

Abstract
Rationale
Endothelial adherens junction proteins constitute an important element in the control of microvascular permeability. Platelet-activating factor (PAF) increases permeability to macromolecules via translocation of eNOS to cytosol and stimulation of eNOS-derived NO signaling cascade. The mechanisms by which NO signaling regulates permeability at adherens junctions are still incompletely understood.
Objective
We explored the hypothesis that PAF stimulates hyperpermeability via S-nitrosation (SNO) of adherens junction proteins.
Methods and Results
We measured PAF-stimulated S-nitrosation of β-catenin and p120-catenin (p120) in three cell lines: ECV-eNOSGFP, EAhy926 (derived from human umbilical vein) and CVEC (derived from bovine heart endothelium) and in the mouse cremaster muscle in vivo. SNO correlated with diminished abundance of β-catenin and p120 at the adherens junction and with hyperpermeability. TNF-α increased NO production and caused similar increase in S-nitrosation as PAF. To ascertain the importance of eNOS subcellular location in this process, we used ECV-304 cells transfected with cytosolic eNOS (GFPeNOSG2A) and plasma membrane eNOS (GFPeNOSCAAX). PAF induced S-nitrosation of β-catenin and p120 and significantly diminished association between these proteins in cells with cytosolic eNOS but not in cells wherein eNOS is anchored to the cell membrane. Inhibitors of NO production and of S-nitrosation blocked PAF-induced S-nitrosation and hyperpermeability whereas inhibition of the cGMP pathway had no effect. Mass spectrometry analysis of purified p120 identified cysteine 579 as the main S-nitrosated residue in the region that putatively interacts with VE-cadherin.
Conclusions
Our results demonstrate that agonist-induced SNO contributes to junctional membrane protein changes that enhance endothelial permeability.
Keywords: S-nitrosation, adherens junction, microvascular permeability
INTRODUCTION
Cell life depends on adequate supply of nutrients and removal of catabolites via blood circulation. This important exchange function is controlled mainly by the microvascular endothelium. Inflammation and wound healing require specific regulation of microvascular permeability to macromolecules to enable tissue repair. The regulation of permeability occurs primarily at postcapillary venules in vivo through activation of endothelial nitric oxide synthase (eNOS) to produce nitric oxide (NO), which has been identified as a key signaling element in eliciting hyperpermeability1, 2, 3, 4, 5. The mechanisms by which eNOS-derived NO induces hyperpermeability include the internalization of eNOS indicating that eNOS location and exact delivery of NO regulate the development of hyperpermeability2, 6.
Endothelial cells from postcapillary venules display adherens junctions, which contain VE-cadherin, forming a complex with the cytosolic proteins α-catenin, β-catenin, plakoglobin, and p120-catenin (p120)7, 8, 9, 10, 11. Pro-inflammatory agents stimulate signaling cascades that by phosphorylation of adherens junction proteins lead to their internalization, which results in increased endothelial paracellular permeability12, 13.
Despite advances in the research of hyperpermeability regulation, the mechanisms by which NO signaling influences processes occurring at the adherens junctions remain unknown. The classic dogma in NO signaling establishes that all of the actions of NO are mediated via soluble guanylate cyclase (sGC) and protein kinase G (PKG). However, increases in permeability have been described in some cellular models regardless of PKG activation but still requiring NO production14. Recently, S-nitrosation (SNO) has emerged as an important NO-dependent posttranslational modification of free-thiol cysteines that alters the function of proteins, and requires proximity between eNOS and the target proteins for appropriate NO delivery15, 16, 17. We report here experiments testing the hypothesis that NO regulates endothelial permeability to macromolecules by SNO of β-catenin and p120, two important and integral components of the adherens junction. We determined that platelet-activating factor (PAF) induces SNO of β-catenin and p120 and their internalization away from the plasma membrane. These processes are associated with the onset of hyperpermeability. Furthermore, we demonstrate that eNOS located in the cytosol, but not the plasma membrane-anchored eNOS, causes SNO of β-catenin and p120. This advance in knowledge may serve as a basis for the development of therapeutic agents in the treatment of vascular diseases having a phase characterized by inflammation (for example: ischemia-reperfusion injury, stroke and atherosclerosis).
MATERIALS AND METHODS
Reagents
Platelet activating factor (PAF) and N-acetyl-L-cysteine (NAC) were obtained from Calbiochem. FITC-labeled dextran 70 (FITC-Dx-70; molecular weight: 70,000 Da), Acetylcholine (ACh), 1H-[1,2, 4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), N-ethylmaleimide (NEM) and NG-methyl-L-arginine (L-NMA) were from Sigma. Tumor necrosis factor-alpha (TNF-α) was from Roche.
Antibodies
Mouse anti-p120 catenin was from BD Transduction Laboratories. Rabbit anti-β-catenin and mouse anti-β-actin, were from Sigma.
Plasmids and cell transfections
GFPeNOS-G2A and GFPeNOS-CAAX constructs were kindly provided by Dr. David Fulton (Georgia Health Sciences University, Augusta, GA). ECV-304 cells were transfected with fluorescently labeled eNOS mutant constructs using Lipofectamine according to manufacturers’ instructions (Invitrogen). Transfected cells were selected for growth in medium including 1 mg/mL of geneticin (G418) (Invitrogen). Resistant colonies were maintained in the complete Dulbecco’s modified Eagle’s medium containing G418 (400ug/mL).
Cell culture
ECV-304, ECV-eNOSGFP, ECV-GFPeNOS-G2A, ECV-GFPeNOS-CAAX, EAhy296 and postcapillary venular endothelial cells (CVEC) were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % (v/v) fetal bovine serum (Invitrogen), 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/mL streptomycin. ECV-eNOSGFP, ECVG2A-eNOSGFP, ECVCAAX-eNOSGFP were additionally supplemented with geneticin (G418) 400μg/mL. ECV-304 and ECV-eNOSGFP were kindly donated by Dr. William Sessa (Yale University, New Haven, CT). EAhy926 cells were kindly provided by Dr C. J. S. Edgell, (University of North Carolina, Chapel Hill, NC, USA).
Immunoprecipitation
Cell lysates from control and agonist-treated cells were incubated with specific antibodies overnight at 4°C. Protein A/G beads were added to samples for 2 h at 4°C; then pelleted by centrifugation and washed with lysis buffer. Proteins of interest were detected using Western blotting and chemiluminescence. Quantification of changes from control was evaluated by densitometric analysis of Western blots using the NIH Image J Program.
Biotin-Switch Assay
Proteins were denatured with sodium dodecyl sulfate (SDS) in the presence of methyl methanethiosulfonate (MMTS)18. After acetone precipitation to remove excess MMTS, 1 mmol/L ascorbate and biotin-HPDP (N-[6-(biotinamido) hexyl]-3′-(2′-pyridyldithio) propionamide) were added to reduce the S-NO bond and label the reduced thiol with biotin, respectively. Biotinylated proteins were captured with streptavidin–agarose beads and then separated by SDS-PAGE and detected with specific antibodies. The biotin-switch assay was performed on 100 μg of lysates from control and agonist-treated cells. Proteins of interest were detected by Western blotting.
Immunofluorescence Microscopy
We followed established protocols19, 20. Cells were cultured on glass coverslips and treated with agonist and then fixed and permeabilized in 100% ethanol for 10 min. at -20C. The secondary Alexa Fluor conjugated antibodies were added after incubation with corresponding primary antibodies for 1h at room temperature. Coverslips were mounted and visualized by fluorescence microscopy. Images were obtained using an epifluorescence microscope (Axioscop; Carl Zeiss) equipped with a 100x oil immersion objective lens and Axio Vision Rel. software (Zeiss, Germany). 8-bit images were prepared for illustration in Photoshop (Adobe).
Endothelial Permeability Assay
Monolayer permeability was determined as described previously2, 21. Cells were grown on fibronectin-coated polycarbonate membranes, for 5-6 days to achieve confluence. On the day of the experiment, the membranes were placed in a Navicyte system (San Diego, CA). Luminal) and abluminal chambers were filled with DMEM without phenol red. After a 15-minute equilibration period, the luminal chamber was loaded with FITC-Dx-70 (final concentration 13.3 mg/mL). Samples for baseline permeability were obtained every 5 minutes for a period of 30 minutes. After addition of PAF, samples were obtained for an additional 30- minute period. In the experiments using NAC (2.5 mmoles/L), this agent was added for 1.5 hour to the cells in the plates prior to the addition of agonists. Fresh NAC was applied for 1 more hour to the cells when they were transferred to the Navicyte system. In the experiments using L-NMA (300 umoles/L), the NOS inhibitor was added for 1 hour while the cells were in the Navicyte system before PAF addition. The permeability to FITC-Dx-70 was determined according to the Fick equation. FITC-Dx-70 was measured using a spectrofluorometer.
In vivo cremaster preparation
Male wild-type (C57BL/6J, Jackson Laboratory, Bar Harbour, MA, USA), were anesthetized with Ketamine (90 mg/Kg)-Xylazine (10 mg/Kg). The cremaster muscle was exposed via a scrotal incision and gently separated from the subcutaneous interstitial tissue under continuous superfusion with intravital buffer solution equilibrated with 95% N2 5% CO2, pH 7.4, 35°C. The whole cremaster sac, containing the testis, was positioned in a plastic container and superfused. After 15 min equilibration, 10-7 moles/L PAF was applied topically, bathing the whole muscle for 3 min. The cremaster and the testis were ligated and quickly excised. The muscle was immediately separated from the testis and homogenized in 500μL buffer containing an antiprotease cocktail. The contralateral cremaster was treated similarly, except that buffer was applied instead of PAF. Both homogenates were processed simultaneously for biotin switch, followed by streptavidin pull down and Western blotting. All experiments on animals were carried out at the P. Universidad Católica de Chile, approved by the Institutional Bioethics and Biosecurity Committee, and conducted according to NIH Guidelines for the use of animals in research.
Identification of S-nitrosated cysteines in p120-catenin
The sites of p120 SNO were identified by MS/MS mass spectrometry. One μg recombinant p120 was treated with 100 μmol/L GSNO 37°C for 30 min. The protein was then precipitated four times with ice cold acetone (50 μg lysozyme as a carrier) and washed with ice cold acetone. Free thiols were blocked with 20 mM MMTS followed by acetone precipitation and washed with 80% ice-cold acetone for 3 times. SNO cysteines were reduced with 10 mmol/L ascorbate and labeled with biotin HPDP. The biotinylated sample (about 0.1 μg p120) was resolved in 8 mol/L urea after acetone precipitation and diluted 10 times using 50 mmol/L NH4HCO3 and 0.01 μg trypsin was added for in solution digestion. The peptides were desalted with a C18 ziptip, and identified by Orbitrap Velos MS. The MS/MS spectra were searched against a Swissprot human database using Thermo proteome Discoverer 1.3.0.339 software with biotin HPDP as a variable modification. Peptides identified with 95% confidence interval were displayed in Scaffold3 software. Protein and peptide False Discovery Rates was less than 1%.
Statistical Analysis
Experiments were conducted in groups with minimum n = 3. Data were expressed as mean ± standard error. Apparent differences were assessed for statistical significance using paired Student’s t test and/or the Newman-Keuls test. Significance was accepted at values of p < 0.05.
RESULTS
SNO reduces the integrity of the endothelial barrier
NO derived from eNOS has been largely recognized as a regulatory signal for endothelial permeability; however, the exact mechanisms by which this regulation is achieved are incompletely understood. In order to investigate how NO modifies proteins of the endothelial barrier to increase microvascular permeability, we applied 10-7 mol/L PAF to confluent monolayers of bovine coronary venular postcapillary endothelial cells (CVEC)21, 22 and assessed whether or not PAF-induced release of NO causes SNO of junctional proteins and loss of morphological integrity of the intercellular junctions. We used CVEC initially because these cells are derived from the main site where in vivo regulation of microvascular permeability to macromolecules occurs. We examined the impact of PAF on β-catenin and p120, two proteins that are constituents of the adherens junction. Figure 1 shows that in control conditions β-catenin and p120 appear as continuous belt-like structures on the plasma membrane, as revealed by indirect immunofluorescence microscopy. PAF induced significant increment of SNO of β-catenin and p120 as shown by representative Western blot and statistical analysis. PAF-induced SNO was associated with re-distribution of these proteins, away from the plasma membrane, and the appearance of a discontinuous pattern on the cell membrane (Figure 1). The increase in SNO was significant at 1 minute after application of PAF, a time-lapse that is compatible with a mechanistic causal effect, in accordance with the time-course of PAF-induced hyperpermeability2, 6, 21, 23.
Figure 1.
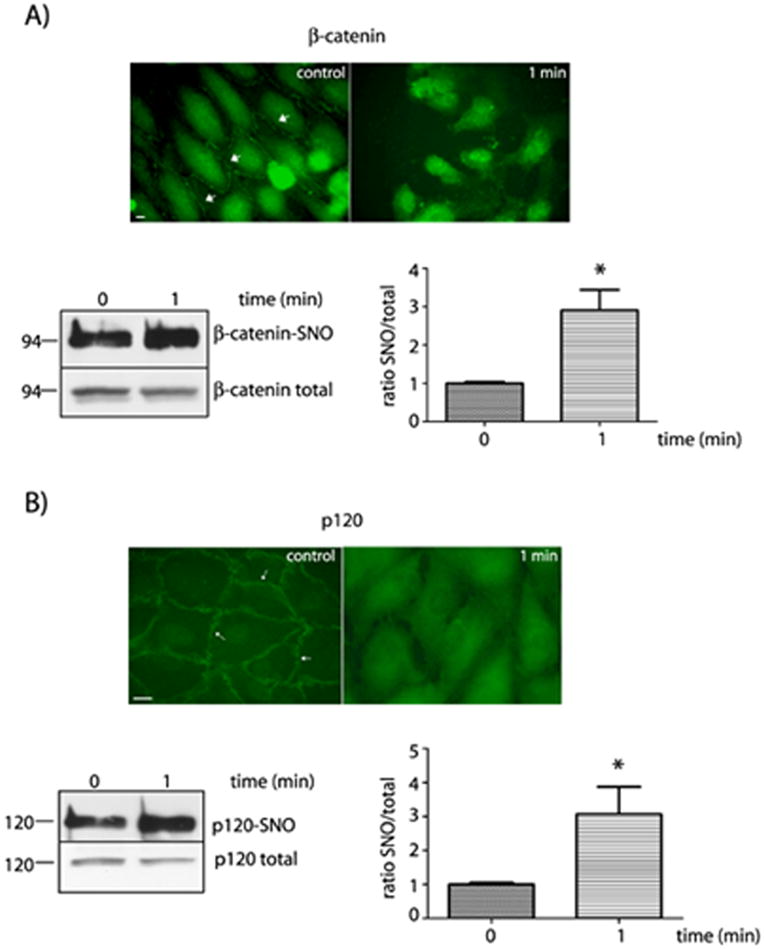
Correlation between protein organization at the cell membrane and SNO of β-catenin and p120 in CVEC. A) Upper Panel: Indirect immunofluorescence staining for β-catenin in control conditions and after stimulation with 10-7 mol/L PAF. Arrows indicate the presence of β-catenin in the cell plasma membrane. Bar represents 10 μm. Lower Panel: SNO of β-catenin as measured by biotin switch. The quantification is shown as the ratio of the S-nitrosylated β-catenin to the total β-catenin on the right lower panel. * P < 0.05 compared to control; n = 3. B) Upper Panel: Indirect immunofluorescence staining for p120 in control conditions and after stimulation with 10-7 mol/L PAF. Left lower panel: SNO of p120 detected by biotin switch. The quantification is shown as the ratio of the S-nitrosylated p120 to the total p120 on the right lower panel. *P < 0.05 as compared with control; n = 3.
SNO is a widespread regulatory mechanism for the re-organization of endothelial junctional proteins
To test whether or not the regulation of endothelial permeability to macromolecules by SNO is a widespread mechanism, we tested PAF-induced effects in EAhy926 cells and in ECV-304 cells transfected with a “wild-type” eNOS construct (ECVeNOSGFP), in which eNOS achieves the same distribution found in native endothelial cells23. The characteristics of these cells are described in Supplemental Data. In control EAhy926 cells we observed a clear label of β-catenin at the plasma membrane, which tended to disappear with PAF treatment. Likewise the strong fluorescence due to p120 at the plasma membrane observed in control conditions decreased as the result of PAF application (Figure 2A). A similar response pattern was observed in ECVeNOSGFP cells, i.e., the immunofluorescence signal for β-catenin and p120 was predominantly at the cell border in control conditions (Figure 2B, arrows) and decreased after 1 minute of PAF treatment. The pattern of both proteins redistribution was maintained at least for 5 min during PAF treatment.
Figure 2.
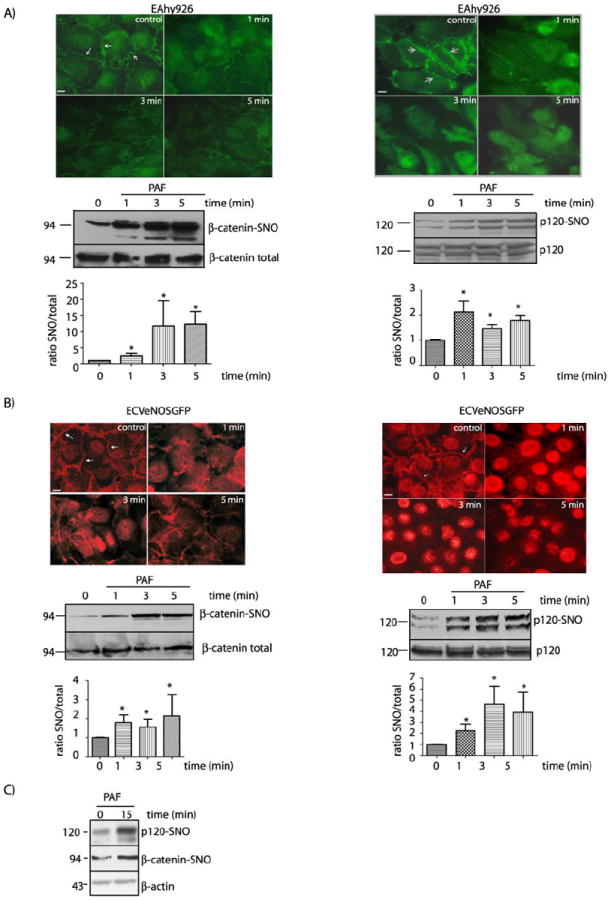
PAF induces internalization and SNO of β-catenin and p120 in EAhy926 cells and ECVeNOS-GFP. A) Experiments in EAhy926 cells. Upper Panel: Indirect immunofluorescence staining of EAhy926 cells for β-catenin and p120 after stimulation with 10-7 mol/L PAF. Middle Panel: SNO of β-catenin (left side) and p120 (right side) as detected by biotin switch. Lower panel: Bar graph showing the quantification of the SNO signal for β-catenin (left side) and p120 (right side). * P < 0.05 compared with control; n = 3. B) Experiments in ECVeNOS-GFP. Upper Panel: Indirect immunofluorescence staining of ECVeNOS-GFP for β-catenin (left side) and p120 (right side) in control and at different times after stimulation with 5×10-7 mol/L PAF. Middle Panel: SNO of β-catenin (left side) and p120 (right side) as detected by biotin switch. Lower Panel: Bar graph reporting the quantification of the SNO signal in Western blots for β-catenin (left side) and p120 (right side). * P < 0.05 as compared with control; n = 3. C) Prolonged application of PAF induces SNO of β-catenin and p120 in EAhy926 cells. PAF at 10-7 mol/L was applied for 15 minutes. Protein extracts were processed for biotin switch assay and probed with anti β-catenin and anti p120 antibodies. β-actin was used as a load control. Bars in microphotographs represent 10μm.
The changes in the localization of β-catenin and p120 were correlated with SNO. In control conditions, we detected a basal level of SNO of both proteins in EAhy926 and ECV-eNOSGFP cells (Figures 2A and B, lower panels). As early as after 1 minute of administration, PAF induced a marked and significant increase in SNO of β-catenin and p120 in both cell models. PAF-induced SNO was associated with re-organization of the distribution of β-catenin and p120 at the plasma membrane in both cell types. Furthermore, these modifications were maintained until the end of the observation period. These observations are consistent with a regulatory association between PAF-induced SNO and PAF-induced hyperpermeability. Interestingly, SNO of β-catenin and p120 was observed also after 15 min of continuous application of PAF (Figure 2C).
To confirm that induction of SNO is a common property of pro-inflammatory agents, we tested whether or not tumor necrosis factor-alpha (TNF-α), a well-known pro-inflammatory agent that increases permeability, recapitulates PAF-induced SNO. Figure 3A shows that TNF-α increased NO production from 8.4 ± 3.7 pmoles/min to 26.9 ± 3.2 pmoles/min at 1 minute after its application. At this time, TNF-α also increased SNO of β-catenin and p120 indicating that SNO is a mechanism shared among agents that cause inflammatory hyperpermeability. To evaluate whether or not release of NO induces nitrosation of junctional proteins independently of changes in permeability, we applied acetylcholine (ACh), an agonist that activates eNOS but does not increase permeability23, 24. Figure 3B illustrates that ACh does not enhance SNO of β-catenin and p120. In addition, to test whether or not nitrosation-induced hyperpermeabilty is associated with activation of soluble guanylyl cyclase (sGC), we pretreated the cells with 1H-[1,2,4] oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) prior to PAF application. Figure 3C shows that PAF caused a significant increase in permeability to macromolecules in the presence of ODQ, suggesting that PAF-induced SNO of junctional proteins is sufficient to cause hyperpermeability.
Figure 3.
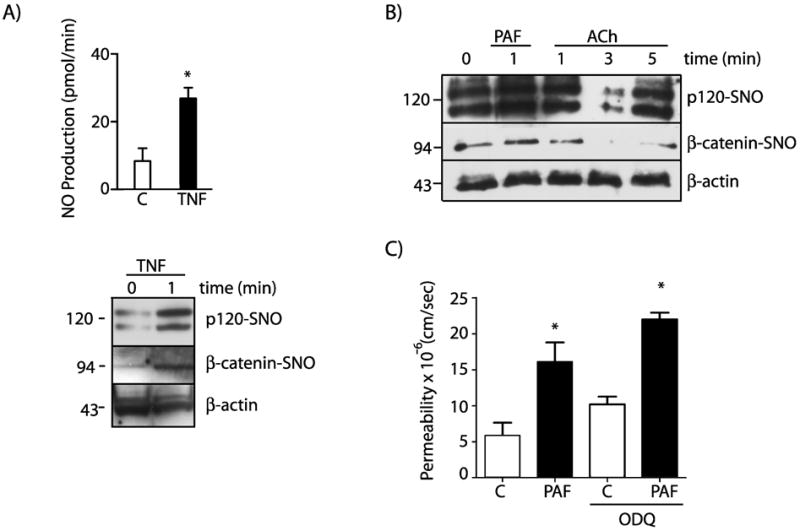
SNO is a widespread regulatory mechanism specific for permeability and independent of sGC and PKG. A) TNF-α induces NO production and SNO of β-catenin and p120 in EAhy926 cells. TNF-α at 50 ng/mL was applied for 1 minute. Protein extracts were processed for biotin switch assay and probed with anti β-catenin and anti p120 antibodies. β-actin was used as a load control. B) ACh at 10-5 moles/L does not induce SNO of β-catenin and p120 in EAhy926 cells. C) PAF at 10-7 moles/L increases permeability to FITC-dextran-70 across confluent EAhy926 monolayers. PAF-stimulated hyperpermeability is not affected by inhibition of sGC with ODQ. * P < 0.05 compared with control; n=3.
eNOS location determines SNO of junctional proteins
Vasoactive agents induce molecular movement or translocation of eNOS in vitro and in vivo 25. Recently, we demonstrated the importance of localized NO production in hyperpermeability using ECV cells transfected with eNOS constructs that target eNOS to different subcellular locations, GFPeNOS-G2A (targets to the cytosol) and GFPeNOS-CAAX (targets to the plasma membrane)6, 26, 27. Even though both cell lines are able to produce NO in response to PAF, only GFPeNOS-G2A, showing eNOS expression in the cytosol, was able to increase permeability in response to PAF6. On the other hand, ECV-GFPeNOS-CAAX cells, expressing eNOS at the plasma membrane, did not increase permeability in response to PAF despite producing NO at the same magnitude as ECV-GFPeNOSG2A and ECV cells transfected with wild type eNOS. To explore whether or not SNO of junctional proteins depends on eNOS location, we tested PAF-induced SNO of β-catenin and p120 in ECV-GFPeNOS-G2A and ECV-GFPeNOS-CAAX cells. Figure 4 displays our results. In agreement with our hypothesis, PAF stimulated SNO of β-catenin and p120 in cells with eNOS targeted to the cytosol (ECV-GFPeNOS-G2A; Figure 4A) but not in cells with eNOS targeted to the plasma membrane (ECV-GFPeNOS-CAAX; Figure 4B). PAF caused significant SNO of junctional proteins only in cells containing eNOS targeted to the cytosol when applied for 1, 3 and 5 minutes, which correlates well with the PAF-induced re-organization of β-catenin and p120 in adherens junctions in ECV-eNOSGFP (see Fig 2). Interestingly, targeting of eNOS to the plasma membrane precluded stimulation of SNO of β-catenin and p120 and is in agreement with our report demonstrating that PAF fails to cause hyperpermeability in these cells6.
Figure 4.
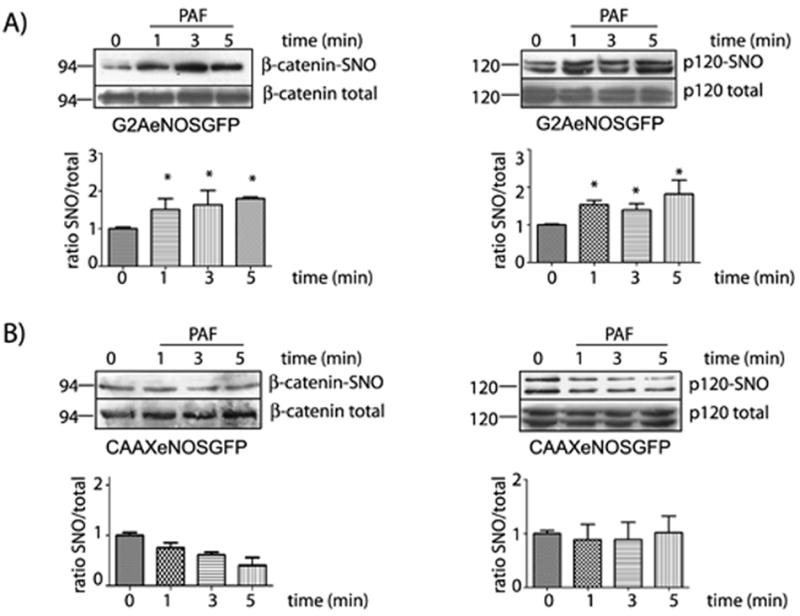
Subcellular location of eNOS determines SNO of β-catenin and p120. Protein extracts from ECV-GFPeNOS-G2A and ECV-GFPeNOS-CAAX cells treated with 5×10-7 mol/L PAF for different times were processed for biotin switch assay and probed with anti β-catenin and anti p120 antibodies. A) Upper Panel: PAF-induced SNO of β-catenin and p120 in ECV-GFPeNOS-G2A cells. Lower Panel: Bar graph showing the quantification of the Western blots of β-catenin (left side) and p120 (right side). * P < 0.05 compared to control; n = 3. B) Upper Panel: PAF-induced SNO of β-catenin and p120 in ECV-GFPeNOS-CAAX cells. Lower Panel: Bar graph showing the quantification of the Western blots of β-catenin (left side) and p120 (right side). * P < 0.05 compared to control; n = 3.
SNO disrupts the association between β-catenin and p120
We investigated whether or not PAF-induced SNO impacts on the association between β-catenin and p120 in ECV-GFPeNOS-G2A and ECV-GFPeNOS-CAAX cells. We tested this concept by immunoprecipitation using an antibody against β-catenin and probing for the presence of p120 in the precipitate. The data were quantified as a ratio to the total content of β-catenin in the input and compared to the levels seen at time 0. Figure 5 demonstrates that PAF significantly decreased the association between β-catenin and p120 in ECV-GFPeNOS-G2A cells (cytosolic eNOS cells) when applied for 1, 3 and 5 minutes (Figure 5A). Interestingly, PAF failed to alter the association between β-catenin and p120 in ECV-GFPeNOS-CAAX cells (plasma membrane eNOS; Figure 5B). These data confirm the importance of eNOS location in regards to the ability of PAF to elicit NO-dependent functional reactions, and support the concept that increases in permeability are related to modifications in the association between proteins that make up the junctional complex.
Figure 5.

SNO disrupts the association between β-catenin and p120. Protein extracts from ECV-GFPeNOS-G2A and ECV-GFPeNOS-CAAX cells, treated with 5×10-7 mol/L PAF for different times, were immunoprecipitated (IPP) with anti β-catenin antibodies and probed by Western blot with anti-p120 antibodies. A) Experiments in ECV-GFPeNOS-G2A cells. A representative Western blot assessing the association between β-catenin and p120 in ECV-GFPeNOS-G2A cells treated with PAF for different times is shown on the upper section, with corresponding statistical analysis illustrated below. Time indicates the duration of PAF application. * P < 0.05 as compared with control; n = 3. B) Experiments in ECV-GFPeNOS-CAAX cells. The upper section shows a representative Western blot assessing whether or not PAF stimulates the disruption of the association between β-catenin and p120 in ECV-GFPeNOS-CAAX cells. The statistical evaluation is illustrated in the lower section. No statistical significance was observed.
SNO of junctional proteins increases permeability to macromolecules
To assess directly the cause-effect relationship between SNO and hyperpermeability to macromolecules, we measured PAF-induced transport of FITC-dextran 70 (molecular weight: 70 kDa) across confluent monolayers of EAhy926 cells under control conditions as well as under inhibition of eNOS by L-NMA (L-N-methyl arginine) and inhibition of SNO by NAC (N-acetyl-L-cysteine). Figure 6A shows that both NAC and L-NMA significantly inhibit PAF-induced hyperpermeability to FITC-dextran 70. The abolition of hyperpermeability was associated with an effective reduction of PAF-induced SNO of β-catenin and p120 in strong support of a cause-effect relationship between PAF-induced SNO and PAF-induced hyperpermeability to macromolecules (Figure 6B). Pretreatment of the cells for 15 minutes with 500umol/L NEM (N-ethylmaleimide; which blocks SH-groups impairing NO binding) strongly blocks S-nitrosation of β-catenin and p120 in response to PAF (Figure 6C). In further support of our hypothesis, we showed that NAC and L-NMA prevented the PAF-induced disruption of the association between β-catenin and p120 (Figure 6C). Taken together, these data demonstrate SNO-mediated disruption of the interaction between β-catenin and p120 at the adherens junction is a relevant mechanism by which eNOS-derived NO regulates endothelial permeability.
Figure 6.
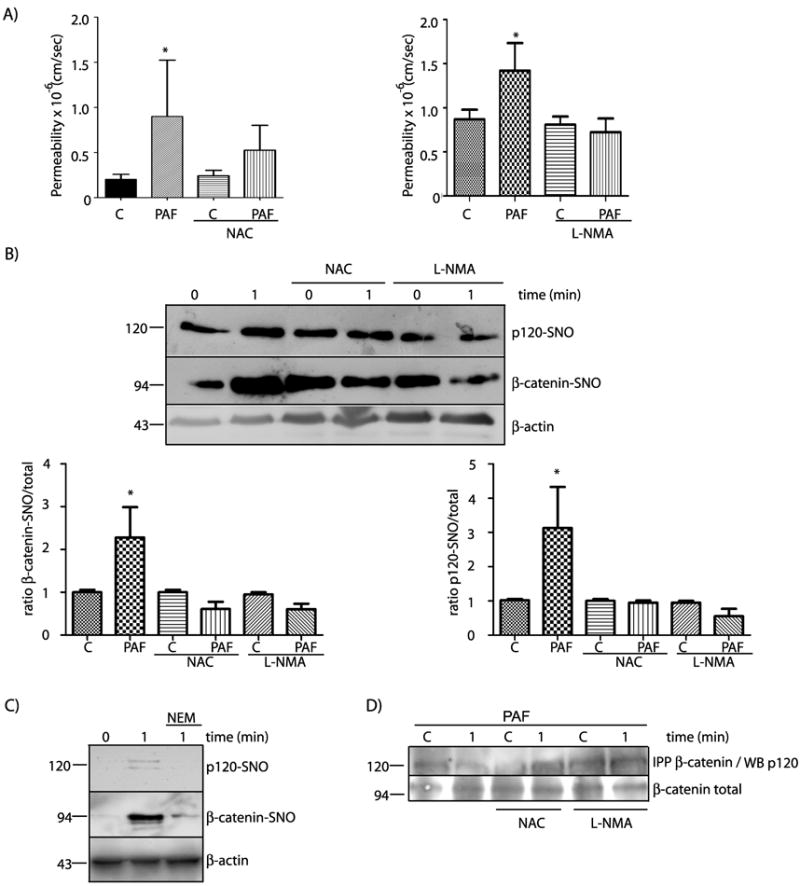
PAF-induced hyperpermeability to macromolecules correlates strongly with SNO of β-catenin and p120 in EAhy926 cells. A) PAF-induced hyperpermeability in EAhy926 cells is blocked by inhibition of SNO with NAC (left side) and by inhibition of eNOS with L-NMA (right side). C= control. Data are expressed as mean permeability ± SEM. * P < 0.05 relative to control, n = 3. B) PAF-induced SNO of β-catenin and p120 is prevented by NAC, a competitive inhibitor of SNO, and by L-NMA, an inhibitor of eNOS. * P < 0.05 as compared with control; n =3. C) PAF-induced SNO of β-catenin and p120 is prevented by NEM, a blocker of SH groups. D) Protein extracts from EAhy926 cells treated with 10-7 moles/L PAF for 1 minute, in the presence of NAC or L-NMA, were immunoprecipitated for β-catenin and probed by Western blot with anti-p120 antibodies. The representative blot shows that NAC and L-NMA blocked PAF-induced disruption of the association between β-catenin and p120.
PAF induces SNO in vivo
To assess whether or not SNO is a mechanism that causes hyperpermeability in vivo, we administered 10-7 mol/L PAF to the mouse cremaster muscle and evaluated nitrosation by the biotin switch assay. PAF application resulted in significant SNO of β-catenin and p120 (Figure 7). The data, obtained at 3 minutes of continuous PAF application, are in agreement with previous reports demonstrating eNOS-derived NO induced hyperpermeability1.
Figure 7.
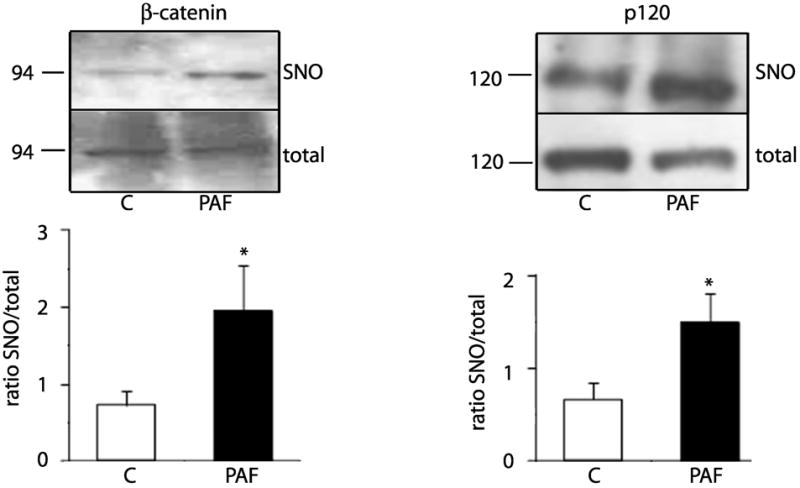
PAF induces S-nitrosation of β-Catenin and p120 in the in vivo mouse cremaster muscle. PAF, at 10-7 moles/L, was applied to one cremaster muscle for 3 minutes while buffer was applied to the other cremaster (control) in the same animal. Homogenized tissues were prepared for biotin switch assay and streptavidin pull down to determine β-catenin and p120 S-nitrosation. The top panels show representative Western blot. The lower panels display the ratio of SNO to total protein (input) for each junctional protein. * P < 0.05 as compared with control; n =6.
Identification of S-nitrosated cysteines in p120
To determine the cysteines nitrosated by NO in p120, we exposed 1 μg of purified p120 to 100 μmol/L S-nitrosoglutathione (GSNO) at 37C for 30 minutes (see Suppl. Data for a detailed account). Analysis by mass spectroscopy identified five cysteines that can be S-nitrosated: C429, C450, C579, C618 and C692 (Figure 8). Of these aminoacids, C579 is the only residue that was S-nitrosated in 100% of the assayed peptides. C579 locates at helix 3 of repeat 5 (R5H3) of the Armadillo domain, a region that is important for the interactions of p120 with E-cadherin42, 43, 28. Therefore, the modification of this cysteine could affect the interaction with VE-cadherin.
Figure 8.

Mapping of S-nitrosation sites in p120. Protein sequence from human p120. Purified p120 was S-nitrosated with GSNO for 30 minutes, subjected to biotin switch assay followed by in-solution trypsin digest. The region that potentially interacts with VE-cadherin is shown in gray background. Cysteine 579 (white letter, red background) was 100% S-nitrosated according to mass spectrometry. Other cysteines (red background) were not S-nitrosated in 100% of the assays. Unlabeled cysteines were not S-nitrosated. (See Supplemental Data for experimental details).
DISCUSSION
Our results advance the novel concept that location of eNOS is important for effective S-nitrosation of endothelial junctional proteins by NO, in particular β-catenin and p120. Our data provide strong support for the hypothesis that SNO of junctional proteins leads to hyperpermeability. Importantly, we show that SNO of β-catenin and p120 is a powerful mechanism that contributes to PAF-stimulated hyperpermeability in vivo.
Increases in microvascular permeability to macromolecules via paracellular pathways have been attributed to two main mechanisms: a) cytoskeletal contraction mediated by myosin light chain29 and b) phosphorylation of adherens junction proteins that causes their internalization away from the adherens junctions30, 12. These processes lead to re-organization of the adherens junctional complex and allow transport of macromolecules across endothelial monolayers. The process of permeability regulation is more complex as modifications of cyclic nucleotides by phosphodiesterases and changes in eNOS activity (leading to nitrosation and/or nitration) may play a role in reducing and enhancing the barrier properties of the endothelium32, 33. While nitric oxide has been reported as an agent that contributes to decrease microvascular permeability34 as well as to increase it3, 35, the importance of NO in promoting the onset of endothelial permeability in response to pro-inflammatory agents has been unequivocally demonstrated using eNOS knockout mouse1, 36 and cells treated with siRNA for eNOS2.
There are no reports of direct effects of NO in the adherens junction in response to PAF in the literature. Using three different cell models (CVEC, EAhy926 and ECV-eNOSGFP cells), we demonstrate S-nitrosation of β-catenin and p120 in response to PAF. This modification leads to changes in the localization and interaction between these proteins at the adherens junction, which finally leads to hyperpermeability. This is the first report linking NO signaling directly to p120 as a target at the adherens junction. Furthermore, we demonstrate that this mechanism does occur in vivo as PAF caused S-nitrosation of β-catenin and p120 in the mouse cremaster muscle at times corresponding to the onset of the hyperpermeability response.
The classic dogma in NO signaling establishes that the actions of NO are mediated via soluble guanylate cyclase (sGC) and protein kinase G (PKG). However, increases in permeability occur in some cellular models regardless of PKG activation but still require NO production14. Our results after inhibition of sGC with ODQ agree with the findings that synthesis of cGMP and activation of PKG are not essential for the development of hyperpermeability in response to PAF (see Figure 3). Recently, S-nitrosation has emerged as an important NO-dependent posttranslational modification (independent of sGC/PKG pathway) of free-thiol cysteines that alters the function of proteins, affecting processes of intracellular trafficking and phosphorylation and requires proximity between eNOS and the target proteins for appropriate NO delivery15, 16, 17. Our results showing S-nitrosation of β-catenin and p120 in cells with cytosolic eNOS strongly confirm the requirement of proximity in order to induce the modification. The fact that plasma membrane-anchored eNOS fails to induce S-nitrosation of these proteins despite that it efficiently produces NO strongly supports the concept that S-nitrosation depends on NO local concentration and the relevance of directed SNO as a mechanism to increase permeability. In support of these concepts, we demonstrate that ACh - an agonist that localizes eNOS to Golgi, stimulates production of NO and induces vasodilation24, 37, 23 - does not cause S-nitrosation. PAF may cause enhanced permeability by formation of peroxynitrite 38, the reaction product of NO and superoxide anion 39. In the hamster cheek pouch, SIN-1 (3-morpholinosydnonimine N-ethylcarbamide) 40, a peroxynitrite generator, induces hyperpermeability 41. However, we did not pursue this line of inquiry because peroxynitrite preferentially leads to protein nitration 42, i.e., protein modification via NO binding to tyrosine rather than by protein nitrosation of cysteines.
Our combined data make a compelling case for SNO of key junctional proteins induced by eNOS-derived NO as a novel mechanism for regulation of permeability to macromolecules across endothelial cell monolayers and the microvasculature. Dissociation between β-catenin and p120 from VE-cadherin disrupts the junctional complex and leads to loss of barrier integrity and enhanced transport of molecules across endothelial monolayers and/or postcapillary venules31. We focused on β-catenin and p120 as relevant targets. The interactions of β-catenin and p120 with VE-cadherin are required for the barrier function. In fact, the absence of p-12043 and β-catenin44, 45, 43 causes loss of barrier integrity and an increase in permeability even in the presence of VE-cadherin. Recently, SNO of β-catenin was associated with changes in permeability in response to 15-minute application of vascular endothelial growth factor46. While confirming SNO of β-catenin, we demonstrate the novel observation that SNO occurs at early times that are consistent with the onset of hyperpermeability in response to pro-inflammatory agonists. In addition, we provide novel evidence showing that SNO of p120 is an important molecular mechanism in PAF-induced hyperpermeability. The relationships and interactions between S-nitrosation and phosphorylation, an established regulatory mechanism, remain to be elucidated. Recent evidence suggests that S-nitrosation plays an important role since mutation of C619, which is subject to S-nitrosation, was sufficient to inhibit VEGF-induced hyperpermeability even though phosphorylation increased in response to the agonist41. In the case of p120 the phosphorylation sites are in the regulatory domain and tail domain, whereas the cysteines that are available for SNO are in the region of possible interactions between p120-VE-cadherin. From these observations, it is possible to speculate that parallel synergism may exist between phosphorylation and S-nitrosation in the process of internalization of junctional proteins.
Based on our mass spectrometry results, we propose that C579 is a strong candidate for SNO-regulation by NO in vivo because it is located in the molecular stretch that interacts with E-cadherin47, 48. Because of the high homology between E-cadherin and VE-cadherin, it is plausible that S-nitrosation of this cysteine affects the interactions of p120 with VE-cadherin. Modification of cysteines by NO is a new chemical mechanism that alters protein-protein interactions at the cell junction and causes enhanced permeability.
Our data demonstrating S-nitrosation of β-catenin and p120 in response to PAF and TNF-α confirm and expand the observation that eNOS-derived NO causes nitrosation of β-catenin in response to VEFG46. These combined data suggest that S-nitrosation of juncional proteins may be a universal mechanism shared by pro-inflammatory agonists. Importantly, we advance the field by demonstrating that S-nitrosation of junctional proteins occurs at times compatible with the onset of hyperpermeability. The NO modification of the cysteines is a new mechanism that alters protein-protein interactions at the cell junction and causes enhanced permeability.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What is known?
Nitric oxide (NO), is a key factor in regulating microvascular permeability.
Platelet-activating factor (PAF) increases permeability via translocation of endothelial nitric oxide synthase (eNOS) to cytosol.
Increases in permeability have been described in cellular models regardless of protein kinase G (PKG) activation.
What new information does this article contribute?
Increase in permeability in response to PAF is independent of PKG activation.
PAF induced S-nitrosation (SNO) of junctional proteins β-catenin and p120 and their internalization away from the plasma membrane.
eNOS located in the cytosol, but not plasma membrane-anchored eNOS, causes SNO of β-catenin and p120.
SNO of β-catenin and p120 significantly diminished association between these proteins.
Mass spectrometry analysis of purified p120 identified cysteine 579 as the main S-nitrosated residue.
During acute inflammation many mediators act on endothelial cells increasing microvascular permeability. Endothelial adherens junction constitute an important element in the control of microvascular permeability. The mechanisms by which NO signaling regulates permeability at adherens junctions are still incompletely understood. We demonstrate S-nitrosation of β–catenin and p120 in response to PAF. This modification leads to changes in the localization and interaction between these proteins at the adherens junction, which finally leads to hyperpermeability. This is the first report linking NO signalling directly to p120 as a target at the adherens junction. Furthermore, we demonstrate that this mechanism does occur in vivo as PAF caused S-nitrosation of β-catenin and p120 in the mouse cremaster muscle at times corresponding to the onset of the hyperpermeability response. We identify cysteine 579 in p120 as the main S-nitrosated residue. We anticipate that our results demonstrating the significance of S-nitrosation in microvascular permeability may serve as a basis for the development of new therapeutic strategies in the treatment of vascular diseases characterized by inflammation (for example: ischemia-reperfusion injury, stroke, cancer and atherosclerosis).
Acknowledgments
This work was supported by grant FONDECYT 1100569, Anillos-ACT71, NIH grants 5RO1 HL070634 and 5RO1 HL088479. The mass spectrometry data were obtained using an Orbitrap instrument funded in part by NIH grant NS046593 for the support of the UMDNJ Neuroproteomics Core Facility.
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- ECV-eNOSGFP
ECV-304 cells transfected with eNOSGFP
- EAhy926
endothelial cell line derived from HUVEC
- CVEC
bovine coronary venular postcapillary endothelial cells
- GFPeNOSG2A
eNOS construct that targets eNOS to the cytosol
- GFPeNOSCAAX
eNOS construct that targets eNOS to the plasma membrane
- MMTS
methyl methanethiosulfonate
- DMEM
Dulbecco’s Modified Eagle Medium
- FITC-dx 70
fluorescein isothiocyanate labeled dextran 70; molecular weight: 70,000 Da
- PAF
Platelet activating factor
- NAC
N-acetyl-L-cysteine
- L-NMA
L-NG-methyl-L-arginine
Footnotes
DISCLOSURES
None.
References
- 1.Hatakeyama T, Pappas PJ, Hobson RW, 2nd, Boric MP, Sessa WC, Duran WN. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. The Journal of physiology. 2006;574:275–281. doi: 10.1113/jphysiol.2006.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanchez FA, Kim DD, Duran RG, Meininger CJ, Duran WN. Internalization of eNOS via caveolae regulates PAF-induced inflammatory hyperpermeability to macromolecules. American journal of physiology. Heart and circulatory physiology. 2008;295:H1642–1648. doi: 10.1152/ajpheart.00629.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramirez MM, Quardt SM, Kim D, Oshiro H, Minnicozzi M, Duran WN. Platelet activating factor modulates microvascular permeability through nitric oxide synthesis. Microvascular research. 1995;50:223–234. doi: 10.1006/mvre.1995.1055. [DOI] [PubMed] [Google Scholar]
- 4.Yuan SY. New insights into eNOS signaling in microvascular permeability. American journal of physiology. Heart and circulatory physiology. 2006;291:H1029–1031. doi: 10.1152/ajpheart.00509.2006. [DOI] [PubMed] [Google Scholar]
- 5.Mayhan WG. Role of nitric oxide in modulating permeability of hamster cheek pouch in response to adenosine 5’-diphosphate and bradykinin. Inflammation. 1992;16:295–305. doi: 10.1007/BF00917622. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez FA, Rana R, Gonzalez FG, Iwahashi T, Duran RG, Fulton DJ, Beuve AV, Kim DD, Duran WN. Functional significance of cytosolic endothelial nitric-oxide synthase (eNOS): regulation of hyperpermeability. J Biol Chem. 2011;286:30409–14. doi: 10.1074/jbc.M111.234294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 8.Dejana E. Endothelial adherens junctions: implications in the control of vascular permeability and angiogenesis. The Journal of clinical investigation. 1996;98:1949–1953. doi: 10.1172/JCI118997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duran WN. The double-edge sword of TNF-alpha in ischemia-reperfusion injury. American journal of physiology. Heart and circulatory physiology. 2008;295:H2221–2222. doi: 10.1152/ajpheart.01050.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geiger B, Ayalon O. Cadherins. Annu Rev Cell Biol. 1992;8:307–332. doi: 10.1146/annurev.cb.08.110192.001515. [DOI] [PubMed] [Google Scholar]
- 11.Weis WI, Nelson WJ. Re-solving the cadherin-catenin-actin conundrum. The Journal of biological chemistry. 2006;281:35593–35597. doi: 10.1074/jbc.R600027200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. Journal of cell science. 1998;111:1853–1865. doi: 10.1242/jcs.111.13.1853. [DOI] [PubMed] [Google Scholar]
- 13.Shasby DM, Ries DR, Shasby SS, Winter MC. Histamine stimulates phosphorylation of adherens junction proteins and alters their link to vimentin. American journal of physiology. Lung cellular and molecular physiology. 2002;282:L1330–1338. doi: 10.1152/ajplung.00329.2001. [DOI] [PubMed] [Google Scholar]
- 14.Lakshminarayanan S, Antonetti DA, Gardner TW, Tarbell JM. Effect of VEGF on retinal microvascular endothelial hydraulic conductivity: the role of NO. Invest Ophthalmol Vis Sci. 2000;41:4256–4261. [PubMed] [Google Scholar]
- 15.Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann RJ, Shah VH, Sessa WC. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19777–19782. doi: 10.1073/pnas.0605907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- 17.Ozawa K, Whalen EJ, Nelson CD, Mu Y, Hess DT, Lefkowitz RJ, Stamler JS. S-nitrosylation of beta-arrestin regulates beta-adrenergic receptor trafficking. Molecular cell. 2008;31:395–405. doi: 10.1016/j.molcel.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Science’s STKE : signal transduction knowledge environment. 2001;2001:pl1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 19.Aramoto H, Breslin JW, Pappas PJ, Hobson RW, 2nd, Duran WN. Vascular endothelial growth factor stimulates differential signaling pathways in in vivo microcirculation. American journal of physiology. Heart and circulatory physiology. 2004;287:H1590–1598. doi: 10.1152/ajpheart.00767.2003. [DOI] [PubMed] [Google Scholar]
- 20.Xiao K, Allison DF, Kottke MD, Summers S, Sorescu GP, Faundez V, Kowalczyk AP. Mechanisms of VE-cadherin processing and degradation in microvascular endothelial cells. The Journal of biological chemistry. 2003;278:19199–19208. doi: 10.1074/jbc.M211746200. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez FA, Rana R, Kim DD, Iwahashi T, Zheng R, Lal BK, Gordon DM, Meininger CJ, Duran WN. Internalization of eNOS and NO delivery to subcellular targets determine agonist-induced hyperpermeability. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6849–6853. doi: 10.1073/pnas.0812694106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meininger CJ, Schelling ME, Granger HJ. Adenosine and hypoxia stimulate proliferation and migration of endothelial cells. The American journal of physiology. 1988;255:H554–562. doi: 10.1152/ajpheart.1988.255.3.H554. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez FA, Savalia NB, Duran RG, Lal BK, Boric MP, Duran WN. Functional significance of differential eNOS translocation. American journal of physiology. Heart and circulatory physiology. 2006;291:H1058–1064. doi: 10.1152/ajpheart.00370.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim DD, Kanetaka T, Duran RG, Sanhez FA, Bohlen HG, Dura WN. Independent regulation of periarteriolar and perivenular nitric oxide mechanisms in the in vivo hamster cheek pouch microvasculature. Microcirculation. 2009;16:323–330. doi: 10.1080/10739680902734876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erwin PA, Lin AJ, Golan DE, Michel T. Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. The Journal of biological chemistry. 2005;280:19888–19894. doi: 10.1074/jbc.M413058200. [DOI] [PubMed] [Google Scholar]
- 26.Fulton D, Babbitt R, Zoellner S, Fontana J, Acevedo L, McCabe TJ, Iwakiri Y, Sessa WC. Targeting of endothelial nitric-oxide synthase to the cytoplasmic face of the Golgi complex or plasma membrane regulates Akt- versus calcium-dependent mechanisms for nitric oxide release. The Journal of biological chemistry. 2004;279:30349–30357. doi: 10.1074/jbc.M402155200. [DOI] [PubMed] [Google Scholar]
- 27.Sessa WC, Garcia-Cardena G, Liu J, Keh A, Pollock JS, Bradley J, Thiru S, Braverman IM, Desai KM. The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. The Journal of biological chemistry. 1995;270:17641–17644. doi: 10.1074/jbc.270.30.17641. [DOI] [PubMed] [Google Scholar]
- 28.Ishiyama N, Lee SH, Liu S, Li GY, Smith MJ, Reichardt LF, Ikura M. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141:117–128. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 29.Yuan SY. Signal transduction pathways in enhanced microvascular permeability. Microcirculation. 2000;7:395–403. [PubMed] [Google Scholar]
- 30.Alexander JS, Alexander BC, Eppihimer LA, Goodyear N, Haque R, Davis CP, Kalogeris TJ, Carden DL, Zhu YN, Kevil CG. Inflammatory mediators induce sequestration of VE-cadherin in cultured human endothelial cells. Inflammation. 2000;24:99–113. doi: 10.1023/a:1007025325451. [DOI] [PubMed] [Google Scholar]
- 31.Kevil CG, Payne DK, Mire E, Alexander JS. Vascular permeability factor/vascular endothelial cell growth factor-mediated permeability occurs through disorganization of endothelial junctional proteins. The Journal of biological chemistry. 1998;273:15099–15103. doi: 10.1074/jbc.273.24.15099. [DOI] [PubMed] [Google Scholar]
- 32.Rentsendorj O, Damarla M, Aggarwal NR, Choi JY, Johnston L, D’Alessio FR, Crow MT, Pearse DB. Knockdown of lung phosphodiesterase 2A attenuates alveolar inflammation and protein leak in a two-hit mouse model of acute lung injury. American journal of physiology. Lung cellular and molecular physiology. 301:L161–170. doi: 10.1152/ajplung.00073.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siddiqui MR, Komarova YA, Vogel SM, Gao X, Bonini MG, Rajasingh J, Zhao YY, Brovkovych V, Malik AB. Caveolin-1-eNOS signaling promotes p190RhoGAP-A nitration and endothelial permeability. The Journal of cell biology. 193:841–850. doi: 10.1083/jcb.201012129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kubes P, Granger DN. Nitric oxide modulates microvascular permeability. The American journal of physiology. 1992;262:H611–615. doi: 10.1152/ajpheart.1992.262.2.H611. [DOI] [PubMed] [Google Scholar]
- 35.Yuan Y, Granger HJ, Zawieja DC, DeFily DV, Chilian WM. Histamine increases venular permeability via a phospholipase C-NO synthase-guanylate cyclase cascade. The American journal of physiology. 1993;264:H1734–1739. doi: 10.1152/ajpheart.1993.264.5.H1734. [DOI] [PubMed] [Google Scholar]
- 36.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, Buerk DG, Huang PL, Jain RK. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Figueroa XF, Gonzalez DR, Martinez AD, Duran WN, Boric MP. ACh-induced endothelial NO synthase translocation, NO release and vasodilatation in the hamster microcirculation in vivo. The Journal of physiology. 2002;544:883–896. doi: 10.1113/jphysiol.2002.021972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klabunde RE, Anderson DE. Role of nitric oxide and reactive oxygen species in platelet-activating factor-induced microvascular leakage. J Vasc Res. 2002;39:238–245. doi: 10.1159/000063689. [DOI] [PubMed] [Google Scholar]
- 39.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dairou J, Atmane N, Rodrigues-Lima F, Dupret JM. Peroxynitrite irreversibly inactivates the human xenobiotic-metabolizing enzyme arylamine N-acetyltransferase 1 (NAT1) in human breast cancer cells: a cellular and mechanistic study. The Journal of biological chemistry. 2004;279:7708–7714. doi: 10.1074/jbc.M311469200. [DOI] [PubMed] [Google Scholar]
- 41.González F. Acción del óxido nítrico sobre distintas vías de señalización. Santiago, Chile: Pontificia Universidad Católica de Chile; 2009. Participación del óxido nítrico en la hiperpermeabilidad a macromoléculas inducida por el factor activador de plaquetas. [Google Scholar]
- 42.Xiong Y, Rabchevsky AG, Hall ED. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. Journal of neurochemistry. 2007;100:639–649. doi: 10.1111/j.1471-4159.2006.04312.x. [DOI] [PubMed] [Google Scholar]
- 43.Herron CR, Lowery AM, Hollister PR, Reynolds AB, Vincent PA. p120 regulates endothelial permeability independently of its NH2 terminus and Rho binding. American journal of physiology. Heart and circulatory physiology. 2011;300:H36–48. doi: 10.1152/ajpheart.00812.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, Czupalla CJ, Reis M, Felici A, Wolburg H, Fruttiger M, Taketo MM, von Melchner H, Plate KH, Gerhardt H, Dejana E. Wnt/beta-catenin signaling controls development of the blood-brain barrier. The Journal of cell biology. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cattelino A, Liebner S, Gallini R, Zanetti A, Balconi G, Corsi A, Bianco P, Wolburg H, Moore R, Oreda B, Kemler R, Dejana E. The conditional inactivation of the beta-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. The Journal of cell biology. 2003;162:1111–1122. doi: 10.1083/jcb.200212157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, Gratton JP. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Molecular cell. 2010;39:468–476. doi: 10.1016/j.molcel.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 47.Ireton RC, Davis MA, van Hengel J, Mariner DJ, Barnes K, Thoreson MA, Anastasiadis PZ, Matrisian L, Bundy LM, Sealy L, Gilbert B, van Roy F, Reynolds AB. A novel role for p120 catenin in E-cadherin function. The Journal of cell biology. 2002;159:465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu H, Komiya S, Shimizu M, Fukunaga Y, Nagafuchi A. Involvement of p120 carboxy-terminal domain in cadherin trafficking. Cell Struct Funct. 2007;32:127–137. doi: 10.1247/csf.07023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.