

Abstract
The World Trade Center (WTC) collapse on September 11, 2001 released copious amounts of particulate matter into the atmosphere of New York City. Follow-up studies on persons exposed to the dusts have revealed a severely increased rate for asthma and other respiratory illnesses. There have only been a few studies that have sought to discern the possible mechanisms underlying these untoward pathologies. In one study, an increased cytokine release was detected in cells exposed to WTC fine dusts (PM2.5 fraction or WTC2.5). However, the mechanism(s) for these increases has yet to be fully defined. Because activation of the MAPK signaling pathways is known to cause cytokine induction, the current study was undertaken to analyze the possible involvement of these pathways in any increased cytokine formation by lung epithelial cells (as BEAS-2B cells) exposed to WTC2.5. Our results showed that exposure to WTC2.5 for 5 hr increased IL-6 mRNA expression in BEAS-2B cells, as well as its protein levels in the culture media, in a dose-dependent manner. Besides IL-6, Cytokine Multiplex analyses revealed that formation of IL-8 and -10 was also elevated by the exposure. Both ERK and p38, but not JNK, signaling pathways were found to be activated in cells exposed to WTC2.5. Inactivation of ERK signaling pathways by PD98059 effectively blocked IL-6, -8, and -10 induction by WTC2.5; the p38 kinase inhibitor SB203580 significantly decreased induction of IL-8 and -10. Together, our data demonstrated activation of MAPK signaling pathway(s) likely played an important role in the WTC2.5-induced formation of several inflammatory (and, subsequently, anti-inflammatory) cytokines. The results are important in that they help to define one mechanism via which the WTC dusts may have acted to cause the documented increases in asthma and other inflammation-associated respiratory dysfunctions in the individuals exposed to the dusts released from the WTC collapse.
Keywords: PM2.5, interleukin (IL)-6, IL-8, IL-10, ERK, p38
Introduction
On September 11, 2001, the World Trade Center (WTC) in the metropolis of New York was attacked and subsequently collapsed, discharging copious amounts of smoke and dust into the air above and around Ground Zero, especially to the south and east (Lioy et al., 2002; McGee et al., 2003). The dust eventually settled and accumulated to depths of several inches in the streets, on roofs of buildings and cars, as well as permeated into the rooms of surrounding residential and business structures. This exposed a great number of people to the potentially toxic and harmful dust, especially police officers, firefighters, and civilians who were at or near Ground Zero at the times of the attacks, as well as rescue workers, volunteers, and emergency response crews who later travelled to the scene.
Studies have been performed to evaluate effects of the WTC dusts on these individuals exposed during/after the building collapses. Several of these studies reported increases in the incidence of respiratory problems, including a persistent cough (WTC Cough; Kelly et al., 2007; Prezant, 2008), upper and lower respiratory symptoms (CDC, 2004; Salzman et al., 2004; Herbert et al., 2007), and shortness of breath/wheezing (Buyantseva et al., 2007) in previously-healthy individuals (specifically, First Responders). Another adverse health effect that paralleled lower airway disorders was an onset of asthma in ~45% of New York City firefighters (FDNY) and other rescue workers within the first year after the collapse (CDC, 2004). In a study conducted three years after the collapse, 926 of 25,748 total previously-healthy workers (3.6%) were now found to have been diagnosed with new-onset asthma (Wheeler et al., 2007), an incidence 12-times above the rate (0.3%) in the general population (Reed, 2006). Prezant (2008), in a recent summary of the clinical/epidemiological findings, stated “To date, the main respiratory health consequence from the collapse of the World Trade Center (WTC) on September 11, 2001 has been the ‘WTC Cough Syndrome’ (chronic rhinosinusitis, asthma, and/or bronchitis, often complicated by gastro-esophageal reflux dysfunction); syndrome incidence and severity have been linked to WTC dust exposure intensity”.
A few in vitro and animal model studies have been performed during the eight years post-collapse to understand mechanisms by which WTC dusts could induce adverse respiratory outcomes in the exposed responders and others. One key study showed that human primary (resident) alveolar macrophages (AM) and Type II epithelial cells, when exposed to WTC dust particles, demonstrated time- and dose-related increases in the formation/release of pro-inflammatory cytokines/chemokines (e.g., interleukin [IL]-6, IL-8, tumor necrosis factor [TNF]-α) (Payne et al., 2004). These findings revealed that exposure of these key lung cell populations to WTC dusts could cause the release of several factors that contribute to inflammation and airway remodeling processes. The results of that investigation are also important in the context of the observations about asthma in responders/others. These cytokines/chemokines both promote inflammation of the airways and attract other cytokine-releasing cells, thereby giving rise to states of chronic inflammation that is considered as a major cause of the onset of asthma (Chung and Barnes, 1999). Lastly, the in vitro findings also help to explain the results from the earliest animal model studies performed with the WTC dusts. In those studies, Gavett et al. (2003) found that a single oropharyngeal aspiration of fine WTC dust (i.e., WTC2.5) induced significant neutrophilic inflammation (without a concurrent macrophage influx) in the lungs of the exposed mice and a significant increase in airway hyper-responsiveness to methacholine aerosol.
While these studies have yielded key information about extracellular outcomes from exposure of lung populations to the WTC dusts, details about intracellular mechanisms of these effects remain absent. There have been reports in the literature describing connections between the activation of mitogen-activated protein kinase (MAPK) pathways and the production of cytokines (Chi et al., 2006). MAPK signalings that include ERK, JNK and p38 protein kinase pathways are stimulated by many outside environmental stresses, including airborne particulate matter (PM) (Johnson and Lapadat, 2002). These pathways up-regulate transcription factors that, in turn, promote the production of pro-inflammatory cytokines (Scherle and Jones, 1998; Johnson and Lapadat, 2002). Therefore, the potential effects of WTC dusts on the activation of MAPK pathways and the release of cytokines/chemokines by human bronchial epithelial BEAS-2B cells were investigated here. This study may help better understand the mechanism(s) that might underlay the increase in asthma attack in the responders/other individuals who were exposed to WTC dusts in the days following the collapse.
Methods and Materials
World Trade Center (WTC) dusts and sources
The WTC2.5 dust samples (fine, sized sub-fractions of the parent WTC dusts) used in this experiment were collected on 9/12/01 and 9/13/01 at the intersection of Liberty and Church Streets (the southeastern corner of Ground Zero). The parent WTC dust samples were first sieved at 53 μm; particles < 53 μm in diameter were then further separated into fractions of 10–53, 2.5–10, and ≤−2.5 μm aerodynamic diameters using standard protocols (see Gavett et al. [2002] for details). For the studies here, ≤2.5 μm particles were suspended in distilled and deionized water to stock concentrations of 700 μg WTC2.5/ml and stored at −80°C until use.
Chemicals/Reagents
Trizol reagent, deoxynucleoside triphosphates (dNTPs), RNase-free H2O, and UltraPure agarose powder were obtained from Invitrogen (Carlsbad, CA). Primers for PCR were designed using Primer3 program (freely available at workbench.sdsc.edu) and synthesized by Invitrogen. Fetal bovine serum (FBS) was purchased from Atlas Biologicals (Fort Collins, CO). Dulbecco’s Modified Eagle’s Medium (DMEM), phosphate-buffered saline (PBS), trypsin-EDTA, and purified H2O were obtained from Gibco, Invitrogen (Carlsbad, CA). The DC protein assay, non-fat dry milk powder, Immuno-Blot PVDF membranes, Tris-buffered saline (TBS), as well as pre-cast gels, were obtained from Bio-Rad Laboratories (Hercules, CA). Stripping buffer and ECL reagents were obtained from Thermo Scientific (Rockford, IL). Chloroform, 2-propanol, ethanol, dimethyl sulfoxide (DMSO), Tween-20, and BSA (bovine serum albumin) powder were purchased from Sigma (St. Louis, MO). Anti-mouse and anti-rabbit secondary antibodies were obtained from Chemicon (Temecula, CA). Taq polymerase was purchased from Roche (Branford, CT). MAPK inhibitors (SB203580 for p38, and PD98059 for ERK) were obtained from Calbiochem (Gibbstown, NJ). Titanium oxide particles (TiO2, fine form [≤2.5 μm]) were purchased from Alfa Aesar (Ward Hill, MA).
Cells and exposures
BEAS-2B cells (a human bronchial epithelial cell line; ATCC, Manassas, VA) were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C and 5% CO2 until a 90% confluent state was reached. One day before exposure, cells were harvested and then seeded (at 6 × 105 in 2.5 ml per well) in 6-well plates. Prior to the exposure, the medium was removed and the cells washed twice with PBS. The PBS was then removed from the dish and particle solution (as a 0.5 ml total volume) applied evenly over the cells. Prior to exposure, the particle stock solutions were sonicated for ~20 min, then diluted in DMEM (without addition of 10% FBS) to the desired final concentrations before being provided to the cells. The cells were then incubated at 37°C (under 5% CO2) for the defined amount of time before the culture medium or cells were collected. In some experiments, cells were treated with fine TiO2 as a control (comparison of effect) particle; this agent was chosen due to its limited ability to cause a temporal induction of IL-6 release by BEAS-2B cells, i.e., an initial spike within 2–3 hr of exposure that dissipates to control value levels over the period from 8–24 hr post-exposure (see Park et al., 2008; Veranth et al., 2008; Val et al., 2009). Pilot studies revealed that under the experimental conditions used herein, the range of doses of WTC2.5 (i.e., 25 – 200 μg/ml) did not cause overt changes in cell survival (i.e., there were no significant observable changes in cell adherence over the 5 hr period of exposure [data not shown]).
Western blotting
Western blotting was used to determine the extent of pathway activation, i.e., by measuring the amounts of phosphorylated kinases (p-p38, p-ERK, and p-JNK in the cells after treatments with vehicle or the test particles solution). Following exposures, cells were lysed using a boiling lysis buffer (1% SDS, 1.0 mM sodium orthovanadate, 10 mM Tris [pH 7.4]). The lysate was then collected, transferred to an Eppendorf tube, heated at 95°C for 5 min, and then placed at −80°C. At the time of the protein assays, each lysate sample was sonicated (Sonifier, Branson Ultrasonics, Danbury, CT) with 10 strokes (1 second per stroke) in order to decrease sample viscosity. The material was then centrifuged (14,000 × g, 15 min, 4°C), the resultant supernatant collected, and protein concentration in the lysate determined using the Biorad DC protein assay.
For each treatment condition, the lysate containing 50 μg protein was then loaded into one well in a 7.5% SDS-PAGE pre-cast gel and electrophoresed at 100 V for 90 min. Proteins in the gel were then transferred to a PVDF membrane at 25 V overnight. After the transfer and blocking, primary antibodies specific to each kinase (anti-p-p38 at 1:1000 dilution and both anti-p-ERK and anti-p-JNK at 1:2000 dilution) were then placed with the membrane in a solution of 5% BSA (for p-p38 and p-ERK) or 5% non-fat dry milk (for p-JNK) in TBST (TBS buffer/0.1% Tween-20) and incubated at 4°C overnight on a rocking platform. The membrane was then washed three times with TBST (5 min/wash), and incubated with secondary anti-rabbit (for p-p38 or p-ERK) or anti-mouse (for p-JNK) antibody (each at 1:5000 dilution in 5% milk-TBST) at room temperature for 1 hr on the rocking platform. The membrane was then washed three times with TBST (5 min/wash). ECL reagents were then added and the chemical fluorescence on the membrane was detected with X-ray film. Next, to measure the expression levels of total p38, ERK, and JNK, the membrane was then processed with stripping buffer (room temperature, with gentle rocking) for 10 min. This buffer was then decanted and the membrane washed with deionized water and blocked with 5% milk TBST. New primary antibodies for p38, ERK, and JNK were then added (dilutions of 1:1000, 1:1000, and 1:5000, respectively), and the immunoblotting carried out again as above.
Activation analyses using specific pathway inhibitors
Based on findings in preliminary Western blot studies (performed in the same manner as noted above), exposures in the presence of chemical inhibitors of the ERK and p38 pathways were performed to confirm any potential role of each of these in any WTC2.5 dust-induced effects on the BEAS-2B cells. In these experiments, the cells were exposed to 30 μM PD98059 or 1 μM SB203580 (each value was the final concentration in culture) 1 hr prior to exposure to the WTC dust. Because the ERK (PD98059) and p38 (SB203580) pathway inhibitors were dissolved in dimethyl sulfoxide (DMSO), this vehicle was added in another set of samples as a control in this experiment. Immediately before the actual exposure, the inhibitor-bearing medium was removed from each dish. The WTC particles were then added (along with fresh medium ± inhibitors [at their original concentrations]) and the cells incubated at 37°C (under 5% CO2) for defined amounts of time.
RNA extraction
To ascertain if the WTC2.5 affected the BEAS-2B cell production of cytokines at the transcription level, post-exposure measures of RNA levels were performed. Based upon pilot studies, IL-6 was selected as the model cytokine for analyses. Here, after cell exposures were performed as outlined above, the culture medium in the well was removed and Trizol reagent added to lyse the cells. The lysed cell suspension was mixed with chloroform (200 μl per 1 ml Trizol), vortexed, incubated at room temperature for 2–3 min, and then centrifuged (12,000 × g, 15 min, 4°C). The resulting upper-most layer in the sample (consisting of RNA) was removed and transferred into a new tube. 2-Propanol was added to this extract to precipitate the RNA (over a 10-min period at room temperature). The samples were then centrifuged (12,000 × g, 10 min, 4°C), after which the supernatant was poured off, leaving only the RNA pellet in the tube. This pellet was washed with 80% ethanol (prepared with DEPC-treated H2O) and then air-dried. The pellet was then solubilized in RNase-free H2O, heated at 50°C for 10 min, and the concentration of RNA present determined by measuring absorbance at 260 nm in a NanoDrop 1000 spectrometer (Thermo Scientific, Rockford, IL).
Reverse transcription and polymerase chain reactions (PCR)
cDNA was synthesized using the SuperScript III first-strand synthesis supermix for qRT-PCR (Invitrogen). After the RNA quantification, RNA samples were diluted to a concentration of 100 ng/μl. In each case, sample (8 μl containing 800 ng total RNA) was mixed with 10 μl of 2X reaction buffer and 2 μl of RT enzyme mix provided in the kit. All the samples were then placed in a thermal cycler (MJ Research Inc., Waltham, MA) and subjected to the following cycles: 25°C for 10 min, 50°C for 30 min, and then 85°C for 5 min. All samples were then placed on ice, mixed with 1 μl RNase H, and then incubated at 37°C for 20 min.
An aliquot (1 μl) of each cDNA sample was mixed with 5 μl of 10X PCR buffer (Roche Applied Science), 2 μl 50 μM primers (1 μl/primer), 0.8 μl 25 mM dNTPs, 41.5 μl purified H2O, and 0.7 μl Taq polymerase (New England Biolabs) to make a total solution of 50 μl for each sample. Sequences of the IL-6 primers were: sense, 5′-GAACTCCTTCTCCACAAGCG-3′; anti-sense, 5′-TTTTCTGCCAGTGCCTCTTT-3′. This PCR mixture was then placed into the thermal cycler and subjected to the following cycles: 95°C for 2 min, 28 cycles of 94°C (30 sec), 55°C (30 sec), and then 72°C (30 sec). The samples were then incubated at 72°C for 5 min before being cooled to 4°C for storage. A total of 15 μl of each sample was then mixed with DNA loading dye and loaded onto a 1.5% agarose gel. The samples were then electrophoresed for 45 min at 100 V, and the DNA in the gel was then visualized using an ultraviolet lamp.
Cytokine measurements by ELISA and Cytokine Multiplex analysis
Culture media from the exposed cells were collected for measurement of IL-6. ELISA kits were purchased from BD Biosciences (San Jose, CA) and all protocols used were as outlined by the manufacturer. Prior to being placed in the ELISA wells, each recovered media sample was centrifuged at 250 rpm for 5 min to remove any cells present; the isolated supernatant was then centrifuged again at 8000 rpm for 5 min to remove any WTC particles that could potentially interfere with cytokine measurements.
Cytokines in the isolated culture samples were also quantified using MesoScale Discovery (Gaithersburg, MD) 96-well multi-array and -spot assays. With this particular assay kit, interferon (IFN)-γ, TNFα, as well as IL-1β, -2, -4, -5, -8, -10, -12 (as p70 form), and -13 were simultaneously analyzed. This methodology employs a sandwich immunoassay technique in which capture antibodies are spotted on the bottom of wells. Standards and samples (as 25 μl aliquots) were incubated in the wells (for 120 min at 25°C), with each cytokine binding to its corresponding capture antibody spot. After washing the wells with Tween solution to remove unbound materials, detection antibody labeled with a SULFO-TAG reagent was added and the well contents then exposed to Read Buffer T; this latter reagent is used to produce an electrochemiluminescent signal that was then measured on a SECTOR Imager (MesoScale Discovery).
Statistical analysis
The results are presented as the mean (± SD) for all variables that were examined. The two-tailed Student t-test was used to determine the significance of differences between two groups, and comparisons among multiple groups were done using ANOVA. All data were tested to assure assumptions of normality and homogeneity of variance were met, and transformations applied as needed. Data were also screened for outliers using Dixon and Grubb’s analyses (Taylor, 1990). The difference was considered significant at p < 0.05.
Results
WTC2.5 induces IL-6 formation in BEAS-2B cells
To test whether WTC2.5 could induce IL-6 expression in BEAS-2B (a human bronchial epithelial cell line) cells, the cells were exposed to different concentrations of WTC2.5 for 5 hr and a reverse transcription polymerase chain reaction was conducted to measure IL-6 mRNA levels. As shown in Figure 1A, significant increases in IL-6 gene expression was observed with the 100 and 200 μg WTC2.5/ml (≈5 and 10 μg/cm2, respectively) doses when compared to the amount of expression seen with the untreated control cells. Next, to measure IL-6 release by the treated cells, ELISA was used to analyze IL-6 levels in the culture media of the exposed cells using the same time intervals and dosages. The ELISA results were found to parallel those in RT-PCR assays, i.e., both 100 and 200 μg WTC2.5/ml doses induced an ~2-fold increase in IL-6 release by treated cells as compared to by control cells (Figure 1B and 1C). Fine TiO2 particles (tested at 100 μg/ml to provide for comparison of potential WTC2.5 effects against weak inducer of IL-6 release [this outcome dependent on dose and length of cell incubation with particles] in BEAS-2B cells) did not produce a statistically significant release of IL-6 from the cells (Figure 1C).
Figure 1.


Effects of WTC2.5 on IL-6 formation by BEAS-2B cells. (A) BEAS-2B cells were treated with varying doses of WTC2.5 (in 0.5 ml total volumes) for 5 hr, and then a reverse transcription polymerase chain reaction was used to assess IL-6 gene expression in the cells. Here, 100 and 200 μg WTC2.5/ml are equivalent to about 5 and 10 μg WTC2.5/cm2, respectively. Expression of β-actin was analyzed in parallel to ensure equal amounts of cDNA for each sample. (B) Corresponding IL-6 release in the BEAS-2B culture medium following the WTC2.5 treatments as in (A). BEAS-2B cells were exposed to WTC2.5 as in (A), and the IL-6 level in culture media was measured using ELISA. (C) BEAS-2B cells were treated by 100 μg/ml WTC2.5 or TiO2 for 5 hr (N = 3), and the levels of IL-6 in the cell media were measured by ELISA. *Indicates a statistically significant change (p < 0.05) when compared to value for untreated samples. Values shown are the mean relative level vs. that in unexposed control cultures (± SD) from four replicate cultures.
Expression of other cytokines in BEAS-2B cells exposed to WTC2.5
A multiplex array assay was used to measure the levels of 10 cytokines in the media of BEAS-2B cells after exposure to different concentrations of WTC2.5 for 5 hr. It was found that WTC2.5 induced significant increases of IL-8 and IL-10 levels in the media (Figure 2), while the concentrations of IL-1β, IL-2, IL-4, IL-5, IL-12, IL-13, interferon (IFN)-γ, and tumor necrosis factor (TNF)-α were not altered significantly (Table 1). In the case of IL-4, though there were near-uniform increases in a dose-independent manner after the 5-hr exposure (i.e., relative values ranging from 2.0–2.7), these increases did not reach the defined significance threshold. In contrast, fine TiO2 particles were found to be more potent in inducing the expression/secretion of several of the assayed cytokines, e.g., IL-1β, IL-4, IL-5 (Table 1). Thus, under the experimental conditions used here, WTC2.5 clearly induced formation of select cytokines in BEAS-2B cells.
Figure 2.
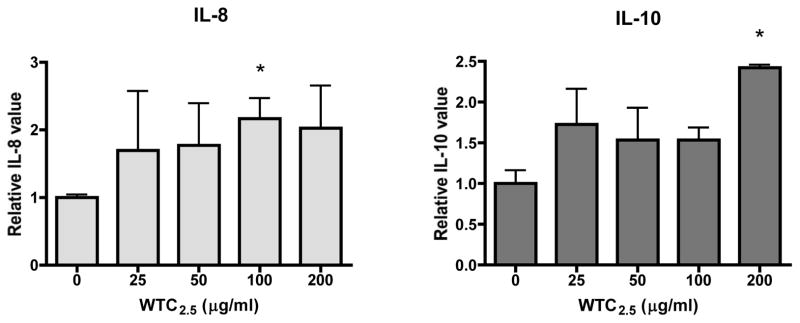
Impact of WTC2.5 on induction of BEAS-2B IL-8 and IL-10 formation. BEAS-2B cells were treated with varying doses of WTC2.5 (in 0.5 ml total volumes) for 5 hr, and the levels of several cytokines in the culture media were measured using a Multiplex array assay. Results of the changes in expression levels of IL-8 and IL-10 are shown. *Indicates statistically significant change (p < 0.05) when compared to value for untreated samples. Value for the untreated control is set to 1.00 for purposes of expressing relative change in cytokine level vs. that in unexposed control cultures. Values are unit-less as they are calculated as relative change in levels (expressed here as mean relative cytokine level vs. that in unexposed control cultures [±SD]).
Table 1.
Levels of various cytokines in BEAS-2B cell media after WTC2.5 exposure for 5 hr.
| Cytokine | 0 | a25 | 50 | 100 | 200 | 100 b TiO2 |
|---|---|---|---|---|---|---|
| IL-1β | c 1.00 | d 3.10 ± 2.70 | 1.00 ± 0.65 | 1.42 ± 0.82 | 1.49 ± 0.27 | 4.90 ± 1.55 |
| IL-2 | 1.00 | 1.87 ± 1.08 | 1.67 ± 0.37 | 1.60 ± 0.99 | 1.62 ± 0.31 | 1.67 ± 0.84 |
| IL-4 | 1.00 | 2.08 ± 3.61 | 2.01 ± 1.81 | 2.05 ± 2.88 | 2.71 ± 2.78 | 12.2 ± 13.6 |
| IL-5 | 1.00 | 1.20 ± 1.02 | 0.92 ± 0.05 | 0.79 ± 0.43 | 0.83 ± 0.40 | 2.52 ± 0.62 |
| IL-8 | 1.00 | 1.73 ± 0.44 | 1.54 ± 0.39 | 1.54 ± 0.15 | 2.42 ± 0.03 | 2.33 ± 0.39 |
| IL-10 | 1.00 | 1.69 ± 0.88 | 1.77 ± 0.62 | 2.16 ± 0.31 | 2.02 ± 0.63 | 2.51 ± 0.44 |
| IL-12 | 1.00 | 0.00 ± 0.00 | 0.95 ± 0.86 | 1.89 ± 0.00 | 3.58 ± 0.84 | 7.58 ± 5.06 |
| IL-13 | 1.00 | 1.48 ± 0.75 | 0.83 ± 0.52 | 1.13 ± 0.48 | 1.29 ± 0.43 | 1.54 ± 0.75 |
| TNF-α | 1.00 | 0.88 ± 0.59 | 0.64 ± 0.39 | 1.55 ± 1.03 | 1.88 ± 1.07 | 2.30 ± 1.25 |
| IFN-γ | 1.00 | 0.29 ± 0.44 | 0.12 ± 0.18 | 0.36 ± 0.33 | 0.51 ± 0.18 | 1.22 ± 0.96 |
Level of WTC2.5 (as μg/ml) used in the 5-hr exposure.
μg/ml used in the 5-hr exposure.
Value set to 1.00 for purposes of expressing relative change in cytokine level vs. that in unexposed control cultures.
Value is unit-less as it is calculated as relative change in levels (expressed here as mean relative cytokine level vs. that in unexposed control cultures [±SD]).
Activation of p38 and ERK kinases in BEAS-2B cells following exposure to WTC2.5
To study whether WTC2.5 activates MAPK signaling pathway, cells were exposed to 100 μg WTC2.5/ml and the phosphorylated forms of different MAPK kinases were measured. The Western blot results indicated that WTC2.5 activated both p38 and ERK kinase pathway in these cells. Specifically, increases in p-p38 levels were noted at 30 and 120 min after exposure, while increases in levels of p-ERK were seen at 5 and 10 min after exposure and were still evident (albeit to a far lesser degree) up to 60 min after initiation of the exposure (Figure 3). Though the p-JNK blot exhibits darkened bands in the 5- and 10-min samples, darker bands were also noted in the portion of the blot used to assess background levels of parent JNK present and the ratios of phosphorylated:total JNK. In follow-on experiments, it was seen again that WTC2.5 exposure clearly led to quantifiable increases in p38 phosphorylation but not that of JNK (Figure 3B). The results of these studies show that WTC2.5 activates both ERK and p38 kinases, but not JNK kinase, in BEAS-2B cells.
Figure 3.


WTC2.5 effects on activation of ERK and p38 kinase pathways. (A) BEAS-2B cells were exposed to 100 μg WTC2.5/ml for varying time intervals, and the levels of phosphorylated form and total MAPK kinases (ERK, p38, and JNK) were assessed from Western blots using specific antibodies. The same membrane was blotted with α-tubulin antibody to verify protein loading in each lane. Because the same amounts of protein lysates from the same samples were used for the ERK, p38 and JNK blots, only one representative α-tubulin blot is shown here. (B) BEAS-2B cells were treated with 100 μg WTC2.5/ml; thereafter, JNK and p38 phosphorylation levels were measured at 10 and 60 min after start of exposure (as these represented the timepoints above where “maximal” phosphorylation appeared to have occurred). Western blot band intensities were quantified at each timepoint using an ImageJ program. Results (expressed as mean ± SD; n=2) shown represent relative intensity values from treated and untreated cells.
Effects of pathway inhibitors on IL-6 expression
To examine if p38 and ERK activation plays any role in IL-6 induction by WTC2.5, specific inhibitors were used to block p38 or ERK pathways in the BEAS-2B cells prior to (and during) 5-hr exposures to WTC2.5. When no inhibitor was added, WTC2.5 (DMSO-WTC2.5) led to ≈5-fold increase of IL-6 levels compared to those seen with the control cultures that received no particles (DMSO) (Figure 4). When the ERK pathway inhibitor PD98059 was present with the particles (PD-WTC2.5), the level of IL-6 was drastically lowered and the IL-6 amount was less than half that of the DMSO-WTC2.5 group. Likewise, when the p38 pathway inhibitor SB203580 was present with the particles (SB-WTC2.5), a decrease in IL-6 levels was observed when compared to the value for the DMSO-WTC2.5 group; however, in this case, the decrease was not found to be statistically significant. These results demonstrated that activation of ERK pathway plays a major role in the WTC2.5-induced expression of IL-6 in BEAS-2B cells.
Figure 4.

Confirmation of ERK/p38 pathway involvement in WTC2.5-induced IL-6 formation. BEAS-2B cells were pre-treated with PD98059 (ERK inhibitor) or SB203580 (p38 kinase inhibitor) for 1 hr and then exposed to WTC2.5 (100 μg/ml) along with the MAPK pathway inhibitors for 5 hr (in 0.5 ml total volumes). Results are expressed as mean ± SD. *Indicates a statistically significant change (p < 0.05) when compared to value for DMSO-treated samples; **indicates significant change (p < 0.05) when compared to value for DMSO-WTC2.5 samples. Values shown are mean relative cytokine level vs. that in DMSO control cultures (± SD) from three replicate cultures.
Effects of pathway inhibitors on other cytokines expression
The roles of p38 and ERK kinases in IL-8 and IL-10 induction by WTC2.5 were also examined in cells incubated with 100 μg WTC2.5/ml for 5 hr along with the above p38 or ERK pathway inhibitors. Both kinase inhibitors effectively blocked induction of IL-8 and IL-10 by the WTC2.5 (Figure 5). These results illustrate that both p38 and ERK signaling pathways appear to have important roles in the induction of IL-8 and -10 formation in BEAS-2B cells by WTC2.5.
Figure 5.

Determination of whether ERK and p38 kinases are involved in WTC2.5-induced IL-8 and IL-10 formation. BEAS-2B cells were treated as in Figure 4, and the levels of IL-8 and IL-10 in the media were measured using Multiplex array analyses. *Indicates a statistically significant change (p < 0.05) when compared to value for DMSO-treated samples; **indicates statistically significant change (p < 0.05) when compared to value for DMSO-WTC2.5 samples. Values shown are mean relative cytokine level vs. that in DMSO control cultures (± SD) from three replicate cultures.
Discussion
The 9/11 attacks on the World Trade Center (WTC) complex resulted in widespread acute and chromic health problems among many of the rescue workers (i.e., First Responders), who were on-site (at Ground Zero) and exposed to significant levels of WTC dusts over the first few days-weeks after the buildings’ collapse. Among the documented health issues in these workers, adverse respiratory symptoms have been among the most commonly reported (recently reviewed in Welden et al., 2010, de la Hoz et al., 2009, and Webber et al., 2009). Among these have included a consistent pattern of increased incidences of: upper and lower airway diseases (UADs and LADs characterized [in the absence of infectious disease] by persistent rhinitis, dyspnea, cough, and wheezing) (de la Hoz et al., 2008b, 2009); new-onset asthma (Wheeler et al., 2007; Brackbill et al., 2009), “sarcoid-like” granulomatous disease (Izbicki et al., 2007); reactive airways dysfunction syndrome (RADS; associated with measurable increases in airway/bronchial hyperreactivity) (Banauch et al., 2005; Welden et al., 2010); and, gastro-esophageal reflux disease (GERD) that appears to be related to lung function abnormalities suggestive of air trapping/diagnoses of lower respiratory disease (de la Hoz et al., 2008a; Webber et al., 2009). The mechanistic base for how WTC dusts may have caused/contributed to each of these pathologies is likely to be severe inflammation in respiratory tracts.
Previous studies have shown that exposure to WTC2.5 collected on-site over the period from 9/12-13/01 caused significant pulmonary inflammation and hyper-responsiveness in mice (Gavett et al., 2003). In vitro studies with these dusts documented that their presence resulted in a release of inflammatory cytokines by primary lung cells (i.e., human alveolar macrophages (AM) and Type II epithelial cells) (Payne et al., 2004). In the study being reported here, the role of MAPK pathway in the induction of inflammatory cytokines by WTC2.5 - events that could significantly contribute to the outcomes described in the signature studies by Gavett and colleagues in 2003 - was examined. In general, the results of our studies revealed that WTC2.5 activated p38 and ERK kinases in BEAS-2B cells and inactivation of these signaling pathways decreased/blocked the production of IL-6, IL-8 and IL-10, indicating that activation of p38 and ERK pathways may both have had important roles in inflammatory responses induced in lungs of hosts who were exposed to WTC dusts.
The doses of WTC2.5 (0 – 200 μg/ml [or 10 μg/cm2]) used in this study were significantly lower than those used in the research by Payne et al. (2004), i.e., 500–2000 μg/ml (or 25–100 μg/cm2). However, these doses were comparable to the levels of exposure that were expected to have been experienced by individuals in a typical urban area or in parts of New York City (NYC) that are relatively “distant” (i.e., Upper Manhattan, the Bronx) from Ground Zero. To date, no previous study has reported that these WTC dusts, when used at such low doses (100 and 200 μg/ml), could cause IL-6 induction. Another difference between this study and Payne’s study is that 5 hr, instead of 24 hr, time interval was used. The current results show that fine WTC dusts could potentially induce the production and release of select inflammatory cytokines in the airways. These outcomes may help to explain why the incidence of asthma and other inflammation-associated diseases were increased in both 9/11 firefighters and rescuers (i.e., First Responders), as well as among Metropolitan area residents 20–30 miles away from Ground Zero. Even so, the events of September 11 exposed the First Responders and residents living near Ground Zero to a much higher concentration of these hazardous particles (Yiin et al., 2004; Geyh et al., 2005). Such a high level of exposure to the WTC dusts may also have caused direct damage to the respiratory tract, based upon our observation that a significant amount of cell death occurred when the BEAS-2B cells were exposed to 1000 μg/ml (50 μg/cm2) WTC PM10 (for 24 hr, data not shown).
There are several potential means by which the fine WTC dusts might have been able to influence the formation/release of pro-inflammatory cytokines (and, subsequently, the anti-inflammatory forms, e.g. IL-10) from these treated cells. In general, MAPK signaling pathway plays an important role in regulating the response of a cell to extracellular stimuli. It has been reported that MAPK pathway activation is essential in regulation of synthesis of IL-6 (Beyaert et al., 1996), IL-8 (Kim and Shi, 2009), and IL-10 (Pilette et al., 2010) upon exposure to extracellular stimuli in many different cell types. With particular regard to urban dusts acting as potential stimuli, a study by Li et al. (2005) that used urban particles collected from St. Louis (i.e., NIST SRM1648, PM10 urban particulate standard) demonstrated a link between pulmonary arterial endothelial cell exposure to particles and activation of ERK and p38 MAPK pathways. A later study using SRM1648 PM10 and a fine PM standard derived from air in Baltimore (i.e., NIST IRM PM2.5) revealed that while each particle differentially affected formation of IL-6 and IL-8 in mouse peritoneal macrophages (with the PM10 having greater inductive effect; IL-10 not analyzed) (Shoenfelt et al., 2009), both apparently required the presence of MyDD88 – an adaptor protein used by toll-like receptors (TLR)-2 and -4 and whose activation results in that of ERK, p38, and JNK pathways in several cell types (Han, 2006). Our current study (using specific pathway inhibitor co-treatments of the cultured cells) also found that the WTC dusts activated the p38 and ERK pathways - but unexpectedly, not JNK pathways. We surmise that the discrepancy between the IRM PM2.5 and the WTC2.5 particles regarding signal pathway activation outcomes is likely due to cell type difference or some as-yet unrecognized differences in particle compositions.
That these disparate sets of particles were able to induce similar outcomes might be explained by the fact that these particulate samples shared the presence of various toxic metal constituents, such as vanadium (V) and chromium (Cr). Specifically, while Li and colleagues reported levels of V and Cr in the SRM1648 at 127 and 403 μg/g, respectively, and the Shoenfelt group reported levels of V and Cr in the IRM PM2.5 at 316 μg/g and 46.8 mg/g, respectively, we found that the levels of these two metals in our WTC2.5 samples averaged 118 and 125 μg/g, respectively (Lioy et al., 2002; McGee et al., 2002; Maciejczyk et al., 2006). There have been numerous publications describing the effects of each of these metal agents (individually or as part of more complex ambient particulate mixtures) on these pathways (Chien et al., 2006; Suzuki et al., 2007; Tessier and Pascal, 2006). Though the results of our studies here indicate that activation of these specific signaling pathways appears to be transient (see Figure 3), nonetheless, each of these short-lived events would still be expected to have activated downstream targets and ultimately cause an increase in the expression of genes for numerous cytokines.
It would not be unreasonable to assume that repeated exposures to, or prolonged pulmonary retention of, the WTC particles would likely lead to a cycle of activation/re-activation of these (and other) signaling pathways in impacted airway cells. In fact, changes in the degree (i.e., an augmentation) of activation of select signal pathways have been documented in airway epithelia in mice repeatedly exposed to particulate insoluble Cr (as basic zinc chromate) (Beaver et al., 2009b). These Authors also found that while a single instillation of these particles induced almost immediate (within 1 hr) increases in Akt phosphorylation, the increase was still evident 24 hr later but the relative intensity (i.e., low, medium, strong) of staining for the phospho-Akt among airway epithelial cells had continued to increase over time (Beaver et al., 2009a).
Beaver and colleagues concluded that the Cr-induced up-regulation of Akt likely promoted inflammation, and affected both cell survival and airway repair after lung injury. In keeping with this hypothesis, we purport that under the exposure scenarios evident at Ground Zero (i.e., repeated exposures to and entrainment of high levels of dust) ultimately, the activation and re-activation of these pathways and resultant induction of cytokine formation and/or release by resident cells eventually led to localized inflammatory changes in the lungs. These changes, in turn, would invariably have contributed to the onset/exacerbation of asthma, RADS, UADs, and LADs documented in the dust/particle-exposed individuals, including WTC First Responders.
The observations here that the a 5-hr treatment with the fine WTC dust resulted in increases in IL-8 and IL-10 formation/release suggest it is quite likely there had also been increases in TNFα and IFNγ expression in the period after exposure initiation and well before the supernatant harvest. It has been reported that an initial induction of TNFα and IFNγ formation (in many cell types) ultimately leads to an up-regulation of IL-10 expression in the same cells (Samarasinghe et al., 2006), and IL-10 acts to down-regulate the expression of TNFα, IL-1β, and IFNs (including IFNγ) (Kim et al., 2009), as well as other cytokines, such as IL-1, IL-2, IL-5, IL-12, and IL-13 (see Kearley et al. [2005] and reviews by Ogawa et al. [2008] and Kaur et al. [2009]). This auto-regulating pattern assures that any local pro-inflammatory response is relatively short-lived. It has also been reported that epithelial cells (like many other types of lung cells) bind and then internalize (and degrade) TNFα and IFNγ. These events may all contribute to the apparent ‘lack of elevated status’ of TNFα and IFNγ (Celada and Schreiber, 1987; Imamura et al., 1987; Bajzer et al., 1989).
The apparent lack of a statistically significant increase in another major anti-inflammatory cytokine measured here, IL-4, remains to be explained. It was expected that the increase in this particular product would more-or-less parallel that of IL-10. However, studies have shown that IL-4 itself is an inducer of IL-10 (at least in macrophages and lymphocytes; it remains to be proven so in epithelial cells) (Schmidt-Weber et al., 1999; Cao et al., 2005). Thus, as with TNFα and IFNγ, it is possible that peak levels of IL-4 had been attained in the cultures prior to the 5-hr timepoint (at which IL-10 levels appear ‘optimal for measure’), but had yet to decline toward basal levels.
In summary, the results from this study demonstrate that exposure of epithelial cells (i.e., BEAS-2B cells) to WTC2.5 increased their formation of inflammatory cytokines through ERK and p38 kinase pathways. This new understanding of one underlying mechanism of effect of the WTC dusts will likely make it possible for investigators/clinicians to begin to better identify how the dusts may have contributed to the increases in asthma and other inflammation-associated respiratory dysfunctions documented in First Responders/other individuals exposed in the days following the buildings’ collapse.
Authors’ addendum
Although we sought to explain our findings here of the effects of the WTC dusts on the BEAS-2B cells in the light of those induced by airborne particulates commonly encountered in NYC or other metropolitan areas, we are cognizant that by their nature the WTC dusts likely represent a “novel” urban dust. It is very possible that other constituent-related factors may have also played a role in the outcomes that were observed here. In fact, several studies that have analyzed WTC dusts (of various sizes found in PM collected during the period starting 1 wk [and up to 5 mo] post-collapse) have reported that, among the myriad of constituents present, levels of total (i.e., organic + elemental) carbonaceous materials, trace element oxides, select man-made vitreous fibers (MMVF; specifically, slag wool), and sulfates were found to exceed those found in background PM from the NYC/Northern New Jersey region (Olson et al., 2004; Rosati et al., 2008; Bern et al., 2009; Lowers et al., 2009). We hope this information provides other potential Investigators a cautionary note when selecting “control” PM for future studies of the toxicologic effects of WTC dusts or, in fact, those associated with other building collapses.
Acknowledgments
The Authors wish to thank Eric Liberda for his help in this study. This work was supported, in part, by NIEHS Grant ES016570 (Q. Qu), NIEHS Center Grant ES00260, and CDC/NIOSH Grant OH008280-01A2 (L. Chen).
Footnotes
Declaration of Interest
The Authors report no conflicts of interest. The Authors are alone responsible for the content and writing of the paper.
References
- Bajzer Z, Myers AC, Vuk-Pavlovic S. Binding, internalization, and intracellular processing of proteins interacting with recycling receptors. A kinetic analysis. J Biol Chem. 1989;264:13623–13631. [PubMed] [Google Scholar]
- Banauch GI, Dhala A, Alleyne D, Alva R, Santhyadka G, Krasko A, Weiden M, Kelly KJ, Prezant DJ. Bronchial hyperreactivity and other inhalation lung injuries in rescue/recovery workers after the World Trade Center collapse. Crit Care Med. 2005;33(S1):S102–106. doi: 10.1097/01.ccm.0000151138.10586.3a. [DOI] [PubMed] [Google Scholar]
- Beaver LM, Stemmy EJ, Constant SL, Schwartz A, Little LG, Gigley JP, Chun G, Sugden KD, Ceryak SM, Patierno SR. Lung injury, inflammation, and Akt signaling following inhalation of particulate hexavalent chromium. Toxicol Appl Pharmacol. 2009a;235:47–56. doi: 10.1016/j.taap.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaver LM, Stemmy EJ, Schwartz AM, Damsker JM, Constant SL, Ceryak SM, Patierno SR. Lung inflammation, injury, and proliferative response after repetitive particulate hexavalent chromium exposure. Environ Health Perspect. 2009b;117:1896–1902. doi: 10.1289/ehp.0900715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern AM, Lowers HA, Meeker GP, Rosati JA. Method development for analysis of urban dust using scanning electron microscopy with energy dispersive X-ray spectrometry to detect the possible presence of World Trade Center dust constituents. Environ Sci Technol. 2009;43:1449–1454. doi: 10.1021/es800865n. [DOI] [PubMed] [Google Scholar]
- Beyaert R, Cuenda A, Vanden Berghe W, Plaisance S, Lee JC, Haegeman G, Cohen P, Fiers W. The p38/RK mitogen-activated protein kinase pathway regulates interleukin-6 synthesis response to tumor necrosis factor. Embo J. 1996;15:1914–1923. [PMC free article] [PubMed] [Google Scholar]
- Brackbill RM, Hadler JL, DiGrande L, Ekenga CC, Farfel MR, Friedman S, Perlman SE, Stellman SD, Walker DJ, Wu D, Yu S, Thorpe LE. Asthma and post-traumatic stress symptoms 5–6 years following exposure to the World Trade Center terrorist attack. JAMA. 2009;302:502–516. doi: 10.1001/jama.2009.1121. [DOI] [PubMed] [Google Scholar]
- Buyantseva LV, Tulchinsky M, Kapalka GM, Chinchilli VM, Qian Z, Gillio R, Roberts A, Bascom R. Evolution of lower respiratory symptoms in New York police officers after 9/11: A prospective longitudinal study. J Occup Environ Med. 2007;49:310–317. doi: 10.1097/JOM.0b013e318032256e. [DOI] [PubMed] [Google Scholar]
- Cao S, Liu J, Song L, Ma X. The proto-oncogene c-Maf is an essential transcription factor for IL-10 gene expression in macrophages. J Immunol. 2005;174:3484–3492. doi: 10.4049/jimmunol.174.6.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (Centers for Disease Control) Physical Health status of World Trade Center rescue and recovery workers and volunteers - New York City, July 2002-August 2004. MMWR. 2004;53:807–812. [PubMed] [Google Scholar]
- Celada A, Schreiber RD. Internalization and degradation of receptor-bound interferon-γ by murine macrophages. Demonstration of receptor recycling. J Immunol. 1987;39:147–153. [PubMed] [Google Scholar]
- Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA. 2006;108:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien PS, Mak OT, Huang HJ. Induction of COX-2 protein expression by vanadate in A549 human lung carcinoma cell line through EGF receptor and p38 MAPK-mediated pathway. Biochem Biophys Res Commun. 2008;339:562–568. doi: 10.1016/j.bbrc.2005.11.045. [DOI] [PubMed] [Google Scholar]
- Chung KF, Barnes PJ. Cytokines in asthma. Thorax. 1999;54:825–857. doi: 10.1136/thx.54.9.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Hoz RE, Christie J, Teamer JA, Bienenfeld LA, Afilaka AA, Crane M, Levin SM, Herbert R. Reflux symptoms and disorders and pulmonary disease in former World Trade Center rescue and recovery workers and volunteers. J Occup Environ Med. 2008a;50:1351–1354. doi: 10.1097/JOM.0b013e3181845f9b. [DOI] [PubMed] [Google Scholar]
- de la Hoz RE, Shohet MR, Chasan R, Bienenfeld LA, Afilaka AA, Levin SM, Herbert R. Occupational toxicant inhalation injury: the World Trade Center (WTC) experience. Int Arch Occup Environ Health. 2008b;81:479–485. doi: 10.1007/s00420-007-0240-x. [DOI] [PubMed] [Google Scholar]
- de la Hoz RE, Shohet MR, Wisnivesky JP, Bienenfeld LA, Afilaka AA, Herbert R. Atopy and upper and lower airway disease among former World Trade Center workers and volunteers. J Occup Environ Med. 2009;51:992–995. doi: 10.1097/JOM.0b013e3181b32093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavett SH, Haykal-Coates N, Highfill JW, Ledbetter AD, Chen LC, Cohen MD, Costa DL. World Trade Center fine particulate matter causes respiratory tract hyper-responsiveness in mice. Environ Health Perspect. 2003;111:981–991. doi: 10.1289/ehp.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyh AS, Chillrud S, Williams DL, Herbstman J, Symons JM, Rees K, Ross J, Kim SR, Lim HJ, Turpin B, Breysse P. Assessing truck driver exposure at the World Trade Center disaster site: Personal and area monitoring for particulate matter and volatile organic compounds during October 2001 and April 2002. J Occup Environ Hyg. 2005;2:179–193. doi: 10.1080/15459620590923154. [DOI] [PubMed] [Google Scholar]
- Han J. MyD88 beyond Toll. Nat Immunol. 2006;7:370–371. doi: 10.1038/ni0406-370. [DOI] [PubMed] [Google Scholar]
- Herbert R, Moline J, Skloot G, Metzger K, Baron S, Luft B, Markowitz S, Udasin I, Harrison D, Stein D, Todd A, Enright P, Stellman JM, Landrigan PJ, Levin SM. The World Trade Center disaster and health of workers: Five-year assessment of a unique medical screening program. Environ Health Perspect. 2006;114:1853–1858. doi: 10.1289/ehp.9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura K, Spriggs D, Kufe D. Expression of tumor necrosis factor receptors on human monocytes and internalization of receptor-bound ligand. J Immunol. 1987;139:2989–2992. [PubMed] [Google Scholar]
- Izbicki G, Chavko R, Banauch GI, Weiden MD, Berger KI, Aldrich TK, Hall C, Kelly KJ, Prezant DJ. World Trade Center “sarcoid-like” granulomatous pulmonary disease in New York City Fire Department rescue workers. Chest. 2007;131:1414–1423. doi: 10.1378/chest.06-2114. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- Kaur K, Slezak DS, Sharma AK, Bajaj A, Singal PK. Biology of TNFα and IL-10, and their imbalance in heart failure. Heart Failure Rev. 2009;14:113–123. doi: 10.1007/s10741-008-9104-z. [DOI] [PubMed] [Google Scholar]
- Kearley J, Barker JE, Robinson DS, Lloyd CM. Resolution of airway inflammation and hyperreactivity after in vivo transfer of CD4+CD25+ regulatory T-cells is IL-10-dependent. J Exp Med. 2005;202:1539–1547. doi: 10.1084/jem.20051166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly KJ, Niles J, McLaughlin MT, Carroll S, Corrigan M, Al-Othman F, Prezant DJ. FDNY WTC Health Effects - a six year assessment. Fire Department of the City of New York; Sep 11, 2007. Available at http://www.nyc.gov/html/om/pdf/2007/wtc_health_impacts_on_fdny_rescue_workers_sept_2007.pdf. Last visit (4/5/2010) [Google Scholar]
- Kim SH, Shi TY. Anti-inflammatory effect of leaves of Eriobotrya japonica correlating with attenuation of p38 MAPK, ERK, and NF-kappaB activation in mast cells. Toxicology In Vitro. 2009;23:1215–9. doi: 10.1016/j.tiv.2009.07.036. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Song M, Ryu JC. Inflammation in methotrexate-induced pulmonary toxicity occurs via the p38 MAPK pathway. Toxicology. 2009;256:183–190. doi: 10.1016/j.tox.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Li Z, Carter JD, Dailey LA. Pollutant particles produce vasoconstriction and enhance MAPK signaling via angiotensin Type I receptor. Environ Health Perspect. 2005;113:1009–1014. doi: 10.1289/ehp.7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy PJ, Weisel CP, Millette JR, Eisenreich S, Vallero D, Offenberg J, Buckley B, Turpin B, Zhong M, Cohen MD, Prophete C, Yang I, Stiles R, Chee G, Johnson W, Porcja R, Alimokhtari S, Hale RC, Weschler C, Chen LC. Characterization of the dust/smoke aerosol that settled east of the World Trade Center (WTC) in lower Manhattan after the collapse of the WTC 11 September 2001. Environ Health Perspect. 2002;110:703–714. doi: 10.1289/ehp.02110703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowers HA, Meeker GP, Lioy PJ, Lippmann M. Summary of the development of a signature for detection of residual dust from collapse of the World Trade Center buildings. J Exposure Sci Environ Epidemiol. 2009;19:325–335. doi: 10.1038/jes.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejczyk P, Zeisler BR, Hwang J, Chen LC. Characterization of size-fractionated WTC dusts and estimation of relative dust concentration to ambient particulate concentrations. ACS Symp Series. 2006;919:114–131. [Google Scholar]
- McGee JK, Chen LC, Cohen MD, Chee G, Prophete C, Haykal-Coates N, Wasson SJ, Conner TL, Costa DL, Gavett SH. Chemical analysis of World Trade Center fine particulate matter for use in toxicologic assessment. Environ Health Perspect. 2003;111:972–980. doi: 10.1289/ehp.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Duru EA, Ameredes BT. Biology of TNFα and IL-10, and their imbalance in heart failure. Curr Mol Med. 2008;8:437–445. doi: 10.2174/156652408785160907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson DA, Norris GA, Landis MS, Vette AF. Chemical characterization of ambient particulate matter near the World Trade Center: Elemental carbon, organic carbon, and mass reconstruction. Environ Sci Technol. 2004;38:4465–4473. doi: 10.1021/es030689i. [DOI] [PubMed] [Google Scholar]
- Ovrevik J, Lag M, Holme JA. Cytokine and chemokine expression patterns in lung epithelial cells exposed to components characteristic of particulate air pollution. Toxicology. 2009;259:46–53. doi: 10.1016/j.tox.2009.01.028. [DOI] [PubMed] [Google Scholar]
- Park EJ, Yi J, Chung KH, Ryu DY, Choi J, Park K. Oxidative stress and apoptosis induced by titanium dioxide nanoparticles in cultured BEAS-2B cells. Toxicol Lett. 2008;180:222–229. doi: 10.1016/j.toxlet.2008.06.869. [DOI] [PubMed] [Google Scholar]
- Payne JP, Kemp SJ, Dewar A, Goldstraw P, Kendall M, Chen LC, Tetley TD. Effects of airborne World Trade Center dust on cytokine release by primary human lung cells in vitro. J Occup Environ Med. 2004;46:420–427. doi: 10.1097/01.jom.0000126021.25149.64. [DOI] [PubMed] [Google Scholar]
- Pilette C, Detry B, Guisset A, Gabriels J, Sibille Y. Induction of interleukin-10 expression through Fcalpha receptor in human monocytes and monocyte-derived dendritic cells: role of p38 MAPKinase. Immunol Cell Biol. 2010;88:486–93. doi: 10.1038/icb.2009.120. [DOI] [PubMed] [Google Scholar]
- Prezant DJ. World Trade Center cough syndrome and its treatment. Lung. 2008;186(S1):S94–102. doi: 10.1007/s00408-007-9051-9. [DOI] [PubMed] [Google Scholar]
- Reed CE. The natural history of asthma. J Allergy Clin Immunol. 2006;118:543–548. doi: 10.1016/j.jaci.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Rosati JA, Bern AM, Willis RD, Blanchard FT, Conner TL, Kahn HD, Friedman D. Multi-laboratory testing of a screening method for world trade center (WTC) collapse dust. Sci Total Environ. 2008;390:514–519. doi: 10.1016/j.scitotenv.2007.10.027. [DOI] [PubMed] [Google Scholar]
- Salzman SH, Moosavy FM, Jiskoff JA, Friedman P, Fried G, Rosen MJ. Early respiratory abnormalities in emergency services police officers at the World Trade Center site. J Occup Environ Med. 2004;46:113–122. doi: 10.1097/01.jom.0000111612.68916.d0. [DOI] [PubMed] [Google Scholar]
- Samarasinghe R, Tailor P, Tamura T, Kaisho T, Akira S, Ozato K. Induction of an anti-inflammatory cytokine IL-10 in cells after toll-like receptor signaling. J Interferon Cytokine Res. 2006;26:893–900. doi: 10.1089/jir.2006.26.893. [DOI] [PubMed] [Google Scholar]
- Scherle PA, Jones EA. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J Immunol. 1998;161:5681–5686. [PubMed] [Google Scholar]
- Schmidt-Weber CB, Alexander SI, Henault LE, James L, Lichtman AH. IL- 4 enhances IL-10 gene expression in murine TH2 cells in the absence of TCR engagement. J Immunol. 1999;162:238–244. [PubMed] [Google Scholar]
- Shoenfelt J, Mitkus RJ, Zeisler R, Spatz RO, Powell J, Fenton MJ, Squibb KA, Medvedev AE. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J Leukocyte Biol. 2009;86:303–312. doi: 10.1189/jlb.1008587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Inageda K, Nishitai G, Matsuoka M. Phosphorylation of p53 at serine 15 in A549 pulmonary epithelial cells exposed to vanadate: Involvement of ATM pathway. Toxicol Appl Pharmacol. 2007;220:83–91. doi: 10.1016/j.taap.2006.12.028. [DOI] [PubMed] [Google Scholar]
- Taylor JK, editor. Statistical Techniques for Data Analysis. Lewis Publishers; Chelsea, MI: 1990. [Google Scholar]
- Tessier DM, Pascal LE. Activation of MAP kinases by hexavalent chromium, manganese and nickel in human lung epithelial cells. Toxicol Lett. 2006;167:114–121. doi: 10.1016/j.toxlet.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Val S, Hussain S, Boland S, Hamel R, Baeza-Squiban A, Marano F. Carbon black and titanium dioxide nanoparticles induce pro-inflammatory responses in bronchial epithelial cells: Need for multi-parametric evaluation due to adsorption artifacts. Inhal Toxicol. 2009;21(S1):115–122. doi: 10.1080/08958370902942533. [DOI] [PubMed] [Google Scholar]
- Veranth JM, Cutler NS, Kaser EG, Reilly CA, Yost GS. Effects of cell type and culture media on IL-6 secretion in response to environmental particles. Toxicol In Vitro. 2008;22:498–509. doi: 10.1016/j.tiv.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber MP, Gustave J, Lee R, Niles JK, Kelly K, Cohen HW, Prezant DJ. Trends in respiratory symptoms of firefighters exposed to the world trade center disaster: 2001–2005. Environ Health Perspect. 2009;117:975–980. doi: 10.1289/ehp.0800291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiden MD, Ferrier N, Nolan A, Rom WN, Comfort A, Gustave J, Zeig-Owens R, Zheng S, Goldring RM, Berger KI, Cosenza K, Lee R, Webber MP, Kelly KJ, Aldrich TK, Prezant DJ. Obstructive airways disease with air trapping among firefighters exposed to World Trade Center dust. Chest. 2010;137:566–574. doi: 10.1378/chest.09-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler K, McKelvey W, Thorpe L, Perrin M, Cone J, Kass D, Farfel M, Thomas P, Brackbill R. Asthma diagnosed after September 11, 2001 among rescue and recovery workers: Findings from the World Trade Center registry. Environ Health Perspect. 2007;115:1584–1590. doi: 10.1289/ehp.10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiin LM, Millette JR, Vette A, Ilacqua V, Quan C, Gorczynski J, Kendall M, Chen LC, Weisel CP, Buckley B, Yang I, Lioy PJ. Comparisons of the dust/smoke particulate that settled inside the surrounding buildings and outside on the streets of southern New York City after the collapse of the World Trade Center. J Air Waste Manage Assoc. 2004;54:515–528. doi: 10.1080/10473289.2004.10470935. [DOI] [PubMed] [Google Scholar]