

Abstract
Elevation of intracellular cAMP and activation of protein kinase A (PKA) lead to activation of large conductance voltage- and Ca2+-activated K+ (BK) channels, thus attenuation of detrusor smooth muscle (DSM) contractility. In this study, we investigated the mechanism by which pharmacological inhibition of cAMP-specific phosphodiesterase 4 (PDE4) with rolipram or Ro-20-1724 (C15H22N2O3) suppresses guinea pig DSM excitability and contractility. We used high-speed line-scanning confocal microscopy, ratiometric fluorescence Ca2+ imaging, and perforated whole-cell patch-clamp techniques on freshly isolated DSM cells, along with isometric tension recordings of DSM isolated strips. Rolipram caused an increase in the frequency of Ca2+ sparks and the spontaneous transient BK currents (TBKCs), hyperpolarized the cell membrane potential (MP), and decreased the intracellular Ca2+ levels. Blocking BK channels with paxilline reversed the hyperpolarizing effect of rolipram and depolarized the MP back to the control levels. In the presence of H-89 [N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide dihydrochloride], a PKA inhibitor, rolipram did not cause MP hyperpolarization. Rolipram or Ro-20-1724 reduced DSM spontaneous and carbachol-induced phasic contraction amplitude, muscle force, duration, and frequency, and electrical field stimulation-induced contraction amplitude, muscle force, and tone. Paxilline recovered DSM contractility, which was suppressed by pretreatment with PDE4 inhibitors. Rolipram had reduced inhibitory effects on DSM contractility in DSM strips pretreated with paxilline. This study revealed a novel cellular mechanism whereby pharmacological inhibition of PDE4 leads to suppression of guinea pig DSM contractility by increasing the frequency of Ca2+ sparks and the functionally coupled TBKCs, consequently hyperpolarizing DSM cell MP. Collectively, this decreases the global intracellular Ca2+ levels and DSM contractility in a BK channel-dependent manner.
Introduction
Ca2+ influx through L-type voltage-gated Ca2+ (CaV) channels initiates detrusor smooth muscle (DSM) spontaneous action potentials and spontaneous phasic contractions (Hashitani et al., 2004; Brading, 2006; Petkov, 2011). Pharmacological manipulation of the large conductance voltage- and Ca2+-activated K+ (BK) channel can effectively modulate DSM cell action potential generations and membrane potential (MP), thereby the Ca2+ influx via CaV channels (Heppner et al., 1997; Sprossmann et al., 2009; Hristov et al., 2011, 2012; Petkov, 2011). Moreover, genetic deletion of BK channels enhances Ca2+ influx and increases DSM excitability and contractility (Meredith et al., 2004; Sausbier et al., 2007; Brown et al., 2008; Sprossmann et al., 2009). Furthermore, impaired BK channel activity has been implicated in neurogenic detrusor overactivity (Hristov et al., 2013). In contrast, the activation of BK channels leads to attenuation of CaV channel activity and intracellular Ca2+ levels, and thereby suppresses DSM contractility (Hristov et al., 2012; Petkov, 2011; Xin et al., 2012a; Xin et al., 2012b). Previous studies have demonstrated that the elevation of intracellular cAMP levels increases BK channel activity and decreases DSM contractions (Petkov and Nelson, 2005; Brown et al., 2008; Hristov et al., 2008; Xin et al., 2012c).
cAMP, via activation of PKA, plays a critical role in the regulation of the fast and localized Ca2+ releases from the ryanodine receptors (RyRs) of the sarcoplasmic reticulum (SR), known as Ca2+ sparks along with the functionally coupled spontaneous transient BK currents (TBKCs) (Herrera et al., 2001; Petkov and Nelson, 2005). The rise in cellular cAMP levels increases the frequency of Ca2+ sparks and TBKCs in DSM cells and consequently reduces the global cellular Ca2+ concentration inducing DSM relaxation (Petkov and Nelson, 2005; Brown et al., 2008; Hristov et al., 2012; Xin et al., 2012b). However, in neurogenic detrusor overactivity associated with overactive bladder, the excessive nerve-released ACh increases the activity of Gαi protein-coupled M2 muscarinic receptors, leading to a reduction in cellular cAMP levels and a consequent increase in DSM contractility (Sellers and Chess-Williams, 2012). This suggests that impaired cAMP signals might contribute to urinary bladder dysfunction, such as overactive bladder.
Moreover, adenylyl cyclases that synthesize cAMP and phosphodiesterases (PDEs) that hydrolyze cAMP are constitutively active in excitable cells, such as cardiomyocytes and DSM cells; thus, pharmacological inhibition of PDEs can lead to a rapid increase in cellular cAMP levels (Beca et al., 2011; Xin et al., 2012c,d). Recent studies have demonstrated that nonselective pharmacological inhibition of PDEs with 3-isobutyl-1-methylxanthine or selective inhibition of PDE1 with 8-methoxymethyl-3-isobutyl-1-methylxanthine increases cAMP levels, stimulates BK channel activity in a RyR-dependent manner, hyperpolarizes the DSM cell MP, and effectively suppresses DSM contractility (Xin et al., 2012b,c).
Among the 11 known PDE families, the cAMP-specific PDE4 has more than 20 different splice variants encoded by four genes (PDE4A–PDE4D) (Bender and Beavo, 2006). Many of the PDE4 isoforms have organ- and cell type-specific expression profile (Houslay et al., 2005; Bender and Beavo, 2006). In addition, different PDE4 isoforms are localized in particular cellular compartments via scaffold proteins, and the pharmacological inhibition of a particular PDE4 isoform might achieve localized activation of cAMP/PKA to regulate specific cellular functions (Houslay et al., 2005; Richter et al., 2005; Horner et al., 2012). Recent studies revealed that the PDE4D mRNA levels in human bladder are the highest among 24 different human tissues, including tissues from central nervous system and heart (Lakics et al., 2010). It is noteworthy that PDE4 plays only a minor role in controlling human cardiomyocyte cellular cAMP levels and has little inotropic and lusitropic effects in the human ventricle (Johnson et al., 2012; Molenaar et al., 2013). Therefore, selective PDE4 isoform inhibition might potentially stimulate the cAMP/PKA pathway in DSM cells and thus regulate DSM function selectively without significant impact on cardiac function. However, the mechanism by which PDE4 regulates DSM function is currently unknown.
In this study, we tested the hypothesis that selective pharmacological inhibition of PDE4 increases the frequency of Ca2+ sparks and their functionally coupled TBKCs, which leads to the attenuation of DSM excitability and contractility. We used high-speed line-scanning confocal microscopy, ratiometric fluorescence imaging, and perforated whole-cell patch-clamp techniques on guinea pig freshly isolated DSM cells, along with isometric tension recordings on guinea pig DSM-isolated strips. To achieve this goal, PDE4 was inhibited pharmacologically with rolipram or Ro-20-1724. The study revealed a novel mechanism by which PDE4 regulates DSM excitability and contractility.
Materials and Methods
DSM Tissue Collection.
A total of 45 male Hartley Albino guinea pigs (Charles River Laboratories, Raleigh, NC) with an average weight of 451 ± 11 g was used in this study. Guinea pigs were euthanized with CO2 inhalation followed by thoracotomy, according to animal use protocol number 1747 reviewed and approved by the Institutional Animal Care and Use Committee of the University of South Carolina. DSM strips (∼2–3-mm wide and ∼5–7-mm long) were prepared by removing the mucosa and were used for single DSM cell isolation and isometric DSM tension recordings.
DSM Single-Cell Isolation.
DSM single cells were freshly isolated, as described previously (Parajuli et al., 2012; Xin et al., 2012a,c). In brief, one to two DSM strips were incubated at 37°C for 12–18 minutes in 2-ml dissection solution supplemented with 1 mg/ml bovine serum albumin (BSA), 1 mg/ml papain, and 1 mg/ml dl-dithiothreitol. DSM tissues were then transferred to 2 ml of dissection solution supplemented with 1 mg/ml BSA, 0.5 mg/ml type II collagenase, 0.5 mg/ml trypsin inhibitor, and 100 μM CaCl2 and incubated at 37°C for 12–15 minutes. The tissues were then washed three times with dissection solution supplemented with 1 mg/ml BSA and gently triturated with a fire-blunted Pasteur pipette to disperse single DSM cells.
Electrophysiological Recordings.
The amphotericin B-perforated whole-cell patch-clamp technique was used for electrophysiological recordings from freshly isolated DSM single cells using the method described previously (Xin et al., 2012a, c). In brief, a few drops of the DSM cell suspension were placed into a recording chamber, and the cells were allowed to adhere to the glass bottom for ∼20 minutes. Patch-clamp recordings were conducted using an Axopatch 200B amplifier system and Digidata 1440A controlled with pCLAMP 10.2 software (Molecular Devices, Union City, CA). The currents were low-pass filtered at 1 kHz using an eight-pole Bessel filter model 900CT/9L8L (Frequency Devices, Ottawa, IL) and sampled at a rate of 10 kHz. The glass pipettes used for the patch-clamp experiments were made from borosilicate glass (Sutter Instruments, Novato, CA), pulled using a PP-830 vertical puller (Narishige Group, Tokyo, Japan), and polished with a Micro Forge MF-830 fire polisher (Narishige Group). Pipette resistance was 4–6 MΩ. TBKCs were recorded at a holding potential of −40 mV, a potential very close to the physiologic resting membrane potential of intact guinea pig DSM preparations (Petkov et al., 2001). DSM cell MP was recorded using the current-clamp mode of the patch-clamp technique without any current input (Ih = 0). The cell MP was measured as the average of the last 5-minute recording under each experimental condition. Average TBKCs’ frequency and amplitude under control conditions were taken to be 100%, respectively. All patch-clamp experiments were conducted at room temperature (22–23°C).
Ca2+ Spark Recordings by High-Speed Line-Scanning Confocal Microscopy.
Ca2+ sparks were detected in cells loaded with the fluorescent Ca2+ probe fluo-4-AM. A suspension of freshly isolated DSM cells was added into a 35-mm glass-bottom dish coated with poly-l-lysine and kept for 30 minutes to allow cells to adhere to the bottom of the dish, after which the supernatant was removed. The extracellular solution (see “Solutions and Drugs”) containing fluo-4-AM at a concentration of 2 µM was added into the dish and stored in the dark for 1 hour. Fluo-4-AM solution was then removed, and cells were washed three times with extracellular solution. Ca2+ sparks were detected using a high-speed line-scanning LSM700-Meta confocal microscope equipped with a 63× oil immersion objective (Carl Zeiss MicroImaging, Jena, Germany). Argon laser (488 nm) was used as the excitation light, and the emission was recorded at 510 nm. A line (1 pixel/0.033-μm wide) was drawn across the cell through potentially active Ca2+ spark sites. The line was scanned for 4 seconds, with a scanning speed of 528 lines/seconds in each cycle, and repeated 10 times at 1-minute intervals. Ca2+ sparks along the line were analyzed for each cycle using ImageJ with SparkMaster plug-in (Picht et al., 2007). The background fluorescence F0 was taken as the resting fluorescence, which did not have Ca2+ sparks. Ca2+ sparks were individually analyzed for amplitude (difference between spark peak fluorescence and background fluorescence). The threshold for the detection of events was 3.8 times the standard deviation of the background noise over the mean value of the background. Ca2+ spark frequency and amplitude were averaged for the last five scanning cycles under control or test compound-treated conditions. Ca2+ spark frequency was expressed as sparks/100 µm × seconds (Picht et al., 2007; Porta et al., 2011). All Ca2+ spark experiments were conducted at room temperature (22–23°C).
Recording Changes of Global Intracellular Ca2+ Levels in Freshly Isolated DSM Cells.
The global intracellular Ca2+ levels were monitored using a ratiometric fluorescent Ca2+ probe fura-2-AM, as previously described (Hristov et al., 2012; Xin et al., 2012c). Freshly isolated DSM cells were loaded with 2 µM fura-2-AM using the same procedure as for fluo-4-AM. Cells were imaged with an OLYMPUS IX81 inverted research microscope equipped with a 40× oil objective and MetaFluor 7.7.2.0 software (Molecular Devices, Sunnyvale, CA). Fura-2-AM was excited for 15 milliseconds every 3 seconds at wavelengths of 340 nm and 380 nm light; the relative intracellular Ca2+ level was expressed as the ratio of F340/F380 emission intensities at 510 nm. All Ca2+-imaging experiments were carried out at room temperature (22–23°C).
Isometric DSM Tension Recordings.
Isometric DSM contractions were measured, as previously described (Parajuli et al., 2012; Xin et al., 2012a,c). In brief, DSM strips were secured to isometric force-displacement transducers and were suspended in temperature-controlled (37°C) water-jacketed tissue baths containing 10 ml of physiologic saline solution aerated with 95% O2 and 5% CO2, pH 7.4. The DSM strips were initially tensioned to 10 mN and washed with fresh physiologic saline solution every 15 minutes during an equilibration period of 45–60 minutes. To minimize the effects of neurotransmitters released from neurons in the DSM, the spontaneous phasic and carbachol-induced contractions were recorded in the presence of 1 µM tetrodotoxin (TTX), a selective inhibitor of the neuronal voltage-gated Na+ channels.
Nerve-evoked contractions were induced by electrical field stimulation (EFS) using a pair of platinum electrodes mounted in the tissue baths parallel to the DSM strips. The EFS pulses were generated using a PHM-152I stimulator (MED Associates, St. Albans, VT). The EFS pulse parameters were as follows: 0.75-millisecond pulse width, 20-V pulse amplitude, 3-second stimulus duration, and polarity reversed for alternating pulses. After the equilibration period, the DSM strips were subjected to continuous repetitive EFS with a frequency of 20 Hz at 1-minute intervals. DSM contractions were recorded using a Myomed myograph system (MED Associates).
Data Analysis and Statistics.
The frequency and amplitude of TBKCs were analyzed using MiniAnalysis (Synaptosoft, Decatur, GA). Data were further analyzed with GraphPad Prism 4.03 software (GraphPad Software, La Jolla, CA). The MP was analyzed using Clampfit 10.2 (Molecular Devices, Union City, CA). Data were summarized using a paired Student’s t test to compare the MP in the presence of a testing compound with the MP before the addition of the compound in the same cell. The five parameters of the DSM phasic and tonic contractions, phasic contraction amplitude, muscle force integral, contraction duration, contraction frequency, and muscle tone, were also analyzed using MiniAnalysis. The contraction parameters for spontaneous phasic and 20-Hz EFS-induced contractions were normalized to the control (taken to be 100%) and expressed as percentages. Net muscle force (muscle force integral) was determined by integrating the area under the phasic contraction force-time baseline curve. The relative change of the muscle tone was determined by measuring changes of the phasic contraction baseline curve. Data were expressed as means ± SEM; n = the number of cells or strips, and N = the number of guinea pigs. The IC50 values are expressed as means (95% confidence interval, CI). Statistical significance was tested using paired Student’s t test or one-way ANOVA, followed by Dunnett's multiple comparison test, and P < 0.05 was considered significant.
Solutions and Drugs.
The nominally Ca2+-free dissection solution contained the following: 80 mM monosodium glutamate, 55 mM NaCl, 6 mM KCl, 10 mM glucose, 10 mM HEPES, and 2 mM MgCl2; pH was adjusted to 7.3 with NaOH. The extracellular solution for whole-cell patch-clamp and Ca2+-imaging experiments contained the following: 134 mM NaCl, 6 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose, and 10 mM HEPES; pH was adjusted to 7.4 with NaOH. The pipette solution contained the following: 110 mM potassium aspartate, 30 mM KCl, 10 mM NaCl, 1 mM MgCl2, 10 mM HEPES, and 0.05 mM EGTA; pH was adjusted to 7.2 with NaOH and supplemented with freshly dissolved 200 µg/ml amphotericin-B. The Ca2+-containing physiologic saline solution was prepared daily and contained the following: 119 mM NaCl, 4.7 mM KCl, 24 mM NaHCO3, 1.2 mM KH2PO4, 2.5 mM CaCl2, 1.2 mM MgSO4, and 11 mM glucose and was aerated with 95% O2–5% CO2 to obtain pH 7.4. Trypsin inhibitor, BSA, and amphotericin-B were from Thermo Fisher Scientific (Fair Lawn, NJ); papain was from Worthington Biochemical (Lakewood, NJ); and collagenase (type II) and tetrodotoxin citrate were purchased from Sigma-Aldrich (St. Louis, MO). All were dissolved in deionized double-distilled water. Paxilline, fura-2-AM (Sigma-Aldrich), rolipram (C16H21NO3), Ro-20-1724 (C15H22N2O3), N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide dihydrochloride (H-89, C20H20BrN3O2S.2HCl) (Tocris Bioscience, Toronto, ON, Canada), and fluo-4-AM (Invitrogen, Carlsbad, CA) were dissolved in dimethylsulfoxide. The maximal dimethylsulfoxide concentration was less than 0.1% in bath solution.
Results
Selective Pharmacological PDE4 Inhibition with Rolipram Increases Ca2+ Spark Frequency in Freshly Isolated DSM Cells.
Ca2+ sparks were detected using high-speed line-scanning confocal microscopy. The average Ca2+ spark frequency under control conditions was 38.2 ± 12.1 sparks/100 µm × seconds (n = 15, N = 9). Rolipram (10 μM) significantly increased the Ca2+ spark frequency to 514 ± 152% of the control (n = 15, N = 9; P < 0.05; Fig. 1), without a significant effect on Ca2+ spark amplitude (95.3 ± 11.6% of the control; n = 15, N = 9; P > 0.05; Fig. 1). This indicates that the inhibition of PDE4 can increase the localized Ca2+ releases from the RyRs in DSM cells.
Fig. 1.

Selective phosphodiesterase 4 (PDE4) inhibition with rolipram increases Ca2+ spark frequency in freshly isolated detrusor smooth muscle (DSM) cells. (A) An image of a freshly isolated DSM cell loaded with fluo-4-AM. The white line passing the active site a is the laser beam scanning pathway (1-pixel width). (B) A three-dimensional view of the recordings illustrating the relative fluorescence intensity profiles of the Ca2+ sparks. The color scale indicates the relative fluorescence intensity F/F0. (C) Summary data illustrating that rolipram (10 µM) increased the Ca2+ spark frequency without a significant change of Ca2+ spark amplitude (n = 15, N = 9; *P < 0.05). Amp, amplitude; Freq, frequency.
Selective Pharmacological PDE4 Inhibition with Rolipram Causes an Increase in Frequency of the TBKCs in Freshly Isolated DSM Cells.
Ca2+ sparks transiently activate BK channels and generate TBKCs. TBKCs were measured using the perforated whole-cell voltage-clamp technique at a holding potential of −40 mV. Rolipram (10 µM) significantly increased TBKC frequency to 239.8 ± 43.3% of the control value (n = 7, N = 6; P < 0.05; Fig. 2), without a significant effect on the TBKC amplitude (131.7 ± 73.0% of control; n = 7, N = 6; P > 0.05; Fig. 2). These data demonstrate that the effects of selective PDE4 inhibition with rolipram on Ca2+ sparks are functionally coupled to TBKCs.
Fig. 2.

Selective PDE4 inhibition with rolipram increases spontaneous transient BK current (TBKC) frequency in freshly isolated DSM cells. (A) An original recording illustrating that rolipram (10 µM) increased the frequency of TBKCs in a single DSM cell. A portion of the recording is shown on an expanded time scale before and after rolipram application. (B) Summary data illustrating that rolipram (10 µM) increased the TBKCs’ frequency without significant change in TBKCs’ amplitude (n = 7, N = 6; *P < 0.05).
Selective PDE4 Inhibition with Rolipram Hyperpolarizes the MP of Freshly Isolated DSM Cells.
Rolipram (10 μM) hyperpolarized DSM cell MP by an average of 8.6 ± 2.9 mV (n = 8, N = 6; P < 0.05; Fig. 3). Blocking the BK channels with paxilline (1 µM) following rolipram application repolarized the cell MP back to the control levels (−20.6 ± 3.5 mV; n = 8, N = 6; P < 0.05 versus rolipram; Fig. 3, A and B). The role of BK channels in DSM cell MP hyperpolarization induced by PDE4 inhibition was further examined by applying paxilline, a selective BK channel inhibitor, before the addition of rolipram. Paxilline (1 µM) blocked the spontaneous transient hyperpolarizations of DSM cell MP and depolarized the MP from −28.6 ± 2.9 to −18.2 ± 2.1 mV (n = 7, N = 5; P < 0.05 versus control; Fig. 3, C and D). In the presence of paxilline (1 µM), the subsequent addition of rolipram (10 µM) did not significantly change the MP measured as −18.1 ± 2.7 mV (n = 7, N = 5; P > 0.05, rolipram versus paxilline; Fig. 3, C and D). These data demonstrate that BK channel mediates the PDE4-hyperpolarizing effects on cell MP.
Fig. 3.

Selective PDE4 inhibition with rolipram hyperpolarizes DSM cell resting membrane potential (MP) in a BK channel-dependent manner. (A) An original current-clamp recording illustrating that rolipram (10 µM) hyperpolarized cell MP. The subsequent addition of the BK channel blocker, paxilline (1 µM), inhibited the spontaneous transient hyperpolarizations and depolarized the MP. (B) Summary data illustrating that rolipram (10 µM) hyperpolarized the MP. The subsequent addition of 1 µM paxilline reversed the rolipram-induced hyperpolarization and further depolarized the cell MP back to the control level (n = 8, N = 6; *P < 0.05). (C) An original current-clamp recording illustrating that paxilline (1 µM) abolished the spontaneous transient hyperpolarizations. Rolipram (10 µM) did not exhibit a hyperpolarizing effect on the MP in the presence of 1 µM paxilline. (D) Summary data illustrating that rolipram (10 µM) did not have any effect on the MP in DSM cells pretreated with 1 µM paxilline (n = 7, N = 5; P > 0.05; NS, nonsignificant).
PKA Inhibitor H-89 Abolishes the Hyperpolarizing Effect of Rolipram on DSM Cell MP.
The role of PKA in the rolipram-induced DSM cell MP hyperpolarization was investigated. H-89 (10 µM), a PKA inhibitor, depolarized the average DSM cell MP from −38.9 ± 11.5 mV under control conditions to −33.3 ± 10.0 mV (n = 6, N = 4; P < 0.05). In the presence of H-89 (10 µM), the subsequent addition of rolipram (10 μM) did not change the MP, which averaged −33.1 ± 10.4 mV (n = 6, N = 4; P > 0.05 versus H-89; Fig. 4). These data support the concept that PDE4 regulates DSM excitability via a PKA-dependent mechanism.
Fig. 4.

Inhibition of PKA with H-89 abolishes the hyperpolarizing effect of rolipram in DSM cells. (A) An original current-clamp recording illustrating that, in the presence of H-89 (10 µM), rolipram (10 µM) did not have any effect on the MP. (B) Summary data illustrating that rolipram (10 µM) did not have any effect on the average MP when PKA was inhibited (n = 6, N = 4; P > 0.05; NS, nonsignificant).
Selective PDE4 Inhibition with Rolipram Decreases Global Intracellular Ca2+ Levels in Freshly Isolated DSM Cells.
The effect of PDE4 inhibition on global intracellular Ca2+ levels in DSM cells was further investigated by measuring the fluorescent emission intensity ratio F340/F380 of fura-2. The average F340/F380 ratio was 0.73 ± 0.06 under control conditions, and rolipram (10 μM) treatment of the DSM cells caused a significant decrease in the F340/F380 ratio to an average of 0.66 ± 0.04 (n = 8, N = 5; P < 0.05; Fig. 5). These results coincide with the observations that the increase in localized Ca2+ sparks and their functionally coupled TBKCs leads to a MP hyperpolarization and decrease in global intracellular Ca2+ levels.
Fig. 5.

Selective PDE4 inhibition with rolipram reduces the global intracellular Ca2+ levels in freshly isolated DSM cells. (A) An original recording illustrating that rolipram (10 µM) decreased the global intracellular Ca2+ level in a single DSM cell shown as a ratio of fura-2 fluorescence emission intensities at 510 nm with excitation at 340 and 380 nm. (B) Summary data indicating that rolipram (10 µM) significantly decreased the average intracellular Ca2+ levels (n = 8, N = 5; *P < 0.05).
Selective PDE4 Inhibition with Rolipram Reduces Spontaneous Phasic (Myogenic) Contractions of DSM-Isolated Strips.
In DSM-isolated strips, rolipram reduced the spontaneous phasic contraction amplitude, muscle force integral, duration, frequency, and muscle tone in a concentration-dependent manner with IC50 values of 18 nM (95% CI 7.9–39.5 nM), 8.2 nM (95% CI 3.4–19.9 nM), 148 nM (95% CI 4.9–4436 nM), 70 nM (95% CI 22.3–222 nM), and 23 nM (95% CI 6.6–77.8 nM), respectively (n = 12, N = 8; P < 0.05; Fig. 6). At the maximal concentration used (10 μM), rolipram inhibited the spontaneous phasic contraction amplitude to 24.9 ± 7.1%, muscle force integral to 11.8 ± 3.8%, duration to 36.4 ± 9.5%, frequency to 41.8 ± 11.2%, and the muscle tone to 70.4 ± 7.1% of the control, respectively (n = 12, N = 8; P < 0.05; Fig. 6). These data support the concept that DSM hyperpolarization leads to its relaxation.
Fig. 6.

Selective PDE4 inhibition with rolipram significantly reduces DSM spontaneous phasic contractions in a concentration-dependent manner. (A) A representative recording of a DSM strip depicts the concentration-dependent inhibitory effects of rolipram (0.1 nM–10 μM) on spontaneous phasic contractions. Post-treatment of the DSM strip with 1 μM paxilline in the presence of rolipram (10 μM) recovered DSM contractility. (B–F) Cumulative concentration-response curves illustrating the inhibitory effects of rolipram (0.1 nM–10 μM) on the amplitude (B), muscle force integral (C), duration (D), frequency (E), and muscle tone (F) of DSM spontaneous contractions (n = 12, N = 8; *P < 0.05 versus control). (G) Summary data illustrating that addition of 1 μM paxilline following application of 10 µM rolipram significantly recovered the phasic contraction amplitude, muscle force integral, and frequency of DSM-isolated strips (n = 11, N = 7; #P < 0.05 paxilline versus 10 μM rolipram; P > 0.05; NS, nonsignificant). TTX (1 μM) was present throughout the experiments. Amp, amplitude; Dur, duration; Freq, frequency.
Blocking BK Channels with Paxilline Attenuates the Inhibitory Effect of Rolipram on DSM Spontaneous Contractility.
To determine the role of BK channels in rolipram-induced DSM relaxation, we applied paxilline. In the presence of 10 μM rolipram, paxilline led to a recovery of DSM phasic contraction amplitude, muscle force integral, frequency, and tone to 95.2 ± 21.3%, 64.4 ± 18.9%, 158.9 ± 21.3%, and 91.8 ± 9.0% of the control, respectively (n = 11, N = 7; P < 0.05; paxilline versus rolipram; Fig. 6G). This suggests that BK channels mediate DSM relaxation induced by rolipram.
To investigate further the role of BK channels in DSM relaxation effect of rolipram, we pretreated DSM strips with paxilline (1 µM) before cumulative application of rolipram. As illustrated in Fig. 7, the relaxation effect of rolipram on paxilline-induced DSM phasic contractions was dramatically inhibited compared with spontaneous phasic contractions (Fig. 6). These data support the concept that BK channel activation is critical in mediating rolipram-induced inhibition of DSM phasic contractions.
Fig. 7.

Blocking BK channels with paxilline significantly attenuates rolipram-induced relaxation in DSM-isolated strips. (A) A representative recording of a DSM strip depicts the inhibitory effects of rolipram (0.1 nM–10 µM) on 1 μM paxilline-induced DSM spontaneous contractions. (B–F) Summary data illustrating the effect of rolipram on the amplitude (B), muscle force integral (C), duration (D), frequency (E), and tone (F) of paxilline-induced contractions of DSM strips pretreated with paxilline (n = 10, N = 4; *P < 0.05, versus control). (G) Summary data illustrating that pretreatment of DSM strips with 1 μM paxilline significantly reduced the maximal relaxation effect of 10 µM rolipram on the contraction amplitude, muscle force integral, frequency, and tone in DSM strips. #P < 0.05, paxilline pretreatment (n = 10, N = 4) versus control (without paxilline) (n = 12, N = 8). TTX (1 μM) was present throughout the experiments. Amp, amplitude; Dur, duration; Freq, frequency.
Selective PDE4 Inhibition with Rolipram Attenuates Carbachol-Induced DSM Phasic Contractions in a Concentration-Dependent Manner.
Rolipram reduced the carbachol-induced phasic contraction amplitude, muscle force integral, duration, frequency, and muscle tone in a concentration-dependent manner with IC50 values of 34 nM (21.1–56.0 nM), 34 nM (24.4–48.4 nM), 72 nM (27.2–192.9 nM), 121.7 nM (9.6–1535 nM), and 36 nM (12.0–108.1 nM), respectively (n = 13, N = 7; P < 0.05; Fig. 8). At the maximal concentration (10 μM), rolipram reduced the carbachol-induced phasic contraction amplitude to 25.2 ± 5.4%, muscle force integral to 17.4 ± 3.3%, duration to 64.2 ± 7.0%, frequency to 58.1 ± 7.3%, and the muscle tone to 69.8 ± 6.9% of the control, respectively (n = 13, N = 7; P < 0.05; Fig. 8). Blocking the BK channels with 1 μM paxilline recovered the phasic contraction amplitude, duration, muscle force, frequency, and tone to 52.5 ± 8.1%, 62.8 ± 5.7%, 36.7 ± 7.0%, 88.8 ± 9.6%, and 81.6 ± 6.6% of the control values, respectively (n = 12, N = 6; P < 0.05; paxilline versus rolipram; Fig. 8G). These data support that rolipram indirectly stimulates BK channel activity and counteracts MR activation, which is known to inhibit TBKCs in DSM (Parajuli and Petkov, 2013).
Fig. 8.

Selective PDE4 inhibition with rolipram significantly reduces carbachol-induced contractions of DSM strips in a concentration-dependent manner. (A) A representative recording of a DSM strip depicts the concentration-dependent inhibitory effects of rolipram (0.1 nM–10 µM) on 0.1 µM carbachol-induced contractions. Post-treatment of the DSM strip with paxilline (1 μM) reversed the inhibitory effect of rolipram. (B–F) Cumulative concentration-response curves illustrating the effect of rolipram on the amplitude (B), muscle force integral (C), duration (D), frequency (E), and tone (F) of carbachol-induced contractions in DSM strips (n = 13, N = 7; *P < 0.05 versus control). (G) Summary data illustrating that addition of 1 μM paxilline following application of 10 µM rolipram significantly recovered contraction amplitude, muscle force integral, frequency, and tone in DSM-isolated strips (n = 12, N = 6; #P < 0.05, paxilline versus 10 μM rolipram; P > 0.05; NS, nonsignificant). TTX (1 μM) was present throughout the experiments. Amp, amplitude; Dur, duration; Freq, frequency; Rol, rolipram.
Selective PDE4 Inhibition with Rolipram Reduced the EFS-Induced DSM Contractions.
EFS causes a release of the excitatory neurotransmitter, ACh, which activate MRs, and consequently increases the contractility in DSM-isolated strips. All EFS experiments were conducted in the absence of TTX. Rolipram inhibited the 20-Hz EFS-induced contraction amplitude, muscle force integral, and muscle tone in a concentration-dependent manner with IC50 values of 110 nM (46.4–260.1 nM), 101 nM (53.6–191.6 nM), and 365 nM (1 nM–1 M), respectively (n = 10, N = 6; P < 0.05; Fig. 9). At the maximal concentration used (10 μM), rolipram reduced the contraction amplitude, muscle force integral, and muscle tone to 40.9 ± 5.9%, 36.4 ± 5.3%, and 77.3 ± 8.6% of the control values, respectively (n = 10, N = 6; P < 0.05; Fig. 9). Subsequent inhibition of the BK channels with paxilline (1 μM) restored the contraction amplitude and muscle force back to 47.6 ± 5.5% and 47.5 ± 6.3% of the control values, respectively (n = 8, N = 5; P < 0.05 versus 10 μM rolipram; Fig. 9D), without significant effects on contraction duration and tone (n = 8, N = 5; P > 0.05 versus 10 μM rolipram; Fig. 9D). This indicates that selective PDE4 inhibition can oppose the contractile effects of excitatory neurotransmitters on DSM contractility, and this process is facilitated by BK channel activation.
Fig. 9.

Selective PDE4 inhibition with rolipram significantly reduces 20 Hz EFS-induced contractions of DSM-isolated strips in a concentration-dependent manner. (A) A representative recording of a DSM strip showing the concentration-dependent inhibitory effects of rolipram (0.1 nM-10 µM) on EFS-induced contractions. Post-treatment of the DSM strip with paxilline (1 μM) recovered the EFS-induced contractions. (B–D) Cumulative concentration-response curves for the inhibitory effects of rolipram on DSM contraction amplitude (B), muscle force integral (C), and muscle tone (D) (n = 10, N = 6; *P < 0.05, versus control). (E) Summary data illustrating that addition of 1 μM paxilline following application of 10 µM rolipram significantly recovered the EFS-induced contraction amplitude and muscle force in DSM-isolated strips (n = 8, N = 5; #P < 0.05 paxilline versus 10 μM rolipram; P > 0.05; NS, nonsignificant). Amp, amplitude; pax, paxilline; Rol, rolipram.
Selective PDE4 Inhibition with Ro-20-1724 Leads to an Inhibition of DSM Spontaneous, Carbachol-, and EFS-Induced Contractions.
To confirm that the relaxation effect of rolipram on DSM was due to the inhibition of PDE4 activity, we investigated the effect of Ro-20-1724, another PDE4 selective inhibitor, on DSM contractility. Ro-20-1724 inhibited DSM spontaneous phasic contractions (n = 7, N = 6; P < 0.05; Fig. 10, A and D), carbachol-induced phasic contractions (n = 12, N = 5; P < 0.05; Fig. 10, B and E), and EFS-induced contractions (n = 10, N = 5; P < 0.05; Fig. 10, C and F) in a concentration-dependent manner. In the presence of Ro-20-1724, post-treatment of DSM strips with paxilline (1 µM) recovered DSM spontaneous (n = 7, N = 6; P < 0.05; Fig. 10, A and D), carbachol-induced (n = 12, N = 5; P < 0.05; Fig. 10, B and E), and EFS-induced contractions (n = 10, N = 5; P < 0.05; Fig. 10, C and F). The mean values of all contraction parameters following PDE4 inhibition by Ro-20-1724 (1 μM) are listed in Table 1. These data confirm that selective pharmacological inhibition of PDE4 is highly effective in suppressing DSM spontaneous, carbachol-induced phasic contractions, and EFS-induced contractions, and that BK channels mediate the regulatory effects of PDE4 on DSM contractility.
Fig. 10.
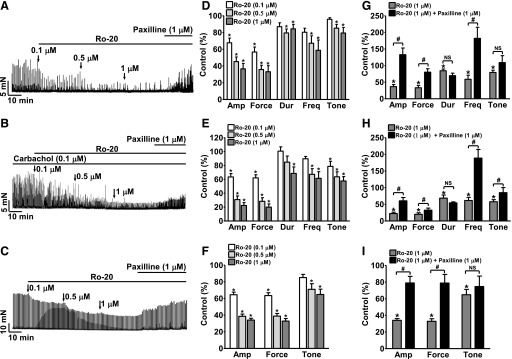
Selective PDE4 inhibition with Ro-20-1724 reduces the spontaneous, carbachol-, and EFS-induced contractions of DSM-isolated strips in a concentration-dependent manner. (A–C) Original recordings of DSM strips showing the concentration-dependent inhibitory effects of Ro-20-1724 (0.1 µM–1 µM) on spontaneous phasic, carbachol-induced, and EFS-induced contractions. (D–F) Summary data showing the concentration-dependent inhibitory effect of Ro-20-1724 on the spontaneous phasic (n = 7, N = 6), carbachol-induced (n = 12, N = 5), and EFS-induced (n = 10, N = 5) contractions in DSM-isolated strips (*P < 0.05 versus control). (G–I) Summary data illustrating that addition of 1 μM paxilline following application of 1 µM Ro-20-1724 significantly recovered the spontaneous phasic (n = 7, N = 6), carbachol-induced (n = 12, N = 5), and EFS-induced (n = 9, N = 5) contractions (*P < 0.05 versus control; #P < 0.05 paxilline versus 1 μM Ro-20-1724; P > 0.05; NS, nonsignificant). TTX (1 μM) was present throughout the experiments except the EFS-induced contractions. Ro-20 refers to Ro-20-1724. Amp, amplitude; Dur, duration; Freq, frequency.
TABLE 1.
Selective pharmacological inhibition of PDE4 with Ro-20-1724 (1 μM) reduces DSM contractions and the post-treatment with paxilline (1 μM) recovered DSM contractility
Data are expressed as means ± S.E.M. in percentage of control.
| Condition | Compound | Amplitude | Force | Duration | Frequency | Tone | n/N |
|---|---|---|---|---|---|---|---|
| Spontaneous contraction | Ro-20-1724 | 36.6 ± 6.5 | 33.2 ± 7.2 | 84.4 ± 5.3 | 58.8 ± 12.3 | 79.5 ± 6.6 | 7/6 |
| P < 0.05 | P < 0.05 | P < 0.05 | P < 0.05 | P < 0.05 | |||
| Ro-20-1724 | 133.0 ± 20.1 | 80.3 ± 10.1 | 69.7 ± 7.5 | 182.1 ± 33.5 | 109.1 ± 20.8 | 7/6 | |
| + Paxilline | P < 0.05 | P < 0.05 | P < 0.05 | P < 0.05 | P < 0.05 | ||
| Carbachol-induced contraction | Ro-20-1724 | 22.4 ± 4.0 | 19.9 ± 4.6 | 68.6 ± 9.3 | 61.5 ± 9.5 | 57.6 ± 6.4 | 12/5 |
| P < 0.05 | P < 0.05 | P < 0.05 | P < 0.05 | P < 0.05 | |||
| Ro-20-1724 | 60.4 ± 9.9 | 32.5 ± 6.1 | 54.3 ± 3.0 | 188.7 ± 25.9 | 84.9 ± 15.2 | 12/5 | |
| + Paxilline | P < 0.05 | P < 0.05 | P < 0.05 | P < 0.05 | P < 0.05 | ||
| EFS-induced contraction | Ro-20-1724 | 34.0 ± 2.1 | 32.9 ± 3.1 | 99.4 ± 5.3 | N/A | 64.8 ± 6.3 | 10/5 |
| P < 0.05 | P < 0.05 | P > 0.05 | P < 0.05 | ||||
| Ro-20-1724 | 78.9 ± 7.9 | 78.8 ± 10.4 | 98.4 ± 4.8 | N/A | 74.6 ± 12.5 | 9/5 | |
| + Paxilline | P < 0.05 | P < 0.05 | P > 0.05 | P > 0.05 |
EFS, electrical field stimulation; N/A, not applicable. The P value less than 0.05 is considered statistically significant.
Discussion
The present study reveals a novel mechanism by which PDE4 regulates DSM excitability and contractility and the role that BK channels serve in this process. Specifically, the study demonstrates that the constitutively active PDE4 modulates spontaneous and nerve-evoked DSM contractions and that pharmacological inhibition of PDE4 reduces DSM contractility by increasing the frequency of Ca2+ sparks and the functionally coupled TBKCs. This hyperpolarizes the DSM cell MP, thus attenuating CaV channel activity and lowering the global intracellular Ca2+ concentration.
Ca2+ sparks and TBKC frequency increases when intracellular cAMP level is elevated in DSM cells (Petkov and Nelson, 2005; Xin et al., 2012c). Inhibition of the cAMP-specific PDE4 can raise cellular cAMP levels (Bender and Beavo, 2006), and our data showed that PDE4 inhibition with rolipram (10 μM) increased Ca2+ spark frequency (Fig. 1). This is consistent with the observations that Ca2+ spark frequency significantly increased in smooth muscle cells treated with forskolin or isoproterenol, which increase cellular cAMP levels via the activation of adenylyl cyclases (Wellman et al., 2001; Petkov and Nelson, 2005). As a consequence, the increase in Ca2+ spark frequency led to an increase in TBKC frequency (Fig. 2). In turn, TBKCs hyperpolarized DSM cell MP (Fig. 3) and decreased the global intracellular Ca2+ (Fig. 5). Additionally, it is also possible that the elevated cAMP level increases Ca2+ uptake into the sarcoplasmic reticulum and this increase in Ca2+ reuptake would enhance Ca2+ spark frequency and reduce cytosolic Ca2+. In addition, the effect of rolipram on DSM cell MP was abolished by H-89, a PKA inhibitor (Fig. 4), which supports the concept that PKA is important in regulating TBKC activity, and thus DSM excitability (Petkov and Nelson, 2005; Brown et al., 2008; Hristov et al., 2008). This signal transduction pathway is depicted in Fig. 11. The elevation in PKA activity (Fig. 11A) leads to an increase in Ca2+ spark and TBKC activities (Fig. 11B), DSM cell hyperpolarization, and a decrease in global Ca2+ levels (Fig. 11C).
Fig. 11.

Proposed novel signaling pathway by which PDE4 regulates BK channel activity in DSM cells. This diagram illustrates that the pharmacological inhibition of PDE4 leads to activation of PKA (A), increase in Ca2+ spark and TBKC activities (B), and hyperpolarization of DSM cell membrane and a consequent decrease in global Ca2+ levels (C). AC, adenylyl cyclase; BK, large conductance voltage- and Ca2+-activated K+ channel; CaV, L-type voltage-gated Ca2+ channel; pPLB, phosphorylated phospholamban; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; TBKC, transient BK current.
Before this study, the mechanism of PDE4 regulation of DSM contractility has not been fully investigated (Longhurst et al., 1997; Nishiguchi et al., 2007; Oger et al., 2007). Our data revealed that PDE4 inhibition attenuated the myogenic, carbachol-induced, and EFS-induced contractility of DSM-isolated strips by a BK channel-dependent mechanism (Figs. 6–10). Specifically, the current study shows that the inhibition of PDE4 reduced the global Ca2+ levels and consequently attenuated DSM spontaneous contractions in a BK channel-dependent manner (Figs. 5, 6, and 10). Figure 11 illustrates that inhibition of PDE4 and the subsequent decrease in the global Ca2+ levels (Fig. 11, A–C) lead to relaxation of DSM. These data further demonstrate the functional interactions among PDE4, cellular Ca2+ signals, and the myogenic DSM contractility.
In contrast to the inhibition of PDE4, the activation of MRs by carbachol, a chemically stable MR agonist, or ACh released from parasympathetic nerves increases DSM phasic contractions (Hashitani et al., 2004; Tsai et al., 2012). Specifically, activation of Gi-coupled M2Rs leads to an inhibition of adenylyl cyclases and a reduction in cAMP production and PKA activity (Shahab et al., 2012). Meanwhile, activation of Gq-coupled M3Rs evokes Ca2+ release via inositol trisphosphate receptors, increases protein kinase C activity, inhibits TBKCs, and depolarizes DSM cell MP (Parajuli and Petkov, 2013). As a consequence, activation of MRs, particularly M3R activation, increases Ca2+ influx, elevates intracellular Ca2+ levels, and enhances DSM contractility (Shahab et al., 2012; Tsai et al., 2012; Parajuli and Petkov, 2013; Yu et al., 2013). The current study indicates that the selective inhibition of PDE4 counteracts the activation of MRs and suppresses carbachol- and EFS-induced DSM contractions (Figs. 8–10). However, the ability of PDE4 to regulate DSM contractility is dramatically diminished upon BK channel blockade (Figs. 6–10). This BK channel dependency of DSM regulation by PDE4 indicates that PDEs, BK channels, and MR signaling pathways functionally interact with each other. Therefore, these results further demonstrate that BK channels are important in regulating DSM excitability and contractility consistent with the findings that genetic deletion of BK channels in mice leads to detrusor overactivity (Meredith et al., 2004; Brown et al., 2008; Sprossmann et al., 2009).
In this study, the functional importance of PDE4 in regulating DSM contractility was demonstrated by using two selective PDE4 inhibitors, rolipram and Ro-20-1724. Rolipram and Ro-20-1724 have IC50 values of ~1–2 μM and ∼3 μM, and their IC50 values are more than 300 and 100 µM for other major cAMP-PDEs, respectively (Reeves et al., 1987; Rybalkin et al., 2002). Both rolipram and Ro-20-1724 significantly inhibited DSM contractions in a concentration-dependent manner, and their inhibitory effects were mediated by the BK channels (Figs. 6–10).
It is particularly important to note that PDE4D mRNA expression levels are high in the human bladder and low in the heart (Lakics et al., 2010; Johnson et al., 2012). Furthermore, rolipram (10 μM) inhibits less than 15% of the total cAMP-PDE activity in human ventricular myocytes (Johnson et al., 2012). In addition, the inhibition of PDE4 does not affect isolated human cardiomyocyte contractility, and therefore, selective PDE4 inhibitors are unlikely to cause cardiac side effects (Johnson et al., 2012; Molenaar et al., 2013). Furthermore, different PDE4 isoforms are localized in distinct cellular compartments by binding different scaffold proteins, such as PKA-anchoring proteins, to form a localized signal transduction complex (Stefan et al., 2007; Blackman et al., 2011). Taken together, drugs targeting bladder-specific PDE4 isoforms could have high DSM specificity with limited side effects on cardiac function.
In summary, the current study reveals a novel BK channel-dependent mechanism of PDE4 regulation of DSM function. Selective pharmacological inhibition of PDE4 leads to an increase in Ca2+ spark activity and a decrease in the global Ca2+ levels leading to DSM relaxation. This novel signaling pathway is depicted in Fig. 11. The current study indicates that PDE4 isoforms might be valuable therapeutic targets for the treatment of overactive bladder.
Acknowledgments
The authors thank Dr. John Malysz, Dr. Shankar P. Parajuli, Dr. Kiril L. Hristov, Amy Smith, Aaron Provence, and Vitor S. Fernandes for the critical evaluation of the manuscript.
Abbreviations
- ACh
acetylcholine
- BK
large conductance voltage- and Ca2+-activated K+
- AM
acetoxymethyl ester
- BSA
bovine serum albumin
- CaV
L-type voltage-gated Ca2+
- CI
confidence interval
- DSM
detrusor smooth muscle
- EFS
electrical field stimulation
- H-89
N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide dihydrochloride
- MP
membrane potential
- MR
muscarinic receptor
- PDE
phosphodiesterase
- PKA
protein kinase A
- Ro-20-1724
C15H22N2O3
- RyR
ryanodine receptor
- TBKC
transient BK current
- TTX
tetrodotoxin
Authorship Contributions
Participated in research design: Xin, Li, Petkov.
Conducted experiments: Xin, Li, Cheng, Petkov.
Performed data analysis: Xin, Li, Cheng, Petkov.
Wrote or contributed to the writing of the manuscript: Xin, Petkov.
Footnotes
This work was supported by the National Institutes of Health [Grant R01-DK084284 to G.V.P.].
References
- Beca S, Helli PB, Simpson JA, Zhao D, Farman GP, Jones PP, Tian X, Wilson LS, Ahmad F, Chen SR, et al. (2011) Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ Res 109:1024–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender AT, Beavo JA. (2006) Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58:488–520 [DOI] [PubMed] [Google Scholar]
- Blackman BE, Horner K, Heidmann J, Wang D, Richter W, Rich TC, Conti M. (2011) PDE4D and PDE4B function in distinct subcellular compartments in mouse embryonic fibroblasts. J Biol Chem 286:12590–12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brading AF. (2006) Spontaneous activity of lower urinary tract smooth muscles: correlation between ion channels and tissue function. J Physiol 570:13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SM, Bentcheva-Petkova LM, Liu L, Hristov KL, Chen M, Kellett WF, Meredith AL, Aldrich RW, Nelson MT, Petkov GV. (2008) β-adrenergic relaxation of mouse urinary bladder smooth muscle in the absence of large-conductance Ca2+-activated K+ channel. Am J Physiol Renal Physiol 295:F1149–F1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashitani H, Brading AF, Suzuki H. (2004) Correlation between spontaneous electrical, calcium and mechanical activity in detrusor smooth muscle of the guinea-pig bladder. Br J Pharmacol 141:183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner TJ, Bonev AD, Nelson MT. (1997) Ca2+-activated K+ channels regulate action potential repolarization in urinary bladder smooth muscle. Am J Physiol 273:C110–C117 [DOI] [PubMed] [Google Scholar]
- Herrera GM, Heppner TJ, Nelson MT. (2001) Voltage dependence of the coupling of Ca(2+) sparks to BK(Ca) channels in urinary bladder smooth muscle. Am J Physiol Cell Physiol 280:C481–C490 [DOI] [PubMed] [Google Scholar]
- Horner A, Goetz F, Tampé R, Klussmann E, Pohl P. (2012) Mechanism for targeting the A-kinase anchoring protein AKAP18δ to the membrane. J Biol Chem 287:42495–42501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houslay MD, Schafer P, Zhang KY. (2005) Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today 10:1503–1519 [DOI] [PubMed] [Google Scholar]
- Hristov KL, Afeli SA, Parajuli SP, Cheng Q, Rovner ES, Petkov GV. (2013) Neurogenic detrusor overactivity is associated with decreased expression and function of the large conductance voltage- and Ca(2+)-activated K(+) channels. PLoS One 8:e68052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristov KL, Chen M, Kellett WF, Rovner ES, Petkov GV. (2011) Large-conductance voltage- and Ca2+-activated K+ channels regulate human detrusor smooth muscle function. Am J Physiol Cell Physiol 301:C903–C912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristov KL, Cui X, Brown SM, Liu L, Kellett WF, Petkov GV. (2008) Stimulation of β3-adrenoceptors relaxes rat urinary bladder smooth muscle via activation of the large-conductance Ca2+-activated K+ channels. Am J Physiol Cell Physiol 295:C1344–C1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristov KL, Parajuli SP, Soder RP, Cheng Q, Rovner ES, Petkov GV. (2012) Suppression of human detrusor smooth muscle excitability and contractility via pharmacological activation of large conductance Ca2+-activated K+ channels. Am J Physiol Cell Physiol 302:C1632–C1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WB, Katugampola S, Able S, Napier C, Harding SE. (2012) Profiling of cAMP and cGMP phosphodiesterases in isolated ventricular cardiomyocytes from human hearts: comparison with rat and guinea pig. Life Sci 90:328–336 [DOI] [PubMed] [Google Scholar]
- Lakics V, Karran EH, Boess FG. (2010) Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 59:367–374 [DOI] [PubMed] [Google Scholar]
- Longhurst PA, Briscoe JA, Rosenberg DJ, Leggett RE. (1997) The role of cyclic nucleotides in guinea-pig bladder contractility. Br J Pharmacol 121:1665–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. (2004) Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem 279:36746–36752 [DOI] [PubMed] [Google Scholar]
- Molenaar P, Christ T, Hussain RI, Engel A, Berk E, Gillette KT, Chen L, Galindo-Tovar A, Krobert KA, Ravens U, et al. (2013) PDE3, but not PDE4, reduces β₁- and β₂-adrenoceptor-mediated inotropic and lusitropic effects in failing ventricle from metoprolol-treated patients. Br J Pharmacol 169:528–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi J, Kwon DD, Kaiho Y, Chancellor MB, Kumon H, Snyder PB, Yoshimura N. (2007) Suppression of detrusor overactivity in rats with bladder outlet obstruction by a type 4 phosphodiesterase inhibitor. BJU Int 99:680–686 [DOI] [PubMed] [Google Scholar]
- Oger S, Behr-Roussel D, Gorny D, Denys P, Lebret T, Alexandre L, and Giuliano F (2007) Relaxation of phasic contractile activity of human detrusor strips by cyclic nucleotide phosphodiesterase type 4 inhibition. Eur Urol 51:772–780; discussion 780–781. [DOI] [PubMed]
- Parajuli SP, Petkov GV. (2013) Activation of muscarinic M3 receptors inhibits large-conductance voltage- and Ca2+-activated K+ channels in rat urinary bladder smooth muscle cells. Am J Physiol Cell Physiol 305:C207–C214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parajuli SP, Soder RP, Hristov KL, Petkov GV. (2012) Pharmacological activation of small conductance calcium-activated potassium channels with naphtho[1,2-d]thiazol-2-ylamine decreases guinea pig detrusor smooth muscle excitability and contractility. J Pharmacol Exp Ther 340:114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkov GV. (2011) Role of potassium ion channels in detrusor smooth muscle function and dysfunction. Nat Rev Urol 9:30–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkov GV, Heppner TJ, Bonev AD, Herrera GM, Nelson MT. (2001) Low levels of K(ATP) channel activation decrease excitability and contractility of urinary bladder. Am J Physiol Regul Integr Comp Physiol 280:R1427–R1433 [DOI] [PubMed] [Google Scholar]
- Petkov GV, Nelson MT. (2005) Differential regulation of Ca2+-activated K+ channels by β-adrenoceptors in guinea pig urinary bladder smooth muscle. Am J Physiol Cell Physiol 288:C1255–C1263 [DOI] [PubMed] [Google Scholar]
- Picht E, Zima AV, Blatter LA, Bers DM. (2007) SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol 293:C1073–C1081 [DOI] [PubMed] [Google Scholar]
- Porta M, Zima AV, Nani A, Diaz-Sylvester PL, Copello JA, Ramos-Franco J, Blatter LA, Fill M. (2011) Single ryanodine receptor channel basis of caffeine’s action on Ca2+ sparks. Biophys J 100:931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves ML, Leigh BK, England PJ. (1987) The identification of a new cyclic nucleotide phosphodiesterase activity in human and guinea-pig cardiac ventricle: implications for the mechanism of action of selective phosphodiesterase inhibitors. Biochem J 241:535–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter W, Jin SL, Conti M. (2005) Splice variants of the cyclic nucleotide phosphodiesterase PDE4D are differentially expressed and regulated in rat tissue. Biochem J 388:803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalkin SD, Rybalkina I, Beavo JA, Bornfeldt KE. (2002) Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ Res 90:151–157 [DOI] [PubMed] [Google Scholar]
- Sausbier M, Zhou XB, Beier C, Sausbier U, Wolpers D, Maget S, Martin C, Dietrich A, Ressmeyer AR, Renz H, et al. (2007) Reduced rather than enhanced cholinergic airway constriction in mice with ablation of the large conductance Ca2+-activated K+ channel. FASEB J 21:812–822 [DOI] [PubMed] [Google Scholar]
- Sellers DJ, Chess-Williams R. (2012) Muscarinic agonists and antagonists: effects on the urinary bladder. Handbook Exp Pharmacol 208:375–400 [DOI] [PubMed] [Google Scholar]
- Shahab N, Kajioka S, Seki N, and Naito S (2012) Functional role of muscarinic receptor subtypes in calcium sensitization and their contribution to rho-kinase and protein kinase C pathways in contraction of human detrusor smooth muscle. Urology 79:1184.e7–e13. [DOI] [PubMed]
- Sprossmann F, Pankert P, Sausbier U, Wirth A, Zhou XB, Madlung J, Zhao H, Bucurenciu I, Jakob A, Lamkemeyer T, et al. (2009) Inducible knockout mutagenesis reveals compensatory mechanisms elicited by constitutive BK channel deficiency in overactive murine bladder. FEBS J 276:1680–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan E, Wiesner B, Baillie GS, Mollajew R, Henn V, Lorenz D, Furkert J, Santamaria K, Nedvetsky P, Hundsrucker C, et al. (2007) Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J Am Soc Nephrol 18:199–212 [DOI] [PubMed] [Google Scholar]
- Tsai MH, Kamm KE, Stull JT. (2012) Signalling to contractile proteins by muscarinic and purinergic pathways in neurally stimulated bladder smooth muscle. J Physiol 590:5107–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman GC, Santana LF, Bonev AD, Nelson MT. (2001) Role of phospholamban in the modulation of arterial Ca(2+) sparks and Ca(2+)-activated K(+) channels by cAMP. Am J Physiol Cell Physiol 281:C1029–C1037 [DOI] [PubMed] [Google Scholar]
- Xin W, Cheng Q, Soder RP, Petkov GV. (2012a) Inhibition of phosphodiesterases relaxes detrusor smooth muscle via activation of the large-conductance voltage- and Ca²⁺-activated K⁺ channel. Am J Physiol Cell Physiol 302:C1361–C1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin W, Cheng Q, Soder RP, Rovner ES, Petkov GV. (2012b) Constitutively active phosphodiesterase activity regulates urinary bladder smooth muscle function: critical role of KCa1.1 channel. Am J Physiol Renal Physiol 303:F1300–F1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin W, Soder RP, Cheng Q, Rovner ES, Petkov GV. (2012c) Selective inhibition of phosphodiesterase 1 relaxes urinary bladder smooth muscle: role for ryanodine receptor-mediated BK channel activation. Am J Physiol Cell Physiol 303:C1079–C1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin W, Yang X, Rich TC, Krieg T, Barrington R, Cohen MV, Downey JM. (2012d) All preconditioning-related G protein-coupled receptors can be demonstrated in the rabbit cardiomyocyte. J Cardiovasc Pharmacol Ther 17:190–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Sun X, Robson SC, Hill WG. (2013) Extracellular UDP enhances P2X-mediated bladder smooth muscle contractility via P2Y(6) activation of the phospholipase C/inositol trisphosphate pathway. FASEB J 27:1895–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]