

Abstract
Thanatophoric dysplasia (TD) is caused by mutation of the gene that encodes fibroblast growth factor 3 (FGFR3). Owing to the poor prognosis for TD, prenatal diagnosis is critical to optimal perinatal management. We report here a case of TD in twin pregnancy, which was prenatally diagnosed by DNA analysis following amniocentesis at 15 weeks, and was managed by selective fetal termination. Prenatal ultrasonography and molecular analysis to detect TD-specific mutations enable accurate diagnosis of FGFR3-related TD in utero and appropriate obstetrical management at early gestation during twin pregnancy.
Keywords: Fibroblast growth factor receptor 3 gene, Selective termination, Thanatophoric dysplasia, Twins
Introduction
Skeletal dysplasias are a highly heterogeneous group of inherited connective tissue disorders. To date, approximately 400 distinct clinical entities belong to this group, and the molecular basis of the disorders have been unraveled in approximately 50% of cases [1]. Among these, fibroblast growth factor 3 (FGFR3)-related skeletal dysplasias are caused by mutations in the FGFR3 gene, which increase the activation of receptors that alter endochondral ossification in all long bones. This class of dysplasias include achondroplasia, hypochondroplasia, and thanatophoric dysplasia (TD) [2].
TD is the most frequent lethal skeletal dysplasia caused by the FGFR3 gene mutation, with an incidence of 3 to 6 births per 100,000 births [1]. Although ultraonography has been used for prenatal diagnosis of TD, occasionally TD fetuses in utero cannot be clearly distinguished from other osteochondrodysplasias. When we searched MEDLINE and Google Scholar extensively for articles on FGFR3-related TD through August 2013, reports of prenatal diagnosis of FGFR3-related TD are not rare. However, to our knowledge, there have been no previous reports of prenatal diagnosis of FGFR3-related TD in a twin pregnancy. We here report such a case, which was prenatally diagnosed by DNA analysis after amniocentesis at 15 weeks.
Case report
A 30-year-old, nulliparous woman with a twin pregnancy was referred to our hospital during the 15th gestational week for a routine prenatal check. She had suffered from primary infertility secondary to bilateral salpingectomy, and underwent in vitro fertilization. She had conceived triplets and had chosen selective embryo reduction during the 8th gestational week. The couple was healthy, and neither the history of teratogenic exposure nor the family history were remarkable. Initial ultrasonographic examination (Accuvix XQ, Medison Co. Ltd., Seoul, Korea) with a 2-6-MHz transabdominal probe) during the 15th gestational week revealed a thick dividing membrane and two separated placentas, indicating dichorionic placentation. The amount of amniotic fluid of both fetuses was appropriate for gestational age. Whereas twin A showed normal growth, in twin B, all of the long bones appeared to be abnormally short (the long bone measurements with the femur measuring 11.6 mm, tibia 7.2 mm, humerus 11.5 mm, radius 7.9 mm, all compatible with <5th percentile). Curved femurs with a telephone-receiver-like appearance, short ribs, and a narrow thorax with prominent abdomen were also observed (Fig. 1). The femur length (FL)/abdominal circumference (AC) ratio was 0.12. The vertebral and skull ossification patterns were normal. No overt cloverleaf skull deformity was observed. The nasal bone was not visible but there are no other abnormalities in fetal face.
Fig. 1.
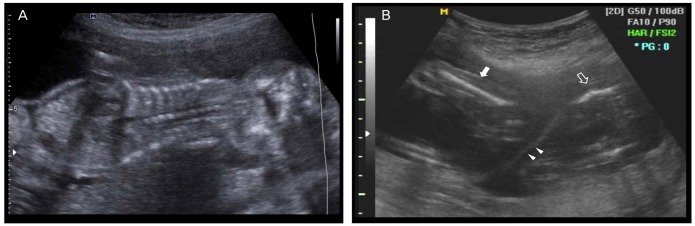
(A) Two-dimensional ultrasonographic images at the 15th gestational week showing that the thorax was small and that the ratio of the thoracic/abdominal circumference is below the mean. Such a low ratio is strongly suggestive of pulmonary hypoplasia. (B) Two-dimensional ultrasonographic images of each femur of the two fetuses at the 19th gestational week, showing a normal femur of twin A (solid arrow), a curved and short femur of affected twin B (open arrow), and a divided amniotic membrane (arrowheads).
The couple was counseled about the increased risk of chromosomal abnormality or lethal TD. Thereafter, they decided to undergo an assessment of specific FGFR3 mutations by routine cytogenetic analysis. Amniocentesis of the twins performed during the 15th gestational week showed normal karyotypes (46, XY and 46, XY) for both babies. However, nucleotide sequence analysis of cDNA during the screen for mutations in FGFR3 for twin B revealed a C to T transition at nucleotide 742 (C742T) within exon 6. This heterozygous 742C->T (R248C) mutation in the FGFR3 causes an Arg248Cys (CGC->TGC) substitution. Molecular analysis of the same locus in twin A showed normal finding (Fig. 2).
Fig. 2.

Electropherogram of the fibroblast growth factor 3 gene from both fetuses, demonstrating a normal sequence in normal fetus (A) and the c.742C>T change resulting in the p.Arg248Cys mutation in the affected fetus (B).
We counseled the couple extensively, and explained that this genetic condition causes type-1 TD. Thereafter, they decided to proceed with selective fetal termination with the hope of proceeding with a normal singleton pregnancy. At the 19th gestational week, selective fetal termination was performed under ultrasonographic guidance using a 20-gauge needle and injection of potassium chloride into the fetal heart. The remainder of the pregnancy was uneventful and the patient delivered a healthy male infant weighing 3,575 g and a mummified, macerated fetus weighing 91 g during the 39th gestational week by Cesarean section due to persistent occiput posterior position.
Discussion
TD was first described by Maroteaux et al. [3] in 1967. The term is derived from the Greek word 'thanatophoros,' which means 'death bearing.' Accordingly, it is the most common lethal skeletal disorder, and the affected neonate usually dies shortly after birth owing to profound pulmonary hypoplasia. There are two main forms of TD based on the bone deformity pattern. Type-1 TD is characterized by short and curved femurs with a telephone-receiver-like appearance, together with very flat vertebral bodies. It is rarely associated with a cloverleaf skull deformity. Type-2 TD has straighter femora, but they are just as short. Type-2 TD is also characterized by flatter vertebral bodies and is almost always associated with a cloverleaf skull [4]. Prenatal diagnosis of TD by ultrasonography has been based on these characteristic shortened limbs or other skeletal defects consistent with TD [5].
The typical ultrasonographic findings in our case with curved short femurs, short ribs, and narrow thorax shape enabled to suspect a FGFR3-related skeletal dysplasia, especially TD. However, individual cases exhibit a wide variance in measurements, and there is considerable phenotypic variability between cases, with certain features shared in common with TD and other severe osteochondrodysplasias but with a lower rate of lethality. Camptomelic dysplasia and achondrogenesis have similar clinical characteristics with an early prenatal-onset and lethal skeletal dysplasia. However, camptomelic dysplasia is differentiated from TD by showing mesomelia and the immature ossification of the tubular bones [6]. And the prevalence is 0.5 per 100,000 (a 10th of TD). Achondrogenesis is differentiated from TD by a short trunk and poor ossification in vertebrae, cranium, and pelvic bone [7]. FGFR3-related skeletal dysplasias include achondroplasia and hypochondroplasia other than TD [2]. Achondroplasia is a non-lethal skeletal dysplasia with the normal fetal growth until the late second trimester, in which the shortening of the long bone is less prominent than in TD. Hypochondroplasia is characterized by a similar but milder phenotype compared to achondroplasia and its main features are rhizomelia, short stature, and lumbar lordosis. The rarity of prenatal diagnosis of hypochondroplasia is attributed to the normal long bone growth throughout pregnancy.
Molecular analysis is therefore essential to enable a definitive diagnosis of TD, which is caused by mutations in the FGFR3 gene located at 4p16 [8]. Several mutations are reported to cause type-1 TD, including p.Arg248Cys (55%), p.Tyr373Cys (24%), p.Ser249Cys (6%), or mutations in the stop codon (10%) [2]. In contrast, type-2 TD is caused by a single recurrent mutation (p.Lys650Glu) [8,9]. With the advent of molecular technology, Sawai et al. [10] first described the prenatal diagnosis of TD by mutational analysis of the FGFR3 gene following amniocentesis. We also detected an p.Arg248Cys mutation (c.742C>T), which is the most common mutation of type-1 TD.
Skeletal dysplasia has been thought to be lethal if there is a hypoplastic thorax, a FL/AC ratio less than 0.16, or certain distinguishing characteristics diagnostic of a known lethal disorder [11]. A hypoplastic thorax is suspected on the basis of a thoracic circumference of less than 5% at the level of the four-chamber view of the heart, or a thoracic-AC ratio of less than 0.79 [12]. The FL/AC ratio was 0.12 in our current case.
The increased incidence of multiple pregnancies and the higher incidence of fetal malformations in multiple pregnancies compared with singletons have increased the prevalence of pregnancies with one anomalous twin. We are thus likely to be increasingly confronted with the choice of either continuing or aborting abnormal fetuses. Nonetheless, with increasing experience and better procedures, selective termination for fetal lethal anomalies has recently become safer and more effective, thereby allowing patients with multiple pregnancies and fetal anomalies to continue the pregnancy without having to maintain the problem [13,14]. There are several reports on multiple pregnancies concordant for TD [15]. However, these cases had been evaluated on the basis of clinical features alone, without molecular analysis. Our current case report may thus be the first description of prenatal diagnosis by molecular analysis of TD in a multiple pregnancy at early gestation.
In conclusion, the typical findings of prenatal ultrasonography enable to suspect TD. Whenever TD is suspected, molecular investigations of TD-specific mutations should be included as part of standard cytogenetic analysis to establish an accurate diagnosis of FGFR3-related TD in utero , and to facilitate early parental counseling and fetal termination during early gestation.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Superti-Furga A, Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 2.Hatzaki A, Sifakis S, Apostolopoulou D, Bouzarelou D, Konstantinidou A, Kappou D, et al. FGFR3 related skeletal dysplasias diagnosed prenatally by ultrasonography and molecular analysis: presentation of 17 cases. Am J Med Genet A. 2011;155A:2426–2435. doi: 10.1002/ajmg.a.34189. [DOI] [PubMed] [Google Scholar]
- 3.Maroteaux P, Lamy M, Robert JM. Thanatophoric dwarfism. Presse Med. 1967;75:2519–2524. [PubMed] [Google Scholar]
- 4.Langer LO, Jr, Yang SS, Hall JG, Sommer A, Kottamasu SR, Golabi M, et al. Thanatophoric dysplasia and cloverleaf skull. Am J Med Genet Suppl. 1987;3:167–179. doi: 10.1002/ajmg.1320280521. [DOI] [PubMed] [Google Scholar]
- 5.Schild RL, Hunt GH, Moore J, Davies H, Horwell DH. Antenatal sonographic diagnosis of thanatophoric dysplasia: a report of three cases and a review of the literature with special emphasis on the differential diagnosis. Ultrasound Obstet Gynecol. 1996;8:62–67. doi: 10.1046/j.1469-0705.1996.08010062.x. [DOI] [PubMed] [Google Scholar]
- 6.Mansour S, Hall CM, Pembrey ME, Young ID. A clinical and genetic study of campomelic dysplasia. J Med Genet. 1995;32:415–420. doi: 10.1136/jmg.32.6.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krakow D, Lachman RS, Rimoin DL. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genet Med. 2009;11:127–133. doi: 10.1097/GIM.0b013e3181971ccb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tavormina PL, Shiang R, Thompson LM, Zhu YZ, Wilkin DJ, Lachman RS, et al. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet. 1995;9:321–328. doi: 10.1038/ng0395-321. [DOI] [PubMed] [Google Scholar]
- 9.Chen CP, Chern SR, Shih JC, Wang W, Yeh LF, Chang TY, et al. Prenatal diagnosis and genetic analysis of type I and type II thanatophoric dysplasia. Prenat Diagn. 2001;21:89–95. doi: 10.1002/1097-0223(200102)21:2<89::aid-pd21>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 10.Sawai H, Komori S, Ida A, Henmi T, Bessho T, Koyama K. Prenatal diagnosis of thanatophoric dysplasia by mutational analysis of the fibroblast growth factor receptor 3 gene and a proposed correction of previously published PCR results. Prenat Diagn. 1999;19:21–24. doi: 10.1002/(sici)1097-0223(199901)19:1<21::aid-pd457>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 11.Rahemtullah A, McGillivray B, Wilson RD. Suspected skeletal dysplasias: femur length to abdominal circumference ratio can be used in ultrasonographic prediction of fetal outcome. Am J Obstet Gynecol. 1997;177:864–869. doi: 10.1016/s0002-9378(97)70284-9. [DOI] [PubMed] [Google Scholar]
- 12.Johnson A, Callan NA, Bhutani VK, Colmorgen GH, Weiner S, Bolognese RJ. Ultrasonic ratio of fetal thoracic to abdominal circumference: an association with fetal pulmonary hypoplasia. Am J Obstet Gynecol. 1987;157:764–769. doi: 10.1016/s0002-9378(87)80046-7. [DOI] [PubMed] [Google Scholar]
- 13.Evans MI, Goldberg JD, Horenstein J, Wapner RJ, Ayoub MA, Stone J, et al. Selective termination for structural, chromosomal, and mendelian anomalies: international experience. Am J Obstet Gynecol. 1999;181:893–897. doi: 10.1016/s0002-9378(99)70321-2. [DOI] [PubMed] [Google Scholar]
- 14.Eddleman KA, Stone JL, Lynch L, Berkowitz RL. Selective termination of anomalous fetuses in multifetal pregnancies: two hundred cases at a single center. Am J Obstet Gynecol. 2002;187:1168–1172. doi: 10.1067/mob.2002.127456. [DOI] [PubMed] [Google Scholar]
- 15.Vidaeff AC, Lucas MJ, Strassberg MB, Spooner KI, Ramin SM. Dichorionic twins discordant for thanatophoric dysplasia managed with selective reduction at 20 weeks' gestation: a case report. J Reprod Med. 2005;50:638–642. [PubMed] [Google Scholar]