

Abstract
The Epidermal Growth Factor Receptor is frequently expressed in triple-negative breast cancer and is a marker of poor prognosis in this patient population. Because activating mutations in this kinase are very rare events in breast cancer, we screened breast tumor gene expression profiles to examine the distribution of EGFR ligand expression. Of the six known EGFR ligands, TGFalpha was expressed more highly in triple-negative breast tumors than in tumors of other subtypes. TGFalpha is synthesized as a transmembrane precursor requiring TACE/ADAM17-dependent proteolytic release in order to activate its receptor. In this study we show that an inhibitor of this proteolytic release blocks invasion, migration and colony formation by several triple-negative breast cancer cell lines. Each of the effects of the drug was reversed upon expression of a soluble TGFalpha mutant which does not require TACE activity, implicating this growth factor as a key metalloproteinase substrate for these phenotypes. Together, these data demonstrate that TACE-dependent TGFalpha shedding is a key process driving EGFR activation and subsequent proliferation and invasion in triple-negative breast cancer cell lines.
Keywords: Triple-negative breast cancer, Epidermal growth factor receptor, TACE/ADAM17, Transforming growth factor alpha
Introduction
Breast tumors are classified based on the expression of estrogen receptor (ERα), progesterone receptor (PR) and the overexpression/amplification status of HER2/ERBB2. Cancers lacking expression of these three proteins have been referred to as “triple-negative breast cancer” (TNBC), and are associated with a poor prognosis. TNBC is a heterogeneous disease, although about 80% of all TNBC are associated with a “basal” pattern of gene expression.1 TNBCs are not sensitive to targeted therapeutics used for HER2-positive (Trastuzumab and Lapatinib) and ERα-positive (Tamoxifen and aromatase inhibitors) tumors.2 A better understanding of the biology of this disease will be essential to improve clinical outcomes.
The Epidermal Growth Factor Receptor (EGFR) participates in the control of cell survival, differentiation and proliferation of the mammary gland, and plays a key role in tumorigenesis of this tissue.3 The ligands for the EGFR include Epidermal Growth Factor (EGF), transforming growth factor-alpha (TGFα), amphiregulin, betacellulin, heparin-binding EGF (HB-EGF) and epiregulin.4 Each of these ligands is produced as a type I transmembrane precursor protein requiring proteolytic release of the mature growth factor (“ectodomain shedding”) which can then bind to and activate EGFR.5 Metalloproteinases, such as members of the matrix metalloprotease (MMP) or “a disintegrin and metalloprotease” (ADAM) family are responsible for this sheddase activity and therefore control EGFR ligand bioavailability.6
EGFR is over-expressed in more than half (54%) of triple negative breast tumors and is associated with poor clinical outcome.7 Mutations in EGFR are frequent events in tumors of some tissues, e.g. non-small cell lung cancer 8, however they are very rare in breast cancer.9-11 Accordingly, if EGFR activity plays a role in TNBC, it is likely that ligand-driven activation of this receptor is necessary. In agreement with this, there is a correlation between high EGFR activation and high ADAM17/TACE levels.12 We have previously demonstrated that TACE-dependent EGFR ligand shedding is an important proliferative signal in breast cancer and that the expression of its ligand, TGFα, is associated with poor prognosis.13
Given the invasive nature of triple-negative tumors and their distinct pattern of local and metastatic spread, the present study examines the distribution of EGFR ligand expression in these tumors and evaluates the requirement for TACE-dependent EGFR ligand shedding for the motility and invasion of triple-negative breast cancer cells, as well as to their ability to form colonies in 3D culture.
Materials and Methods
Cell culture
A panel of 4 basal-like breast cancer cell lines was cultured as follows: HCC70 in RPMI 1640 (Cellgro) with 10% FBS (Hyclone); MDA-MB-468 in L-15 (Hyclone) with 10% FBS; MDA-MB-231 in DMEM (Cellgro) with 10% FBS and T4-2 in H14 medium (DMEM/F12 supplemented with 5 μg/ml prolactin, 250 ng/ml insulin, 1.4 × 10−6 M hydrocortisone, 2.6 ng/ml sodium selenite, 10 μg/ml transferrin and 10−8 M β-estradiol as previously described.14 The HCC70, MDA-MB-468 and MDA-MB-231 cell lines were obtained from ATCC which validates their correct identity. The identity of the T4-2 cell line (which is available from Sigma) was further confirmed by detecting the specific mutation in codon 179 of p53 previously reported for this cell line.15 The metalloproteinase inhibitor, TAPI-2 (Calbiochem), was used at 20 μM. INCB3619 was a generous gift from Incyte Corporation and was used at concentrations up to 10 μM. For all experiments, a negative vehicle control consisting of an equivalent volume of ethanol was used. Breast cancer cell lines were stably infected with pBM-IRES-Puro retroviruses containing a C-terminal TGFα truncation mutant consisting of amino acids 1-98 which lacks the transmembrane and cytosolic domains as we have previously described13 or an empty vector control. Suppression of TGFα expression was achieved by lentiviral infection with two independent pGIPZ constructs with the following sequences (shTGFA1 CCATGGATTCCAGGCTATA and shTGFA2 GAGATGATGTCTTATTTAT) or a non-silencing control (CTCGCTTGGGCGAGAGTAA).
3D laminin-rich extracellular matrix cultures16 were prepared by trypsinization of cells from tissue culture plastic, seeding of single cells on top of a thin layer of Growth Factor-Reduced Matrigel (BD Biosciences), and the addition of a medium containing 5% Matrigel. The cell lines were seeded at the following densities; T4-2: 1.0×104 cells per cm2; MDA-MB-468 and HCC70: 2.1×104 cells per cm2; and MDA-MB-231: 1.6×104 cells per cm2. All 3D cell cultures were performed in H14 medium with 1% fetal bovine serum, with the exception of T4-2, which cultured in H14 medium without serum. 3D cultures were maintained with media changes every second day.
Expression studies
TGFA gene expression was assessed in the NKI-295 dataset consisting of the microarray profiles of 295 primary breast tumors.17 Raw data are available from the NCBI Gene Expression Omnibus (Accession number: GSE2845). Tumors were assigned to molecular subtypes based on their gene expression profiles and those matching to the basal-like, HER2-overexpressing, luminal A and luminal B subtypes were analyzed for TGFA gene expression. Samples matching to the “Normal-Like” subtype were excluded from the analysis as these are believed to be substantially contaminated by normal tissue. Statistical significance was evaluated using the Kruskal-Wallis test with Dunn's Post-test. The expression of this family of ligands, the four ERBB receptors and the two ligand-releasing proteases was also evaluated in a panel of luminal and basal-like cell lines. 18 Raw data are available from ArrayExpress (#E-TABM-244). Antibodies used for western blot analysis were against phospho-EGFR, total EGFR, phospho-MAPK and Total MAPK (Cell Signaling Technology).
Monolayer Wound Healing Assay
Cells were grown to confluence in 12-well plates (BD Falcon) and scraped with a sterile micropipette tip. The wound was marked and imaged immediately after scraping and again 24 hours later. The area of the wound was measured at the 0 hour and 24 hour time points and results are presented as percentage wound closure at 24 hours. Recombinant TGFα (1 ng/ml) was added in rescue experiments to confirm the specificity of the shRNA phenotype.
Transwell Invasion assay
For the transwell invasion assay, cell culture inserts (8 μm pore, 12-well format; BD Falcon) were coated with 100 μl dilute Matrigel (0.5 mg protein/ml) in serum-free medium which was allowed to solidify overnight in a humidified cell culture incubator and was rehydrated with warm serum-free medium for three hours prior to the experiments. Cells were grown to 75% confluence and then starved for 24 hours in growth medium containing 0.1% FBS. 1.0×105 cells were seeded in 1 ml serum-free medium in the upper chamber and the lower chamber was filled with 1 ml of growth medium with 10% FBS as a chemoattractant. The cultures were maintained for the following time periods which were empirically determined for each cell line due to inherent differences in relative invasive ability: MDA-MB-231: 5 hours; T4-2: 24 hours; and MDA-MB-468 and HCC70: 48 hours. Invaded cells were fixed and visualized by staining with 0.2% crystal violet and counted using a 4X objective in each of three randomly chosen fields. Each experiment was performed in duplicate.
Statistics
All data analysis was performed using GraphPad Prism version 5.03. The non-parametric Kruskal-Wallis test was used to test differences between median cross-sectional area of colonies in 3D culture. Student's t-test was used for all other comparisons.
Results
EGFR expression has been associated with triple-negative breast cancer7, but it remains unclear which EGFR ligand(s) are important for receptor activation in this disease. We examined the expression of six EGFR ligands – Amphiregulin, Betacellulin, EGF, Epiregulin, HB-EGF and TGFα – in the NKI-295 gene expression dataset 17 which includes 46 basal-like tumors. When compared to the other tumor subtypes (Fig 1A), there was a very strong enrichment of TGFα expression in basal-like tumors (P < 0.01 versus the ERBB2 subtype and P < 0.001 versus the Luminal subtypes). HB-EGF was higher in basal-like tumors than in either of the luminal subtypes (P < 0.001), but was not significantly different from the levels found in the ERBB2 subtype. In all cases, the magnitude of the median expression differences between the basal-like tumors and the other subtypes was much greater for TGFα than for HB-EGF. Accordingly, we investigated the role of TGFα in the pathobiology of basal-like/triple-negative breast cancer.
Figure 1. Distribution of EGFR ligand expression in breast cancer subtypes.

(a) The gene expression profiles of each EGFR ligand was evaluated in Luminal A (n = 88), Luminal B (n = 81), ERBB2-overexpressing (n = 49) and Basal-like (n = 46) breast tumors. The most significantly elevated ligand in Basal-like tumors was TGFα. (b) Heatmap of microarray analysis of the relative expression levels of ERBB receptors, ligands and ligand-shedding proteases in 10 basal-like and 13 luminal breast cancer cell lines.
To select suitable cell line models, we examined the expression of all of the ERBB family of receptors and ligands, as well as the proteases responsible for ligand mobilization in a panel of 10 basal-like and 13 luminal breast cancer cell lines grown in 3D culture.18 The TACE/ADAM17 protease was highly expressed in the basal-like cell lines (Fig 1B). Consistent with the tumor data (Fig 1A), the majority of the basal-like cell lines expressed TGFα (Fig 1B). A minority of the basal-like cell lines expressed HB-EGF and Epiregulin. We selected four TGFα-expressing basal-like cell lines (MDA-MB-231, MDA-MB-468, HCC70 and T4-2) for our experiments which were broadly representative of the observed expression patterns. Each of these cell lines lacks expression of ER, PR and ERBB2.
TGFα is one of many signaling proteins requiring metalloproteinase-dependent shedding for activity. To specifically evaluate the role of TGFα among all sheddase substrates, we expressed a soluble mutant of TGFα lacking the transmembrane and cytosolic domains (Fig 2a, TGFαΔTM) in this panel of triple-negative breast cancer cell lines. Because TGFαΔTM lacks a membrane anchor, it is efficiently secreted without requiring proteolytic shedding.13 Each of these cell lines expresses endogenous full length TGFαΔ(Fig 1B and references 13, 18, 19). This system allowed us to suppress the shedding of endogenous TGFα as well as other known and unknown TACE/MMP substrates and restore the function of soluble TGFα in this background to determine the specific contribution of this growth factor to the phenotypes under investigation. Analysis of soluble TGFα by ELISA in conditioned medium of MDA-MB-231 cells indicates that TGFα shedding is reduced by the TACE/MMP inhibitor, TAPI-2, in the vector control cell line, while the elevated levels of TGFα in the TGFαΔTM cell line are mostly insensitive to TACE inhibition (Fig 2b).
Figure 2. A soluble TGFα mutant allows specific activation of EGFR signaling when cleavage of other TACE substrates is suppressed.

(a) Schematic representation of full length TGFα and the ΔTM truncation mutant used in this study. SP: signal peptide; Pro: pro-peptide; EGF-like: EGFR-binding signaling domain ; TM: transmembrane domain. (b) Inhibition of TACE activity reduces TGFα shedding in control MDA-MB-231 cells (P <0.01), while cells expressing the soluble TGFα mutant are largely resistant to TAPI-2. (c) Western blot analysis of activated and total EGFR levels in MDA-MB-231 cells expressing the soluble TGFα mutant and the parental cell line control, treated 20 μM TAPI-2 or vehicle control.
We then analyzed EGFR activation in these cell lines in the presence and absence of TAPI-2, an inhibitor of TACE and some other metalloproteinases. As expected, phosphorylated EGFR levels were reduced in the control cell line after addition of TAPI-2 while this drug had no effect on the cell line secreting the soluble TGFα mutant (Fig 2c), indicating that activity of TACE or a TACE-like protease is required for EGFR activity in this cell line.
We evaluated the impact of TAPI-2 treatment on tumor cell motility using a scratch assay and observed a significant suppression of tumor cell motility in three triple-negative breast cancer cell lines. In contrast, breast cancer cells expressing the soluble TGFα mutant efficiently migrated to fill the wound in the presence of TAPI-2. Representative experimental data for T4-2 cells are shown in Fig 3a, and quantitative data for all cell lines is shown in Fig 3b. The HCC70 cell line, which we used in other experiments in this study, does not form the confluent monolayers necessary for performing this assay. To confirm the contribution of TGFα to this phenotype, we suppressed endogenous TGFα expression in the parental MDA-MB-231 cell line using two independent shRNAs (Fig 3c). MDA-MB-231 cell lines with TGFα suppression had a significant reduction in cell motility, which was rescued by addition of recombinant TGFα (Fig 3d). These data indicate that, among all TACE substrates, TGFα plays a key role in the motility of these triple-negative breast cancer cell lines.
Figure 3. A soluble TGFα mutant rescues the motility suppression induced by TACE/MMP inhibition.

(a) Wound healing motility assay showing that TAPI-2 efficiently prevents migration of T4-2 cells over a 24 hour period. This impaired motility was completely overcome by expression of the soluble TGFα mutant. Dashed lines indicate the position of the boundaries of the scratched wound at 0 hours. (b) Quantification of the wound healing motility assay for all cell lines in the presence and absence of TAPI-2 (* P<0.05, ** P<0.01, *** P<0.001). Soluble TGFα rescues motility in all cases. HCC70 does not form confluent monolayers and was not included in this experiment. (c) qRT-PCR analysis of MDA-MB-231 cells stably infected with independent shRNAs against TGFA. (d) shRNA against TGFα suppresses motility in the wound healing assay, and the phenotype is rescued by addition of soluble recombinant TGFα.
To evaluate a more physiologically-relevant mode of tumor cell migration, we next tested the ability of these cell lines to migrate through Boyden chambers coated with a layer of extracellular matrix proteins. Treatment with TAPI-2 reduced invasion in four triple-negative breast cancer cell lines by 70 to 88% (Fig 4a). Restoration of TGFα using the secreted mutant was sufficient to overcome the effect of the protease inhibitor. Finally, we examined the effect of TAPI-2 on colony formation in 3D extracellular matrix culture, a commonly used surrogate for the malignant phenotype.18, 20 As with the previous examples, inhibition of TACE significantly reduced colony growth in 3D culture and this was reversed when the soluble TGFα mutant was expressed (Fig 4b). The contribution of endogenous TGFa expression to these phenotypes in the parental MDA-MB-231 cell line was confirmed using shRNA (Fig 4c and 4d).
Figure 4. A soluble TGFα mutant rescues the suppression of both invasion through extracellular matrix and growth in 3D culture induced by TACE/MMP inhibition.

(a) Quantification of relative invasion through Matrigel-coated 8 μm pore Boyden chambers for four triple-negative breast cancer cell lines. TAPI-2 suppresses invasion in all cases, and this is rescued by expression of the soluble TGFα mutant. (b) Quantification of relative colony size for four triple-negative breast cancer cell lines grown in 3D culture. Each point represents the relative cross-sectional area of a single colony and each bar represents the median for that condition. TAPI-2 strongly suppresses colony formation, and this is rescued by expression of the soluble TGFα mutant. Suppression of TGFα expression in MDA-MB-231 cells blocks (c) invasion through extracellular matrix and (d) colony growth in 3D culture. (* P<0.05, ** P<0.01, *** P<0.001)
The data thus far are consistent with a model in which TGFα expression is upregulated in basal-like and triple-negative breast cancers, and is liberated by TACE or a TACE-like protease to activate the EGFR pathway leading to increased migration, proliferation and invasion. To further confirm the key regulatory role of proteolytic growth factor shedding, we evaluated the activity of a more specific TACE inhibitor, INCB3619. As we found earlier with the more broad-spectrum inhibitor, INCB3619 suppressed shedding of TGFα (Fig 5a), attenuated MAPK pathway activation (Fig 5b) and suppressed the growth of T4-2 cell colonies in 3D culture (Fig 5c). Finally, to confirm the central role of the TGFα receptor in these phenotypes, we inhibited EGFR with gefitinib (Iressa) and found that this blocked motility, invasion and migration (Figs 6 a, b, c – left panels). In contrast to the previous experiments in which the soluble TGFα mutant overcame the effect of the TAPI-2 (Figs 3b, 4a and 4b), expression of this mutant did not rescue motility, invasion and colony formation in the presence of gefitinib (Fig 6a, b, c – right panels). These data demonstrate that EGFR activation downstream of TGFα shedding is essential for these phenotypes.
Figure 5. Validation of the role of proteolytic growth factor shedding using an alternative TACE inhibitor.
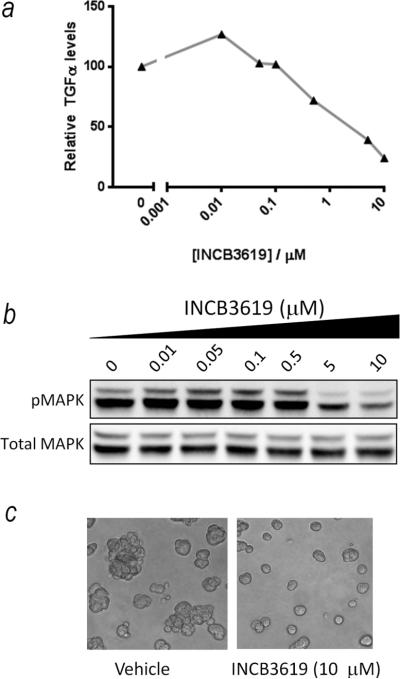
(a) INCB3619 causes a dose-dependent reduction in TGFα shedding in T4-2 cells. (b) INCB3619 causes a dose-dependent reduction in MAPK activity in T4-2 cells. (c) INCB3619 suppresses the growth of T4-2 cells in 3D culture.
Figure 6. Inhibition of the Epidermal Growth Factor Receptor blocks cell migration, invasion and colony growth in 3D culture and is not rescued by the soluble TGFα mutant.

Quantification of (a) cell migration, (b) invasion through extracellular matrix and (c) colony growth in 3D culture in the presence of gefitinib (Iressa) or a vehicle control in the control cell lines (left panels) and cell lines expressing the soluble TGFα mutant (right panels).
Discussion
TAPI-2, which inhibits a number of metalloproteases including TACE/ADAM17, exerted a strong anti-migratory effect on the four triple-negative breast cancer cell lines in this study. This finding is consistent with the well-established role of metalloproteases in mediating invasion by degrading a variety of extracellular matrix proteins.21 Colony formation by each of the cell lines in 3D culture was also strongly suppressed by TAPI-2. The effects on cell proliferation are consistent with our previous work 13 and with a recent study from McGowan, Duffy and colleagues which examined the effect of a different TACE inhibitor on the proliferation of triple-negative breast cancer cells.19 Importantly, all of the phenotypes suppressed by TAPI-2 in our study were reversed upon restoration of a single TACE substrate, TGFα. Thus, even in a context with a significant suppression of cleavage of many ADAM and MMP target proteins, a functional TGFα is sufficient to drive key aspects of the pathobiology of triple-negative breast cell lines. This is consistent with the upregulation of TGFα we observed in these tumors (Fig 1A) and with the association between high levels of TGFα and reduced survival we noted in a previous report.13
Although the protease inhibitors used in this study do not discriminate between ADAM10 and ADAM17/TACE, they do demonstrate that activity of a TACE-like protease is essential for the phenotypes investigated. We have previously reported that siRNA mediated suppression of TACE/ADAM17 reduces the shedding of TGFα to below the threshold of detection of a sensitive ELISA assay.13 Taking these prior data with studies showing that ES cells deficient in ADAM17 but not ADAM10 have a defect in TGFα processing,22 and the demonstration that TACE23 and TGFα24 knockout mice share similar phenotypes, we consider it most likely that TACE/ADAM17 is the key sheddase for TGFα but we cannot exclude the possibility that ADAM10 or other proteases may contribute under some circumstances.
The modes of invasion employed by individual tumor cells can be broadly classified into protease-dependent and protease-independent mechanisms.25 Proteases with roles in invasion include metalloproteinases, serine proteases and cathepsins which can clear a path for migration through the extracellular matrix. In protease-independent migration, the cancer cells move through the existing matrix in an amoeboid fashion. Our results indicate that TGFα can induce the invasion of triple-negative breast cancer cells in the absence of MMP-dependent proteolysis, however whether this migratory mechanism is fully protease-independent or whether cathepsins or serine proteases make a contribution remains to be resolved. Our data do indicate that constraining EGFR ligand mobilization rather than simple suppression of extracellular matrix degradation may make a significant contribution to the widely observed effects of MMP inhibitors on tumor cell invasion.
It is now well recognized that the suite of activities orchestrated by MMP and ADAM proteases extends far beyond the degradation of extracellular matrix proteins and include roles in cell migration, bone remodeling, angiogenesis, cell proliferation, signal transduction and inflammation.26 Consequently, suppression of MMP activity with broad spectrum inhibitors necessarily affects many pathways and cellular processes. Our experiments, using a TGFα mutant which does not require metalloproteinase activity to be functional, emphasize the important role that metalloproteinase-dependent EGFR activation plays in controlling key features of triple-negative breast cancer cell behavior. Because of their broad specificities, the use of inhibitors to assess the requirement for MMP activity for biological functions may be difficult to interpret. However, the experimental restoration of individual MMP insensitive substrates in the presence of an inhibitor, as we have done here, may provide a valuable approach to identifying key substrates for these important enzymes. This may be particularly valuable as the number of reported MMP substrates is continually growing, yet many of those identified in biochemical assays are not necessarily physiological substrates in vivo. 26
Previous clinical studies of small molecule EGFR inhibitors in unselected breast cancer patients have been generally negative.27, 28 Clinical trials of EGFR inhibition in triple-negative breast cancer are ongoing. The EGFR blocking antibody, Cetuximab, has led to improved response rates in small numbers of TNBC patients when added to various other chemotherapies 29, indicating a dependence on EGFR signaling in some tumors of this disease subtype. It is notable that higher levels of TGFα have been associated with Cetuximab resistance in colorectal cancer 30, suggesting that approaches to limit TGFα production (e.g. by TACE inhibition) may improve responses to this EGFR inhibitor.
Phase I trials of a TACE inhibitor, INCB7839, demonstrated that it had in vivo activity and led to disease stabilization in a subgroup of patients with Trastuzumab-refractory breast cancer.31 A subsequent trial in women with HER2-positive cancer, showed that adding INCB7839 to Trastuzumab improved clinical response rate and time to progression.32 Although the clinical development of this compound has been discontinued for HER2+ patients, the fact that it has been shown to be well tolerated and to effectively inhibit TACE in vivo suggests that it may have some utility in treating tumors dependent on TACE-driven processes if these can be identified using appropriate biomarkers. In an alternative approach, which will likely provide more specificity than small molecule inhibitors, Yamamoto and co-workers have recently described an approach using bi-specific (TACE and CD3) antibodies to promote T-cell mediated killing of TACE-expressing cancer cells.33
In conclusion, our study suggests that TGFα is the dominant EGFR ligand in triple-negative breast cancer and that it makes an important contribution to tumor cell proliferation and invasion which are key features of the pathobiology of this disease. These data suggest that blocking TGFα/EGFR signaling using inhibitors of either TACE or EGFR may have clinical utility in patients whose tumors are dependent on this autocrine loop.
What's New?
Expression of the Epidermal Growth Factor Receptor has been frequently associated with basal-like and triple-negative breast cancer, however the mechanism by which it is activated is unclear. Giricz and colleagues implicate TGF-alpha as the most strongly upregulated ligand for this receptor in these tumors and demonstrate that TACE-dependent proteolytic shedding of this ligand makes an important contribution to the invasion and growth of triple-negative breast cancer cell lines.
Acknowledgments
This work was supported by Susan G. Komen for the Cure (KG091136 to OG and PK, and KG100888 to PK) and the American Cancer Society (RSG-TBE-123001). VC was supported by a postdoctoral fellowship from the Fundacion Alfonso Martin Escudero. EAP was supported by an IRACDA K12 training grant (1K12GM102779-01). The Einstein shRNA Core Facility was supported by the Albert Einstein Cancer Center (3P30CA013330). We gratefully acknowledge Jordan Fridman (Incyte Corporation) for providing the INCB3619 compound and the Integrative Cancer Biology Program of the National Cancer Institute for provision of cell lines from the ICBP45 kit for use in this study.
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N. Engl. J. Med. 2010;363:1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Carlson RW, Allred DC, Anderson BO, Burstein HJ, Carter WB, Edge SB, Erban JK, Farrar WB, Goldstein LJ, Gradishar WJ, Hayes DF, Hudis CA, et al. Breast cancer. Clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2009;7:122–92. doi: 10.6004/jnccn.2009.0012. [DOI] [PubMed] [Google Scholar]
- 3.Eccles SA. The epidermal growth factor receptor/Erb-B/HER family in normal and malignant breast biology. Int. J. Dev. Biol. 2011;55:685–96. doi: 10.1387/ijdb.113396se. [DOI] [PubMed] [Google Scholar]
- 4.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 5.Kenny PA. TACE: a new target in epidermal growth factor receptor dependent tumors. Differentiation. 2007;75:800–8. doi: 10.1111/j.1432-0436.2007.00198.x. [DOI] [PubMed] [Google Scholar]
- 6.Harris RC, Chung E, Coffey RJ. EGF receptor ligands. Exp Cell Res. 2003;284:2–13. doi: 10.1016/s0014-4827(02)00105-2. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004;10:5367–74. doi: 10.1158/1078-0432.CCR-04-0220. [DOI] [PubMed] [Google Scholar]
- 8.Han C, Ma J, Zhao J, Zhou Y, Jing W, Zou H. EGFR mutations, gene amplification, and protein expression and KRAS mutations in primary and metastatic tumors of nonsmall cell lung cancers and their clinical implications: a meta-analysis. Cancer Invest. 2011;29:626–34. doi: 10.3109/07357907.2011.621914. [DOI] [PubMed] [Google Scholar]
- 9.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–9. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, Ng S, Lin L, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–60. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borrell-Pages M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-[alpha] in tumors. EMBO J. 2003;22:1114–24. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kenny PA, Bissell MJ. Targeting TACE-dependent EGFR ligand shedding in breast cancer. J. Clin. Invest. 2007;117:337–45. doi: 10.1172/JCI29518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Briand P, Petersen OW, Van Deurs B. A new diploid nontumorigenic human breast epithelial cell line isolated and propagated in chemically defined medium. In Vitro Cell. Dev. Biol. 1987;23:181–8. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- 15.Moyret C, Madsen MW, Cooke J, Briand P, Theillet C. Gradual selection of a cellular clone presenting a mutation at codon 179 of the p53 gene during establishment of the immortalized human breast epithelial cell line HMT-3522. Exp. Cell Res. 1994;215:380–5. doi: 10.1006/excr.1994.1355. [DOI] [PubMed] [Google Scholar]
- 16.Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359–65. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 18.Kenny PA, Lee GY, Myers CA, Neve RM, Semeiks JR, Spellman PT, Lorenz K, Lee EH, Barcellos-Hoff MH, Petersen OW, Gray JW, Bissell MJ. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Molecular Oncology. 2007;1:84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGowan PM, Mullooly M, Caiazza F, Sukor S, Madden SF, Maguire AA, Pierce A, McDermott EW, Crown J, O'Donovan N, Duffy MJ. ADAM-17: a novel therapeutic target for triple negative breast cancer. Ann. Oncol. 2012 doi: 10.1093/annonc/mds279. [DOI] [PubMed] [Google Scholar]
- 20.Kenny PA. Three-dimensional extracellular matrix culture models of EGFR signalling and drug response. Biochem. Soc. Trans. 2007;35:665–8. doi: 10.1042/BST0350665. [DOI] [PubMed] [Google Scholar]
- 21.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8:221–33. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004;164:769–79. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, et al. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–4. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 24.Luetteke NC, Qiu TH, Peiffer RL, Oliver P, Smithies O, Lee DC. TGF alpha deficiency results in hair follicle and eye abnormalities in targeted and waved-1 mice. Cell. 1993;73:263–78. doi: 10.1016/0092-8674(93)90228-i. [DOI] [PubMed] [Google Scholar]
- 25.Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011;21:736–44. doi: 10.1016/j.tcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 27.Baselga J, Albanell J, Ruiz A, Lluch A, Gascon P, Guillem V, Gonzalez S, Sauleda S, Marimon I, Tabernero JM, Koehler MT, Rojo F. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J. Clin. Oncol. 2005;23:5323–33. doi: 10.1200/JCO.2005.08.326. [DOI] [PubMed] [Google Scholar]
- 28.Dickler MN, Rugo HS, Eberle CA, Brogi E, Caravelli JF, Panageas KS, Boyd J, Yeh B, Lake DE, Dang CT, Gilewski TA, Bromberg JF, et al. A phase II trial of erlotinib in combination with bevacizumab in patients with metastatic breast cancer. Clin. Cancer Res. 2008;14:7878–83. doi: 10.1158/1078-0432.CCR-08-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gelmon K, Dent R, Mackey JR, Laing K, McLeod D, Verma S. Targeting triple-negative breast cancer: optimising therapeutic outcomes. Ann. Oncol. 2012;23:2223–34. doi: 10.1093/annonc/mds067. [DOI] [PubMed] [Google Scholar]
- 30.Tabernero J, Cervantes A, Rivera F, Martinelli E, Rojo F, von Heydebreck A, Macarulla T, Rodriguez-Braun E, Eugenia Vega-Villegas M, Senger S, Ramos FJ, Rosello S, et al. Pharmacogenomic and pharmacoproteomic studies of cetuximab in metastatic colorectal cancer: biomarker analysis of a phase I dose-escalation study. J. Clin. Oncol. 2010;28:1181–9. doi: 10.1200/JCO.2009.22.6043. [DOI] [PubMed] [Google Scholar]
- 31.Infante J, Burris HA, Lewis N, Donehower R, Redman J, Friedman S, Scherle P, Fridman JS, Li J, Emm T, Troy S, Eckhardt SG. A multicenter phase Ib study of the safety, pharmacokinetics, biological activity and clinical efficacy of INCB7839, a potent and selective inhibitor of ADAM10 and ADAM17. Breast Cancer Res. Treat. 2007;106:S269. [Google Scholar]
- 32.Newton R, Bradley E, Levy R, Doval D, Bondarde S, Sahoo T, Lokanatha D, Julka P, Nagarkar R, Friedman S. Clinical benefit of INCB7839, a potent and selective ADAM inhibitor, in combination with trastuzumab in patients with metastatic HER2+ breast cancer. J Clin Oncol. 2010;28:3025. meeting abstract. [Google Scholar]
- 33.Yamamoto K, Trad A, Baumgart A, Huske L, Lorenzen I, Chalaris A, Grotzinger J, Dechow T, Scheller J, Rose-John S. A novel bispecific single-chain antibody for ADAM17 and CD3 induces T-cell-mediated lysis of prostate cancer cells. Biochem. J. 2012;445:135–44. doi: 10.1042/BJ20120433. [DOI] [PubMed] [Google Scholar]