 2007;282:28874–28883. doi: 10.1074/jbc.M703472200. [DOI] [PubMed] [Google Scholar]
- 53.Terry-Lorenzo RT, Elliot E, Weiser DC, Prickett TD, Brautigan DL, Shenolikar S. Neurabins recruit protein phosphatase-1 and inhibitor-2 to the actin cytoskeleton. J Biol Chem. 2002;277:46535–46543. doi: 10.1074/jbc.M206960200. [DOI] [PubMed] [Google Scholar]
- 54.Wang H, Brautigan DL. A novel transmembrane Ser/Thr kinase complexes with protein phosphatase-1 and inhibitor-2. J Biol Chem. 2002;277:49605–49612. doi: 10.1074/jbc.M209335200. [DOI] [PubMed] [Google Scholar]