

Abstract
The ability to extract somatic cells from a patient and reprogram them to pluripotency opens up new possibilities for personalized medicine. Induced pluripotent stem cells (iPSCs) have been employed to generate beating cardiomyocytes from a patient's skin or blood cells. Here, iPSC methods were used to generate cardiomyocytes starting from the urine of a patient with Duchenne muscular dystrophy (DMD). Urine was chosen as a starting material because it contains adult stem cells called urine-derived stem cells (USCs). USCs express the canonical reprogramming factors c-myc and klf4, and possess high telomerase activity. Pluripotency of urine-derived iPSC clones was confirmed by immunocytochemistry, RT-PCR and teratoma formation. Urine-derived iPSC clones generated from healthy volunteers and a DMD patient were differentiated into beating cardiomyocytes using a series of small molecules in monolayer culture. Results indicate that cardiomyocytes retain the DMD patient's dystrophin mutation. Physiological assays suggest that dystrophin-deficient cardiomyocytes possess phenotypic differences from normal cardiomyocytes. These results demonstrate the feasibility of generating cardiomyocytes from a urine sample and that urine-derived cardiomyocytes retain characteristic features that might be further exploited for mechanistic studies and drug discovery.
Keywords: Nuclear reprogramming, Induced pluripotent stem cells, Myocytes, Cardiac, Muscular Dystrophy, Duchenne, Cell differentiation
1.1 Introduction
Mutations in the dystrophin gene cause Duchenne Muscular Dystrophy (DMD), an X-chromosome inherited disorder affecting 1:3500 male births. Considering the vast number of dystrophin gene mutations identified and the variability in disease phenotype in patients makes this a disease amendable to the benefits of personalized medicine. We propose that induced pluripotent stem cells (iPSCs) will provide a platform for personalized medicine in DMD.
The reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) can provide a limitless source of cells that can be terminally differentiated into a variety of cell types and faithfully retain the donor's genotype as well as phenotypic traits. These features make iPSCs a highly desirable basic research tool for the purpose of disease modeling, screening for therapeutic compounds and providing seed cells for autologous cell replacement therapy. However, iPSC derivation is still an expensive and time-consuming process (usually months) with relatively low efficiency, in most cases less than 1% of the starting cell number. An ideal cell source for cellular reprogramming would be collected non-invasively, expanded easily in culture, and reprogrammed both rapidly and efficiently.
Previously, Zhou T et al. reported that human urine could be a novel source for iPSCs derivation(Zhou et al., 2012). Here we show that iPSCs can be generated from progenitor cells present in human urine and be differentiated into cardiomyocytes in monolayer culture. In our hands, USCs exhibited faster reprogramming kinetics with higher efficiency than human dermal fibroblasts or adult adipose derived mesenchymal stem cells (MSCs). USC-derived iPSCs (USC-iPSCs) expressed multiple pluripotent markers and formed teratomas upon engraftment into immune-compromised mice, indicating they are bona-fide pluripotent stem cells. In vitro, USC-iPSCs derived from either healthy volunteers or from a patient harboring a dystrophin mutation efficiently differentiated into cardiomyocytes. Only cardiomyocytes derived from healthy volunteers stained positive for dystrophin, whereas the cardiomyocytes derived from a patient with a dystrophin deletion were dystrophin negative. Physiological assays including Ca++ handling, oxidative stress, and oxygen consumption and hypotonic stress all support the feasibility that drug-discovery assays can be developed using urine-derived cardiomyocytes as a biological reagent.
1.2 Materials and methods
1.2.1 Human subjects
Participants gave informed consent as required by the Institutional Review Board (IRB). Healthy males (N=3) and one patient with Duchenne muscular dystrophy (DMD) harboring a large dystrophin deletion provided ∼100 cc of a “clean catch” urine sample.
1.2.2 Urine cell culture
Urine cells were isolated and expanded from urine specimens as described(Zhang et al., 2008). In brief, cell pellets were collected from whole urine samples (15 - 400ml) via centrifugation, washed in PBS, and plated as single cell suspensions in 10 cm tissue culture dishes with a cocktail of keratinocyte serum-free medium (KSFM; Invitrogen, Carlsbad, CA) and DMEM/10%FBS (USC medium).
1.2.3 Telomerase activity (TA)
2 × 105 cells were assayed for telomerase activity using Telo TAAGG ELISA kit (Roche Applied Sciences, Upper Bavaria, Germany), according to the manufacturer's instructions. HEK-293 cell lysates were used as positive controls and heat-inactivated (85°C for 10 minutes) HEK-293 cell lysates served as the negative control. Samples were considered to be positive for telomerase activity when the difference in absorbance was at least twice that of the negative control.
1.2.4 Reprogramming vector
A polycistronic lentiviral vector encoding human Oct-3/4, Sox2, Klf4 and c-Myc (OSKM)(Warlich et al., 2011) was used to transduce the urine-derived cells. The fluorescent reporter d-tomato gene was linked by an IRES element in the construct to serve as a real-time readout of viral transgene expression. To generate high titer viral supernatants suitable for urine cell transduction, HEK 293T cells were transfected with the OKSM plasmid, psPAX2 (Addgene #12260) and pMD2.G (Addgene #12259) using the FuGENE HD Transfection Reagent (Roche, Mannheim, Germany) according to the manufacturer's instructions. After transfection, medium was changed daily. Supernatants from day 2 and 3 were pooled together and concentrated in a 100 kD Amicon Ultra Centrifugal Filter tube (100 kD, Millipore, Billerica, MA) and frozen at -80°C until future use.
1.2.5 Cellular reprogramming
Urine-derived cells were seeded onto Matrigel (BD, San Jose, California) coated 12 well plates at 50,000 cells/well and allowed to attach overnight (day 0). On day two, cells were transduced with high-titer OSKM viral supernatants in the presence of 8 μg/ml polybrene for three hours. Viral supernatants were replaced with fresh USC medium and after three days, replaced with mTeSR1 medium (StemCell Technology, Vancouver, BC) and changed daily. As iPSC-like colonies appeared over time, they were picked using glass Pasteur pipettes under a stereo dissection microscope (Leica M205C, Buffalo Grove, IL) and transferred to new Matrigel-coated plates for further expansion.
1.2.6 Flow cytometry
Three days after viral transduction, d-tomato fluorescence expression was assayed to assess transduction efficiency. Briefly, cells were detached by TrypLE (Invitrogen, Grand Island, NY) and washed three times in PBS. The fluorescence expression was detected using a FACSCalibur flow cytometer (BD, San Jose, CA) and that data was analyzed using FlowJo vX software (Tree Star, Ashland, OR).
1.2.7 Immunohistochemistry and alkaline phosphatase (AP) staining
iPSCs were stained with live staining antibody Tra-1-81 (Stemgent, Cambridge, MA) and SSEA4 (BD, San Jose, CA) for 90 minutes at 37°C. Cells were imaged in mTeSR1 medium after being washed 3 times with DMEM/F12(Chan et al., 2009). For fluorescence immunocytochemistry of iPSCs and cardiomyocytes, cells were passaged onto glass coverslips coated with Matrigel. After being fixed with 4% paraformaldehyde and permeabilized by 0.2% Triton X-100, cells were incubated with the following primary antibodies at 4°C overnight: Sox2 (R&D systems, Minneapolis, MN), SSEA4, Oct4, Tra-1-81 and Tra-1-60 (all from Stemgent, Cambridge, MA), cardiac myosin heavy chain (abcam, Cambridge, MA), sarcomeric α-actinin (thermo scientific, Rockford, IL) connexin43 (Cell Signaling, Danvers, MA) and Cav1.3(Hell et al., 1993). For dystrophin staining, differentiated cardiomyocytes were plated onto glass coverslips coated with Matrigel. After being fixed with acetone for 10 minutes, cardiomyocytes were incubated with dystrophin antibody (Leica Microsystems Inc., Buffalo Grove, IL). Fluorochrome conjugated secondary antibodies were added the second day for 1 hour at room temperature. After counterstaining nuclei with DAPI, coverslips were mounted with Prolong Gold antifade reagent (Invitrogen, Grand Island, NY). Confocal images were acquired using a Nikon A1R confocal microscope. For alkaline phosphate (AP) staining, iPSCs were fixed with ice cold ethanol. Color was developed by incubating with AP staining solution (400 ul Naphthol AS-MX Phosphate Alkaline Solution, 2.4 mg Fast Red TR in 9.6 ml Water) for 1 hour in the dark. All images were analyzed with ImageJ (version 1.47n, National Institutes of Health) with standard plugin.
1.2.8 Teratoma assay of iPSCs
All animal procedures were approved by the University's Institution Animal Care and Use Committee. Eight to twelve week-old female NOD/SCID mice were purchased from Jackson Laboratory (Bar Harbor, Maine). Kidney capsule injections were performed as described(Ritner & Bernstein, 2010). To summarize, 1×106 cells were injected under the kidney capsule via a catheter connected to a Hamilton syringe. Tumors were excised after 8-12 weeks and fixed with 4% paraformaldehyde in PBS and embedded in paraffin. Sample blocks were sectioned at 5 μm and Hematoxylin & Eosin (H&E) staining was performed on tumor sections.
1.2.9 Cardiomyocyte differentiation
Urine-derived iPSCs were differentiated to cardiomyocytes following an established protocol with modifications(Laflamme et al., 2007). Briefly, iPSC colonies were detached by 10 minute incubation with Versene (Life technologies, Carlsbad, CA), triturated to a single-cell suspension and seeded onto Matrigel-coated plastic dishes at a density of 250,000 cells/cm2 in mTeSR1 medium and cultured for 4 more days. Differentiation was then initiated by switching the medium to RPMI-1640 medium supplemented with 2% insulin reduced B27 (Life Technologies) and fresh L-glutamine.
1.2.10 RT-PCR
Total RNA was extracted from undifferentiated iPSC clones and their corresponding cardiomyocytes using a Qiagen RNeasy kit. 1 μg of total RNA was reverse transcribed to cDNA using an RT2 First Strand Synthesis Kit (SA Biosciences, Valencia, CA), following the manufacturer's instructions. cDNAs were amplified to the level of detection using RT2 SYBR Green Master Mixes (SA Biosciences, Valencia, CA) and individual iPSC (Cat # IPSH-001) or cardiac (Cat # IPSH-102) markers were assayed using prefabricated arrays (SA Biosciences, Valencia, CA). All RT-PCR data was collected on a 7300 Real Time PCR system (Applied Biosystems, Carlsbad, CA).
1.2.11 Western Blot
Dystrophin proteins were visualized by western blot analysis using method as previously described (Wang et al., 2012). 15μg of cell lysates were loaded in designated lanes. DYS2 monoclonal antibody (1:50 Novocastra) was used as the primary antibody, horse-radish peroxidase conjugated anti mouse antibody (1:1000, Cell Signaling) was used as the secondary antibody. A mouse monoclonal antibody to GAPDH (Millipore) was used as protein loading control. Western blots were developed using ECL Plus Western Blotting Detection System (GE Healthcare).
1.2.12 Electrophysiological recording of beating cardiomyocytes
Clusters of beating iPS-CM were dissociated into single cells using Accutase (Sigma-Aldrich, St. Louis, MO) per manufacturer instructions and plated on Matrigel-coated coverslips (BD, San Jose, CA). Action potentials (AP) were recorded using an Axopatch 200B amplifier in current-clamp mode. The amplifier was under the control of pClamp 10.2 software (Axon instrument, USA). APs were recorded while iPS-CM were superfused with a solution containing (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 Glucose, 1 Na-Pyruvate, adjusted to pH 7.4 with NaOH. The patch pipettes were filled with a solution containing (in mM): 5 NaCl, 140 KCl, 7 MgATP, 15 HEPES, adjusted to pH 7.2 with KOH. Pipette resistances ranged from 3 to 6 MΩ.
1.2.13 Confocal imaging of calcium flux
To monitor [Ca2+]i, iPS-CMs were loaded for 30 min with 5 μM of the acetoxymethyl ester form of the Ca2+ indicator Fluo-4 (Fluo4-AM, Invitrogen; Kd = 345 nM). Cells were superperfused with a Tyrode's solution. APs were evoked via field stimulation at a frequency of 1 Hz using an Ion-Optix Myopacer (IonOptix Corp) delivering 4-ms square voltage pulses with an amplitude of 20 V via two platinum wires placed on each side of the perfusion chamber base (0.5 cm separation). AP-evoked [Ca2+]i transients were imaged using a Nikon (Eclipse TE2000-S) Swept Field confocal system equipped with a Plan Apo 60x 1.45 N.A. oil immersion objective, controlled with Elements software. Fluo-4 was excited with a 488 nm laser. Images were analyzed using Image J.
1.2.14 Oxygen Consumption Rate (OCR)
Cardiomyocytes differentiated from normal and DMD iPSC were seeded at 3×104 cells/well into 0.1% gelatin pre-coated SeaHorse™ plates in cardiomyocyte media (RPMI supplemented with B27 with insulin and antibiotics). Culture media was switched to base media (unbuffered DMEM, Sigma D5030) supplemented with sodium pyruvate (Gibco, 1mM) and with 25 mM glucose 1 hour before the assay and for the duration of the measurement. Selective inhibitors were injected during the measurements to achieve final concentrations of 4-(trifluoromethoxy) phenylhydrazone (FCCP, 1 μM), oligomycin (2.5 μM), antimycin (2.5 μM) and rotenone (2.5 μM). Mitochondrial stress protocol starts with the measurement of baseline oxygen consumption rate (OCR) following by measurement of OCR changes in response to injection of oligomycin, FCCP and finally antimycin and rotenone. The OCR values were further normalized to the number of cells present in each well, quantified by the Hoechst staining (HO33342; Sigma-Aldrich) as measured using fluorescence at 355 nm excitation and 460 nm emission.
1.2.15 Mitochondrial Permeability Transition Pore (mPTP) opening time
Cardiomyocytes derived from normal (n=6) and DMD iPSCs (n=6) were loaded with tetramethylrhodamine ethyl ester (TMRE), a fluorescent indicator that accumulates in the mitochondria proportionally to the ΔΨm, and exposed to controlled and narrowly-focused laser-induced oxidative stress until mPTP opening occurs(Pravdic et al., 2009). To ensure equal delivery of oxidative stress among experimental groups, laser excitation settings remain consistent within the experimental groups. Mitochondrial PTP opening was detected by a decrease in TMRE fluorescence which indicates loss of ΔΨm. Arbitrary mPTP opening time was determined as the time of the loss of average TMRE fluorescence intensity by one half between initial and residual fluorescence after mPTP opening.
1.2.16 Cardiomyocyte hypotonic stress assay
Hypotonic solutions were made by diluting DPBS solution to ½ (145 mOsm) and ¼ (73 mOsm) osmolarity with water(Musch, Davis-Amaral, Vandenburgh, & Goldstein, 1998). Normal and DMD iPS-CM were incubated in hypotonic solutions (145 mOsm or 73 mOsm) for 30 minutes at room temperature. Then osmolarity was adjusted back to normal by adding equal volume of 435 mOsm or 507 mOsm hypertonic solutions, for 5 minutes. The supernatants were collected and analyzed for human cardiac troponin I (cTnI) and creatine kinase-MB (CK-MB), using Meso Scale Discovery (MSD) 96 well custom human cardiac I assay (MSD, Maryland) following the manufacturer's instructions. The plate was imaged by SECTOR Imager 2400 (MSD, Maryland).
1.2.17 Statistics
Data are expressed as mean ± SD unless stated otherwise. For the transduction efficiency study, mesenchymal cell lines (n=4) and USC lines (n=7) were aggregated separately for a Mann-Whitney U test by using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Hypotonic stress assay was analyzed using Two-Way ANOVA. A p value less than 0.05 was considered significant.
1.3 Results
1.3.1 Isolated urine cells express c-Myc and Klf4 in addition to high telomerase activity
USCs isolated from urine samples provided by healthy volunteers (n=3) and a DMD patient all displayed a mesenchymal stem cell phenotype, including spindle-shaped morphology (Fig 1A) and expression of cell surface markers CD44, CD73, CD90, CD105 and CD146. In addition, USCs did not express the hematopoietic stem cell markers CD25, CD31, CD34 and CD45 (Supplemental Fig 1). Prior to iPSC reprogramming, four different USC clones were assayed for endogenous expression of four classical reprogramming factors c-Myc, Klf4, Oct4 and Sox2. Quantitative RT-PCR analysis revealed that USCs expressed high levels of c-Myc and Klf4 relative to human ES cells (normalized as a control), IMR90 (fetal human lung cells) and NCCIT (mixed germ cell tumor) while expression of Oct4 and Sox2 were negligible compared to hESC and NCCIT (Fig 1B). A high telomerase activity clone and a low telomerase activity clone were examined using RT-PCR from two different patients (labeled “A” and “K”). Young adult donors (20-40 years old) produced a greater proportion of telomerase high USC clones (75% TA high vs 25% TA low; n=10 clones/group) than donors older than 50 years (50% TA high and 50% low; n=10 clones/group)(Shi, Liu, Shantaram, Atala, & Zhang, 2012). In addition, USCs express the kidney glomerular podocyte markers podocin and synaptopodin (unpublished data) suggesting a mesodermal origin of the isolated cells, consistent with their MSC-like morphology and marker profile. Examination of USCs isolated from the DMD patient demonstrated a dystrophin deletion of exon 50 (complete male DMD evaluation, test 181, Athena Diagnostics) resulting in a frameshift mutation that halts production of normal dystrophin protein (Supplemental Fig 1).
Figure 1. Characterization of Urine-derived stem cells (USCs).
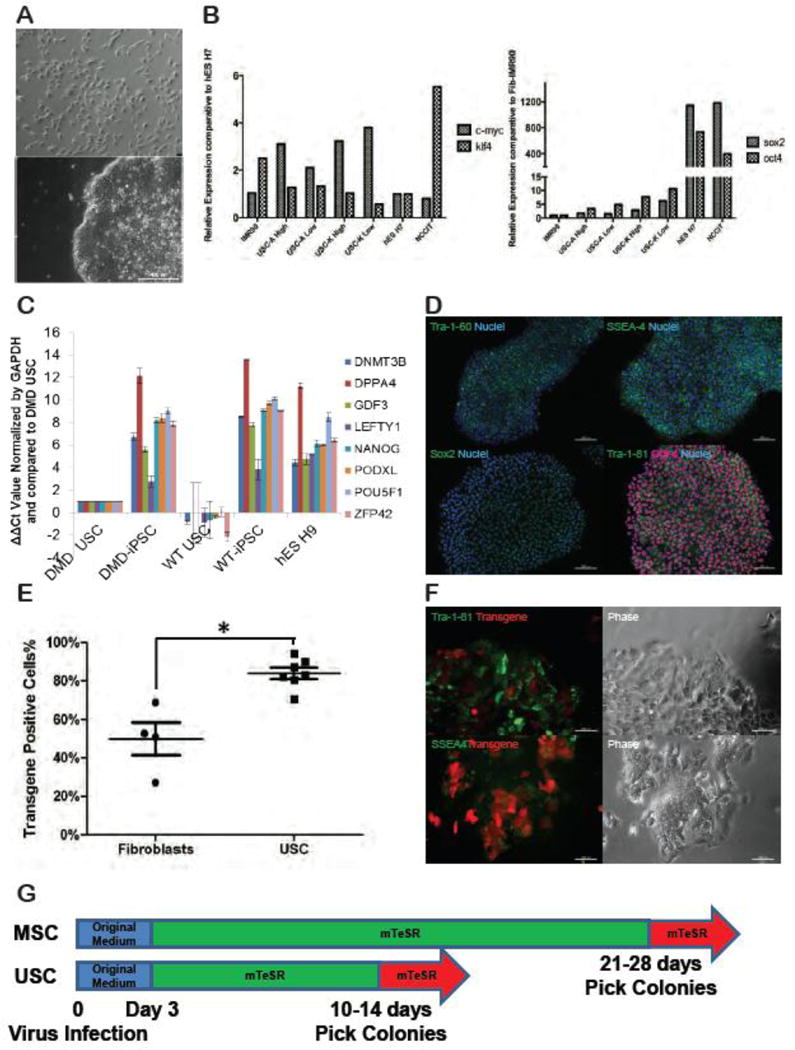
(A) A representative phase-contrast microscopic image of cells isolated from the urine (top) and the morphology of its derivative iPSC colony after reprogramming (bottom); (B) Quantitative RT-PCR of USC clones assayed for expression of the reprogramming factors Oct4, Sox2, Klf4 and c-Myc. Expression of Klf4 and c-Myc are shown relative to expression of the same genes in the human embryonic stem (hES) cell line, H7 (marked at 1, y-axis) while Oct4 and Sox2 expression is shown relative to IMR90 fibroblasts (also arbitrarily standardized to 1); (C) To evaluate pluripotency, quantitative RT-PCR for several genes highly expressed in human embryonic stem cells was compared between several iPSC clones (n=4) derived from both healthy donor and DMD patient USC clones and compared to hESC H9. All data were shown relative to the expression of DMD USC (set at 1, y-axis). Relative values (2ΔΔCt) were normalized against the housekeeping gene GAPDH and plotted for the genes DNMT 3B, DPPA4, GDF3, LEFTY1, NANOG, PODXL, POU5F1 and ZFP42.; (D) Representative iPSC colonies derived from normal human urine cells and probed with antibodies highly expressed on human embryonic stem cells (Tra-1-60, SSEA4, Sox2, Tra-1-81 and Oct4). Scale bar, 200 μm; (E) Flow cytometric analysis of viral transduction efficiency compared between fibroblasts and cells isolated from urine (USC) three days after the initiation of reprogramming. The asterisk denotes a p-value of 0.0061 (Mann-Whitney U test); (F) Early up-regulation of pluripotent surface markers SSEA4 and Tra-1-81 at day 7, accompanied by the down-regulation of viral transgenes, indicated by red fluorescence; (G) Overall kinetics of lentiviral-mediated reprogramming for USCs and MSCs. Both USCs and MSCs were kept in original medium for 3 days post viral transduction. hESC medium mTeSR was added on the fourth day. Colonies were picked at indicated time.
1.3.2 Urine cells from healthy volunteers and a DMD patient reprogram to bona fide iPSCs
The iPSC colonies derived from USCs displayed typical pluripotent stem cell morphology (Fig 1A). Cells appeared tightly packed within colonies, while individual cells displayed prominent nucleoli with an elevated nuclei/cytoplasm ratio. Individual colonies (each representing a unique clone) were manually picked and subcultured for 2 years without notable senescence or deterioration. Quantitative RT-PCR confirmed that iPSCs generated from urine cells expressed a panel of pluripotency-related genes comparable to those expressed by hESC H9 (Fig 1C). Immunofluorescent staining of iPSCs demonstrated characteristic localization of several key pluripotent markers, including Oct4, Sox2, SSEA4, Tra-1-60 and Tra-1-81 (Fig 1D). To verify in vivo pluripotency, four different clones of USC-iPSCs were implanted under the kidney capsule of NOD/SCID mice (N=4/iPSC clone), three derived from the urine of healthy donors and one from a DMD patient. All iPSC clones formed multi-differentiated teratomas in at least 3 of the 4 mice injected. Teratomas showed tissue structures indicative of all three germ layers (ectoderm, endoderm, and mesoderm) and comparable to teratomas formed from the hESC H9, thus confirming that USC-iPSCs were truly pluripotent (Supplemental Fig 1).
1.3.3 Urine-derived cells reprogram to iPSCs more rapidly than fibroblasts or MSCs
During early experiments, it was noticed that USCs formed iPSC colonies faster than fibroblasts that were also being reprogrammed in parallel. To test the hypothesis that USCs reprogram faster than commonly used starting cell lines, the efficiency and kinetics of reprogramming were compared between USCs and mesenchymal cell lines, including a human foreskin fibroblast line (BJ), a human fetal lung fibroblast line (IMR90), an adipose derived MSC line (MSC-A1), and a human skin fibroblast line (Coriell GM 04422). The efficiency of transduction was based on the initial percentage of transduced cells which was achieved using a multiplicity of infection (MOI) of 5. Figure 1E shows that 80% of the USC clones (n=7) were successfully transduced as indicated by the expression of a red fluorescence reporter. In contrast, at the same MOI, only 50% of mesenchymal cell lines (n=4) were transduced (Fig 1E).
To evaluate the speed of reprogramming, morphological changes, expression of pluripotency cell surface markers and alkaline phosphatase staining were assessed. As early as 3 days after viral transduction, USC cell lines demonstrated morphological changes indicative of reprogramming (reduced cell size, increased nuclear/cytoplasmic ratio with prominent nucleoli) while MSCs did not . By day 7, only the USC cell lines developed individual colonies with defined borders and expressed SSEA4 and Tra-1-81, two surface glycoproteins that characterize the somatic to pluripotency transition (Fig 1F). Expression of SSEA4 and Tra-1-81 was accompanied by silencing of viral transgenes, manifested by the gradual decrease of d-tomato red fluorescence (Fig 1F). MSC lines did eventually go through similar morphological changes and expression of pluripotency markers, but at later time points compared to USC lines. USC-derived iPSC colonies were mature enough to be manually picked between days 10-14, whereas MSC-derived iPSC colonies required 28 days (Fig 1G). Together, these data indicate that, compared to the mesenchymal lines tested, USCs are more receptive to lentiviral transduction and reprogram more rapidly than MSCs.
1.3.4 Telomerase activity is associated with improved reprogramming efficiency
USC clones were generated by limiting dilution and assayed for TA activity individually. A pair of USC clones derived from three different donors (USC-A, USC-B and USC-K) were tested (Fig 2A). Each donor produced both telomerase high and low clones that were subsequently tested to determine whether telomerase activity is positively correlated with reprogramming kinetics. 50,000 cells of each USC clone were infected with same amount of the reprogramming lentivirus (MOI 5). Even though iPSC colonies start to appear around day 12, the reprogramming process was evaluated over 17 days to maximize the colony yield for all clones. The reprogramming kinetics were comparable as all clones gave rise to alkaline phosphatase and SSEA4 positive, transgene-silenced colonies at day 17 (Fig 2B and D). However, TA exhibited a positive correlation with reprogramming efficiency (Fig 2C). Though low TA clones universally manifested a higher viral transduction rate at day 3, they generated fewer colonies at day 17 (with calculated efficiency of 0.002% to 0.007% compared to 0.1% to 0.5% for high TA clones) (Table 1).
Figure 2. Association between telomerase activity and USC reprogramming efficiency.
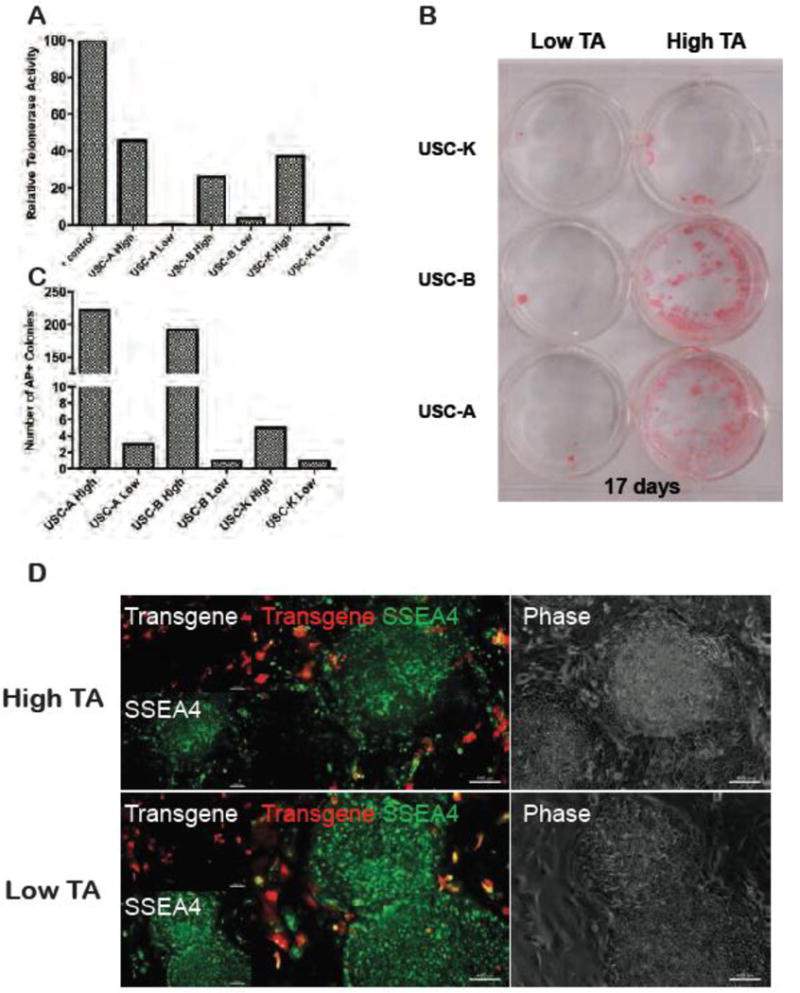
(A) Relative telomerase activity (TA) of various USCs clones relative to TS8, the internal positive control from the TRAP assay kit. Telomerase high and telomerase low clones were derived from three independent donors (A, B and K); (B) Alkaline phosphatase (AP) staining of reprogrammed USC clones from (A) at day 17. Higher telomerase expression was associated with marked increase in reprogramming efficiency, indicated by the number of AP positive colonies; (C) Quantitation of photographs shown in panel B; (D) Mature day 17 iPSC colonies resulting from both high telomerase and low telomerase UC clones. Reprogrammed colonies express only SSEA4 (uniform green staining), while the surrounding non-reprogrammed single cells still express the transgene (red fluorescence).
Table 1. Summary of transduction and reprogramming efficiency of various USC clones.
| Input Cells | Viral Transduction Efficiency | Colony Number | iPS Generation Efficiency(Input Cells*Viral transduction efficiency/Colony #) | |
|---|---|---|---|---|
|
UC-A High |
5000 0 |
80.4% | 222 | 0.5522% |
|
UC-A Low |
5000 0 |
87.3% | 3 | 0.0069% |
|
UC-B High |
5000 0 |
70.5% | 192 | 0.5447% |
|
UC-B low |
5000 0 |
83.2% | 1 | 0.0024% |
|
UC-K High |
5000 0 |
82.1% | 5 | 0.0122% |
|
UC-K low |
5000 0 |
94.2% | 1 | 0.0021% |
1.3.5 Urine cells reprogram into functional cardiomyocytes
The overall goal of this work was to determine if cardiomyocytes derived from reprogrammed urine cells donated by a patient with a single-gene disease, can recapitulate aspects of the disease phenotype. Therefore, urine-derived iPSC clones were generated from a DMD patient with a dystrophin mutation, and subsequently differentiated into beating cardiomyocytes via in vitro monolayer culture. Sporadic contracting cardiomyocytes were observed 8-20 days after initiating differentiation. The beating loci expanded over several days and synchronized to form a beating cell sheet (Supplemental online video 1 & 2). Immunostaining confirmed that beating cells were positive for cardiac markers sarcomeric α-actinin, cardiac α and β myosin heavy chain (MHC), as well as membrane localized connexin43 (Fig 3A). Both normal and DMD cardiomyocytes were assessed by quantitative RT-PCR and demonstrated upregulation of a series of cardiac genes (Fig 3B). To investigate functionality, differentiated cardiomyocytes were subjected to patch clamp recording. These cells exhibited spontaneous action potentials (AP). Based on the amplitude, depolarization speed (dV/dt) and 50% AP duration (APD50) differentiated cardiomyocytes were further characterized as nodal (33%), ventricular (59%) and atrial (9%) subtypes (Fig 3C).
Figure 3. Cardiac induction from urine-derived iPSCs and characterization.
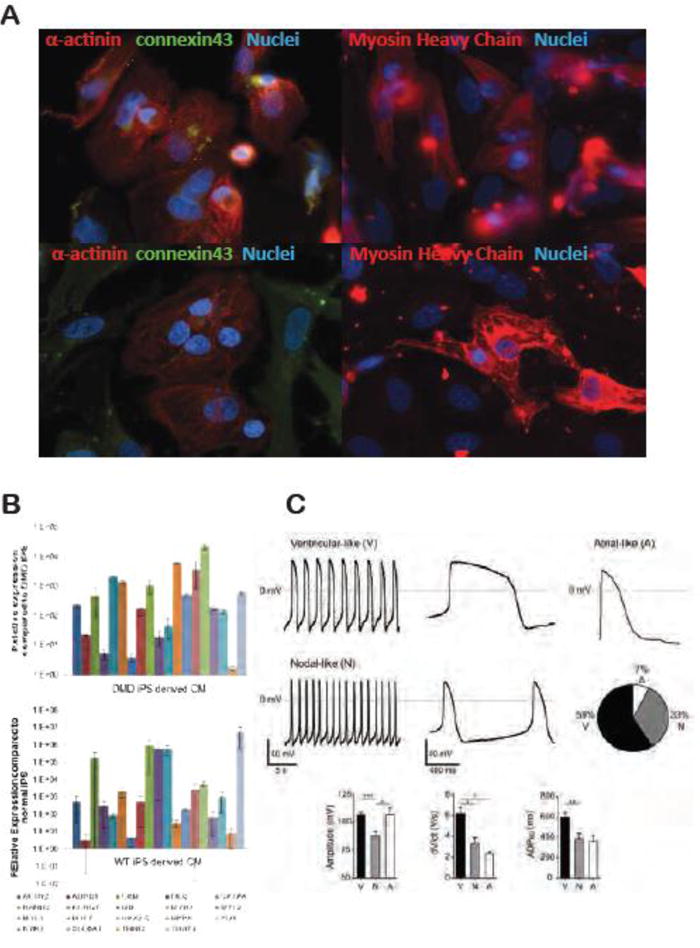
(A) Immunofluorescent staining of differentiated cardiomyocytes. Both normal and DMD cardiomyocytes stained positive for cardiac sarcomeric α-actinin, cardiac Myosin Heavy Chain (MHC), and membrane localized connexin 43; (B) Quantitative RT-PCR of cardiomyocytes differentiated from both DMD and normal USC-iPSCs. Data shown in relative to the expression of parental iPSCs. Relative values were normalized against the housekeeping gene GAPDH and plotted for the genes ACTN2, CKM, DES, GATA4, HAND2, KCNQ1, MB, MYH7, MYL2, MYL3, MYL7, NKX2-5, NPPA, PLN, RYR2, SLC8A1, TNNI3, TNNT2. (C) Action Potential (AP) recordings of normal iPS derived cardiomyocytes. Representative traces of the spontaneous firing of ventricular-like and nodal-like cells and the evoked AP of a quiescent atrial-like cell. Percentage distribution of ventricular, atrial, and nodal phenotypes (n=30). Bar plots summarize the characteristics of the AP for the three phenotypes (Mean ± SEM).
1.3.6 Cardiomyocytes derived from the urine of a DMD patient were dystrophin negative
Normal and dystrophin-deficient DMD cardiomyocytes were probed with antibodies against dystrophin and cardiac-specific markers Nkx2.5 and α-actinin. In cardiomyocytes derived from hES H9 and normal USC iPSC, dystrophin expression was localized to the plasma membrane. In contrast, no dystrophin expression was detected in DMD iPSC cardiomyocytes by either immunohistochemistry (Fig 4A) or by immunoblot, e.g., full-length 426kDa dystrophin band not detected (Fig4B). These findings support that DMD cardiomyocytes maintained their dystrophin-deficient phenotype.
Figure 4. Immunohistochemical staining of cardiomyocytes derived from the urine of a DMD patient and a normal volunteer.
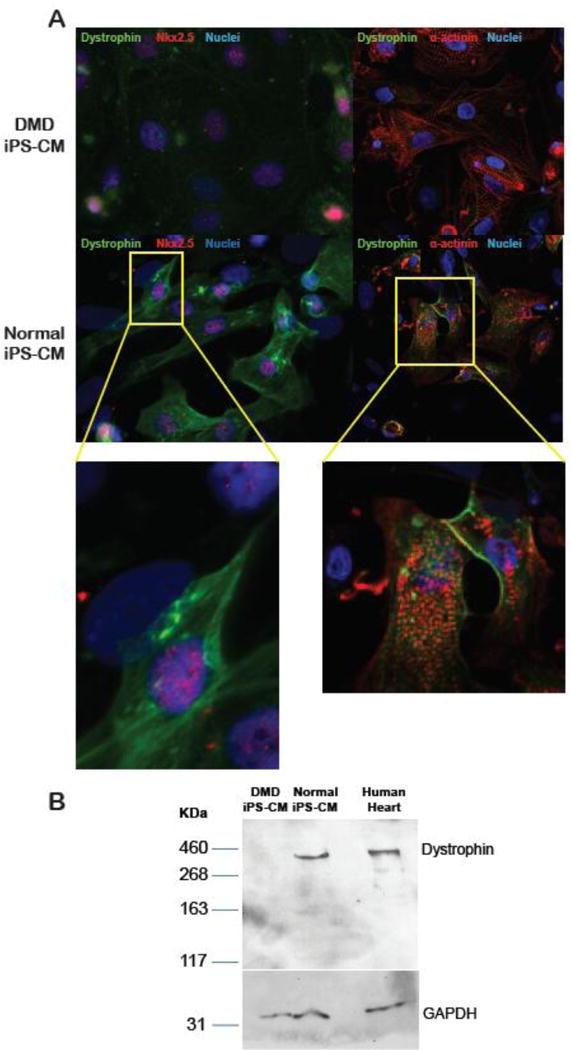
(A) Cardiomyocytes derived from normal and DMD USC-iPSCs were probed with antibodies against dystrophin, together with cardiac specific protein Nkx2-5 or sarcomeric α-actinin. Dystrophin staining is only demonstrated in normal cardiomyocytes, but not in cardiomyocytes derived from the DMD patient; (B) Western blot for full length dystrophin detected in the lysates from normal iPS-derived cardiomyocytes (lane 2) and human heart tissue (lane 3) but absent in the lysate from DMD cardiomyocytes (lane 1). Molecular weight markers (in kDa) are shown on the left.
1.3.7 Physiological consequences of dystrophin deficiency in cardiomyocytes
Several domains of cardiac function in DMD and normal iPS-derived cardiomyocytes are demonstrated in Figure 7, including calcium handling, mitochondrial permeability pore (mPTP) opening, cellular metabolism and susceptibility to mechanical stress. Significant (p<0.05) differences between DMD and normal cells were detected in calcium handling: The duration of recovery (T50) of DMD calcium transient (Fig 5A) was prolonged compared to the normal control (629.7 ± 20 ms vs 311.8 ± 3 ms, respectively). Mitochondrial permeability pore opening (Fig 5B) occurred earlier in DMD compared to normal controls (62 ± 14 msec vs. 163 ± 13 sec). No differences between the groups were detected in cellular metabolism (Fig 5C). In the hypotonic stress experiment (Fig 5D), DMD cardiomyocytes responded with an increase of the cardiac injury marker CK-MB and cTnI, with both markers inversely correlated with osmolarity. In DMD cells, both cardiac injury markers were significantly (p<0.05) higher than normal cardiomyocytes (145 mOsm: CK-MB = 69.6 ± 8.6 μg/ml; cTnI = 1.64 ± 0.3 μg/ml; 73 mOsm: CK-MB = 127.2 ± 11 μg/ml. cTnI = 6.09 ± 0.4 μg/ml and 145 mOsm: CK-MB = 5.8 ± 0.1 μg/ml. cTnI = 0. 73 mOsm: CK-MB = 4.3 ± 0.02 μg/ml, cTnI = 0, respectively).
Figure 5. Potential physiological readouts for high throughput screening in iPS derived cardiomyocytes.
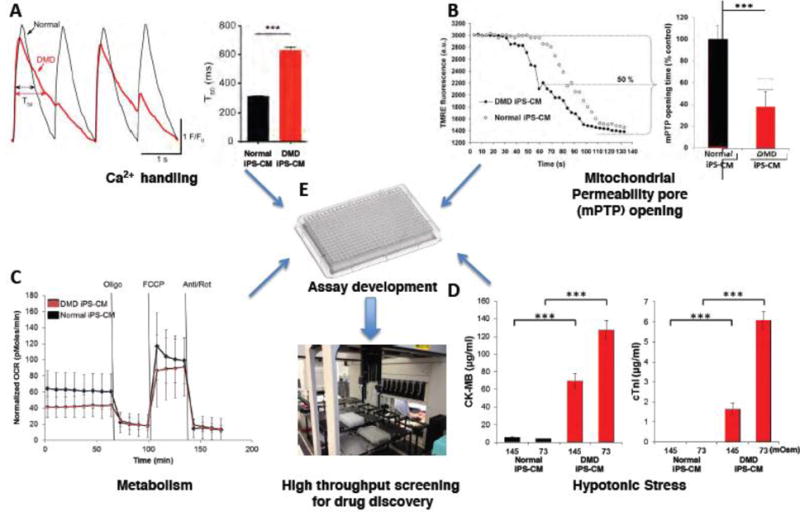
(A) Single cell tracing of evoked Ca2+ transients from a normal and a dystrophin-deficient iPS-derived cardiomyocyte (Normal and DMD iPS-CM) loaded with a Ca2+ indicator. Individual cells were paced at 1 Hz and a confocal microscope recorded the resulting Ca2+ signal. Typical tracing of normal and DMD iPSC-CM Ca2+ transients are displayed on the left. Bar plots summarize the duration of the recovery of the calcium transient (T50) (Normal iPSC-CM, n = 3; DMD iPSC-CM, n = 3).
(B) Individual cardiomyocytes derived from normal and DMD iPS were loaded with an inner mitochondria membrane potential (ΔΨm) dye Tetramethylrhodamine ethyl ester (TMRE). Oxidative stress, induced by controled laser exposure, causes mitochondria permeability transition pore (mPTP) opening which leads to a decrease in TMRE fluorescence, indicating the loss of ΔΨm. A representative readout of the TMRE fluorescence decay is shown between a normal and DMD iPS-CM (open and closed circles, respectively). The mean calculated mPTP opening time, determined as the half decay time of the average initial TMRE fluorescence intensity, is presented by bar graph for (n=6) experiments.
(C) Oxygen consumption rate (OCR) of normal and DMD iPS-CMs (n=6 plates of cells) measured using the Seahorse™ XF96 Extracellular Flux analyzer. Selective inhibitors were injected during the measurements as indicated. OCR was measured at baseline and following injection of various inhibitors, and values were normalized to the number of cells present in each well.
(D) Cardiac damage following hypotonic stress. Normal and DMD iPS-CM were incubated in hypotonic solutions as indicated for 30 minutes. The supernatants were analyzed for human cardiac troponin I (cTnI) and creatine kinase-MB (CK-MB). DMD cells released show markedly elevated levels of both injury markers whereas only negligible amounts were detected from normal CMs.
(E) Physiological assays described above (A-D) could be adapted to a high throughput format to serve as initial readouts or as confirmation of “hits” for drug discovery.
Legend: Bar plots: Mean ± SD. ***P<0.001; **0.001 < P < 0.01; *0.01 < P < 0.05. Oligo = oligomycine; FCCP = 4-(trifluoromethoxy) phenylhydrazone; Anti/Rot = antimycin and rotenone; Osm = osmolar
1.4 Discussion
The isolation of a highly proliferative cell population from human urine samples was described previously by our group(Bharadwaj et al., 2011) as well as others(Dorrenhaus et al., 2000; Zhou et al., 2011). Early-passage USC cultures contain highly motile cells with the distinctive mesenchymal morphology and an MSC-like cell surface marker profile. In this study, USCs derived from the urine of healthy volunteers and a DMD patient were successfully reprogrammed to bona fide iPSCs using a conventional Yamanaka-factor, lentiviral-mediated delivery method. More importantly, the whole reprogramming process only took approximately three weeks from urine sample collection to iPSC colonies, comparable to the iPSC reprogramming kinetics reported for human hepatocytes(Liu, Ye, Kim, Sharkis, & Jang, 2010).
The endogenous expression of the reprogramming factors c-Myc and Klf4 and high telomerase activity in USCs led to the hypothesis that USCs may be more easily reprogrammed compared to skin fibroblasts, the more typical starting material for iPSC generation. To test this idea, the reprogramming kinetics among several mesenchymal cell lines, including fibroblasts were compared to the kinetics observed for USCs. In all of the mesenchymal lines tested, reprogramming required at least 4 weeks, in contrast to only 2 weeks for USCs. Although USCs intrinsically express c-Myc and Klf4, this is not likely to be the sole reason for the observed fast kinetics. The fibroblast IMR90 has also been shown to express comparable levels of these two genes as USCs, but reprograms with slower kinetics. Previous reports indicated that these two reprogramming factors could be detected in several in vitro culture-adapted cell types, such as human keratinocytes(Aasen et al., 2008) and fibroblasts(I.-H. Park et al., 2008). Another possible contributing factor to the overall success of this approach was that USCs were particularly receptive to lentiviral transduction. However, this was not likely to contribute to rapid iPSC conversion. A wide range of lentiviral MOIs on these were tested on the mesenchymal cells with little variance in reprogramming kinetics (data not shown).
Although the addition of hTERT (increasing telomerase activity) to the conventional four reprogramming factors can facilitate the dedifferentiation of otherwise refractory human cells,(Park, Lerou, Zhao, Huo, & Daley, 2008) the high TA clones in the present study failed to display faster reprogramming kinetics compared to low TA clones derived from the same donors. Moreover, various clones from different donors all gave rise to iPSC colonies at a comparable rate, suggesting that the fast reprogramming kinetics of USCs is independent of telomerase activity, as well as to the donor's genetic background. Nevertheless, high TA clones did manifest a 5- to 192-fold increase of reprogramming efficiency, determined by the number of alkaline phosphatase-positive colonies. Together, these observations suggest that the effect of high telomerase activity primarily functions to improve reprogramming efficiency, rather than accelerating the reprogramming kinetics. , The mechanism behind the fast reprogramming kinetics of USC-iPSCs remains unclear and large-scale transcriptome and epigenetic studies may be necessary to elucidate the underlying mechanism.
iPSC technology permits the preservation of the individual's unique genotype in reprogrammed pluripotent stem cells, which can be further differentiated into various specialized somatic cells. Our findings are in keeping with a recent report(Dick et al., 2013) where iPSCs derived from DMD patients harboring various dystrophin mutations maintained these unique mutations following cardiomyocyte differentiation. Due to the cardiomyopathy that develops in patients with DMD, we chose to differentiate USC-iPSCs into cardiomyocytes. Exon analysis of urine cell genomic DNA confirmed the exon 50 deletion in the dystrophin gene. Since this is an out-of-frame mutation, the cells should not be able to translate the dystrophin protein. Immunofluorescence for dystrophin revealed that DMD cardiomyocytes did not express the protein thus maintaining the disease-causing condition in the terminally differentiated cells suitable for study.
The majority of DMD patients develop cardiac abnormalities, with congestive heart failure (CHF) and sudden cardiac death directly account for 10-20% of the mortality in these young patients. Cardiac manifestations include rhythmic disturbance, heart structural alteration and hemodynamics abnormalities. Although the pathogenesis of dystrophin deficient cardiomyopathy is still not fully understood, evidence supports that disruption of the dystroglycan complex predisposes dystrophin-deficient cardiomyocytes to load-induced sarcolemma damage. The subsequent cascade of abnormal signaling events eventually lead to cell death, triggering inflammation further exacerbating tissue pathology. Several pathways have been suggested in the disease process, but none of them have been confirmed in human DMD cardiomyocytes. The lack of information can largely be explained by the risk associated with a patient heart biopsy, as well as the fact that cardiac cells derived from patients cannot be well maintained and expanded in culture. iPSCs from DMD patients can be a source of cardiac tissue in which to base experiments.
In the present study, we explored four cellular physiological domains using established assays (Fig 5) to assess potential phenotype abnormalities associated with dystrophin deficiency. While our preliminary data must be interpreted cautiously until additional biological replicates with isogenic controls are completed, the strong signal obtained from the “stress assay” (Fig 5D) supports the idea that dystrophin deficiency renders the cardiomyocyte abnormally vulnerable to mechanical stress. This idea is supported by a line of evidence in dystrophin-deficient skeletal muscle(Allen, Zhang, & Whitehead, 2010; Childers et al., 2002; Childers et al., 2001; Childers, Staley, Kornegay, & McDonald, 2005; Deconinck & Dan, 2007; Grounds, 2008) and to a lesser extent in cardiac muscle(De Pooter, Vandeweghe, Vonck, Loth, & Geraedts, 2012; Phillips & Quinlivan, 2008). While the concept of increased susceptibility to stress might be considered a foregone conclusion, to our knowledge, stress assays conducted in iPS-derived DMD cardiomyocytes have not yet been reported. Moreover, questions remain about the underlying pathophysiology of cardiomyopathy in DMD patients(Judge, Kass, Thompson, & Wagner, 2011). Evidence in mice(Crisp et al., 2011) suggests that cardiac dysfunction results from the heart developing in the face of progressively fatal respiratory muscle failure. In other words, heart failure in DMD patients develops secondary to respiratory failure and not as a direct consequence of dystrophin deficiency. However, this concept has been debated in the literature(Wasala, Bostick, Yue, & Duan, 2013) and until now, this hypothesis has never been directly tested. Invention of iPS technology allows us to conduct physiological tests in patient-derived heart cells developed completely outside of the body. Our initial readouts from iPS-derived cardiomyocytes support the idea that dystrophin deficiency directly results in an increased susceptibility to mechanical damage and liberation of cardiac-specific injury markers. These markers, CK-MB and cTnI are widely used by clinicians for the detection of myocardial damage in the face of a suspected heart attack(Adams et al., 1993; Hoogerwaard et al., 2001). DMD patients, in particular, are abnormally vulnerable to cardiac injury as demonstrated by elevated cTnI and CK-MB levels(Ramaciotti, Iannaccone, & Scott, 2003; Townsend et al., 2010). Furthermore, several observations have been made that these cardiac markers are powerful predictors of cardiac events in patients with heart failure and in the general population(Arenja et al., 2012). Cardiac injury markers are also used to assess the efficacy of cardioprotective treatments in general(Mangiacapra et al., 2013). Thus, the iPS-derived cardiomyocytes generated from patient urine samples provide a novel biological resource for personalized medicine. This new resource might further be exploited using one or more of the assays reported here to discover new compounds or test existing drugs that can protect dystrophin-deficient heart cells from stress-induced damage. Compounds identified in this way have a higher likelihood of working in the patient since they were tested in the patient's own cells.
1.5 Conclusions
We have shown the feasibility of rapid iPSC generation from human urine samples that can subsequently differentiate into beating cardiomyocytes. The cells found in human urine manifest unique features for iPSC generation, including ease of collection, intrinsic expression of reprogramming factors c-Myc and Klf4, and high telomerase activity. Our findings also support the idea that cardiomyocytes derived from the urine of a dystrophin-mutant DMD patient maintain the dystrophin-deficient phenotype and display unique features which might be further exploited in mechanistic studies or in drug discovery assays.
Supplementary Material
Supplemental figure: (A) USC surface antigens measured by flow cytometry; (B) Diagram of the dystrophin mutation found in USCs isolated from a patient with Duchenne muscular dystrophy (DMD). The deletion of exon 50 results in an out-of-frame mutation and eliminates expression of normal dystrophin protein. (C) H&E staining of representative teratomas arising after kidney capsule engraftment of urine-derived iPSC clones and hESC H9 control cells. Scale bar, 100 μm.
Supplemental online video 1: The video demonstrates the beating monolayer of cardiomyocytes derived from hESC H9.
Supplemental online video 2: The video demonstrates the beating monolayer of cardiomyocytes derived from DMD iPSC.
Highlights.
Human induced pluripotent stem cells (iPSCs) can be derived from urine samples, with fast reprogramming kinetics;
Telomerase activity promotes reprogramming efficiency instead of reprogramming kinetics;
Duchenne Muscular Dystrophy (DMD) disease specific iPSCs were also derived from urine. Cardiomyocytes generated from DMD iPSCs phenotypically differ from normal cardiomyocytes, thus proven to be a useful reagent that can be further exploited for mechanistic studies and drug discovery.
Acknowledgments
This work was supported by grants from the National Institutes of Health (K18HL102884-01) and the Muscular Dystrophy Association (MDA201127). MWL is supported by NIH grants (K08 AR059750 & L40 AR057721). JM is supported by a fellowship from the American Heart Association. HRB is supported by NIH grants (R01GM097372, R01GM083867 and 1P01GM081619). We acknowledge the Children's Research Institute Histology Core Facility at the Medical College of Wisconsin for the sectioning and staining of tumor specimens and Dr. Didier Trono for providing the lentiviral packaging vectors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, Izpisua Belmonte JC. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26(11):1276–1284. doi: 10.1038/nbt.1503. doi: nbt.1503 [pii] 10.1038/nbt.1503. [DOI] [PubMed] [Google Scholar]
- Adams JE, 3rd, Bodor GS, Davila-Roman VG, Delmez JA, Apple FS, Ladenson JH, Jaffe AS. Cardiac troponin I. A marker with high specificity for cardiac injury. Circulation. 1993;88(1):101–106. doi: 10.1161/01.cir.88.1.101. Comparative Study Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S. [DOI] [PubMed] [Google Scholar]
- Allen DG, Zhang BT, Whitehead NP. Stretch-induced membrane damage in muscle: comparison of wild-type and mdx mice. Advances in experimental medicine and biology. 2010;682:297–313. doi: 10.1007/978-1-4419-6366-6_17. Research Support, Non-U.S. Gov't Review. [DOI] [PubMed] [Google Scholar]
- Arenja N, Reichlin T, Drexler B, Oshima S, Denhaerynck K, Haaf P, Mueller C. Sensitive cardiac troponin in the diagnosis and risk stratification of acute heart failure. J Intern Med. 2012;271(6):598–607. doi: 10.1111/j.1365-2796.2011.02469.x. Comparative Study. [DOI] [PubMed] [Google Scholar]
- Bharadwaj S, Liu G, Shi Y, Markert C, Andersson KE, Atala A, Zhang Y. Characterization of urine-derived stem cells obtained from upper urinary tract for use in cell-based urological tissue engineering. Tissue Eng Part A. 2011;17(15-16):2123–2132. doi: 10.1089/ten.TEA.2010.0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Ratanasirintrawoot S, Park IH, Manos PD, Loh YH, Huo H, Schlaeger TM. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol. 2009;27(11):1033–1037. doi: 10.1038/nbt.1580. doi: nbt.1580 [pii] 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]
- Childers MK, Okamura CS, Bogan DJ, Bogan JR, Petroski GF, McDonald K, Kornegay JN. Eccentric contraction injury in dystrophic canine muscle. Arch Phys Med Rehabil. 2002;83(11):1572–1578. doi: 10.1053/apmr.2002.35109. doi: S000399930200254X [pii] [DOI] [PubMed] [Google Scholar]
- Childers MK, Okamura CS, Bogan DJ, Bogan JR, Sullivan MJ, Kornegay JN. Myofiber injury and regeneration in a canine homologue of Duchenne muscular dystrophy. Am J Phys Med Rehabil. 2001;80(3):175–181. doi: 10.1097/00002060-200103000-00004. [DOI] [PubMed] [Google Scholar]
- Childers MK, Staley JT, Kornegay JN, McDonald KS. Skinned single fibers from normal and dystrophin-deficient dogs incur comparable stretch-induced force deficits. Muscle Nerve. 2005;31(6):768–771. doi: 10.1002/mus.20298. [DOI] [PubMed] [Google Scholar]
- Crisp A, Yin H, Goyenvalle A, Betts C, Moulton HM, Seow Y, Wood MJ. Diaphragm rescue alone prevents heart dysfunction in dystrophic mice. Human molecular genetics. 2011;20(3):413–421. doi: 10.1093/hmg/ddq477. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- De Pooter J, Vandeweghe J, Vonck A, Loth P, Geraedts J. Elevated troponin T levels in a female carrier of Duchenne muscular dystrophy with normal coronary angiogram: a case report and review of the literature. Acta cardiologica. 2012;67(2):253–256. doi: 10.1080/ac.67.2.2154220. Case Reports Review. [DOI] [PubMed] [Google Scholar]
- Deconinck N, Dan B. Pathophysiology of duchenne muscular dystrophy: current hypotheses. Pediatric neurology. 2007;36(1):1–7. doi: 10.1016/j.pediatrneurol.2006.09.016. Review. [DOI] [PubMed] [Google Scholar]
- Dick E, Kalra S, Anderson D, George V, Ritso M, Laval SH, Denning C. Exon Skipping and Gene Transfer Restore Dystrophin Expression in Human Induced Pluripotent Stem Cells-Cardiomyocytes Harboring DMD Mutations. Stem Cells Dev. 2013 doi: 10.1089/scd.2013.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrenhaus A, Muller JI, Golka K, Jedrusik P, Schulze H, Follmann W. Cultures of exfoliated epithelial cells from different locations of the human urinary tract and the renal tubular system. Arch Toxicol. 2000;74(10):618–626. doi: 10.1007/s002040000173. [DOI] [PubMed] [Google Scholar]
- Grounds MD. Two-tiered hypotheses for Duchenne muscular dystrophy. Cellular and molecular life sciences : CMLS. 2008;65(11):1621–1625. doi: 10.1007/s00018-008-7574-8. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123(4):949–962. doi: 10.1083/jcb.123.4.949. Research Support, Non-U.S. Gov't Research Support, U.S. Gov't, P.H.S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogerwaard EM, Schouten Y, van der Kooi AJ, Gorgels JP, de Visser M, Sanders GT. Troponin T and troponin I in carriers of Duchenne and Becker muscular dystrophy with cardiac involvement. Clin Chem. 2001;47(5):962–963. [PubMed] [Google Scholar]
- Judge DP, Kass DA, Thompson WR, Wagner KR. Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. American journal of cardiovascular drugs : drugs, devices, and other interventions. 2011;11(5):287–294. doi: 10.2165/11594070-000000000-00000. Research Support, Non-U.S. Gov't Review. [DOI] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Murry CE. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25(9):1015–1024. doi: 10.1038/nbt1327. doi: nbt1327 [pii] 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- Liu H, Ye Z, Kim Y, Sharkis S, Jang YY. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology. 2010;51(5):1810–1819. doi: 10.1002/hep.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiacapra F, Peace AJ, Di Serafino L, Pyxaras SA, Bartunek J, Wyffels E, Barbato E. Intracoronary EnalaPrilat to Reduce MICROvascular Damage During Percutaneous Coronary Intervention (ProMicro) study. J Am Coll Cardiol. 2013;61(6):615–621. doi: 10.1016/j.jacc.2012.11.025. Randomized Controlled Trial. [DOI] [PubMed] [Google Scholar]
- Musch MW, Davis-Amaral EM, Vandenburgh HH, Goldstein L. Hypotonicity stimulates translocation of ICln in neonatal rat cardiac myocytes. Pflugers Arch. 1998;436(3):415–422. doi: 10.1007/s004240050651. [DOI] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451(7175):141–146. doi: 10.1038/nature06534. 10.1038/nature06534. http://www.nature.com/nature/journal/v451/n7175/suppinfo/nature06534_S1.html. [DOI] [PubMed] [Google Scholar]
- Park IH, Lerou PH, Zhao R, Huo H, Daley GQ. Generation of human-induced pluripotent stem cells. Nat Protoc. 2008;3(7):1180–1186. doi: 10.1038/nprot.2008.92. doi: nprot.2008.92 [pii] 10.1038/nprot.2008.92. [DOI] [PubMed] [Google Scholar]
- Phillips MF, Quinlivan R. Calcium antagonists for Duchenne muscular dystrophy. The Cochrane database of systematic reviews. 2008;(4):CD004571. doi: 10.1002/14651858.CD004571.pub2. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravdic D, Sedlic F, Mio Y, Vladic N, Bienengraeber M, Bosnjak ZJ. Anesthetic-induced preconditioning delays opening of mitochondrial permeability transition pore via protein Kinase C-epsilon-mediated pathway. Anesthesiology. 2009;111(2):267–274. doi: 10.1097/ALN.0b013e3181a91957. In Vitro Research Support, N.I.H., Extramural. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaciotti C, Iannaccone ST, Scott WA. Myocardial cell damage in Duchenne muscular dystrophy. Pediatr Cardiol. 2003;24(5):503–506. doi: 10.1007/s00246-002-0408-9. Case Reports Research Support, U.S. Gov't, P.H.S. [DOI] [PubMed] [Google Scholar]
- Ritner C, Bernstein HS. Fate Mapping of Human Embryonic Stem Cells by Teratoma Formation. J Vis Exp. 2010;(42):e2036. doi: 10.3791/2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Liu G, Shantaram B, Atala A, Zhang Y. Urine derived stem cells with high telomerase activity for cell based therapy in urology. Journal of Urology. 2012;187(4, Supplement):1. doi: 10.1016/j.juro.2012.02.821. abstract. [DOI] [Google Scholar]
- Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120(4):1140–1150. doi: 10.1172/JCI41329. doi: 41329 [pii] 10.1172/JCI41329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Storb R, Halbert CL, Banks GB, Butts TM, Finn EE, Tapscott SJ. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol Ther. 2012;20(8):1501–1507. doi: 10.1038/mt.2012.111. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warlich E, Kuehle J, Cantz T, Brugman MH, Maetzig T, Galla M, Schambach A. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol Ther. 2011;19(4):782–789. doi: 10.1038/mt.2010.314. doi: mt2010314 [pii] 10.1038/mt.2010.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasala NB, Bostick B, Yue Y, Duan D. Exclusive skeletal muscle correction does not modulate dystrophic heart disease in the aged mdx model of Duchenne cardiomyopathy. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McNeill E, Tian H, Soker S, Andersson KE, Yoo JJ, Atala A. Urine derived cells are a potential source for urological tissue reconstruction. J Urol. 2008;180(5):2226–2233. doi: 10.1016/j.juro.2008.07.023. doi: S0022-5347(08)01820-X [pii] 10.1016/j.juro.2008.07.023. [DOI] [PubMed] [Google Scholar]
- Zhou T, Benda C, Dunzinger S, Huang Y, Ho JC, Yang J, Esteban MA. Generation of human induced pluripotent stem cells from urine samples. Nat Protoc. 2012;7(12):2080–2089. doi: 10.1038/nprot.2012.115. [DOI] [PubMed] [Google Scholar]
- Zhou T, Benda C, Duzinger S, Huang Y, Li X, Li Y, Esteban MA. Generation of induced pluripotent stem cells from urine. J Am Soc Nephrol. 2011;22(7):1221–1228. doi: 10.1681/ASN.2011010106. doi: ASN.2011010106 [pii] 10.1681/ASN.2011010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure: (A) USC surface antigens measured by flow cytometry; (B) Diagram of the dystrophin mutation found in USCs isolated from a patient with Duchenne muscular dystrophy (DMD). The deletion of exon 50 results in an out-of-frame mutation and eliminates expression of normal dystrophin protein. (C) H&E staining of representative teratomas arising after kidney capsule engraftment of urine-derived iPSC clones and hESC H9 control cells. Scale bar, 100 μm.
Supplemental online video 1: The video demonstrates the beating monolayer of cardiomyocytes derived from hESC H9.
Supplemental online video 2: The video demonstrates the beating monolayer of cardiomyocytes derived from DMD iPSC.