

Summary
Genetic engineering of human induced pluripotent stem cells (hiPSCs) via customized designer nucleases has been shown to be significantly more efficient than conventional gene targeting, but still typically depends on the introduction of additional genetic selection elements. In our study, we demonstrate the efficient nonviral and selection-independent gene targeting in human pluripotent stem cells (hPSCs). Our high efficiencies of up to 1.6% of gene-targeted hiPSCs, accompanied by a low background of randomly inserted transgenes, eliminated the need for antibiotic or fluorescence-activated cell sorting selection, and allowed the use of short donor oligonucleotides for footprintless gene editing. Gene-targeted hiPSC clones were established simply by direct PCR screening. This optimized approach allows targeted transgene integration into safe harbor sites for more predictable and robust expression and enables the straightforward generation of disease-corrected, patient-derived iPSC lines for research purposes and, ultimately, for future clinical applications.
Highlights
-
•
Footprintless gene editing in hiPSCs using ssODNs without any preselection
-
•
Targeted clone isolation simply via PCR screening
-
•
Targeted reporter integration into safe harbor locus AAVS1 with up to 1.6% efficiency
-
•
hiPSC reporter assay for gene editing via customized designer nucleases
This study by Merkert et al. demonstrates efficient nonviral and selection-independent gene targeting in human pluripotent stem cells. High efficiencies of up to 1.6% of gene-targeted hiPSCs eliminated the need for antibiotic or FACS selection, and allowed the use of short donor oligonucleotides for footprintless gene editing and clone establishment via direct PCR screening.
Introduction
To date, it is extremely difficult to perform site-specific transgenesis and gene targeting in patient-specific cells due to the inability to sufficiently expand most primary cell types or adult stem and progenitor cell lineages in vitro. However, the availability of human induced pluripotent stem cells (hiPSCs), with their far-reaching potential for proliferation and differentiation, now offers novel opportunities for biomedical research and ultimately the development of tailored cellular therapies. The ability to genetically modify pluripotent stem cells (PSCs) through the introduction of reporter and selection genes or for the overexpression of disease-related transgenes would further broaden their usefulness for drug screening, disease modeling, and cellular therapies. Moreover, the possibility to genetically and functionally correct inherited gene defects in patient-specific iPSCs may pave the way for novel concepts of ex vivo gene therapy.
Clearly, conventional viral and nonviral gene transfer technologies resulting in the random integration of the introduced genetic elements and more or less unpredictable integration-site-dependent expression of the transgene are not in accordance with the requirements of current biomedical research. It has also been shown in animal experiments and clinical studies that random integration and insertional mutagenesis can result in the malignant transformation of stem cell transplants (Hacein-Bey-Abina et al., 2003; Modlich et al., 2009; Stein et al., 2010). It is therefore of the utmost importance to develop more precise techniques that enable efficient site-specific gene editing and safe long-term transgene expression at well-defined genomic integration sites in human PSCs (hPSCs) and especially iPSCs.
In murine embryonic stem cells (mESCs), gene targeting through homologous recombination (HR) has been utilized over the last 25 years to generate thousands of knockout mice, which has led to major advances in our basic understanding of mammalian biology, gene function, and disease mechanisms. Although the frequencies of HR are rather low in classical approaches (10−4 to 10−6 in mESCs) (Doetschman et al., 1988; Reid et al., 1991), such techniques have so far represented the standard approach for producing gene knockouts in mESCs and mice due to the relative robustness of mESC culture and high transfection rates in ESCs. Although two papers reported frequencies of HR (1.5–4 × 10−6) in a range similar to that seen in mESCs (Di Domenico et al., 2008; Zwaka and Thomson, 2003), conventional gene targeting in human ESCs (hESCs) is still considered to be more difficult and less successful due to challenging culture characteristics and lower transfection rates (Elliott et al., 2011; Goulburn et al., 2011; Irion et al., 2007). Moreover, until recently, the very low survival rates obtained after dissociation prevented fluorescence-activated cell sorting (FACS) and single-cell cloning. It is only since the invention of the Rho-associated coiled-coil kinase (ROCK) inhibitor Y-27632 that such techniques have become feasible for hPSCs (Zweigerdt et al., 2011).
More recently, however, it has been demonstrated that targeted induction of double-strand breaks (DSBs) by employing tailored designer nucleases, such as zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeat (CRISPR) RNA-guided nucleases greatly enhances HR (Fu et al., 2013; Mussolino and Cathomen, 2012; Rahman et al., 2011). ZFNs and TALENs consist of a target-specific DNA-binding domain fused to an unspecific nuclease domain, which induces a DSB upon activation. A ZFN/TALEN-induced DSB can be repaired either by nonhomologous end joining (NHEJ) or by HR (Shrivastav et al., 2008). Recent reports demonstrated that ZFNs and TALENs allow for not only efficient gene inactivation through NHEJ but also enhanced HR-based gene targeting in hPSCs (Hockemeyer et al., 2009, 2011; Soldner et al., 2011; Zou et al., 2009). Remarkably, ZFN/TALEN-based HR has already been applied for functional correction of genetic diseases either by genotypic correction of the defective gene (Yusa et al., 2011) or by insertion of the functional gene into a safe harbor locus (Zou et al., 2011). The majority of gene-targeting studies in hPSCs directly applied a transgene-based antibiotic selection of targeted clones (Hockemeyer et al., 2009, 2011; Sebastiano et al., 2011; Yusa et al., 2011; Zou et al., 2011). Clearly, further improvements in targeting efficiencies would not only minimize the required screening procedures but would considerably facilitate selection-independent targeting approaches in PSCs, including footprintless restoration of wild-type sequences in disease-specific iPSCs prior to their clinical application.
Although in previous studies by Soldner et al. (2011) and Ding et al. (2013), an initial FACS-based enrichment step of TALEN-expressing cells prior to subsequent PCR-based screening of this enriched cell population had to be included, both groups provided initial evidence that the footprintless correction of disease-specific mutations in hPSCs without the need for an antibiotic selection is generally possible through the application of single-stranded oligodeoxynucleotides (ssODNs).
In this study, we report on an efficient technique for nonviral and selection-independent gene targeting in hESCs and hiPSCs. For the development of our targeting protocol, we first used an iPSC enhanced GFP (eGFP) reporter system in which ZFNs were applied to disrupt the eGFP open reading frame by NHEJ with an overall efficiency of up to 4%, and to integrate a red fluorescent protein into the eGFP locus by HR. Without any antibiotic selection, we achieved HR-targeting efficiencies of up to 1.2% and could show that the ZFN-treated PSCs preserved their pluripotency and chromosomal integrity. Finally, we targeted the endogenous “safe harbor” locus AAVS1, which is known for robust transgene expression in PSCs (Smith et al., 2008). Here, by using ZFNs and TALENs, we obtained targeting efficiencies comparable to those achieved with our eGFP/RedStar reporter system for one hESC and two hiPSC lines, and generated stable transgenic PSC lines by FACS. Moreover, by applying TALENs together with short ssODN donors without any preselection, we show that the high targeting efficiencies obtained actually facilitate direct PCR screening of correctly targeted clones. This should enable footprintless gene correction and transgene-independent isolation of mutation-corrected, disease-specific iPSC clones, and may ultimately lead to novel concepts for iPSC-based ex vivo gene therapies.
Results
Efficient ZFN-Based Reporter Gene Inactivation in Transgenic hiPSCs through NHEJ
To develop a reporter system for gene targeting in hPSCs, we first established transgenic hiPSC clones stably expressing eGFP under control of the ubiquitous cytomegalovirus early enhancer element and chicken beta-actin (CAG) promoter as described in the Experimental Procedures. Optimization of cell transfection included supplementation of the ROCK inhibitor Y-27632 for improved survival after single-cell dissociation (data not shown), the use of feeder-free culture conditions, and a direct comparison of two electroporation devices (Figure S1A available online). Notably, we observed significantly higher transfection rates after microporation using the Neon device as compared with the Amaxa Nucleofection technology, which is commonly considered to be the most efficient technique for transfection of hPSCs. Under optimized conditions, transfection rates of 72% ± 16% were observed in hPSCs (Figure S1A). The application of these optimized transfection conditions followed by antibiotic selection led to the isolation of two cell clones with single eGFP integration (hCBiPS2eGFPC7 and hCBiPS2eGFPC16), an important prerequisite for their use as a reporter cell line (Figures S1B–S1E). To inactivate the eGFP reporter gene (Figure 1A), we applied our protocol to transfect hCBiPS2eGFPC7 cells with increasing amounts of eGFP-specific ZFN expression plasmids. Analysis of the percentage of eGFPneg cells at day 6 or 7 posttransfection via flow cytometry revealed a distinct population of up to 4% eGFPneg cells (Figures 1B and 1E). Comparative experiments with the second eGFP reporter cell line hCBiPS2eGFPC16 revealed eGFP knockout efficiencies of 2% (Figure 1C). Due to the high stability of the eGFP protein, the distinct eGFPneg cell population emerged only at day 6 after transfection (Figure 1D). Notably, the efficiency of eGFP inactivation was clearly dependent on the amount of transfected ZFN encoding plasmids (Figure 1E). To confirm the gene knockout, we established eGFPneg hCBiPS2eGFPC7 cell clones via FACS and prepared coding DNA. Sequencing of the eGFP open reading frame of six randomly selected cell clones showed a deletion of 5, 9, or 15 bp, or an insertion of 4 or 265 bp overlapping with the ZFNs target sites compared with the wild-type sequence (Figure 1F). These sequencing data confirmed that all analyzed eGFPneg clones lost eGFP expression due to ZFN-targeted inactivation through the error-prone NHEJ; consequently, two clones showed the same mutation and may have been derived from the same NHEJ event.
Figure 1.
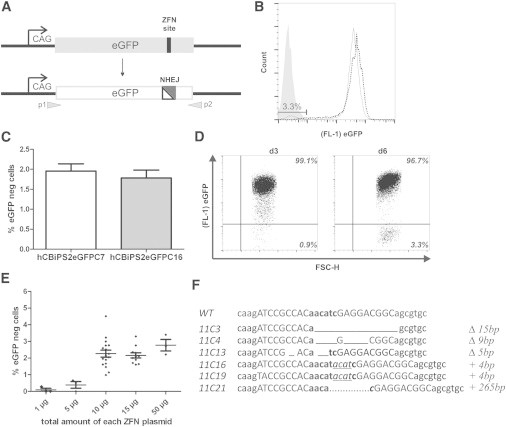
Efficient ZFN-Based Gene Knockout in hPSCs
A hiPSC reporter line carrying a single eGFP copy under control of the CAG promoter was used.
(A) Scheme of eGFP knockout by ZFN-mediated NHEJ. Gray arrows indicate primers used for cDNA sequencing.
(B) Flow-cytometric analysis of eGFP disruption. Expression of eGFP-specific ZFNs resulted in 3.3% eGFPneg iPSCs (continuous line) at day 6 posttransfection as shown by flow cytometry. The dotted line represents the eGFP reporter cell line before transfection. The gray histogram illustrates hCBiPS2 cells without eGFP.
(C) Direct comparison of eGFP inactivation through NHEJ between two hCBiPS2eGFP reporter cell clones (C7 and C16). The proportion of eGFPneg cells on day 6 after transfection with eGFP-ZFN plasmids (mean ± SEM of seven independent experiments for direct comparison) is shown.
(D) Flow cytometry demonstrated a distinct eGFPneg iPSC population only as of day 6 after transfection.
(E) ZFN-dose-dependent gene knockout. After transfection of hCBiPS2eGFPC7 cells with increasing amounts of ZFN-plasmid DNA, the percentage of eGFPneg cells was determined by flow cytometry (depicted is the total amount of each ZFN plasmid; each point is a biological replicate; mean ± SEM).
(F) Sequence analysis of eGFPneg iPSC clones. cDNA of all eGFPneg iPSC clones analyzed showed eGFP mutations surrounding the predicted DSB. ZFN target sites are shown in capital letters. Δ, deletion of basepairs; +, insertion of base pairs.
See also Figure S1.
Efficient HR-Mediated Gene Targeting in hiPSCs
Next, we utilized our reporter cell line to assess the efficiency of HR-mediated gene targeting in hiPSCs. For this purpose, an appropriate eGFP donor vector carrying a 2A-Redstarnuc-poly(A) expression cassette flanked by two eGFP homology arms was constructed (Figure 2A). With application of this vector, successful HR-mediated targeting should result in the functional insertion of the RedStarnuc cassette associated with transcriptional inactivation of the eGFP gene. The utilization of a 2A sequence in our system enables eGFP-locus-specific RedStarnuc expression and should minimize the chance for undesired RedStarnuc expression after random donor insertion.
Figure 2.
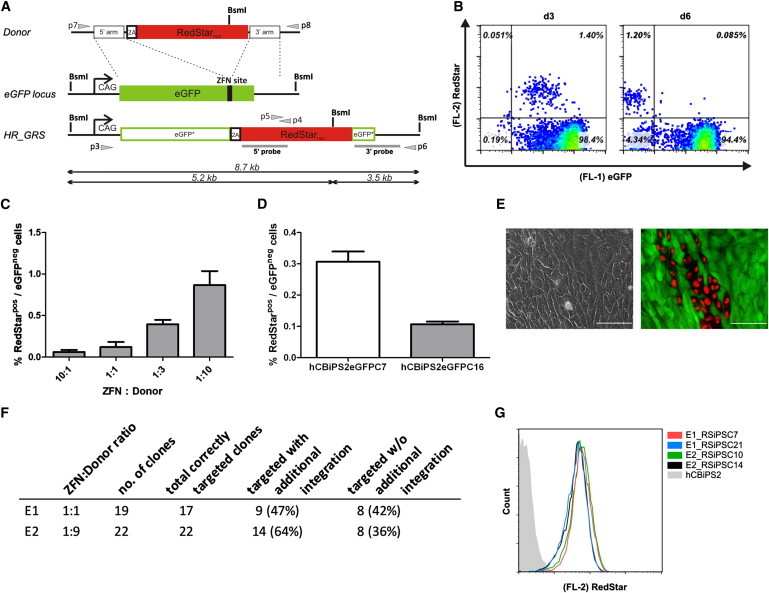
ZFN-Based Gene Targeting in hiPSCs
An iPSC reporter line carrying a single eGFP copy under control of the CAG promoter was used.
(A) Schematic illustration of the targeting strategy. The donor vector contains a 2A-RedStar-poly(A) expression cassette flanked by two arms of 700 bp eGFP locus homology sequences. The anticipated HR product (HR_GRS) is shown below. Southern blot probes are shown as gray boxes. The gray arrows represent primers used to confirm the 5′ and 3′ junctions generated by targeted integration of the respective reporter construct. 2A, self-cleaving peptide sequence; RedStarnuc, variant of red fluorescent protein containing a nuclear membrane location signal; eGFP, enhanced green fluorescence protein; CAG, cytomegalovirus early enhancer element coupled to chicken beta-actin promoter.
(B) Targeted knockin of RedStarnuc. ZFN-based HR resulted in 1.2% targeted eGFPnegRedStarpos hiPSCs at day 6 posttransfection as shown by flow cytometry.
(C) Dose dependency of HR. A molar ratio of 1:10 (ZFN:donor) resulted in the most efficient HR as indicated by the proportion of eGFPnegRedStarpos hiPSCs at day 7 posttransfection. The molar ratio 1:10 is equivalent to 1 mol of each ZFN plasmid to 10 mol of donor plasmid, with a total amount of each ZFN plasmid of 10 μg (mean ± SEM of three independent experiments).
(D) Direct comparison of targeting efficiencies between two hCBiPS2eGFP reporter cell clones. Proportion of eGFPneg/RedStarpos cells on day 6 after transfection with eGFP-ZFN plasmids and 2A-RedStar donor plasmid (mean ± SEM of three independent experiments using a ZFN:donor ratio of 1:3).
(E) Fluorescence microscopy (day 6 after transfection) showing a group of targeted eGFPnegRedStarpos hiPSCs within a monolayer of cells, which show the original eGFPposRedStarneg phenotype. Scale bars represent 100 μm.
(F) Tabular summaries of genomic analyses. PCR/Southern blot analyses of FACS-sorted eGFPnegRedStarpos single hiPSC clones isolated after two targeting experiments (E1 and E2) applying different molecular ratios of ZFN:donor (for details, see Figures S2A and S2B).
(G) RedStar expression profile. Flow cytometry demonstrated comparable intensities of RedStar fluorescence in four different eGFPnegRedStarpos iPSC clones.
See also Figure S2.
Accordingly, hCBiPS2eGFPC7 cells were transfected with different ratios of ZFN versus donor expression plasmids and analyzed on day 3 and day 6 posttransfection. After 3 days, we obtained a distinct population of eGFPposRedStarpos cells, which turned into eGFPnegRedStarpos cells after a further 3 days (Figure 2B). Flow-cytometric analysis revealed up to 1.2% RedStarposeGFPneg hCBiPS2eGFPC7 cells and demonstrated a clear dependency on the ZFN:donor ratio (Figure 2C). Similar experiments with the second eGFP reporter cell line (hCBiPS2eGFPC16) showed robust, but 3-fold lower, eGFP gene-targeting efficiency as compared with the hCBiPS2eGFPC7 cells (Figure 2D). Successfully targeted hCBiPS2eGFPC7 cells expressed bright RedStar protein targeted to the nuclear membrane and completely lost their eGFP expression (Figure 2E). Remarkably, microscopic analyses revealed various cell clusters consisting of eGFPnegRedStarpos cells spread over the plate, which may indicate a multitude of independent HR events.
FACS was applied to sort and establish single-cell clones from RedStarposeGFPneg hCBiPS2eGFPC7 cells for further confirmation of HR-mediated gene targeting. PCR and Southern blot analysis (Figures S2A and S2B) revealed that almost all established clones were correctly targeted (95%) and an unexpectedly high proportion of the clones (36%–42%) did not carry additional nonhomologous integrations (Figure 2F). All clones analyzed showed comparable levels of RedStarnuc expression (Figure 2G). Two representative RedStarposeGFPneg hCBiPS2eGFPC7 cell clones (E1_RSiPSC7 and E2_RSiPSC14) were chosen for further characterization. Both clones retained a normal karyotype (Figure S2C), suggesting the absence of deleterious side effects of the ZFN treatment. Morphologically, both clones resembled hESCs and maintained their RedStarnuc expression for up to 20 passages and during differentiation (Figures S2D and S2E). Furthermore, they expressed pluripotency markers such as OCT4, NANOG, SOX2, SSEA-3, and SSEA-4, and could be differentiated in vitro into derivatives of all three germ layers (Figures S2D and S2E).
Efficient Gene Targeting of the Endogenous AAVS1 Locus in hPSCs using ZFNs and TALENs
Finally, we sought to evaluate gene targeting of an endogenous locus and decided to use the human safe harbor site AAVS1 for the generation of stable transgenic PSC lines. Thus, AAVS1-specific ZFNs, together with one donor plasmid for CAG-promoter-mediated ubiquitous expression of either eGFP or RedStarnuc (Figure 3A), were transfected into two hiPSC lines and the hES3 cell line. The targeting efficiency in terms of transgene (eGFP or RedStarnuc) expressing cells was determined on day 14 after transfection. Control transfections without ZFN plasmids were performed to determine background expression due to random integration and potential long-term transient donor expression. Transfection of the respective donor plasmid together with both AAVS1 ZFNs resulted on average in 0.8% eGFPpos/RedStarpos hiPSCs/ESCs on day 14 after transfection when using a ratio of 1:3 (ZFN:donor). In contrast, control transfection of the donor plasmid alone resulted in an average of 0.08% eGFPpos/RedStarpos cells, presumably representing cells with randomly integrated transgenes (Figure 3B). The observed targeting efficiency was comparable to the results we obtained in targeting the transgenic eGFP locus (see Figure 2) and was verified by PCR analysis of targeted integration in the AAVS1 locus for 97% of all established eGFPpos/RedStarpos cell clones derived from two iPSC lines and one ESC line (Figures 3C and S3A). Consistent with the results we obtained in targeting the GFP reporter locus (see Figure 2F), a very high proportion of AAVS1-targeted clones (25%–79%) did not show any additional random integration of the applied transgenes. Interestingly, 13%–75% of the correctly targeted clones actually underwent HR at both AAVS1 alleles (Figures 3C and S3A). Finally, two iPSC and two ESC clones were characterized in more detail (data for ESC clones not shown). The eGFP as well as the RedStar transgenic clones retained a normal karyotype (Figure S3B) and showed comparable bright fluorescence intensity (Figure 3D). The expression of the respective transgene was also stable in differentiating cells (Figure 3D) and was observed in cell types of all three germ layers (Figures S3C and S3D). In addition to the experiments with the ZFNs, transfection experiments with AAVS1-specific TALENs were performed with one hiPSC line. The targeting efficiencies for TALENs were even higher than those for ZFNs and resulted on average in 1.6% eGFPpos/RedStarpos hiPSCs (Figure 3B).
Figure 3.
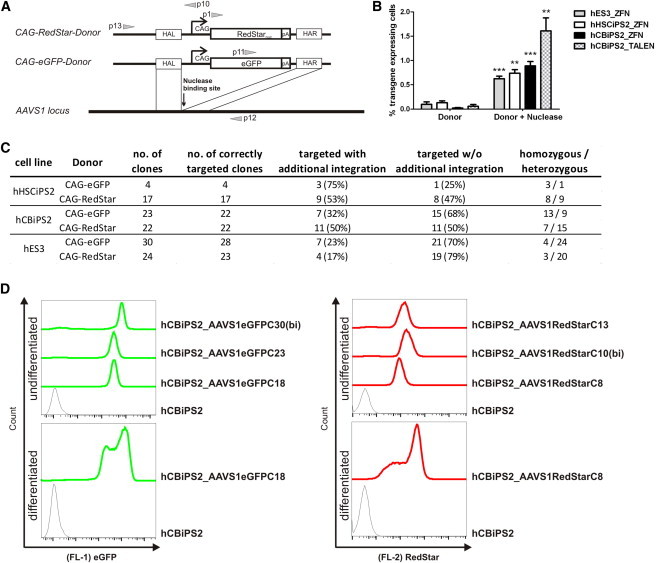
Efficient Gene Targeting at the AAVS1 Locus in hiPSCs and hESCs
(A) Schematic illustration of the targeting strategy. Either a CAG-eGFP donor plasmid or a CAG-RedStarnuc donor plasmid carrying ∼700 bp of homology on both sides was applied. The gray arrows represent primers used to confirm the 5′ and 3′ junctions generated by targeted integration of the respective reporter constructs. RedStarnuc, mutant of red fluorescent protein coupled to nuclear membrane location signal; eGFP, enhanced green fluorescence protein; CAG, cytomegalovirus early enhancer element coupled to chicken β-actin promoter.
(B) Proportion of transgene (eGFP or RedStar) expressing cells on day 14 after transfection. ZFN- or TALEN-based donor integration into the AAVS1 locus is shown in comparison with control transfection without nuclease plasmids (mean ± SEM of four independent experiments).
(C) Summary of AAVS1 targeting and additional random donor integration in single-cell clones.
(D) Flow cytometry demonstrated comparable intensities of eGFP (left) or RedStar (right) fluorescence in three different hCBiPS2_AAVS1-targeted cell clones, respectively (upper histograms). Notably, the biallelic targeted clones showed a slightly elevated transgene expression compared with monoallelic targeted clones. In the lower histograms, the detected stable eGFP or RedStar expression of one chosen transgenic clone is depicted on day 25 of EB-based differentiation (continuous line) as compared with nontargeted hCBiPS2 cells (dashed line).
See also Figure S3.
Efficient Editing of the AAVS1 Locus in hiPSCs using ssODNs
Targeting via designer nucleases typically uses donor plasmids with homology arms of at least 200–800 bp. Recent studies showed that short ssODNs can be used as an alternative template for genome editing in human cells (Chen et al., 2011; Ding et al., 2013; Soldner et al., 2011). Importantly, such donor oligonucleotides would provide a straightforward technique for footprintless gene editing without the need for antibiotic or FACS selection, if targeting could be achieved with similar efficiencies as observed for conventional plasmid donors. We therefore aimed to evaluate whether our efficient targeting protocol would enable direct PCR screening for gene targeting with ssODNs in the AAVS1 locus. As a donor, a 98 bp ssODN carrying a HindIII site flanked by 43 bp of homology on each side of the TALEN cut side was synthesized (Figure 4A). The hCBiPS2 cell line was transfected with AAVS1-specific TALENs together with the ssODN and plated onto 96-well plates for limiting dilution. Up to five cells survived per well, and PCR screening for targeted integration showed successful insertion of the ssODN in 32 out of 480 (7%) analyzed wells (Figure 4B). Limiting dilution from nine positive wells to single-cell clones yielded 33 clones from 144 (23%) with targeted integration (Figure S4), from which we randomly chose seven clones for further evaluation. PCR amplification of their AAVS1 loci, HindIII digestion (Figure 4C), and sequencing of the PCR products revealed correct monoallelic HindIII conversion. Additional karyotype analysis for three clones proved chromosomal integrity (data not shown).
Figure 4.
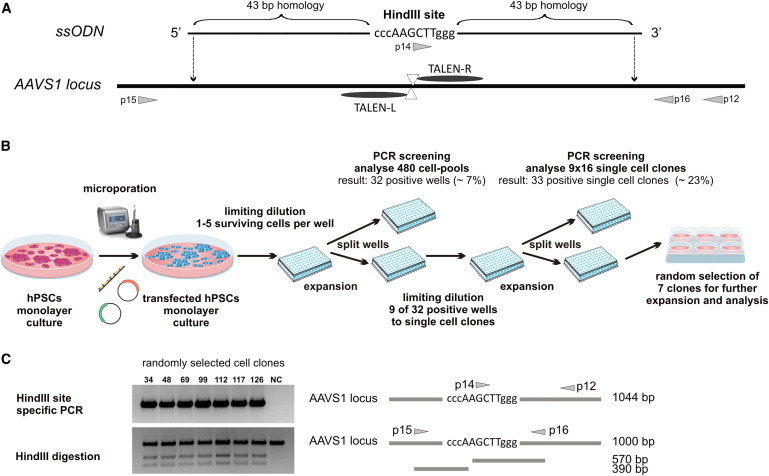
Efficient Editing of the AAVS1 Locus in hiPSCs Using an ssODN
(A) The schematic shows the 98 bp ssODN donor DNA designed to incorporate the HindIII site into the AAVS1 locus. The gray arrows represent primers used to confirm the targeted integration and to amplify the PCR product for HindIII digestion.
(B) Schematic illustration of the targeting procedure and establishment of single-cell clones.
(C) PCR screening with two rounds of single-cell dilution identified 33 positive single iPSC clones (see Figure S4). Seven out of 33 clones were randomly selected for further analyses. All seven clones carried a HindIII site introduced through targeted ssODN integration, as demonstrated by HindIII site-specific PCR (upper picture) and HindIII digestion (lower picture). NC, negative control (hCBiPS2).
See also Figure S4.
Discussion
Genetic engineering techniques based on tailored nucleases, such as ZFNs and TALENs, represent a genuine breakthrough in gene targeting. In particular, these technologies offer exciting opportunities for genetic engineering in patient-specific iPSCs with respect to the introduction of reporter genes as well as the correction and introduction of disease-specific mutations. Apart from two studies (Ding et al., 2013; Soldner et al., 2011), all known publications in this area have applied transgene-based antibiotic selection to isolate targeted cell clones. Obviously, low efficiencies typically continue to hamper the antibiotic-selection-free isolation of correctly targeted clones and impede footprintless correction and introduction of disease-specific mutations.
For the establishment and optimization of our targeting protocol, we used a reporter iPSC line, which enabled us to visualize successful ZFN-based gene targeting in hPSCs. Using this line, we observed a proportion of up to 4% of eGFPneg cells 6 days after transfection, which is similar to what was recently reported for NHEJ-based dTomato reporter inactivation in the active DNMT3b locus in hESCs by using CRISPR RNA-guided nucleases (Hou et al., 2013). Remarkably, the observed proportion of eGFPneg cells does not reflect the actual efficiency of DSB generation. Targeted DSBs are probably induced in a considerably higher proportion of cells, followed by an unknown frequency of error-free repair by NHEJ. In order to extend these findings to HR-based gene targeting, we constructed a donor plasmid with two arms of ∼700 bp of DNA homologous to the sequences enclosing the predicted ZFN target site within the genomically inserted eGFP. Notably, a red fluorescence reporter, RedStarnuc, which is targeted to the nuclear membrane, was chosen in order to ensure discrimination from potential autofluorescent cells. Also of importance, the RedStarnuc was not coupled to a promoter, but was cloned in frame to the eGFP sequence via a 2A sequence to exclude transient, plasmid-based RedStar expression, and to minimize a potential background due to undesired reporter expression after random genomic insertion of the donor plasmid. Three days after transfection of the hCBiPS2eGFPC7 reporter line with the ZFN plasmids and the donor plasmid, a population of up to 1.2% of RedStarnucpos cells was detectable. At this time point, the vast majority of these cells still expressed eGFP, which can be explained by the typical persistence of eGFP mRNA/protein in hPSCs for several days (Hartung et al., 2013). Also, this persistence over a period of at least 3 days correlates perfectly with our observation of the time course of fluorescence loss after NHEJ-based eGFP inactivation. Six days after transfection, however, no eGFPposRedStarpos cells were observed, indicating that almost all RedStarpos cells represent correctly targeted cells. The fact that cell clusters consisting of eGFPneg RedStarpos cells were distributed over the whole plate speaks strongly in favor of a multitude of independent HR events. In this way, we achieved considerably higher targeting rates than those previously reported for the correction of a mutant eGFP reporter gene in 0.24% of hESCs and 0.14% of hiPSCs (Zou et al., 2009).
Targeting experiments with a second eGFP reporter cell line (hCBiPS2eGFPC16) showed similar results. The observed 3-fold lower eGFP gene targeting efficiency for hCBiPS2eGFPC16 as compared with hCBiPS2eGFPC7 cells most likely represents clone-specific variations with respect to survival rates after single-cell dissociation, transfection efficiency, and general growth characteristics under feeder-free monolayer culture conditions. Correct targeting in eGFPnegRedStarpos cells was confirmed through PCR and Southern blot analysis in 39 out of 41 isolated clones derived from two targeting experiments. In the two remaining clones, accurate targeting occurred with respect to the 5′ junction, but the 3′ junction was not correctly formed. As observed for the established eGFP reporter clone, RedStar expression of different targeted clones was almost uniform and persisted to a large extent during differentiation.
Subsequently, we aimed to evaluate gene targeting for another genomic locus. Through a highly efficient transfection of AAVS1-specific ZFN and TALEN plasmids, we obtained targeting efficiencies that were similar, or in the case of TALENs even higher, than the 0.8% reported by Lombardo et al. (2011) for eGFP integration in the same locus using integrase-deficient lentiviral ZFN vectors. In the case of plasmid-donor-based AAVS1 targeting, transgenic clones were identified based on the emergence of eGFP or RedStar expression. Remarkably, only four nontargeted clones among a total of 120 isolated eGFPpos or RedStarpos clones were detected by PCR. This high proportion of ∼97% correctly targeted clones, together with the fact that on average only 40% of correctly targeted clones contained additional random integrations, underlines the efficiency of ZFN-based gene targeting in our protocol. Notably, the frequency of additional (nontargeted) integration was in a range similar to the 24%–38% reported in previous studies that applies transgene-based antibiotic selection after ZFN- or TALEN-mediated integration at AAVS1 in hiPSCs (Hockemeyer et al., 2009, 2011; Zou et al., 2009). Interestingly, compared with these studies, which reported 6%–32% biallelic targeting (Hockemeyer et al., 2009; Zou et al., 2011), we observed an even higher proportion (13%–75%) of ZFN-targeted clones in which both alleles had been edited correctly. Given that correct targeting occurs only in up to 0.8% of transfected cells, one would actually expect a much lower frequency of biallelic targeting. The unexpectedly high proportion of biallelic targeting events observed suggests that in the majority of cells, the intracellular conditions do not allow for efficient targeting, whereas the generation of DSBs and subsequent repair through HR is highly efficient in only a relatively small proportion of cells, resulting in a considerable number of biallelic targeting events. These conditions may include efficient transfection of the respective cell, the appropriate timing of the peak of ZFN expression and adequate cell-cycle phases (S or G2 phase for HR), and the simultaneous expression of essential HR cofactors.
Finally, it is worth mentioning that our targeting protocol did not affect the pluripotent state of the resulting clones, as indicated by the persistent expression of typical pluripotency markers and efficient embryoid body (EB)-based differentiation into derivatives of all three germ layers. Moreover, we were able to exclude any karyotypic abnormalities in our analyzed cell clones and also obtained robust transgene expression from the AAVS1 locus upon differentiation, indicating that ZFN/TALEN treatment and application of a ROCK inhibitor (despite its potential side effects; Chapman et al., 2010) did not affect or impair the PSC properties.
Interestingly, we obtained similar targeting efficiencies using TALENs in combination with short ssODNs with 2 × 43 bp of homology to the AAVS1 locus instead of plasmid donors with 2 × 700 bp of homology. Here, we assume that the lower pairing probability of a relatively short homologous oligonucleotide sequence is compensated for by the much higher number of molecules that enter the transfected cells in comparison with a 5.9 kb plasmid donor. The calculated high targeting frequency of ∼1.6% enabled us to identify correctly targeted single-cell clones after transfection and limiting dilution simply by PCR. To date, the insertion of ssODNs on a clonal level has only been reported by Soldner et al. (2011) and Ding et al. (2013), who introduced point mutations in hESCs. However, in contrast to our study, both of these groups preselected positive transfected cells and subsequently established clones with efficiencies of 0.4%–0.8% (Soldner et al., 2011) and 1.5% (Ding et al., 2013) from the preselected cell population.
Of course, critical aspects and limitations of our approach should be outlined. The targeted locus could be considered as the most problematic, as the targeting efficiency can widely vary among different genomic sites depending on the chromatin state and local gene expression. Furthermore, transfection efficiencies may vary among individual cell clones, and the required dissociation steps may be critical depending on the culture characteristics of the applied PSC clone. In case of low targeting efficiencies and cell clones that are highly sensitive to single-cell dissociation even in the presence of a ROCK inhibitor, it may be easier and less time-consuming to apply a targeting strategy based on antibiotic selection.
On the other hand, selection-free targeting has clear advantages or is even crucial for various applications, including seamless correction and the introduction of disease-specific mutations using ssODNs. When considering a specific application of our approach, one should take into account that the introduction of additional genetic elements may change characteristics of the targeted cell. Even if the subsequent removal of such elements is possible (e.g., through loxP or frt sites), such steps will also require considerable additional efforts. It is important to perform genetic engineering without the use of potentially immunogenic reporter or selection genes if the engineered cells are to be transplanted in animal studies or if a clinical application is considered. The usefulness and the required workload of our approach therefore need to be weighed with regard to the respective application.
In summary, we provide an efficient protocol for selection-independent gene targeting in hESCs and hiPSCs using ZFN and TALEN pairs specific for two different targets. The observed efficiencies enabled antibiotic-selection-free isolation of targeted hPSC clones by FACS. Targeting of the AAVS1 locus demonstrated robust CAG-promoter-mediated transgene expression in undifferentiated PSCs similar to that observed in their differentiated derivatives. Finally, we show that TALEN/ssODN-based footprintless gene editing in hiPSCs is actually feasible without any preselection, and that correctly targeted clones can be isolated simply via PCR.
The established targeting protocols will facilitate footprintless correction or introduction of disease-specific mutations in patient-specific iPSCs for disease modeling, drug screening, and ultimately the generation of clinically useful transgenic iPSC derivatives.
Experimental Procedures
Cell Culture
We used hES-3 cells and hiPSC lines generated in our group by the lentiviral transduction of cord-blood-derived endothelial cells (hCBiPS2) (Haase et al., 2009) and cord-blood-derived CD34+ cells (HSC_F1441_4F_iPS2, termed hHSCiPS2) (Hartung et al., 2013). Unless otherwise indicated, hiPSC lines and hES-3 cells were cultured and expanded on irradiated mouse embryonic fibroblasts (MEFs) in knockout Dulbecco’s modified Eagle’s medium supplemented with 20% knockout serum replacement, 1 mM L-glutamine, 0.1 mM β-mercaptoethanol, 1% nonessential amino acid stock (all from Life Technologies), and 10 ng/ml basic fibroblast growth factor (bFGF; supplied by the Institute for Technical Chemistry, Leibniz University, Hannover, Germany) (Chen et al., 2012).
For generation of the eGFP reporter cell lines (hCBiPS2eGFPC7 and hCBiPS2eGFPC16), a CAG-promoter-driven eGFP expression vector, together with a αMHCneo_PGKhygro selection vector, was stably transfected into hCBiPS2 cells. Hygromycin-based clone selection was initiated 72 hr posttransfection for 4 days. Upcoming colonies were manually picked and transferred onto irradiated feeder cells and expanded clonally.
Plasmid Vectors and Oligonucleotides
For this study, eGFP-specific ZFN (Höher et al., 2012; Osiak et al., 2011), AAVS1-specific ZFN (Hockemeyer et al., 2009), and AAVS1-specific TALEN (Holkers et al., 2013) expression cassettes were placed under control of a CAG promoter. The coding sequences of the AAVS1-specific nucleases are indicated in the Supplemental Experimental Procedures. In brief, all nucleases contained an amino-terminal hemagglutinin tag and an SV40 nuclear localization signal. The codon-optimized, AAVS1-specific zinc-finger arrays were synthesized by GeneArt/Life Technologies and fused to the obligate heterodimeric FokI variant EA-KV (Szczepek et al., 2007) by the 4 AA linker LRGS (Händel et al., 2009). TALENs are based on the NΔ134/C+17 architecture (Mussolino et al., 2011) and contain wild-type FokI nuclease domains. For generation of the eGFP donor plasmid, a 2A protease sequence (Szymczak and Vignali, 2005) was fused in frame at the beginning of an improved version of the red fluorescent protein coupled to a nuclear membrane location signal (RedStarnuc) (Gruh et al., 2005; Okita et al., 2004). Afterward, the resulting 2A-RedStar-poly(A) expression cassette was cloned between two arms of ∼700 bp eGFP locus homology sequences in a pJet1.2 (Thermo Scientific) expression vector backbone. For generation of the AAVS1 donor plasmids, CAG-eGFP and CAG-RedStarnuc expression cassettes were cloned between two arms of ∼700 bp AAVS1 locus homology sequences in a pUC19 expression vector backbone (Life Technologies). All oligonucleotides, including PCR primers (Table S1), were manufactured by Eurofins MWG Operon and dissolved in water at 100 μM.
Transfection, Flow Cytometry, and Clone Establishment
In the optimization experiments and for eGFP targeting, cells were detached from the feeder layer by 0.2% collagenase IV (Life Technologies) followed by an incubation step with TrypLE (Life Technologies) for single-cell dissociation. In all other experiments, cells were expanded as monolayer cultures on Geltrex (Life Technologies) for at least two passages as previously described (Burridge et al., 2011) and harvested by Accutase (PAA).
Transfections using the Amaxa Nucleofection system were performed according to the manufacturer’s protocol. The majority of transfections, however, were performed using the Neon transfection system (Life Technologies) as follows: 1 × 106 dissociated cells were resuspended in 100 μl NEON buffer with various amounts of ZFN/TALEN and donor plasmids or ssODN (eGFP targeting, e.g., 1:3 = 10 μg of each ZFN plasmid + 30 μg donor plasmid; AAVS1 targeting: 5 μg of each ZFN/TALEN plasmid + 15 μg donor plasmid), electroporated with two pulses at 1,000 V for 20 ms, and plated onto Geltrex-coated dishes with MEF conditioned medium (CM) supplemented with 10 μM ROCK inhibitor Y-27632 (RI).
Transfected cells were cultivated as a monolayer prior to flow-cytometric analysis using the FACSCalibur cell analyzer (BD Bioscience) and FlowJo 7.6.5 software (Celeza). For clone generation, cells were harvested from the monolayer culture by either TrypLE or Accutase on day 7 (for eGFP targeting) or day 12 (for AAVS1 targeting) after transfection, and were sorted on the FACSAria IIu (BD Bioscience) or XDP (Beckman-Coulter) for eGFPpos or RedStarpos cells. Sorted populations were plated onto Geltrex-coated dishes. Arising colonies were picked manually and transferred into feeder-based culture conditions. For gene editing using ssODNs, 1 × 106 hiPSCs were resuspended in 100 μl NEON buffer, electroporated with 5 μg of each TALEN-encoding plasmid and 5 μl of the ssODN (5′-GTTCTGGGTACTTTTATCTGTCCCCTCC ACCCCACAGTGGGGCCCCAAGCTTGGGCACTAGGGACAGGATTGGTGACAGAAAAGCCCCATCCTTAGGC-3′), and plated onto Geltrex-coated dishes with CM+RI. On day 3 after transfection, 10 cells per well were seeded into Geltrex-coated 96-well plates. On average, one to five single cells survived per well and were analyzed via direct PCR screening. Subsequent subcloning via limiting dilution (20 cells/cm2) resulted in single-cell clones, which were also analyzed via PCR screening (see schematic illustration in Figure 4B).
PCR Screening and Characterization of Transgenic Clones
Total RNA was prepared with the RNeasy Kit (Macherey-Nagel) and reverse transcribed with Superscript II (Life Technologies) using random primers according to the manufacturer’s instructions. Genomic DNA was prepared using the QIAamp DNA Blood Mini Kit (QIAGEN) according to the manufacturer’s instructions, and 1 μl of cDNA or 100 ng of gDNA was amplified by PCR with GoTaq DNA polymerase (Promega). Sequences and specifications of the primers are shown in Table S1. For gene editing using ssODNs, cell lysis was done in 96-well plates using directPCR Lysis Reagent (PeqLab). Subsequently, 2 μl of the lysate was directly applied for PCR reaction.
For Southern blot analysis, 10 μg of genomic DNA was digested as indicated with EcoRI, ScaI, BglII, or BsmI. Fragments were separated on a 0.8% TAE-agarose gel, transferred onto Biodyne B nylon membrane (PALL Life Sciences), hybridized with radioactive labeled DNA fragments using the DecaLabel DNA Labeling Kit (Fermentas), and stripped using standard procedures (Stocking et al., 1988). The hybridization probe for the copy number of genomic eGFP integrations in the hCBiPS2eGFP reporter cell lines was an EcoRI/BglII fragment of eGFP. For determination of targeted and additional donor integrations, the probes were as follows: a PstI fragment (792 bp) of pJet_2A-RedStar-poly(A) (3′ probe) and a AvrII/MfeI fragment (570 bp) of pJet_2A-RedStar-poly(A) (5′ probe).
Immunocytological Staining
Cells were fixed with 4% paraformaldehyde (w/v) and stained by standard protocols using primary antibodies, as listed in Table S2, and appropriate secondary antibodies (DyLight 488_donkey_anti-mouse_IgG and IgM, DyLight 488_donkey_anti-goat_IgG, DyLight 594_donkey_anti-mouse_IgG; 1:200; Jackson ImmunoResearch Laboratories). The corresponding isotype antibodies were used for negative control staining. Cells were counterstained with DAPI (Sigma) and analyzed with an AxioObserver A1 fluorescence microscope and Axiovision software 4.71 (Zeiss).
In Vitro Differentiation of hPSCs
hPSCs were detached from the feeder layer by collagenase IV, dispersed into small clumps, and cultured in differentiation medium (80% Iscove’s modified Dulbecco’s medium supplemented with 20% fetal calf serum, 1 mM L-glutamine, 0.1 mM β-mercaptoethanol, and 1% nonessential amino acid stock) in ultralow-attachment plates (Corning) for 7 days. Subsequently, EBs were plated onto 0.1% gelatin-coated tissue culture dishes and cultured for a further 13 days before fixation and immunostaining.
Karyotype Analysis
After trypsination, metaphases were prepared according to standard procedures. Fluorescence R-banding using chromomycin A3 and methyl green was performed as previously described in detail (Schlegelberger et al., 1999). At least 15 metaphases were analyzed per clone. Karyotypes were described according to the International System for Human Cytogenetic Nomenclature (ISCN).
Statistical Analysis
Results are reported as mean ± SEM. Statistical analyses were performed with GraphPadPrism5. The significance of two groups was analyzed using the unpaired t test (∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.0005).
Acknowledgments
The authors are grateful to T. Kohrn and M. Götz for providing technical assistance, and to R. Olmer for contributing to fruitful discussions. We also thank I. Gruh and N. McGuinnes for their critical readings of the manuscript, T. Scheper for providing bFGF, A. Kirschning and G. Dräger for providing Y-27632, and the Cell-Sorting Core Facility of Hannover Medical School for cell sorting. Human cord-blood-derived hematopoietic stem cells for generation of hHSCiPS2 were kindly provided by Vita34 (Leipzig). This work was funded by the Mukoviszidose Institut GmbH (S03/11), the German Federal Ministry of Education and Research (CARPuD; 01GM1110A), the German Center for Lung Research (DZL; 82DZL00201), and the German Research Foundation (Cluster of Excellence REBIRTH, EXC 62/3).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplemental Information
References
- Burridge P.W., Thompson S., Millrod M.A., Weinberg S., Yuan X., Peters A., Mahairaki V., Koliatsos V.E., Tung L., Zambidis E.T. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS ONE. 2011;6:e18293. doi: 10.1371/journal.pone.0018293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman S., Liu X., Meyers C., Schlegel R., McBride A.A. Human keratinocytes are efficiently immortalized by a Rho kinase inhibitor. J. Clin. Invest. 2010;120:2619–2626. doi: 10.1172/JCI42297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F., Pruett-Miller S.M., Huang Y., Gjoka M., Duda K., Taunton J., Collingwood T.N., Frodin M., Davis G.D. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat. Methods. 2011;8:753–755. doi: 10.1038/nmeth.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., John J., Lavrentieva A., Mueller S., Tomala M., Zhao Y., Zweigerdt R., Beutel S., Hitzmann B., Kasper C. Cytokine production using membrane adsorbers: Human basic fibroblast growth factor produced by Escherichia coli. Eng. Life Sci. 2012;12:29–38. [Google Scholar]
- Di Domenico A.I., Christodoulou I., Pells S.C., McWhir J., Thomson A.J. Sequential genetic modification of the hprt locus in human ESCs combining gene targeting and recombinase-mediated cassette exchange. Cloning Stem Cells. 2008;10:217–230. doi: 10.1089/clo.2008.0016. [DOI] [PubMed] [Google Scholar]
- Ding Q., Lee Y.K., Schaefer E.A., Peters D.T., Veres A., Kim K., Kuperwasser N., Motola D.L., Meissner T.B., Hendriks W.T. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetschman T., Maeda N., Smithies O. Targeted mutation of the Hprt gene in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA. 1988;85:8583–8587. doi: 10.1073/pnas.85.22.8583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott D.A., Braam S.R., Koutsis K., Ng E.S., Jenny R., Lagerqvist E.L., Biben C., Hatzistavrou T., Hirst C.E., Yu Q.C. NKX2-5(eGFP/w) hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat. Methods. 2011;8:1037–1040. doi: 10.1038/nmeth.1740. [DOI] [PubMed] [Google Scholar]
- Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulburn A.L., Alden D., Davis R.P., Micallef S.J., Ng E.S., Yu Q.C., Lim S.M., Soh C.L., Elliott D.A., Hatzistavrou T. A targeted NKX2.1 human embryonic stem cell reporter line enables identification of human basal forebrain derivatives. Stem Cells. 2011;29:462–473. doi: 10.1002/stem.587. [DOI] [PubMed] [Google Scholar]
- Gruh I., Schwanke K., Wunderlich S., Blomer U., Scherr M., Ganser A., Haverich A., Martin U. Shuttle system allowing simplified cloning of expression cassettes into advanced generation lentiviral vectors. BioTechniques. 2005;38:530. doi: 10.2144/05384BM02. 532, 534. [DOI] [PubMed] [Google Scholar]
- Haase A., Olmer R., Schwanke K., Wunderlich S., Merkert S., Hess C., Zweigerdt R., Gruh I., Meyer J., Wagner S. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell. 2009;5:434–441. doi: 10.1016/j.stem.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Händel E.M., Alwin S., Cathomen T. Expanding or restricting the target site repertoire of zinc-finger nucleases: the inter-domain linker as a major determinant of target site selectivity. Mol. Ther. 2009;17:104–111. doi: 10.1038/mt.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung S., Schwanke K., Haase A., David R., Franz W.M., Martin U., Zweigerdt R. Directing cardiomyogenic differentiation of human pluripotent stem cells by plasmid-based transient overexpression of cardiac transcription factors. Stem Cells Dev. 2013;22:1112–1125. doi: 10.1089/scd.2012.0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R.C., Katibah G.E., Amora R., Boydston E.A., Zeitler B. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D., Wang H., Kiani S., Lai C.S., Gao Q., Cassady J.P., Cost G.J., Zhang L., Santiago Y., Miller J.C. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höher T., Wallace L., Khan K., Cathomen T., Reichelt J. Highly efficient zinc-finger nuclease-mediated disruption of an eGFP transgene in keratinocyte stem cells without impairment of stem cell properties. Stem Cell Rev. 2012;8:426–434. doi: 10.1007/s12015-011-9313-z. [DOI] [PubMed] [Google Scholar]
- Holkers M., Maggio I., Liu J., Janssen J.M., Miselli F., Mussolino C., Recchia A., Cathomen T., Gonçalves M.A. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63. doi: 10.1093/nar/gks1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z., Zhang Y., Propson N.E., Howden S.E., Chu L.F., Sontheimer E.J., Thomson J.A. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irion S., Luche H., Gadue P., Fehling H.J., Kennedy M., Keller G. Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat. Biotechnol. 2007;25:1477–1482. doi: 10.1038/nbt1362. [DOI] [PubMed] [Google Scholar]
- Lombardo A., Cesana D., Genovese P., Di Stefano B., Provasi E., Colombo D.F., Neri M., Magnani Z., Cantore A., Lo Riso P. Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat. Methods. 2011;8:861–869. doi: 10.1038/nmeth.1674. [DOI] [PubMed] [Google Scholar]
- Modlich U., Navarro S., Zychlinski D., Maetzig T., Knoess S., Brugman M.H., Schambach A., Charrier S., Galy A., Thrasher A.J. Insertional transformation of hematopoietic cells by self-inactivating lentiviral and gammaretroviral vectors. Molecular therapy: the journal of the American Society of Gene Therapy. 2009;17:1919–1928. doi: 10.1038/mt.2009.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussolino C., Cathomen T. TALE nucleases: tailored genome engineering made easy. Curr. Opin. Biotechnol. 2012;23:644–650. doi: 10.1016/j.copbio.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Mussolino C., Morbitzer R., Lütge F., Dannemann N., Lahaye T., Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39:9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita C., Sato M., Schroeder T. Generation of optimized yellow and red fluorescent proteins with distinct subcellular localization. Biotechniques. 2004;36:418–422. doi: 10.2144/04363ST01. 424. [DOI] [PubMed] [Google Scholar]
- Osiak A., Radecke F., Guhl E., Radecke S., Dannemann N., Lütge F., Glage S., Rudolph C., Cantz T., Schwarz K. Selection-independent generation of gene knockout mouse embryonic stem cells using zinc-finger nucleases. PLoS ONE. 2011;6:e28911. doi: 10.1371/journal.pone.0028911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S.H., Maeder M.L., Joung J.K., Cathomen T. Zinc-finger nucleases for somatic gene therapy: the next frontier. Hum. Gene Ther. 2011;22:925–933. doi: 10.1089/hum.2011.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid L.H., Shesely E.G., Kim H.S., Smithies O. Cotransformation and gene targeting in mouse embryonic stem cells. Mol. Cell. Biol. 1991;11:2769–2777. doi: 10.1128/mcb.11.5.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegelberger B., Metzke S., Harder S., Zühlke-Jenisch R., Zhang Y., Siebert R. Springer; New York: 1999. Diagnostic Cytogenetics. [Google Scholar]
- Sebastiano V., Maeder M.L., Angstman J.F., Haddad B., Khayter C., Yeo D.T., Goodwin M.J., Hawkins J.S., Ramirez C.L., Batista L.F. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells. 2011;29:1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M., De Haro L.P., Nickoloff J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- Smith J.R., Maguire S., Davis L.A., Alexander M., Yang F., Chandran S., ffrench-Constant C., Pedersen R.A. Robust, persistent transgene expression in human embryonic stem cells is achieved with AAVS1-targeted integration. Stem Cells. 2008;26:496–504. doi: 10.1634/stemcells.2007-0039. [DOI] [PubMed] [Google Scholar]
- Soldner F., Laganière J., Cheng A.W., Hockemeyer D., Gao Q., Alagappan R., Khurana V., Golbe L.I., Myers R.H., Lindquist S. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S., Ott M.G., Schultze-Strasser S., Jauch A., Burwinkel B., Kinner A., Schmidt M., Krämer A., Schwäble J., Glimm H. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- Stocking C., Löliger C., Kawai M., Suciu S., Gough N., Ostertag W. Identification of genes involved in growth autonomy of hematopoietic cells by analysis of factor-independent mutants. Cell. 1988;53:869–879. doi: 10.1016/s0092-8674(88)90329-7. [DOI] [PubMed] [Google Scholar]
- Szczepek M., Brondani V., Büchel J., Serrano L., Segal D.J., Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat. Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- Szymczak A.L., Vignali D.A. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin. Biol. Ther. 2005;5:627–638. doi: 10.1517/14712598.5.5.627. [DOI] [PubMed] [Google Scholar]
- Yusa K., Rashid S.T., Strick-Marchand H., Varela I., Liu P.Q., Paschon D.E., Miranda E., Ordóñez A., Hannan N.R., Rouhani F.J. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature. 2011;478:391–394. doi: 10.1038/nature10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J., Maeder M.L., Mali P., Pruett-Miller S.M., Thibodeau-Beganny S., Chou B.K., Chen G., Ye Z., Park I.H., Daley G.Q. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J., Sweeney C.L., Chou B.K., Choi U., Pan J., Wang H., Dowey S.N., Cheng L., Malech H.L. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117:5561–5572. doi: 10.1182/blood-2010-12-328161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaka T.P., Thomson J.A. Homologous recombination in human embryonic stem cells. Nat. Biotechnol. 2003;21:319–321. doi: 10.1038/nbt788. [DOI] [PubMed] [Google Scholar]
- Zweigerdt R., Olmer R., Singh H., Haverich A., Martin U. Scalable expansion of human pluripotent stem cells in suspension culture. Nat. Protoc. 2011;6:689–700. doi: 10.1038/nprot.2011.318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.