

Abstract
Sulfotransferase (SULT) 2A1 catalyzes sulfonation of drugs and endogenous compounds and plays an important role in xenobiotic metabolism as well as in the maintenance of steroid and lipid homeostasis. A recent study showed that 17β-estradiol (E2) increases the mRNA levels of SULT2A1 in human hepatocytes. Here we report the underlying molecular mechanisms. E2 enhanced SULT2A1 expression in human hepatocytes and HepG2-ER cells (HepG2 stably expressing ERα). SULT2A1 induction by E2 was abrogated by antiestrogen ICI 182,780, indicating a key role of ERα in the induction. Results from deletion and mutation assays of SULT2A1 promoter revealed three cis-elements located within –257/+140 region of SULT2A1 that are potentially responsible for the induction. Chromatin immunoprecipitation assay verified the recruitment of ERα to the promoter region. Electrophoretic mobility shift assays revealed that AP-1 proteins bind to one of the cis-elements. Interestingly, SULT2A1 promoter assays using ERα mutants revealed that the DNA-binding domain of ERα is indispensable for SULT2A1 induction by E2, suggesting that direct ERα binding to the SULT2A1 promoter is also necessary for the induction. Taken together, our results indicate that E2 enhances SULT2A1 expression by both the classical and nonclassical mechanisms of ERα action.
Introduction
17β-Estradiol (E2) is a key regulator of growth, differentiation, and function in a wide array of target tissues, including female reproductive tracts, mammary gland, and cardiovascular systems (Knowlton and Lee, 2012; Villa et al., 2012). Estrogens induce expression of certain hepatic drug-metabolizing enzymes including CYP2A6, CYP2B6, and UGT1A4 (Higashi et al., 2007; Chen et al., 2009; Lo et al., 2010; Koh et al., 2012), and estrogens are potentially responsible for the reported sex differences in drug metabolism or drug–drug interaction involving oral contraceptives (Christensen et al., 2007; Al Koudsi et al., 2010). The effects of estrogen on drug metabolism by other drug-metabolizing enzymes remain unclear.
Results from our recent cDNA microarray experiments revealed that E2 upregulates expression of a phase II enzyme, sulfotransferase (SULT) 2A1, in human hepatocytes (Koh et al., 2012). SULT2A1 is the major SULT2 enzyme expressed in liver and responsible for detoxification of xenobiotics by metabolizing drugs such as tibolone and raloxifene. In addition, SUTL2A1 is involved in maintaining homeostasis of steroid hormones and lipids by sulfonating endogenous compounds, such as dehydroepiandrosterone (DHEA) and bile acids (Falany et al., 1995; He et al., 2004; Labrie et al., 2005; Riches et al., 2009).
Expression of SULT2A1 is regulated at the transcriptional level by multiple nuclear receptors including constitutive androstane receptor and pregnane X receptor (Echchgadda et al., 2007). The role of E2 in the regulation of SULT2A1 expression and the underlying molecular mechanisms remain unknown. Estrogen regulates expression of its target genes by binding to cognate nuclear receptors, estrogen receptor (ER) α and ERβ. While expression of ERβ is localized to the cholangiocytes (Alvaro et al., 2002), ERα is the major isoform expressed in the parenchymal cells in livers (Kuiper et al., 1997). Upon activation by E2, ER regulates the expression of target genes by directly binding to estrogen response element (ERE) (i.e., classical mechanism). Alternatively, ligand-activated ER can be tethered to other transcriptional regulators (such as AP-1 proteins) and modulate expression of target genes (i.e., nonclassical mechanism) (Safe and Kim, 2008).
In the current study, we further characterized the effects of E2 on SULT2A1 expression and examined the role of ERα in the transcriptional regulation of SULT2A1 expression. Our results indicate that E2 enhances SULT2A1 expression by both the classical and nonclassical mechanisms of ERα action.
Materials and Methods
Chemicals and Reagents.
E2, DHEA, DHEA sulfate, 3′-phosphoadenosine 5′-phosphosulfate, mebendazole, cycloheximide, and ICI 182,780 were purchased from Sigma-Aldrich (St. Louis, MO).
Cell Culture.
Freshly isolated human hepatocytes (HHs) from two different donors (i.e., HH1 and HH2) were obtained from Liver Tissue Cell Distribution System (Pittsburgh, PA), which was funded by NIH Contract #N01-DK-7-0004 / HHSN267200700004C. No demographic information was available for HH1, while HH2’s donor was a 76-year old Caucasian. Upon receipt, media were replaced with serum-free Williams’ E media (ThermoScientific, Logan, UT), and the cells were cultured as previously described (Koh et al., 2012). HepG2 cells (from ATCC, Manassas, VA) and HepG2-ER cells (HepG2 cells stably expressing ERα; kindly provided by Dr. David Shapiro, University of Illinois at Urbana-Champaign) were cultured as previously described (Koh et al., 2012).
Quantitative Reverse-Transcription Polymerase Chain Reaction.
Total RNAs were isolated using TRIzol (Life Technologies, Carlsbad, CA). cDNA was synthesized using High Capacity cDNA Archive Kit (Life Technologies). Real-time polymerase chain reaction (PCR) was performed using FastStart Universal Probe Master for TaqMan probe assays (Roche, Indianapolis, IN), and fold changes in mRNA levels of genes were determined after normalizing the gene expression levels by those of GAPDH (2–ΔΔCt method). Commercially available TaqMan primers for human SULT2A1 were used (Hs00234219_m1; Life Technologies). Primers for GAPDH are shown in Supplemental Table 1.
Western Blotting.
Protein extracts (1 μg for human hepatocytes and 30 μg for HepG2-ER cells) were separated by electrophoresis on a 10% denaturing SDS gel and transferred to polyvinylidene fluoride membrane using standard procedures. Blots were blocked with 5% skim milk/Tris buffered saline containing 0.1% Tween 20 (TBST) and incubated overnight at 4°C with antibody against SULT2A1 (1:1500 dilution; Proteintech, Chicago, IL). The membranes were then washed in TBST before incubation with horseradish peroxidase–coupled anti-rabbit IgG or anti-mouse IgG antibodies at room temperature. The membranes were visualized by an enhanced chemiluminescence detection system (SuperSignal West Pico; ThermoScientific, Rockford, IL) and FluorChem E imager (Cell Biosciences, Santa Clara, CA).
Plasmids.
Upstream regulatory region (–826/+140) of SULT2A1 was subcloned from pLightSwitch_Prom_SULT2A1 (SwitchGear Genomics, Menlo Park, CA) into pGL3-Basic (Promega, Wisconsin, MI) at KpnI and XhoI restriction enzyme sites, the resulting plasmid being pGL3-SULT2A1. 5′-Deletion constructs of SULT2A1 promoter were then constructed using the primers shown in Supplemental Table 1. Mutation constructs of pGL3-SULT2A1 were prepared using a QuikChange XL Site-Directed Mutagenesis Kit (Agilent Technologies, La Jolla, CA) following the manufacturer’s protocol and using the primers listed in Supplemental Table 1. pGL3-ERE3 and expression vectors for wild-type or for mutant ERα were previously described (Chen et al., 2009).
Dual-Luciferase Assay.
HepG2 cells were transfected with 0.7 μg plasmid (0.3 μg luciferase construct, 0.3 μg pcDNA3-ERα and pcDNA3, and 0.1 μg pCMV-Renilla) using FuGene HD (Promega, Madison, WI). Luciferase activity was detected by using Dual-Luciferase Reporter Assay System (Promega).
Chromatin Immunoprecipitation Assay.
Chromatin immunoprecipitation (ChIP) assay for ERα protein was performed as previously described (Koh et al., 2012). Briefly, HepG2-ER cells were treated with vehicle (0.1% ethanol) or E2 (100 nM) for 45 minutes, and the cell lysates were collected. After crosslinking, sonication, immunoprecipitation (using IgG or anti-ERα antibody; Santa Cruz Biotechnology, Dallas, TX), and de-crosslinking, the DNA was detected by using quantitative real-time PCR (qRT-PCR) using the primers shown in Supplemental Table 1.
Electrophoretic Mobility Shift Assay.
Electromobility shift assay (EMSA) was performed as previously described (Koh et al., 2012). Briefly, nuclear proteins (15 μg) or recombinant ERα (1 ng; ThermoScientific) were preincubated with binding buffer [4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, 10 mM Tris-HCl (pH 7.5), and 0.05 mg/ml poly(dI-dC)] at room temperature in the presence or absence of nonradioactive DNA probe (for competition) or antibodies against c-Jun, c-Fos, and ERα (Santa Cruz Biotechnology). After 10 minutes, the binding reaction was initiated by adding 0.035 pmol of 5′-end 32P-labeled SULT2A1 probes harboring putative ERα or AP-1-binding sequences (Supplemental Table 1). The reaction mixture was incubated at room temperature for 20 minutes. Protein-bound probes were separated from free probes on 4% (w/v) nondenaturing polyacrylamide gel. The gel was dried, and radioactivity was visualized by using PhosphorImager.
Measurement of SULT2A1 Activity.
HepG2-ER cells were treated with vehicle (0.1% ethanol) or E2 (100 nM) for 72 hours. The cells were harvested and then resuspended in 1.5 ml of ice-cold buffer (50 mM Tris-HCl, 150 mM KCl, 2 mM EDTA, and 250 mM sucrose, pH 7.0), followed by homogenization using a Teflon-glass homogenizer. The homogenate was centrifuged at 9000g for 20 minutes at 4°C, and the supernatants (S9) were collected. The S9 (100 μg) was incubated with DHEA (5 μM) in a buffer (100 mM Tris-HCl, 20 mM MgCl2, and 200 μM 3′-phosphoadenosine 5′-phosphosulfate, pH 7.4). The reactions were terminated after 30 minutes incubation at 37°C by adding 100 μl ice-cold acetonitrile containing mebendazole (1 μM) as internal standard (IS) and kept on ice for 30 minutes, followed by centrifugation at 16,100g for 15 minutes at 4°C. The concentrations of DHEA sulfate in the supernatant were determined using high-performance liquid chromatography (HPLC) (Agilent 1200) coupled to a 5500 QTRAP triple quadrupole mass spectrometer (Applied Biosystems, Foster City, CA). Chromatographic separation was performed on an XTerra MS C18 column (3.5 μm, 2.1 × 50 mm; Waters Corp., Milford, MA). The mobile phase was water containing 5 mM ammonium acetate (A) and acetonitrile (B). The HPLC gradient elution program started at 10% B, ramped linearly to 90% B over 1 minute, held at 90% B for 2 minutes, and then brought back to the initial condition in 0.5 minutes followed by 6.5 minutes equilibration. Ionization was performed using an electrospray ionization (ESI) source, which was operating in the negative mode. The multiple reaction monitoring (MRM) transitions for DHEA sulfate and IS were 367.06→96.90 and 293.99→262.10, respectively.
Statistical Analysis.
All data were presented as means ± SD. Statistical analyses were performed by using one-way analysis of variance (ANOVA) followed by Dunnett’s post-hoc test for multiple comparisons. The student's t-test was used for comparison of two groups. A P value < 0.05 was considered statistically significant.
Results
E2 Upregulates SULT2A1 via ERα.
Previous microarray results indicate that E2 upregulates SULT2A1 expression in human hepatocytes (Koh et al., 2012). To verify the data, two different batches of primary human hepatocytes (HH1 and HH2) were treated with E2 or vehicle control, and SULT2A1 mRNA expression levels were determined by qRT-PCR. The results showed that mRNA expression levels of SULT2A1 were increased by E2 by 1.6- and 1.9-fold in HH1 and HH2, respectively, as compared with those in the vehicle-treated cells (Fig. 1A). The extent of increases in SULT2A1 mRNA expression by E2 was comparable to that by rifampicin, a known inducer of SULT2A1 expression (Echchgadda et al., 2007). Results from Western blot showed a 2-fold increase in SULT2A1 protein level in E2-treated group in human hepatocytes (Fig. 1B). In the following studies, HepG2-ER cells (HepG2 cells stably expressing ER) were used due to the limited availability of primary human hepatocytes. Similar to the results in human hepatocytes, E2-enhanced the mRNA and protein expression of SULT2A1 by ∼3-fold in HepG2-ER cells (Supplemental Fig. S1). Such SULT2A1 induction by E2 was not observed in parental HepG2 cells (data not shown). The activity of SULT2A1, measured by using DHEA as a substrate, exhibited a 2-fold increase after E2 treatment in HepG2-ER cells (Fig. 1C).
Fig. 1.
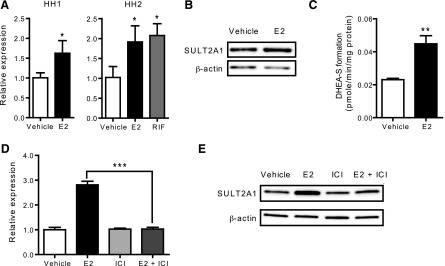
E2 induces SULT2A1 expression via ERα. (A) Primary human hepatocytes from two donors (HH1 and HH2) were treated with vehicle (ethanol), E2 (1 μM), or rifampicin (RIF) (10 μM) for 72 hours. mRNA expression level of SULT2A1 was determined by qRT-PCR. *P < 0.05 versus control. (B) Primary human hepatocytes (HH2) were treated with vehicle (ethanol) or E2 (1 μM) for 72 hours. Protein expression level of SULT2A1 was determined by Western blot. (C) HepG2-ER cells were treated with vehicle (ethanol) or E2 (100 nM) for 72 hours, and S9 fractions were prepared. SULT2A1 activity in the S9 fraction was determined using DHEA as a probe substrate. **P < 0.01 versus control. (D) HepG2-ER cells were treated with vehicle (ethanol) or E2 (100 nM) in the presence or absence of ICI 182,780 (ICI; 10 μM) for 72 hours, and mRNA expression level of SULT2A1 was determined by qRT-PCR. ***P < 0.001. (E) HepG2-ER cells were treated with vehicle (ethanol) or E2 (100 nM) in the presence or absence of ICI 182,780 (10 μM) for 72 hours and protein expression level of SULT2A1 was determined by Western blot.
To determine whether ERα is involved in SULT2A1 induction by E2, HepG2-ER cells were treated with E2 in the presence or absence of ICI 182,780 (an ERα-degrading antiestrogen), and SULT2A1 expression was examined by qRT-PCR. The results showed that ICI 182,780 abrogated the E2-mediated increases in mRNA and protein levels of SULT2A1 (Fig. 1D and 1E, respectively). Together, these data indicate that E2 induces SULT2A1 expression via ERα.
ERα Transactivates SULT2A1 Promoter.
To determine whether E2 upregulates SULT2A1 in a direct manner, we examined the effects of inhibition of de novo protein synthesis on SULT2A1 induction by E2. HepG2-ER cells were treated with E2 (or vehicle) in the presence or absence of cycloheximide (an inhibitor of protein synthesis; 10 μg/ml) for 16 hours, and SULT2A1 expression levels were determined by qRT-PCR. The results showed that SULT2A1 induction by E2 was maintained upon cotreatment with cycloheximide (Fig. 2A), suggesting that SULT2A1 is likely a direct target gene of ERα.
Fig. 2.
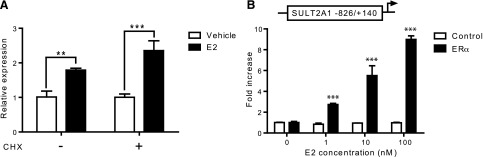
ERα transactivates SULT2A1 promoter. (A) HepG2-ER cells were treated with vehicle (ethanol) or E2 (100 nM) in the presence or absence of cycloheximide (CHX; 10 μg/ml) for 16 hours. SULT2A1 mRNA expression was determined by qRT-PCR. **P < 0.01; ***P < 0.001. (B) HepG2 cells were cotransfected with pGL3-SULT2A1 (–826/+140), ERα expression plasmid (or pcDNA3 empty vector), and pCMV-Renilla. The transfected cells were treated with vehicle (ethanol) or E2 (1–100 nM) for 24 hours, and luciferase assay was performed. ***P < 0.001 versus vehicle treatment.
To determine whether ERα transactivates SULT2A1 promoter, promoter reporter assays were performed in HepG2 cells. To this end, a luciferase vector harboring –826/+140 upstream regulatory region of SULT2A1 (i.e., pGL3-SULT2A1) was constructed. HepG2 cells were cotransfected with pGL3-SULT2A1, an ERα expression vector (or empty vector as control), and pCMV-Renilla (for normalization of transfection efficiency). As a positive control for ERα action, pGL3-ERE3 that harbors thymidine kinase promoter driven by three copies of Xenopus vitellogenin-A2 ERE (Catherino and Jordan, 1995) was used. The transfected cells were treated with E2 for 24 hours, and luciferase assays were performed. E2 increased the ERE3-driven promoter activity (by ∼300 fold) only in ERα-transfected cells (data not shown). Similarly, E2-enhanced SULT2A1 promoter activity in ERα-transfected cells, while such induction was not observed in the 2 empty vector–transfected cells (Fig. 2B). Transactivation of SULT2A1 promoter was E2 concentration–dependent, and the E2 effect did not reach a plateau even at the highest E2 concentration tested in the study (100 nM).
The –257/+140 Is Responsible for SULT2A1 Induction by E2.
To map the cis-elements responsible for SULT2A1 induction by E2, a deletion assay was performed. A series of luciferase vectors each carrying a 5′-deleted upstream regulatory region of SULT2A1 were constructed. HepG2 cells were cotransfected with one of the deletion constructs, ERα expression vector, and pCMV-Renilla. The transfected cells were treated with E2 for 24 hours, and luciferase assay was performed. Deletion of –826 to –258 of SULT2A1 upstream region did not affect the E2-responsiveness (Fig. 3A); however, deletion of –257 to –89 significantly decreased SULT2A1 induction by E2. Further deletion of the regulatory region removed the TATA box (Fig. 3B), abrogating the promoter activity. Together, the results indicate that the cis-element(s) responsible for SULT2A1 induction by E2 lies within –257/+140 region. This SULT2A1 promoter region harbors an inverted repeat (IR) 2 (–109/–98; called S1 hereafter), an imperfect ERE (–38/–24; called S2 hereafter), and a half-ERE (+130/+134; called S3 hereafter) (Fig. 3B). Luciferase constructs carrying mutations at these sites were constructed, and their effects on SULT2A1 induction by E2 were examined using luciferase reporter assays. The SULT2A1 promoter activities of S1, S2, or S3 mutant promoter vectors were higher than that of promoterless pGL3-basic (data not shown), indicating that mutation of these sites does not abrogate the basal promoter activities of SULT2A1. Upon E2 treatment, the mutation of S1 or S3 sites led to modest decreases in the promoter response, whereas S2 mutation led to a significant decrease in the promoter response to E2 (Fig. 3C). Mutations at all three sites completely abrogated the E2-responsiveness of the SULT2A1 promoter (Fig. 3C). These results suggest that while all three sites are involved in mediating the E2 action on SULT2A1 promoter, S2 likely plays a key role.
Fig. 3.
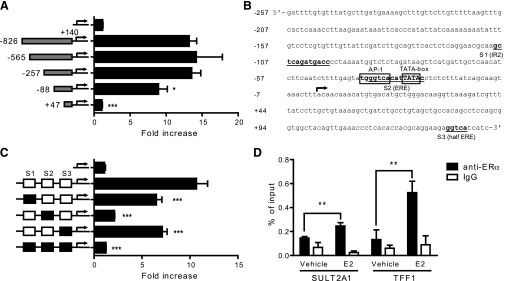
Putative cis-element responsible for SULT2A1 induction by E2 is located within -257/+140. (A) HepG2 cells were cotransfected with a 5′-deletion construct of pGL3-SULT2A1, ERα expression plasmid, and pCMV-Renilla. The transfected cells were treated with vehicle (ethanol) or E2 (100 nM), and luciferase assay was performed. *P < 0.05; ***P < 0.001 versus pGL3-SULT2A1. (B) Putative ERα or AP-1 binding sites within –257/+140 upstream region of SULT2A1 are shown. National Center for Biotechnology Information reference sequence NG_016745.1 was used. (C) HepG2 cells were cotransfected with pGL3-SULT2A1 (–257/+140) harboring mutations in S1, S2, and/or S3 (represented by a black box); ERα expression plasmid, and pCMV-Renilla. The transfected cells were treated with vehicle (ethanol) or E2 (100 nM), and luciferase assay was performed. ***P < 0.001 versus wild-type pGL3-SULT2A1 (–257/+140). (D) HepG2-ER cells were treated with E2 (100 nM) for 45 minutes, and cell lysates were collected. ChIP assays were carried out using rabbit IgG or an antibody against ERα, and the amount of ERα-bound DNA was measured by qRT-PCR using primer sets that amplify –128/–23 of SULT2A1 or –487/–381 of TFF1. **P < 0.01.
To examine whether ERα is recruited to the proximal promoter region of SULT2A1, ChIP assays were carried out using HepG2-ER cells treated with E2. The SULT2A1 promoter region bound to ERα (directly or indirectly) was detected by using a primer set amplifying –128/–23 of SULT2A1. TFF1, a known target gene of ERα, was included as a positive control. The results showed an increase in the ERα recruitment to the promoter region of SULT2A1 after E2 treatment (Fig. 3D), and a similar finding was observed for TFF1. Together, these data indicate that ERα is recruited to the proximal promoter region of SULT2A1.
AP-1 Proteins and ERα Bind to S2.
To determine whether ERα physically binds to the identified cis-elements, EMSA was performed. Recombinant protein ERα was incubated with radiolabeled DNA probes harboring S1, S2, or S3, and the reaction samples were resolved on nondenaturing gels. The results showed a lack of complex formation between ERα and any of the probes (data not shown). These results indicate that ERα does not bind directly to any of the three elements when tested individually in this system. Considering the presence of putative AP-1 binding sequences in S2, binding of AP-1 proteins to the DNA probes was examined using nuclear extracts from HepG2-ER cells. A single shifted band was observed when nuclear extracts from HepG2-ER cells were incubated with a labeled probe harboring the consensus AP-1 binding sequence (Fig. 4, lane 2). E2 treatment of the cells did not affect the extent of AP-1 binding to the probe (Fig. 4, lane 3), consistent with previous results (Murdoch et al., 1990). Unlabeled competitor S2 decreased the signal significantly (Fig. 4, lane 4), whereas mutated S2 (mS2) competitor did not affect the binding (Fig. 4, lane 5), indicating specific interaction between S1 and AP-1 proteins. Antibodies against c-Fos and c-Jun decreased the signals of shifted band, and a supershift band was observed upon incubation with c-Jun antibody (Fig. 4, lanes 6 and 7), indicating binding of c-Fos and c-Jun proteins to the probe. On the other hand, when radiolabeled SULT2A1/S2 probe was incubated with the nuclear extracts, multiple shifted bands were observed (Fig. 4, lanes 9 and 10). The signal of the topmost band was significantly decreased in the presence of unlabeled S2 probe or consensus AP-1 probe (Fig. 4, lanes 11 and lane 13), indicating AP-1 binding to the probe. Interestingly, unlabeled mS2 probe did not completely compete for the AP-1 binding to S2 probe (Fig. 4, lane 12), indicating that AP-1 proteins may bind to the probe different from the mutated site. Adding the antibodies against c-Jun, c-Fos, or ERα to the binding reaction mixture significantly decreased the signal of shifted band (Fig. 4, lanes 14, 15, and 16), suggesting the formation of complex among AP-1 proteins and ERα at S2.
Fig. 4.

AP-1 proteins bind to S2. Nuclear proteins from HepG2-ER cells treated with vehicle or E2 for 45 minutes were prepared. The proteins were incubated with 32P-labeled DNA probes harboring S2 or consensus AP-1 binding sequence (shown at the bottom) in the presence or absence of various antibodies or unlabeled DNA probes as competitors (in 100-fold excess). The mixture was resolved on nondenaturing gel. The lower arrow indicates the location of shifted bands by apparent AP-1 binding to DNA. The upper arrow indicates the super-shift complex.
DNA-Binding Domain of ERα Is Involved in SULT2A1 Induction by E2.
DNA-binding domain (DBD) of ERα is not essential when ERα is tethered to other transcription factors such as AP-1 for regulation of target gene expression (Webb et al., 1995; Kushner et al., 2000). Based on our EMSA results suggesting the formation of AP-1 proteins and ERα complexes at the S2 site, the role of ERα in SULT2A1 regulation was further examined using ERα mutants. HepG2 cells were cotransfected with pGL3-SULT2A1 along with one of the following ERα mutants: 1) a deletion of activation function (AF) 1; 2) point mutations in the DBD; 3) point mutations in AF2; and 4) a deletion of AF1 and point mutations in AF2. The transfected cells were treated with E2 (or vehicle), and luciferase assays were performed. The results showed that SULT2A1 induction by E2 was completely abrogated when both AF1 and AF2 of ERα were mutated (Fig. 5), consistent with the significant roles of the two domains in AP-1–mediated ERα action (Webb et al., 1999). Surprisingly, the mutations in the DBD of ERα led to a significant decrease in the E2 responsiveness of SULT2A1 promoter, suggesting that a direct binding of ERα to the promoter is necessary for SULT2A1 induction by E2 in HepG2 cells. These results suggest that in addition to AP-1–mediated pathway, direct binding of ERα to the SULT2A1 promoter is also involved in SULT2A1 induction by E2 in HepG2 cells.
Fig. 5.

DBD of ERα is essential for SULT2A1 induction by E2. HepG2 cells were cotransfected with pGL3-SULT2A1 and an expression vector for an ERα mutant along with pCMV-Renilla. The ER domain whose function is inactivated by point mutations is shown in black. The transfected cells were treated with vehicle (ethanol) or E2 (100 nM), and luciferase assay was performed. ***P < 0.001 versus wild-type ERα.
Discussion
SULT2A1 plays an important role in the sulfonation of a variety of endogenous and exogenous substrates responsible for the detoxification of drugs and the maintenance of lipid homeostasis. Results from a previous study indicated that E2 enhances SULT2A1 expression in human hepatocytes (Koh et al., 2012). In this study, the underlying molecular mechanisms for SULT2A1 induction by E2 were investigated.
Transient transfection and luciferase reporter assays revealed that ERα transactivates SULT2A1 promoter. In this, S1, S2, and S3 sites located within –257/+140 of SULT2A1 are likely involved. S1, S2, and S3 harbor putative IR2, AP-1 binding sequence/imperfect ERE, and half-ERE, respectively, that can bind to ERα; however, results from EMSA indicate that the recombinant ERα protein does not bind directly to any of the three sites. Instead, AP-1 proteins were found to bind to the S2 site, suggesting that ERα may exert its action on SULT2A1 promoter in part by tethering to AP-1 proteins.
Direct binding of ERα to the DNA is not required for its activity through the nonclassical mechanism involving AP-1 proteins (Webb et al., 1995; Kushner et al., 2000). Interestingly, however, transfection of DBD-mutated ERα led to a significant decrease in E2-mediated SULT2A1 induction compared with the cells transfected with wild-type ERα. This suggests that in addition to the AP-1 pathway, the classical mechanism may be involved in SULT2A1 induction by E2. The presence of tandem half-EREs in SULT2A1 promoter may provide potential explanation. Promoters of most estrogen target genes do not harbor a perfect ERE (IR3, 5′-GGTCANNNTGACC-3′; Gruber et al., 2004), indicating that gene-specific promoter context and variations of consensus EREs may accommodate ER binding to the promoters. Indeed, it has been previously reported that ERα binds to three tandem copies of half-ERE, but not to a single or two tandem copies of half-ERE (Klinge et al., 1997). Supporting the idea, the promoters of corticotropin-releasing hormone and prothymosin α harbor three tandem half-EREs that are activated by E2 (Vamvakopoulos and Chrousos, 1993; Martini and Katzenellenbogen, 2001). It appears plausible that the three tandem repeats of half-ERE in SULT2A1 promoter (i.e., S1, S2, and S3) allow direct binding of ERα to the DNA, leading to transactivation of SULT2A1 promoter in cells. The short (25-bp) DNA probes used in our EMSA, on the other hand, would not detect such cooperativity in ERα binding to tandem half-EREs. Together, our results indicate that E2 enhances SULT2A1 expression via both direct and indirect binding of ERα to the promoter region.
Estrogen enhanced SULT2A1 expression in a concentration-dependent manner, suggesting its differential biologic actions under the conditions of different estrogen concentrations. During menstrual cycle, plasma E2 concentration ranges from 0.2–1 nM. The E2 concentration increases gradually, reaching up to 100 nM during human pregnancy (Tulchinsky et al., 1972; Mathur et al., 1980; Stricker et al., 2006). After menopause, the concentration decreases below 100 pM (Baird and Guevara, 1969). During pregnancy, the enhanced SULT2A1 expression and activity may be necessary to meet the increased need of steroid hormone biosynthesis at the placenta. In fact, the enhanced expression of DHEA-sulfonating enzymes by estrogen appears to be conserved in rodents. For example, mouse hepatic Sult2a1/a2 is female-predominant, and estrogen replacement in gonadectomized female mice restored the expression of hepatic Sult2a1/a2 (Alnouti and Klaassen, 2011). After menopause, the lack of estrogen-mediated regulation of SULT2A1 expression may potentially play a role in the development of symptoms associated with menopause. Menopause is associated with worsening lipid profiles as well as increased risks for developing cardiovascular diseases such as atherosclerosis (Ouyang et al., 2006). These changes are accompanied by increased hepatic expression of genes involved in lipid synthesis, many of which are the targets of liver X receptor (LXR) (Li et al., 1996; Villa et al., 2012). Of note, SULT2A1 inactivates oxysterols such as 25-hydroxycholesterol that are activators of LXR signaling and hepatic lipid synthesis. Furthermore, SULT2A1 inactivates 7-ketocholesterol, a cytotoxic oxysterol that plays a major role in development of atherosclerosis (Fuda et al., 2007). Whether estrogen-mediated SULT2A1 regulation plays a key role in the worsening lipid profiles or increased risk of cardiovascular diseases in menopausal women remains to be determined.
In conclusion, this study demonstrated that SULT2A1 is a direct target of estrogen and ERα and that estrogen action on SULT2A1 is mediated by both classical and nonclassical mechanisms of ERα action. Together, our results should shed light on physiologic and molecular basis of estrogen pharmacology.
Supplementary Material
Acknowledgments
The authors thank Yan-Yan Zhang and Xian Pan for technical assistance.
Abbreviations
- AF
activation function
- ChIP
chromatin immunoprecipitation
- DBD
DNA-binding domain
- DHEA
dehydroepiandrosterone
- E2
17β-estradiol
- EMSA
electromobility shift assay
- ER
estrogen receptor
- ERE
estrogen response element
- HH
human hepatocytes
- qRT-PCR
quantitative reverse-transcription polymerase chain reaction
- SULT
sulfotransferase
Authorship Contributions
Participated in research design: Li, Ning, Jeong.
Conducted experiments: Li, Ning, Koh, Kim.
Performed data analysis: Li, Ning.
Wrote or contributed to the writing of the manuscript: Li, Ning, Jeong.
Footnotes
This work was supported by the National Institutes of Health Eunice Kennedy Shriver National Institute of Child Health and Human Development [Grant HD065532].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Al Koudsi N, Hoffmann EB, Assadzadeh A, Tyndale RF. (2010) Hepatic CYP2A6 levels and nicotine metabolism: impact of genetic, physiological, environmental, and epigenetic factors. Eur J Clin Pharmacol 66:239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alnouti Y, Klaassen CD. (2011) Mechanisms of gender-specific regulation of mouse sulfotransferases (Sults). Xenobiotica 41:187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvaro D, Alpini G, Onori P, Franchitto A, Glaser SS, Le Sage G, Folli F, Attili AF, Gaudio E. (2002) Alfa and beta estrogen receptors and the biliary tree. Mol Cell Endocrinol 193:105–108 [DOI] [PubMed] [Google Scholar]
- Baird DT, Guevara A. (1969) Concentration of unconjugated estrone and estradiol in peripheral plasma in nonpregnant women throughout the menstrual cycle, castrate and postmenopausal women and in men. J Clin Endocrinol Metab 29:149–156 [DOI] [PubMed] [Google Scholar]
- Catherino WH, Jordan VC. (1995) Increasing the number of tandem estrogen response elements increases the estrogenic activity of a tamoxifen analogue. Cancer Lett 92:39–47 [DOI] [PubMed] [Google Scholar]
- Chen H, Yang K, Choi S, Fischer JH, Jeong H. (2009) Up-regulation of UDP-glucuronosyltransferase (UGT) 1A4 by 17beta-estradiol: a potential mechanism of increased lamotrigine elimination in pregnancy. Drug Metab Dispos 37:1841–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J, Petrenaite V, Atterman J, Sidenius P, Ohman I, Tomson T, Sabers A. (2007) Oral contraceptives induce lamotrigine metabolism: evidence from a double-blind, placebo-controlled trial. Epilepsia 48:484–489 [DOI] [PubMed] [Google Scholar]
- Echchgadda I, Song CS, Oh T, Ahmed M, De La Cruz IJ, Chatterjee B. (2007) The xenobiotic-sensing nuclear receptors pregnane X receptor, constitutive androstane receptor, and orphan nuclear receptor hepatocyte nuclear factor 4alpha in the regulation of human steroid-/bile acid-sulfotransferase. Mol Endocrinol 21:2099–2111 [DOI] [PubMed] [Google Scholar]
- Falany CN, Comer KA, Dooley TP, Glatt H. (1995) Human dehydroepiandrosterone sulfotransferase. Purification, molecular cloning, and characterization. Ann N Y Acad Sci 774:59–72 [DOI] [PubMed] [Google Scholar]
- Fuda H, Javitt NB, Mitamura K, Ikegawa S, Strott CA. (2007) Oxysterols are substrates for cholesterol sulfotransferase. J Lipid Res 48:1343–1352 [DOI] [PubMed] [Google Scholar]
- Gruber CJ, Gruber DM, Gruber IML, Wieser F, Huber JC. (2004) Anatomy of the estrogen response element. Trends Endocrinol Metab 15:73–78 [DOI] [PubMed] [Google Scholar]
- He DN, Meloche CA, Dumas NA, Frost AR, Falany CN. (2004) Different subcellular localization of sulphotransferase 2B1b in human placenta and prostate. Biochem J 379:533–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi E, Fukami T, Itoh M, Kyo S, Inoue M, Yokoi T, Nakajima M. (2007) Human CYP2A6 is induced by estrogen via estrogen receptor. Drug Metab Dispos 35:1935–1941 [DOI] [PubMed] [Google Scholar]
- Klinge CM, Bodenner DL, Desai D, Niles RM, Traish AM. (1997) Binding of type II nuclear receptors and estrogen receptor to full and half-site estrogen response elements in vitro. Nucleic Acids Res 25:1903–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton AA, Lee AR. (2012) Estrogen and the cardiovascular system. Pharmacol Ther 135:54–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KH, Jurkovic S, Yang K, Choi SY, Jung JW, Kim KP, Zhang W, Jeong H. (2012) Estradiol induces cytochrome P450 2B6 expression at high concentrations: implication in estrogen-mediated gene regulation in pregnancy. Biochem Pharmacol 84:93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuiper GG, Carlsson B, Grandien K, Enmark E, Häggblad J, Nilsson S, Gustafsson JA. (1997) Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138:863–870 [DOI] [PubMed] [Google Scholar]
- Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. (2000) Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol 74:311–317 [DOI] [PubMed] [Google Scholar]
- Labrie F, Luu-The V, Bélanger A, Lin SX, Simard J, Pelletier G, Labrie C. (2005) Is dehydroepiandrosterone a hormone? J Endocrinol 187:169–196 [DOI] [PubMed] [Google Scholar]
- Li ZL, McNamara JR, Fruchart JC, Luc G, Bard JM, Ordovas JM, Wilson PWF, Schaefer EJ. (1996) Effects of gender and menopausal status on plasma lipoprotein subspecies and particle sizes. J Lipid Res 37:1886–1896 [PubMed] [Google Scholar]
- Lo R, Burgoon L, Macpherson L, Ahmed S, Matthews J. (2010) Estrogen receptor-dependent regulation of CYP2B6 in human breast cancer cells. Biochim Biophys Acta 1799:469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini PGV, Katzenellenbogen BS. (2001) Regulation of prothymosin alpha gene expression by estrogen in estrogen receptor-containing breast cancer cells via upstream half-palindromic estrogen response element motifs. Endocrinology 142:3493–3501 [DOI] [PubMed] [Google Scholar]
- Mathur RS, Landgrebe S, Williamson HO. (1980) Progesterone, 17-hydroxyprogesterone, estradiol, and estriol in late pregnancy and labor. Am J Obstet Gynecol 136:25–27 [DOI] [PubMed] [Google Scholar]
- Murdoch FE, Meier DA, Furlow JD, Grunwald KA, Gorski J. (1990) Estrogen receptor binding to a DNA response element in vitro is not dependent upon estradiol. Biochemistry 29:8377–8385 [DOI] [PubMed] [Google Scholar]
- Ouyang P, Michos ED, Karas RH. (2006) Hormone replacement therapy and the cardiovascular system lessons learned and unanswered questions. J Am Coll Cardiol 47:1741–1753 [DOI] [PubMed] [Google Scholar]
- Riches Z, Stanley EL, Bloomer JC, Coughtrie MWH. (2009) Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie”. Drug Metab Dispos 37:2255–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S, Kim K. (2008) Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol 41:263–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker R, Eberhart R, Chevailler MC, Quinn FA, Bischof P, Stricker R. (2006) Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT analyzer. Clin Chem Lab Med 44:883–887 [DOI] [PubMed] [Google Scholar]
- Tulchinsky D, Hobel CJ, Yeager E, Marshall JR. (1972) Plasma estrone, estradiol, estriol, progesterone, and 17-hydroxyprogesterone in human pregnancy. I. Normal pregnancy. Am J Obstet Gynecol 112:1095–1100 [DOI] [PubMed] [Google Scholar]
- Vamvakopoulos NC, Chrousos GP. (1993) Evidence of direct estrogenic regulation of human corticotropin-releasing hormone gene expression. Potential implications for the sexual dimophism of the stress response and immune/inflammatory reaction. J Clin Invest 92:1896–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa A, Della Torre S, Stell A, Cook J, Brown M, Maggi A. (2012) Tetradian oscillation of estrogen receptor α is necessary to prevent liver lipid deposition. Proc Natl Acad Sci USA 109:11806–11811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb P, Lopez GN, Uht RM, Kushner PJ. (1995) Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol 9:443–456 [DOI] [PubMed] [Google Scholar]
- Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, Katzenellenbogen BS, Enmark E, Gustafsson JA, Nilsson S, et al. (1999) The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol 13:1672–1685 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.