

Abstract
This perspective article highlights the leukocyte-cancer cell hybrid theory as a mechanism for cancer metastasis. Beginning from the first proposal of the theory more than a century ago and continuing today with the first proof for this theory in a human cancer, the hybrid theory offers a unifying explanation for metastasis. In this scenario, leukocyte fusion with a cancer cell is a secondary disease superimposed upon the early tumor, giving birth to a new, malignant cell with a leukocyte-cancer cell hybrid epigenome.
Keywords: Bone marrow-derived cell-cancer cell hybrids, metastasis, cell fusion
Background and Research Results
The cancer cell fusion theory
In 1911, Prof. Aichel[1], a German pathologist, first proposed the fusion theory of tumor metastasis, the deadly ability of malignant cells to migrate to distant vital organs and form destructive tumors[1]–[4]. From a biological viewpoint, the theory is quite simple. On one hand, white blood cells have the ability to undergo chemotactic migration around the body and “home” to distant organs, but they rarely divide and have a limited life span. On the other hand, early-stage cancer cells have a low propensity for migration but under proper growth conditions go through unlimited cell divisions. Leukocytes are attracted to the chemical signals released into the blood stream by tumor cells. They follow the signals to the tumor, and if they are phagocytes like macrophages or neutrophils, they attempt to phagocytize cancer cells. How or why fusion occurs is not understood. Perhaps a phagocyte fails to ingest the cancer cell and the two cells fuse, pooling chromosomes and forming a white blood cell–tumor cell hybrid. At least some of these hybrids become metastatic, exhibiting both motility and continuous cell division. The model is simple— white blood cell + non-metastatic cancer cell = metastatic cancer cell—yet it provides a profound and unifying explanation for metastasis (Figure 1)[4]. Highlights of Aichel's hypothesis and ensuing supportive evidence are presented in Table 1.
Figure 1. The bone marrow-derived cell (BMDC)-cancer cell fusion hypothesis.
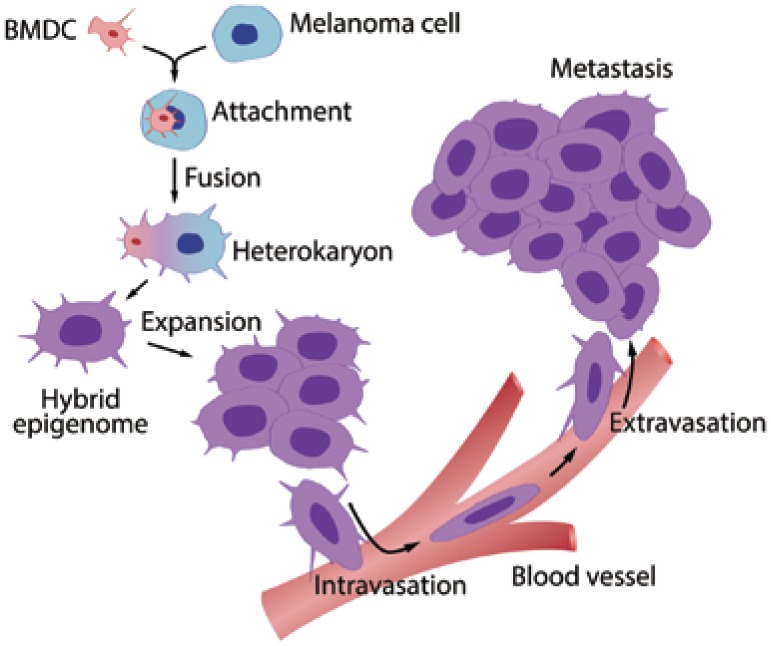
A motile BMDC (red) such as a macrophage or stem cell is drawn to a cancer cell (blue). The outer cell membranes of the two cells become attached. Fusion occurs with the formation of a bi-nucleated heterokaryon having a nucleus from each of the fusion partners. The heterokaryon goes through genomic hybridization creating a BMDC-melanoma hybrid with co-expressed epigenomes, conferring deregulated cell division and metastatic competence to the hybrid. (This figure was first published in reference [2]; used with permission.)
Table 1. Aichel's hypothesis and supporting evidence.
| Hypothesis | Ensuing evidence | Reference(s) |
| Tumor-leukocyte hybrids are generated in vivo. | Numerous cases of spontaneous BMDC-tumor cell fusions in animal models have been reported. | [3],[4] |
| The capacity for cell division of the leukocyte would increase. | Whereas normal leukocytes rarely divide, leukocyte-tumor cell hybrids with unrestricted cell division are seen in culture and animal cancer models, and in a human metastatic melanoma to the brain. | [3],[4], [6],[8],[9] |
| The end product would be what we have learned to understand as a malignant cell. | Macrophage-tumor cell hybrids produced in culture, and spontaneous tumor hybrids in animal models are highly metastatic. | [8] |
At the time of Aichel's proposal, there was no knowledge of DNA-based genetics and no experimental model to test, and for decades the literature was silent. Then in 1971, Meckler[5], a Russian philosopher/scientist, independently proposed a similar theory. At almost the same time, Goldenberg et al.[6] produced the first experimental evidence in hamster tumor models. By 1985, there had been a flurry of cancer fusion studies from several laboratories, confirming cancer cell fusion and metastasis in mouse models[3],[4]. However, this work attracted little attention from the cancer community at large and interest waned. One problem was that at the time there was no way to confirm fusion in humans, the gold standard of cancer research. I was deeply drawn to this theory in 1992, when three Czechoslovakian physicians again proposed it, this time in the context of my research specialty, malignant melanoma[7]. Their arguments totally convinced me, and with no hesitation my lab began experiments.
Experimental macrophage-melanoma cell fusion in culture
Our first step was to experimentally fuse normal mouse or human macrophages with mouse Cloudman S91 melanoma cells that are only weakly metastatic[8]. The majority of these hybrid clones showed markedly enhanced chemotactic motility toward a variety of attractants in two-chambered culture systems, a hallmark of metastatic cells[9]. When implanted in mice, these hybrids showed elevated metastatic potential compared with the implanted parental Cloudman S91 melanoma cells (Figure 2)[8]. Hybrid clones with the highest rate of metastasis also expressed high levels of melanin production (see the darker bars in Figure 2)[8] and autophagy[10]. Underlying this phenotype was the expression of a macrophage-like glycosylation pattern, most notably an increase in oligosaccharide chains conjugated with β1,6-branched oligosaccharides and the responsible glucosyltransferase, β1,6-N-acetylglucosaminyltransferase (GNT-V; E.C.2.4.1.155)[10],[11]. This is important because β1,6-branched oligosaccharides and GNT-V are highly associated with malignant transformation of rodent and human cells and poor prognosis in cancer patients[12]. Subsequent studies in human melanomas, breast carcinomas, and other solid tumors revealed that β1, 6-branched oligosaccharides, autophagy and, in melanoma, melanization are all associated with invasion and metastasis and are predictors of poor patient survival in a variety of solid tumors[12]–[14]. Should these traits be indicators of fusion, the frequency of hybrid tumors would be high indeed.
Figure 2. Metastatic potential of macrophage-melanoma hybrids implanted in DBA/2J mice.

The bar heights indicate the percentage of mice developing metastases by 6 months post-implantation of hybrid clones. The box inset shows the relative pellet colors of the centrifuged cells with the darker the pellet colors indicating higher melanin content. Mice were implanted with parental Cloudman S91 melanoma cells or 35 experimentally derived macrophage-melanoma hybrid cell lines and one spontaneous in vivo hybrid named “PADA.” Unless otherwise noted, 10-20 mice were involved in a single experiment. Arrows denote the parental Cloudman S91 cells and a spontaneous in vivo hybrid of Cloudman S91 cells, PADA. P values for significance vs. parental cells: P≤0.0001 (asterix); P≤0.001 (dagger); P≤0.05 (square). Significance was determined by χ2 analysis. (This figure was first published in reference [8]; used with permission.)
The hybrid epigenome
As above, the metastatic macrophage-melanoma cell hybrids generated in our lab were highly pigmented—a trait of the melanocytes, and also expressed numerous myeloid lineage traits such as enhanced chemotactic motility, autophagy, and macrophage-like glycosylation patterns[4]. Further, when we fused human macrophages with mouse melanoma cells, hybrids expressed both human and mouse secreted protein, acidic, cysteine-rich (SPARC) genes, indicating that the genes of both fusion partners were expressed[15]. In other systems, when fluorescently labeled mouse bone marrow–derived cells were introduced into mice, macrophage-cancer cell hybrids exhibiting gene expression patterns that are characteristic of both parental fusion partners were formed[16]. Likewise, implanting human glioblastoma or Hodgkin lymphoma cells into hamster cheeks resulted in metastatic human-hamster hybrids with coexpression of human and hamster genes[17],[18]. Thus, mouse-mouse, human-mouse, and human-hamster hybrids all coexpressed cancer cell and normal cell genes. These findings are central to the hybrid theory of malignancy and provide an explanation for why malignant cells show marked changes in gene expression patterns from their premalignant counterparts[2]–[4].
Studies on Hybridization in Human Tumors
Preliminary evidence of cancer cell fusion in humans
To prove that a cell is a biological hybrid requires that genes from both fusion partners are present in the hybrid cell. In animal tumor models, detecting hybrids is straightforward because a tumor can be implanted with a genome different from that of the host, and host-tumor hybrid cells can be confirmed genetically. In humans, however, tumor cells and normal leukocytes from the same patient cannot be easily distinguished genetically, making the proof of hybrids difficult. To circumvent this problem, we analyzed tumors arising as secondary malignancies after the patient had undergone an allogeneic bone marrow transplant (BMT)[19],[20]. In these cases, the circulating leukocytes from the donor differ genetically from the patient tumor cells, allowing for genetic confirmation of leukocyte-tumor cell hybrids. In an earlier case, a renal cell carcinoma arose in a patient who had previously received an allogeneic bone marrow from her son[20]. Through fluorescence in situ hybridization (FISH) analyses of the tumor cells, we demonstrated the son's Y chromosome in the nuclei of the mother's tumor cells. In Figure 3, the left panel shows a nucleus with three blue/green dots highlighting three copies of chromosome 17. Trisomy 17 is a common feature of renal cell carcinoma. Essentially all of the nuclei in the tumor cells contained this abnormality. However, the right panel shows a nucleus from the same tumor with trisomy 17 plus an additional red dot showing the donor Y chromosome in the same nucleus. Approximately 1%–10% of the nuclei in the tumor cells showed this pattern. This was consistent with donor cell-patient cell fusion; but unfortunately at that time, we were unable to verify the presence of patient genes, and fusion, though likely, could not be fully proven. For example, we did not rule out the possibility that the entire tumor arose from a transplanted normal donor stem cell that differentiated into a renal cell in the patient's kidney and then transformed into a renal carcinoma cell, with trisomy 17 and Y chromosome but no patient-specific genetic input.
Figure 3. Fluorescence in situ hybridization analyses of a primary renal cell carcinoma from a female patient who had received a male bone marrow transplant.
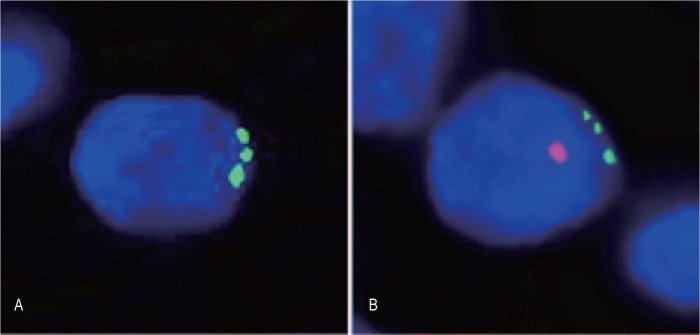
A, a cancer cell nucleus with a trisomy 17 (green) but without a Y chromosome (red). B, a cancer cell nucleus with both a trisomy 17 and a Y chromosome[20]. (This figure was first published in reference [20]; used with permi-ssion.)
Forensic genetics and the first proof of bone marrow-derived cell (BMDC)-tumor cell fusion in humans
To deal with this problem, we turned to forensic genetics, a powerful technique to identify two or more individuals in the same sample using short terminal repeat (STR) analyses[2]. For this, we collaborated with the Denver Police Crime Lab (Denver, Colorado, USA), well-known for their expertise in the field. We obtained a pathologic specimen of a melanoma brain metastasis that had occurred six years after the patient received an allogeneic bone marrow from his brother. Tumor cells were dissected with a laser microscope and analyses of the extracted DNA unequivocally revealed the presence of STR alleles from both the donor and the patient, indicating fusion (Figure 4). We further determined that hybrid cancer cells populated the entire tumor from one end to the other with repeating genetic patterns of donor and patient alleles throughout. This indicated that the tumor was a clone that had arisen from a single fusion event.
Figure 4. Forensic short terminal repeat (STR) analyses of the MH3 melanoma along with donor and patient pre-bone marrow transplant (BMT) lymphocytes.

Shown are “informative” loci exhibiting donor- and patient-specific alleles in pre-BMT lymphocytes. Tumor loci are listed in order of relative abundance of the donor-specific alleles (red asterisk) compared to patient-specific (blue asterisk) and shared alleles (black asterisk). Allele peaks < 50 relative fluorescence units were censored as “no call” (o). Loci with no detectable alleles after polymerase chain reaction amplification were designated as “---”. (This figure was first published in reference [2]; used with permission.)
Figure 4 shows STR alleles from nine regions throughout the tumor compared to the STR alleles in donor and patient pre-transplant normal DNA. Alleles specific to the patient are marked with a blue asterisk and those specific to the donor with a red asterisk. All alleles in the pre-transplant donor and patient blood lymphocytes were also found in tumor cells. For each locus there was at least one allele common to both the donor and the patient, consistent with them being brothers. Eight loci exhibited donor-specific alleles and six of these exhibited both donor- and patient-specific alleles. These results indicated that the tumor cells possessed a donor-patient hybrid genome (Figure 4, Table 2). Of great interest, the allelic ratios of the donor and the patient alleles in tumor cells differed between loci, but for any given locus, they were similar throughout all nine regions sampled. This suggested that the metastasis originated from a single hybrid clone generated from a fusion of a BMDC with a tumor cell. The remaining loci were uninformative regarding fusion as they contained no donor-specific alleles but only alleles common to the donor and the patient or the patient[2].
Table 2. Summary of short terminal repeat (STR)-length polymorphisms.
| Locus | Chromosome | Donora | Patienta | Tumora | Tumor genotype |
| D13S317 | 13 | 9,11 | 9,10 | 9,11 | D/S |
| D19S433 | 19 | 12,13 | 13,14 | 12,13,14 | D/P/S |
| CSF1PO | 5 | 10,11 | 11,12 | 10,11,12 | D/P/S |
| D16S539 | 16 | 9,11 | 9,13 | 9,11,13 | D/P/S |
| FGA | 4 | 20,22 | 20,21 | 20,21,22 | D/P/S |
| D7S820 | 7 | 8,9 | 8,12 | 8,9,12 | D/P/S |
| D8S1179 | 8 | 13,14 | 13,13 | 13,14 | D/S |
| vWA | 12 | 19,20 | 19,19 | 19,20 | D/S |
Thus, it appeared that the initiator of the metastasis was a hybrid cell, acting as a stem cell or tumor-initiating cell. The findings could have important implications for understanding central questions in cancer research today such as the identity of tumor-initiating cells/cancer stem cells and regulation of the hybrid epigenome[2]–[4].
Although this is only one case, it seems highly unlikely that the one tumor we studied was the only hybrid tumor there is or ever was, but this remains for future study. Undoubtedly, we need more cases and more labs joining in this pursuit before we can begin to estimate the frequency of hybridization in cancer.
Discussion and Conclusions
Interpretation of the data
STR analyses of the tumor DNA revealed that donor and patient alleles were present together at multiple loci and that there were widespread allelic imbalances and aneuploidy. One problem in interpreting the results was the possibility that even though we isolated cancer cells free of leukocytes by laser dissection, the samples might still, somehow, contain chimeric mixtures of patient tumor cells and donor BMDCs. However, this is unlikely for a number of reasons[2]. For example, for a given locus, the donor and patient allelic ratios were similar throughout, indicating a clonal origin of the tumor (Figure 4). It is difficult to explain these repeating patterns as being due to chimeric mixtures because this would require that the different cell types existed together in the same ratios throughout the tumor. Notably, while most of the informative tumor loci had patient or shared alleles in greater relative abundance to donor-specific alleles, tumor locus D13S317 was reversed, with the patient-specific allele absent and the donor-specific allele in prominence (Figure 4). Because the initial dose of genomic DNA for each sample was determinative of the intensity of polymerase chain reaction (PCR) products and varied widely between samples, and given the consistency in allelic ratios from sample to sample, the reversal of allelic dosage at locus D13S317 cannot be explained by preferential PCR or donor leukocyte contamination. Moreover, our statistical analyses of the allelic patterns strongly favored BMDC-melanoma cell fusion over leukocyte contamination models (P < 0.005)[2].
Treatments targeting metastasis
A role for BMDC-melanoma cell fusion in metastasis would imply that targeted interference of the fusion process or of rate-limiting post-fusion events might prevent invasion and metastasis. As we learn more of these processes, better strategies should emerge for targeting vulnerable steps in fusion and the generation of hybrids. The molecular mechanics of hybrid formation could present prime targets such as the molecular pathways governing the integration of parental fusion partner genes into hybrid genomes and the regulation of epigenetic expression from the hybrid genomes of both parental fusion partners. Fused cells are also likely to express unique antigenic profiles, making them candidates for immunotherapy.
Future direction
The hybrid theory has long been put forth as a unifying explanation for tumor metastasis[4]. Results from our lab and many others indicate that this theory is not only plausible but likely to be correct to some degree in human cancers. Accordingly, a major goal is to determine the frequency of hybrids in larger cohorts of human cancers and their role(s) in malignant transformation. We hope that more laboratories will begin to focus on the many questions of cancer hybrids. Indeed, in his proposal on leukocyte-tumor hydrids in 1911, Prof. Aichel[1] implored future scientists to “study chromosomes from all angles” for evidence of hybrids in cancer.
Acknowledgments
I am indebted to all the individuals who have contributed to this work over the past 20 years; it could not have been accomplished without them. This work has been supported in part by the Amway Corporation and by institutional funds from Yale School of Medicine, the University of Colorado School of Medicine, and the Denver Police Crime Lab.
References
- 1.Aichel O. About cell fusion with qualitatively abnormal chromosome distribution as cause for tumor formation. In: Roux W, editor. Vorträge und Aufsätze über Entvickelungsmechanik Der Organismen. Leipzig, Germany: Wilhelm Engelmann; 1911. pp. 92–111. [in German] [Google Scholar]
- 2.Lazova R, Laberge GS, Duvall E, et al. A melanoma brain metastasis with a donor-patient hybrid genome following bone marrow transplantation: first evidence for fusion in human cancer. PLoS One. 2013;8:e66731. doi: 10.1371/journal.pone.0066731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pawelek JM. Tumor cell hybridization and metastasis revisited. Melanoma Res. 2000;10:507–514. doi: 10.1097/00008390-200012000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Pawelek JM, Chakraborty AK. Fusion of tumour cells with bone marrow-derived cells: a unifying explanation for metastasis. Nat Rev Cancer. 2008;8:377–386. doi: 10.1038/nrc2371. [DOI] [PubMed] [Google Scholar]
- 5.Mekler LB. Hybridization of transformed cells with lymphocytes as one of the probable courses of the progression leading to the development of metastatic malignant cells. Vestn Akad Med Nauk SSSR. 1971;26:80–89. [in Russian] [PubMed] [Google Scholar]
- 6.Goldenberg DM, Pavia RA, Tsao MC. In vivo hybridization of human tumour and normal hamster cells. Nature. 1974;250:649–651. doi: 10.1038/250649a0. [DOI] [PubMed] [Google Scholar]
- 7.Munzarova M, Lauerova L, Capkova J. Are advanced malignant melanoma cells hybrids between melanocytes and macrophages? Melanoma Res. 1992;2:127–129. doi: 10.1097/00008390-199207000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Rachkovsky MS, Sodi S, Chakraborty A, et al. Enhanced metastatic potential of melanoma x macrophage fusion hybrids. Clin Exp Metastasis. 1998;16:299–312. doi: 10.1023/a:1006557228604. [DOI] [PubMed] [Google Scholar]
- 9.Rachkovsky M, Pawelek J. Acquired melanocyte stimulating hormone-inducible chemotaxis following macrophage fusion with Cloudman S91 melanoma cells. Cell Growth Differ. 1999;10:515–524. [PubMed] [Google Scholar]
- 10.Rupani R, Handerson T, Pawelek J. Co-localization of β1, 6-branched oligosaccharides and coarse melanin in macrophage-melanoma fusion hybrids and human melanoma cells in vitro. Pigment Cell Res. 2004;17:281–288. doi: 10.1111/j.1600-0749.2004.00148.x. [DOI] [PubMed] [Google Scholar]
- 11.Chakraborty AK, Pawelek JM. β1,6-branched oligosaccharides regulate melanin content and motility in macrophage-melanoma fusion hybrids. Melanoma Res. 2007;17:9–16. doi: 10.1097/CMR.0b013e3280114f34. [DOI] [PubMed] [Google Scholar]
- 12.Handerson T, Camp R, Harigopal M, et al. β1-Branched oligosaccharides are associated with metastasis and predict poor outcome in breast carcinoma. Clin Cancer Res. 2005;11:2969–2973. doi: 10.1158/1078-0432.CCR-04-2211. [DOI] [PubMed] [Google Scholar]
- 13.Lazova R, Camp RL, Klump V, et al. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis and poor outcome. Clin Cancer Res. 2012;18:370–379. doi: 10.1158/1078-0432.CCR-11-1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brożyna AA, Jóźwicki W, Carlson JA, et al. Melanogenesis affects overall and disease-free survival in patients with stage III and IV melanoma. Hum Pathol. 2013;44:2071–2074. doi: 10.1016/j.humpath.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakraborty AK, de Freitas Sousa J, Espreafico EM, et al. Human monocyte x mouse melanoma fusion hybrids express human gene. Gene. 2001;275:103–106. doi: 10.1016/s0378-1119(01)00647-3. [DOI] [PubMed] [Google Scholar]
- 16.Powell AE, Anderson EC, Davies PS. Fusion between intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011;71:1497–2005. doi: 10.1158/0008-5472.CAN-10-3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldenberg DM, Zagzag D, Heselmeyer-Haddad KM, et al. Horizontal transmission and retention of malignancy, as well as functional human genes, after spontaneous fusion of human glioblastoma and hamster host cells in vivo. Int J Cancer. 2012;131:49–58. doi: 10.1002/ijc.26327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldenberg DM, Gold DV, Loo M, et al. Horizontal transmission of malignancy: in-vivo fusion of human lymphomas with hamster stroma produces tumors retaining human genes and lymphoid pathology. PLoS One. 2013;8:e55324. doi: 10.1371/journal.pone.0055324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakraborty A, Lazova R, Davies S. Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transplant. 2004;34:183–186. doi: 10.1038/sj.bmt.1704547. [DOI] [PubMed] [Google Scholar]
- 20.Yilmaz Y, Lazova R, Qumsiyeh M, et al. Donor Y chromosome in renal carcinoma cells of a female BMT recipient. Bone Marrow Transplant. 2005;35:1021–1024. doi: 10.1038/sj.bmt.1704939. [DOI] [PubMed] [Google Scholar]