

Abstract
Neutralizing antibodies against influenza viruses have traditionally been thought to provide protection exclusively through their variable region; the contributions of mechanisms conferred by the Fc domain remain controversial. We investigated the in vivo contributions of Fc interactions with their cognate receptors for a collection of neutralizing anti-influenza antibodies. Whereas five broadly neutralizing monoclonal antibodies (bNAbs) targeting the conserved stalk region of hemagglutinin (HA) required interactions between the antibody Fc and Fc receptors for IgG (FcγRs) to confer protection from lethal H1N1 challenge, three strain-specific monoclonal Abs (mAbs) against the variable head domain of HA were equally protective in the presence or absence of FcγR interactions. Although all antibodies blocked infection, only anti-stalk bNAbs were capable of mediating cytotoxicity of infected cells, which accounts for their FcγR dependence. Immune complexes generated with anti–HA stalk mAb efficiently interacted with FcγRs, but anti–HA head immune complexes did not. These results suggest that FcγR binding capacity by anti-HA antibodies was dependent on the interaction of the cognate Fab with antigen. We exploited these disparate mechanisms of mAb-mediated protection to reengineer an anti-stalk bNAb to selectively enhance FcγR engagement to augment its protective activity. These findings reveal a previously uncharacterized property of bNAbs and guide an approach toward enhancing mAb-mediated antiviral therapeutics.
Worldwide influenza epidemics result in substantial morbidity and the deaths of 250,000–500,000 people annually, with the young and elderly representing the majority of this mortality1. Worldwide pandemics can cause even more severe mortality, such as during 1918 when approximately 50 million deaths were attributed to the Spanish flu2. Vaccination is the most effective method to prevent infection, but influenza vaccines must be reformulated annually because of antigenic drift in HA, the immunogenic glycoprotein to which the majority of the influenza immune response is directed. Although mAbs generated against other influenza proteins (such as neuraminidase) may provide varying levels of protection in vivo, antibodies specific for HA can neutralize infection in vivo3 and correlate with protection against viral infection in humans4.
The influenza virus HA is composed of two major domains: the immunodominant globular head (HA1) domain, in which the majority of antigenic variation occurs, and the stalk (HA2) domain, which remains relatively conserved between influenza virus strains5. Neutralizing mAbs to influenza virus HA are thought to act by inhibiting one of the three main functions of HA6. First, studies using anti–HA head mAbs demonstrated that this class of mAbs disrupts virus attachment to sialic acids on the surface of host cells, thereby blocking viral entry7,8. In addition, some anti–HA head mAbs may also prevent the release of progeny virions from infected cells9. Finally, studies showed that anti–HA stalk mAbs that bind to the region containing the fusion peptide neutralize virus by blocking viral fusion with the target cell10–12. Because of the antigenic diversity found within the HA globular head domain, most mAbs targeting this domain usually neutralize only a single specific strain of virus. By contrast, recent studies have described bNAbs that target the conserved stalk domain and neutralize an array of viruses either within or across influenza virus subtypes11–13.
Although the recognition of neutralizing epitopes by the Fab region of an antibody is crucial during antibody-mediated neutralization of a microbial pathogen, as assayed in vitro, it is becoming increasingly clear that coupling Fab recognition to Fc-mediated effector function, such as FcγR binding, has an important role in providing protection in vivo. For example, in vivo protection from Bacillus anthracis infection by anthrax protective antigen–specific mAbs showed an absolute requirement for FcγR engagement14,15. A role for FcγRs has been implicated during in vivo protection from influenza virus infections by antibodies targeting non-HA antigens, such as the viral M2 protein16. Mice that were passively treated with immune serum from H1N1 virus–immunized mice also showed a dependence on FcγRs for protection17. FcγRs may also contribute to in vivo protection by a bNAb that targets HA, which is expressed on the viral membrane13. How these results integrate with the assumption that anti-HA mAbs neutralize virus by blocking viral entry or disrupting fusion is unclear, and the mechanism by which HA-specific antibodies provide protection against virus infection in vivo thus remains controversial.
FcγRs represent a major component of the immune system that both couples and regulates innate and adaptive immunity. The FcγR system contains both activating and inhibitory receptors, whose signals must be appropriately balanced to regulate the outcome of inflammation and immunity18. Mice express two low-affinity activating FcγRs on myeloid cells and dendritic cells, FcγRIII and FcγRIV, as well as the low-affinity inhibitory FcγRIIB, which is widely expressed on mouse hematopoietic cells. The biological activities of mouse IgG subclasses are dependent on their affinities for the activating and inhibitory FcγRs. Thus, an activating/inhibitory (A/I) ratio can be assigned to each IgG subclass on the basis of the subclass’s relative affinities for the activating and inhibitory FcγRs19. IgG2a antibodies are the most potently activating (A/I = 69) and preferentially interact with the activating FcγRs, whereas IgG1 antibodies are the least activating (A/I = 0.1) and preferentially interact with inhibitory FcγRIIB. The balance between activating and inhibitory FcγRs determines the biological effect of circulating immune complexes or antibodies bound to pathogens or cells. A similar FcγR system exists in humans, albeit with considerable differences in the structure, binding affinity and expression patterns of the human activating (FcγRIIA and IIIA) and inhibitory (FcγRIIB) receptors from those of their mouse counterparts.
In this study, we use previously described anti-HA antibodies, including two anti–HA stalk bNAbs that neutralize a panel of H1 or of both H1 and H5 influenza viruses, respectively20; three anti–HA stalk bNAbs that react with all 16 HA subtypes13,21 ; and three anti–HA head antibodies displaying strain-specific neutralization capabilities20,22,23. We also employ a mouse model in which mice express the full array of human FcγRs (huFcγRs) on a genetic background lacking all mouse FcγRs24, thereby facilitating the interpretation of the contribution of human Fc function in a mouse infection model. We observed that the anti-stalk bNAbs required Fc-FcγR interactions for maximum bNAb-mediated neutralization of influenza virus in vivo, whereas the strain-specific anti–HA head mAbs did not. These anti-stalk bNAbs functioned after viral entry in vivo and were superior inducers of antibody-dependent cellular cytotoxicity (ADCC), suggesting a mechanism for their FcγR-dependent function in vivo. Engineering the human IgG1 Fc to enhance binding to its activation receptors and augment antibody effector function may inform the development of future therapies, including vaccination protocols to elicit bNAbs and passive antibody therapies using bNAbs.
RESULTS
FcγRs are required for protection by anti–HA stalk bNAb
The recently described 6F12 bNAb binds to the stalk region of a divergent panel of H1 HAs and effectively neutralizes in vitro a panel of H1 influenza viruses that have arisen over 79 years of antigenic drift, including the 2009 pandemic H1N1 strains20 (Table 1 and Supplementary Table 1). To determine the contributions of Fc-FcγR interactions during influenza virus neutralization in vivo, we generated 6F12 bNAb constructs with different Fc domains that either preferentially engaged activating FcγRs (IgG2a) or the inhibitory FcγRIIB (IgG1) or were null for FcγR binding (DA265 mutant). We compared the 6F12 bNAb constructs for their ability to bind H1 protein (Fig. 1a) and neutralize (Fig. 1b) the influenza virus strain A/Puerto Rico/8/1934 H1N1 (PR8), and we detected no differences between constructs.
Table 1.
Antibody requirements for Fc-FcγR interactions during passive protection from viral infection
| Name | Specificity | Reactivity | FcγR-dependent protection in vivo? | In vitro ADCC potential | Reference |
|---|---|---|---|---|---|
| 6F12 | HA stalk | All H1 viruses | Yes | +++ | 20 |
| FI6 | HA stalk | All group 1 and group 2 viruses | Yes | ++++ | 13 |
| 2G02 | HA stalk | All group 1 and group 2 viruses | Yes | ++++ | 21 |
| 2B06 | HA stalk | All group 1 and group 2 viruses | Yes | ++++ | Unpublished |
| 1F02 | HA stalk | H1 and H5 viruses | Yes | ++++ | 23 |
| PY102 | HA head | A/PR/8/1934 H1N1 | No | − | 22 |
| 7B2 | HA head | 2009 pandemic H1N1 | No | +/− | 20 |
| 4C04 | HA head | 2009 pandemic H1N1 | No | − | 23 |
The + and − scale qualitatively describes the ability of each mAb to activate NK cells during in vitro ADCC assays. Clone 2B06 has not been previously described in the literature.
Figure 1.

c-FcγR interactions are required for protection from viral infection by an anti–HA stalk bNAb in vivo. (a) Binding of 6F12 bNAb variants to PR8 HA. Mouse IgG2a, mouse IgG1 and DA265 mutant 6F12 bNAb and an IgG2a isotype control mAb diluted as indicated and tested for binding to PR8 HA by ELISA. Values represent mean ± s.e.m. relative optical density (OD) values from triplicate wells. (b) In vitro plaque reduction neutralization by 6F12 bNAb variants. Values represent mean % inhibition, calculated by comparing plaque numbers in mAb-treated wells with wells receiving only PBS. (c) Percentage weight change compared to day 0 (left) or percentage survival (right) in wild-type mice treated with mouse IgG2a 6F12 bNAb, mouse IgG1 6F12 bNAb, DA265 mutant 6F12 bNAb or PBS before infection with PR8 virus. Weight change values represent mean ± s.e.m. n ≥ 5 mice per group. (d) PFU per lung in mice treated as in c, with lungs harvested on day 3 and analyzed for viral titers using a plaque assay. Weight change values represent mean ± s.e.m. (n ≥ 4 mice per group). For c,d, significant differences between the indicated sample and PBS-treated sample are shown: *P < 0.05; **P < 0.001.
We next investigated whether Fc-FcγR interactions contributed to bNAb-mediated protection in vivo. We treated wild-type mice with 4 mg per kg body weight (mg/kg) of 6F12 bNAb before infecting them with five 50% mouse lethal doses (mLD50) of the mouse-adapted laboratory H1N1 strain, PR8, and monitored weight loss and survival daily. Mice that received the IgG2a 6F12 bNAb showed minimal weight loss compared to PBS-treated mice (P < 0.0001) at day 7, whereas mice receiving the IgG1 or DA265 bNAb constructs showed weight loss curves similar to that of PBS-treated animals (Fig. 1c). Further, 6F12 IgG2a bNAb–treated mice showed 100% survival, whereas no animals in the IgG1-, DA265- or PBS-treated groups survived (P < 0.0001). We used PBS as a control treatment in this and all subsequent experiments because we did not observe any differences in weight loss or survival curves in mice receiving PBS or isotype control mAb before infection (Supplementary Fig. 1). To verify that lung viral titers mirrored morbidity and mortality in the mice, we harvested lungs from these same groups of mice 3 d after infection. We detected similar viral titers in the lungs of mice receiving IgG1 bNAb, DA265 bNAb or PBS while mice receiving IgG2a 6F12 bNAb had 81% (P < 0.0001) decreased lung titers compared to PBS-treated mice (Fig. 1d). These results were not influenced by antibody isotype–specific differences in in vivo half-lives (t1/2) (Supplementary Table 1). Therefore, although Fc-FcγR interactions are dispensable during in vitro neutralization by 6F12 bNAb, they are required for bNAb-mediated protection during in vivo viral challenge.
Activating FcγRs are required for bNAb-mediated protection
We next sought to confirm the requirement for FcγRs during in vivo viral neutralization and determine which of the FcγRs mediated protection. First, we tested wild-type or Fcγ chain–deficient mice (Fcer1g−/− mice), which lack the activating FcγRs FcγRI, FcγRIII and FcγRIV but retain expression of inhibitory FcγRIIb, for in vivo protection from PR8 challenge after treatment with 4 mg/kg 6F12 IgG2a bNAb. Wild-type mice that received the IgG2a 6F12 bNAb showed minimal weight loss (P < 0.0001) at day 7 and 100% survival (P < 0.0001) compared to Fcer1g−/− mice, which markedly lost weight and did not survive the viral challenge (Fig. 2a). Because deletion of the Fcγ chain in Fcer1g−/− mice may also affect other components of the immune system that use the Fcγ chain, we repeated this experiment in FcRα-null mice, which specifically lack only FcγRI, FcγRIIb, FcγRIII and FcγRIV but retain a functional Fcγ chain, and saw similar results (Supplementary Fig. 2a). Because FcγRIV shows the highest affinity for IgG2a Fc and thus mediates the majority of IgG2a effector function, we tested whether deletion of FcγRIV affected IgG2a bNAb–mediated protection in vivo in Fcgr4−/− mice. Fcgr4−/− mice showed 1.6-fold (P = 0.02) increased weight loss at day 7 and 50% decreased survival (P = 0.04) compared to wild-type mice after 6F12 IgG2a bNAb treatment (Supplementary Fig. 2b). By contrast, we saw no major defects in protection in mice lacking the other activating FcγRs, FcγRI or FcγRIII, compared to wild-type mice after treatment with IgG2a 6F12 mAb (Supplementary Fig. 2c). Collectively, these results demonstrate that activating FcγRs mediate bNAb-mediated protection in vivo; FcγRIV is sufficient to provide mAb-mediated protection from lethal infection, and FcγRI and FcγRIII compensate to provide partial protection in the absence of FcγRIV.
Figure 2.

ctivating FcγRs are required for bNAb-mediated protection from viral infection in vivo. (a) Percentage weight change compared to day 0 and survival over time in wild-type (WT) mice or Fcer1g−/− mice treated with mouse IgG2a 6F12 bNAb or PBS before infection with PR8 virus. n = 6–10 mice per group. (b) Percentage weight change compared to day 0 and survival over time in wild-type mice or Fcgr2b−/− mice treated with mouse IgG1 6F12 bNAb or PBS before infection with PR8 virus. n = 5–10 mice per group. (c) Percentage weight change compared to day 0 and survival over time in wild-type mice treated with mouse IgG2a 6F12 bNAb, IgG1 6F12 bNAb, DA265 mutant 6F12 bNAb or PBS before infection with Neth09 virus. n ≥ 6 mice per group. (d) Percentage weight change compared to day 0 and survival over time in wild-type mice treated with mouse IgG2a 6F12 bNAb, mouse IgG1 6F12 bNAb, DA265 mutant 6F12 bNAb or PBS before infection with PR8 virus. Weight change values represent mean ± s.e.m. in a–d. n ≥ 5 mice per group. For a–c, significant differences between the indicated sample and PBS-treated sample are shown: **P < 0.001.
To determine whether modulating the balance between activating and inhibitory FcγRs affected in vivo protection, we treated wild-type or Fcgr2b−/− mice with 4 mg/kg IgG1 6F12 bNAb, a dose that does not protect wild-type mice from infection (Fig. 1c). IgG1 Fcs interact preferentially with FcγRIIb but also bind FcγRIII with low affinity and do not interact with FcγRIV. Whereas wild-type mice did not survive challenge, Fcgr2b−/− mice treated with IgG1 6F12 bNAb showed decreased weight loss and 55% survival (P = 0.003; Fig. 2b). Thus, removing inhibitory FcγR function enhanced bNAb-mediated protection.
We next tested the generality of the requirement for Fc-FcγR interactions during in vivo protection by bNAb 6F12. Wild-type mice were treated with 4 mg/kg of 6F12 bNAb constructs before infection with 5 mLD50 of the 2009 H1N1 pandemic isolate A/Netherlands/602/2009 (Neth09) (ref. 25). Most mice that received the IgG2a 6F12 bNAb were protected and showed minimal weight loss, whereas mice receiving the IgG1 or DA265 mutant bNAb showed comparably poor survival (P < 0.008; Fig. 2c). Thus, the 6F12 bNAb also protected mice from infection by a current viral strain in an FcγR-dependent manner.
Four other bNAbs protect mice via FcγRs
The bNAb FI6 was recently identified to recognize all 16 HA subtypes and neutralize both group 1 and group 2 viruses13. Although a mutated huIgG1 version of this bNAb (LALA mutant) that abrogates huFcγR binding showed impaired protection in mice, the incompatibility of the antibody and the Fc receptors expressed in mice confounds the interpretation of that result. To address the contribution of Fc-FcγR engagement during in vivo protection by FI6, we tested FI6 mAb with mouse Fcs in the context of the mouse FcγR system. We generated FI6 bNAb constructs with mouse IgG2a, IgG1 or DA265 mutant Fc domains. Each mutant bound HA and neutralized virus in vitro similarly (Supplementary Fig. 3). We treated wild-type mice with 4 mg/kg of these FI6 bNAb constructs before infecting them with 5 mLD50 of PR8 virus. Mice that received the IgG2a FI6 bNAb showed minimal weight loss and 100% survival, whereas no animals in the IgG1-, DA265- or PBS-treated groups survived (P < 0.0001; Fig. 2d). Further, 4 mg/kg of FI6 IgG2a mAb was also unable to protect Fcer1g−/− mice from lethal viral challenge (Supplementary Fig. 4).
We saw similar results with three additional anti–HA stalk bNAbs of human origin that we modified to contain mouse Fcs that are reactive either with all group 1 and group 2 viruses (mAbs 2G02 and 2B06)21 or with H1 and H5 viruses (mAb 1F02)23 (Table 1). 2G02, 2B06 and 1F02 all protected mice from lethal 2009 pandemic Neth09 challenge in an FcγR-dependent manner (Supplementary Fig. 5). Thus, FcγRs are required for protection mediated by five different anti-stalk bNAbs during in vivo viral challenge.
The FcγR requirement during protection is dose dependent
Next, we investigated whether the requirement for activating FcγRs during bNAb-mediated neutralization in vivo is absolute or can be compensated for by increasing the dose of antibody. We treated wild-type mice with increasing doses of IgG2a or DA265 mutant 6F12 bNAb before infecting them with PR8 virus. All mice receiving IgG2a bNAb at 4–16 mg/kg were protected, whereas only 40% of mice receiving 2 mg/kg and no mice receiving 1 mg/kg bNAb survived (Supplementary Fig. 6a). Mice receiving 2–8 mg/kg DA265 6F12 bNAb were not protected from infection in vivo, but 100% of mice receiving 16 mg/kg of this mutant bNAb were protected (Supplementary Fig. 6b).
We also administered increasing doses of 6F12 IgG2a bNAb to Fcer1g−/− mice. High-dose (32 mg/kg) 6F12 bNAb was able to protect Fcer1g−/− mice (Supplementary Fig. 6c). Thus, FcγR engagement increases the potency of 6F12 by approximately a factor of 10, suggesting that FcγR effector mechanisms in vivo have a key role in protection by this anti-stalk bNAb. At high doses of mAb, in vivo protection is FcγR independent.
Three strain-specific anti–HA head mAbs do not require FcγRs
We next sought to determine the generality of FcγR requirements for in vivo protection by anti-HA antibodies by investigating a second class of neutralizing antibodies, those that recognize the HA globular head. Owing to the highly variable nature of the HA globular head between strains, most mAbs with this specificity are not broadly neutralizing and recognize only a single viral strain. We first tested the PR8 strain–specific anti–H1 globular head mAb PY102 (ref. 22) (Supplementary Table 1). We generated PY102 mAb constructs with mouse IgG2a, IgG1 or DA265 Fcs and tested these in vitro. The PY102 mAb constructs bound PR8 HA protein similarly (Supplementary Fig. 7a), showed similar in vivo t1/2 (Supplementary Table 1) and also similarly neutralized infection by PR8 virus, as determined by plaque reduction neutralization assays (Supplementary Fig. 7b).
We next assessed whether Fc-FcγR interactions contributed to PY102 anti–H1 head mAb–mediated protection in vivo. We treated wild-type mice with 0.5 mg/kg of each PY102 mAb construct before infecting them with PR8 virus. We observed similar weight loss and survival curves in mice receiving IgG2a, IgG1 or DA265 PY102 mAb, with no statistical differences observed between groups (Fig. 3a; P = 0.95 for IgG2a versus DA265 survival curvesxs, P = 0.21 for IgG2a versus IgG1 survival curves). We noted no differences in survival between mice receiving IgG2a and DA265 PY102 mAbs at any dose between 0.25 mg/kg and 4 mg/kg (Fig. 3b). Further, we saw no difference in protection between wild-type and Fcer1g−/− mice treated with IgG2a PY102 mAb (Supplementary Fig. 8).
Figure 3.
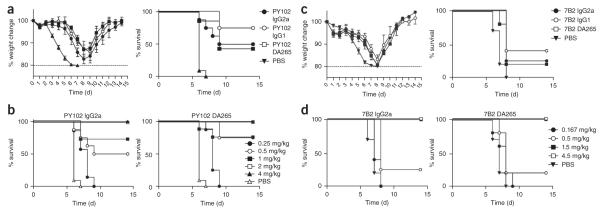
Two strain-specific anti–H1 head mAbs do not require FcγR contributions during protection from viral infection in vivo. (a) Percentage weight change compared to day 0 and survival over time in wild-type mice treated with mouse IgG2a, mouse IgG1 or DA265 mutant PY102 mAb or PBS before infection with PR8 virus. (b) Percentage survival over time in wild-type mice treated with the indicated doses of mouse IgG2a or DA265 mutant PY102 mAb, or PBS, before infection with PR8 virus. (c) Percentage weight change compared to day 0 and survival over time in wild-type mice treated with 0.5 mg/kg of mouse IgG2a, mouse IgG1 or DA265 mutant 7B2 mAb or PBS before infection with Neth09 virus. (d) Percentage survival over time in wild-type mice treated with the indicated doses of mouse IgG2a or DA265 mutant PY102 mAb, or PBS, before infection with Neth09 virus. Weight change data in a,c are expressed as mean ± s.e.m. n = 5–8 mice per group.
To confirm this finding with a second strain-specific anti–globular head mAb, we tested a 2009 H1N1 pandemic strain–specific mAb, 7B2 (ref. 20). We treated wild-type mice with 0.5 mg/kg of IgG2a, IgG1 or DA265 7B2 mAb constructs (Supplementary Fig. 9) before infecting them with Neth09 virus. We observed similar weight loss and survival curves in mice receiving the 7B2 mAb constructs (Fig. 3c). We noted no differences in survival between mice receiving IgG2a and DA265 7B2 mAb constructs at any dose between 0.167 mg/kg and 4.5 mg/kg (Fig. 3d), and we saw no difference in protection between wild-type and FcRα-null mice treated with IgG2a 7B2 mAb (Supplementary Fig. 10a). We obtained similar results with a third anti–HA head mAb of human origin that is reactive with the 2009 pandemic strain, mAb 4C04 (ref. 23) (Supplementary Fig. 10b,c). Thus, Fc-FcγR interactions are not required for in vivo protection by strain-specific anti–HA globular head mAbs, even at suboptimal mAb doses.
FcγRs contribute to in vivo protection after viral entry
To determine the mechanism by which FcγR engagement was operating for the anti-stalk but not the anti-head HA antibodies, we determined whether FcγRs facilitate mAb-mediated protection in vivo during the initial disruption of viral binding, fusion and entry into target cells, or whether they contribute to protection after cells have already been infected. We incubated PR8 virus with various doses of IgG2a and DA265 mutant 6F12 bNAb before using it to infect wild-type mice. Six hours later, before replication was complete and secondary cells were infected, we used collagenase II to digest lungs and stained them for intracellular nucleoprotein (NP) to identify infected cells.
Infection was impaired by preincubation with 100 μg/ml, 50 μg/ml and 25 μg/ml 6F12 mAb, and we saw no differences between IgG2a and DA265 mutant 6F12 mAb constructs (Fig. 4a). Therefore, FcγRs do not substantially contribute to protection during mAb-mediated inhibition of viral entry, which is consistent with the ability of all 6F12 Fc variants to neutralize virus in vitro (Fig. 1b).
Figure 4.
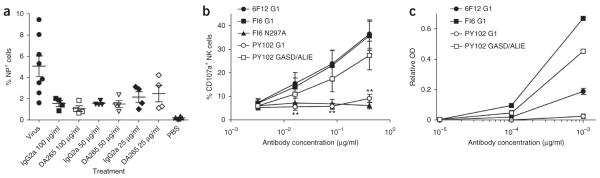
Anti–HA stalk mAbs function through FcγRs after viral entry and induce superior ADCC compared to anti-head mAb. (a) Antibody-mediated inhibition of viral entry is FcγR independent. Mice were infected with PR8 virus alone, virus preincubated with the indicated concentration of IgG2a or DA265 6F12 mAb, or PBS. Values represent mean ± s.e.m. frequency of NP+ cells in the lungs of individual mice 6 h after receiving the indicated treatment. Horizontal bars indicate mean values. (b) Impaired NK cell activation by anti–HA head mAb compared to anti–HA stalk mAb. Values represent the mean ± s.e.m. frequency of CD107a+ cells among NK cells from cultures with target cells bound by the indicated antibody from 4 individual leukocyte donors. Results are representative of 3 independent experiments. Significant differences between the indicated sample and 6F12 huIgG1 sample are shown: **P < 0.01. (c) Immune complexes were generated between huIgG1 anti-stalk 6F12, anti-stalk FI6 or anti-head PY102 mAbs and PR8 HA and assessed for their ability to engage huFcγRIIIa Phe158. Values represent the mean ± s.e.m. relative OD from duplicate ELISA measurements, with background binding by N297A mutant versions of each antibody subtracted. Results are representative of 2 independent experiments.
Anti-stalk mAbs induce ADCC, but anti-head mAbs do not
As FcγRs did not contribute to protection during mAb-mediated inhibition of viral entry by 6F12 bNAb, we speculated that killing of infected cells by ADCC may be responsible for the FcγR-dependent survival of mAb-treated animals. Recent studies have suggested that some anti-influenza antibodies are more potent inducers of ADCC than others, possibly owing to the distinct structures that they recognize26–28, thus favoring productive engagement. Therefore, we determined the relative abilities of anti-stalk and anti–globular head mAbs to activate natural killer (NK) cells in an in vitro ADCC assay. We coated PR8 virus–infected A549 lung epithelial cells with huIgG1 versions of 6F12, FI6 or PY102 mAbs and incubated them with peripheral blood mononuclear cells (PBMCs) from four different donors for 3 h before assessing surface CD107a expression on CD56+CD3− NK cells by flow cytometry. CD107a (LAMP1) is a sensitive NK cell activation marker whose expression levels tightly correlate with cytokine production and cytotoxicity by NK cells29. Both anti-stalk 6F12 and FI6 huIgG1 mAbs similarly induced NK cell CD107a expression, whereas FI6 N297A (a mutant that does not engage FcγRs) was unable to induce CD107a expression on NK cells (Fig. 4b); these results indicate that these anti-stalk antibodies were able to mediate FcγR-dependent NK cell activation. By contrast, cells coated with the anti–globular head PY102 huIgG1 mAb induced little to no CD107a expression on NK cells (P < 0.0001). However, a PY102 mAb Fc mutant that augments interactions with activating FcγRs (GASD/ALIE mutant24) recovered CD107a expression on NK cells to levels comparable with that of anti-stalk mAbs, indicating that the Fc region of the anti-head mAb is still accessible to FcγRs and is not sterically hindered or spatially disrupted in some other way. Notably, PY102 mAb bound to the surface of infected cells similarly to or better than 6F12 and FI6 mAbs (Supplementary Fig. 11), and surface plasmon resonance studies showed that PY102 mAb has an approximately 3.4-fold greater affinity for PR8 HA than does 6F12 (PY102 Kd = 5.6 × 10−10 M; 6F12 mAb Kd = 1.9 × 10−9 M), with a higher ‘on rate’ (PY102 ka = 1.9 × 105 M−1 s−1 ; 6F12 mAb ka = 7.5 × 104 M−1 s−1) and a lower ‘off rate’ (PY102 kd = 1.1 × 10−4 M s−1; 6F12 mAb kd = 1.4 × 10−4 M s−1) compared to 6F12 mAb (Supplementary Fig. 12). Thus, this anti-head mAb is not characterized by a high off rate, ruling out this possibility as a reason for its inferior ADCC induction. We saw similar NK cell activation results with 2009 pandemic strain–infected target cells using all five broadly neutralizing anti-stalk mAbs and three strain-specific anti-head mAbs that we tested in vivo (Supplementary Fig. 13 and Table 1). Thus, these two classes of neutralizing antibodies were markedly different at inducing NK cell activation and thus ADCC.
Anti–HA head mAb immune complexes poorly interact with FcγRs
To mechanistically understand why the broadly neutralizing anti–HA stalk mAbs activate NK cells for ADCC but the strain-specific anti–HA head mAbs do not, we compared the ability of HA-bound anti-head and anti-stalk mAbs to engage FcγR. To study Fc-FcγR interactions the use of immune complexes is essential as activating FcγRs (other than the high-affinity FcγRI) do not engage monomeric IgG30. Thus, we generated immune complexes between PY102 anti–HA head and 6F12 and FI6 anti–HA stalk huIgG1 mAbs and PR8 HA. These immune complexes were similar in size, as they each contained similar amounts of bound IgG (Supplementary Fig. 14). We tested the ability of these immune complexes to engage huFcγRIIIa in an ELISA assay. Whereas FI6 and 6F12 mAb immune complexes efficiently engaged huFcγRIIIa, PY102 mAb immune complexes showed little to no FcγRIIIa binding (Fig. 4c). By contrast, a PY102 mAb Fc point mutant that augments interactions with activating FcγRs (GASD/ALIE mutant) recovered FcγRIIIa binding, again demonstrating that the Fc region of the anti-head mAb is accessible to FcγRs and not sterically hindered or spatially disrupted in some other way. Therefore, strain-specific anti–HA head mAbs do not activate NK cells for ADCC because they are unable to efficiently interact with activating FcγRs. These results indicate that binding by Fabs to specific epitopes on the HA molecule influences the ability of the Fc to engage cognate FcγRs.
Enhanced activating huFcγR engagement augments protection
The observation that anti-stalk antibodies require FcγR engagement for their in vivo protective activity suggested a strategy to enhance the potency of this class of antibodies for use as human therapeutics. However, the huFcγR system differs from that of mice in terms of its expression patterns, the affinities of each IgG Fc subclass for each FcγR and which FcγRs and effector cells dominate in vivo. Therefore, in order to address the functional contributions of huFcγRs during in vivo viral neutralization with a human bNAb, we used the recently described FcγR-humanized mice24. These mice have been bred to express the full array of huFcγRs on a background lacking all mouse FcγRs. FcγR-humanized mice recapitulate the unique expression profile of huFcγRs and function to mediate the inflammatory and cytotoxic activities of huIgG antibodies in vivo24.
The 6F12 heavy chain variable region (VH) and kappa light chain variable region (VK) sequences were cloned upstream of the human IgG1 and human kappa constant regions to generate a huIgG1 version of the 6F12 bNAb (Supplementary Table 1), which bound to PR8 HA and effectively neutralized PR8 virus in vitro (Supplementary Fig. 15). We tested whether huIgG1 6F12 bNAb protected against viral infection in FcγR-humanized mice infected with PR8 after bNAb administration. We saw no in vivo protection at a 2 mg/kg dose of huIgG1 6F12 bNAb but found that 36% of mice survived when receiving 4 mg/kg bNAb and 75% of mice survived after receiving 8 mg/kg bNAb (Fig. 5a). Thus, human 6F12 bNAb binds HA and engages the FcγR system in vivo to effectively protect mice during viral challenge.
Figure 5.

Selectively enhancing huFc-huFcγR interactions augments bNAb-mediated protection from viral infection in vivo. (a) Percentage weight change compared to day 0 and survival over time in FcγR-humanized mice treated with the indicated doses of wild-type huIgG1 6F12 bNAb or PBS before infection with PR8 virus. Weight change values represent mean ± s.e.m. n ≥ 4 mice per group. (b) Percentage weight change compared to day 0 and survival over time in FcγR-humanized mice treated with wild-type huIgG1 6F12 bNAb, GASD/ALIE mutant 6F12 bNAb or PBS before infection with PR8 virus. Weight change values represent mean ± s.e.m. n ≥ 12 mice per group. Significant differences between wild-type IgG1 mAb–treated and GASD/ALIE mAb–treated groups are shown: **P < 0.01.
We and others have described a series of point mutations in the huIgG1 Fc that allow for selective and enhanced engagement of huFcγRs. For example, the GASD/ALIE mutant Fc shows selective and enhanced affinity to the activating huFcγRs and binds to the FcγRIIA and FcγRIIIA allelic variants expressed in FcγR-humanized mice, huFcγRIIA Arg131 and huFcγRIIIA Phe158, with 25- and 30-fold increased affinities compared to wild-type huIgG1, but it only binds to huFcγRIIB with threefold increased affinity24, thereby selectively increasing the A/I ratio for this Fc variant. To test whether protection by an anti-stalk bNAb can be enhanced by selectively increasing the binding of the Fc to activating huFcγRs, we generated a GASD/ALIE mutant 6F12 bNAb. We investigated the ability of this bNAb construct to bind PR8 HA protein and neutralize the virus in vitro, and as expected, we detected no differences when compared with the wild-type IgG1 Fc (Supplementary Fig. 15). We also noted a similar t1/2 for both constructs (Supplementary Table 1).
To test whether we could augment in vivo protection against viral challenge by selectively enhancing engagement by activating huFcγRs, we treated FcγR-humanized mice with a suboptimal 4 mg/kg dose of wild-type huIgG1 6F12 bNAb or GASD/ALIE mutant 6F12 bNAb before challenging them with PR8 virus. Mice receiving wild-type huIgG1 bNAb lost 1.8-fold more weight than GASD/ALIE mutant bNAb–treated mice (P = 0.01; Fig. 5b) at day 7. Further, 73% of FcγR-humanized mice receiving the GASD/ALIE mutant bNAb survived, whereas only 25% of mice receiving wild-type huIgG1 bNAb survived (P = 0.04). Therefore, selectively engaging activating huFcγRIIA and huFcγRIIIA augmented in vivo protection during viral challenge by at least twofold.
DISCUSSION
Historically, influenza virus neutralization by mAbs reactive with the HA surface glycoprotein was thought to be mediated by blocking HA binding to sialic acid residues on the surface of the target cell or by inhibiting the viral fusion process6. These mechanisms may hold true during in vitro assays in which FcγR-expressing cells are not present. However, it is now clear that the in vivo mechanism of viral neutralization is more complex and involves not only Fab recognition of viral epitopes but also Fc interactions with the FcγR system. Our results with anti-stalk bNAbs indicate that both engagement of activating FcγRs and the disruption of the fusion process occur in vivo. It had been unclear at which steps of the viral life cycle FcγRs contribute during mAb-mediated neutralization. Our results now show that although free virus may be opsonized by bNAb that could potentially interact with FcγRs on effector cells for virus clearance, viral binding, fusion and entry are abrogated by bNAb in an FcγR-independent manner. Thus, it is likely that bNAbs recognize HA expressed on the surface of already infected cells, thereby activating FcγR-expressing monocytes, macrophages or NK cells to kill the infected cells through ADCC17,31,32, as has been observed for anti-tumor antibodies such as anti-CD20 (ref. 33), anti-HER2/neu34,35 and anti–epidermal growth factor receptor36 in both animal models and human populations. It is likely that these mechanisms, as well as other potential pathways, are at play during anti–HA stalk bNAb–mediated neutralization in vivo. However, we also demonstrate that at high doses of bNAb, in vivo protection is FcγR independent and thus ADCC independent. Therefore, the mechanisms that underlie in vitro neutralization, i.e., direct binding, blocking entry or blocking fusion, probably dominate at such high doses of mAb. As the majority of vaccination-elicited anti-HA antibodies are strain specific, head reactive and polyclonal, high levels of broadly reactive anti-stalk antibodies are unlikely to be achieved and the low serum concentrations of these antibodies may necessitate FcγR interactions to be effective. Mouse studies may suffer from some limitations because mice differ from humans in their lung physiology, sialic acid expression patterns and ability to transmit virus37. Influenza studies are often performed in ferrets, which are considered a more physiologically relevant model system. However, little to nothing is known about ferret antibody isotypes, the FcγRs expressed in ferrets or ferret FcγR expression patterns and signaling, thereby precluding mechanistic studies in ferrets. However, the wealth of information and genetic tools available to study FcγRs in mice has allowed us to make powerful mechanistic conclusions in this study. The mouse and human FcγR systems are quite similar, and FcγR mechanistic studies in mice have translated to patients with various diseases38,39. Moreover, we have increased the human relevancy of these studies by using FcγR-humanized mice to demonstrate that enhancing human Fc-FcγR interactions may confer augmented protection during lethal influenza challenge.
Notably, there seems to be a differential requirement for FcγR contributions during in vivo neutralization when anti-stalk bNAbs or strain-specific anti-head mAbs are compared. FcγRs were not required for in vivo protection by anti-head mAbs, even at suboptimal mAb doses, indicating that simply blocking viral binding to target cells by some mAbs may be sufficient to provide full protection in vivo. This difference in FcγR requirements may be due to multiple factors. The simplest explanation is that anti-head mAbs function by blocking viral binding and therefore entry to target cells, an efficient process that is sufficient for in vivo protection7,8. Furthermore, the disruption of fusion by anti-stalk mAbs may be an inefficient process in vivo, mandating that FcγR be involved for effective in vivo protection. Although it cannot be ruled out that the difference in the FcγR requirement between head and stalk antibodies may be due to the relative affinities of these mAbs for HA, our results indicate that both head and stalk mAbs show similar binding characteristics to cell surface HA; in fact, the anti-head mAb PY102 demonstrates a greater affinity and slower off rate than the anti-stalk bNAb 6F12. However, the finding that anti-stalk mAbs are superior inducers of ADCC compared to anti-head mAbs is compatible with a model in which neutralizing antibody blocks viral binding, fusion and entry in an FcγR-independent manner but infected cells express cell surface HA, which becomes a target for ADCC-mediating effector cells. We speculate that these two classes of neutralizing mAbs are coselected in response to viral challenge to provide multiple pathways for antiviral protection: those that require the FcγR effector system and induce ADCC of infected cells and those that do not. Whether all strain-specific anti-head mAbs neutralize in vivo independently of Fc-FcγR interactions and all anti-stalk bNAbs induce ADCC and require FcγRs for in vivo protection is a hypothesis with major clinical implications that must be further examined.
Further, we have now mechanistically clarified why some antibodies that bind influenza epitopes efficiently induce ADCC but others do not. We demonstrate that immune complexes between HA and anti–HA stalk bNAbs efficiently engage FcγR, whereas immune complexes between HA and anti–HA head mAbs do not. Our results also indicate that this process of differential FcγR engagement by anti-HA mAbs cannot be simply be explained by steric hindrance of the Fc or by Fc proximity to the surface of the target cell, as the ability of anti-head mAb to engage FcγR and induce NK cell activation and ADCC is rescued by introducing known point mutations into the Fc that augment Fc-FcγR interactions. Thus, the Fc is in an orientation that does not preclude FcγR engagement. Traditionally, the Fc domain of an antibody functions independently of the Fab region, and the ability of an antibody to induce ADCC relies of the affinity of the antibody and the density at which the antibody coats its target. However, because the anti-head and anti-stalk mAbs tested here bind similarly to target cells, it is possible that for certain Fabs, binding to particular epitopes may induce conformational changes in the Fc structure that alter its ability to engage FcγRs. This hypothesis is supported by several earlier studies40–44 and a very recent study in which conjugating small peptides to the Fab domain of an antibody induced Fc conformational changes45. Such induced conformational changes may explain the concept of ‘ADCC epitopes’, in which certain antigens elicit ADCC effector functions more efficiently than others26–28. Alternatively, our data could also be explained by a mechanism in which HA-bound strain-specific anti-head mAbs are unable to efficiently multimerize and stably interact with the low-affinity FcγRs, whereas anti-stalk mAbs are able to cluster so that their Fcs make high-avidity contacts with FcγRs. Future studies, including those analyzing other anti-head mAbs with various antigenic specificities, will allow us to further explore these mechanisms. Regardless of the biophysical mechanism by which FcγR binding is increased, our data indicate that the ability of an antibody to bind to FcγRs is linked to the specific epitope recognized by the Fab. Thus, generating mAbs or combinations of mAbs for use as passive protection treatments should involve not only Fabs optimized for antigen binding but also Fcs optimized to engage the appropriate effector systems. Moreover, future vaccination strategies should be designed to induce not only powerful bNAbs but also bNAbs that optimally elicit maximum effector function.
ONLINE METHODS
Viruses, cell lines and mice
The A/Puerto Rico/8/1934 (PR8) and A/Netherlands/602/2009 (Neth09) H1N1 viruses were grown in 10-d-old specific-pathogen–free embryonated chicken eggs (Charles River Laboratories, Wilmington, MA). MDCK, 293T and A549 cells were maintained in DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% FBS (Life Technologies), 100 units per ml of penicillin and 100 μg/ml of streptomycin (Life Technologies). C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Fcer1g−/− (ref. 46), Fcgr2b−/− (ref. 47), Fcgr1−/− (ref. 48), Fcgr3−/− (ref. 49), Fcgr4−/− (ref. 50) and FcRα-null24 mice on the C57BL/6 genetic background have been previously described. FcγR-humanized mice, which express all huFcγRs on the FcRα-null C57BL/6 genetic background, have been described24. All mice were maintained in a specific-pathogen–free facility at the Rockefeller University, all infections were performed in a BSL-2 facility at the Rockefeller University and all studies were approved by the Rockefeller University Institutional Animal Care and Use Committee. Female mice 6–8 weeks of age were used in all experiments.
Antibodies
To generate 6F12, PY102 and 7B2 mAb constructs, total RNA was obtained from hybridoma cells, and cDNA was generated by using SuperscriptIII reverse transcriptase (Life Technologies) and immunoglobulin gene–specific primers. The VH- and VK-encoding genes were amplified by PCR and cloned in-frame into mammalian expression vectors with mouse IgG2a, mouse IgG1, mouse DA265 mutant, mouse kappa, huIgG1 or human kappa Fc backbones. The FI6 VH- and VK-encoding genes were synthesized by Genewiz (South Plainfield, NJ), and plasmids containing 2G02, 2B06, 1F02 and 4C04 VH and VK genes were provided by P. Wilson (University of Chicago). The human GASD/ALIE Fc mutant was generated by site-directed mutagenesis with PCR amplification of the entire vector using complementary primers containing the desired point mutations, as described51. Antibodies were produced by transient transfection of 293T cells and subsequent protein G purification from culture supernatants, as described52. Alkaline-phosphatase–conjugated anti–mouse IgG (Cat. #0102-04), anti–human IgG (Cat. #2041-04) and anti–mouse kappa (Cat. #1050-04) were from SouthernBiotech (Birmingham, AL; 1:1000 dilution). Flow cytometry antibodies utilized included fluorescent-conjugated anti-huCD56 (Biolegend, 1:200 dilution, clone HCD56), anti-huCD3 (Biolegend, 1:200 dilution, clone SK7), anti-huCD107a (eBiosciences, 1:200 dilution, clone eBioH4A3) and anti–influenza NP mAb (Abcam, 1:100 dilution, clone 431). Mouse IgG2a or huIgG1 versions of an anti–human CD20 (clone CAT13.6E12) mAb were used as the isotype control mAb in ELISA and plaque reduction neutralization assays and in vivo protection experiments (produced via transient transfection of 293T cells).
ELISA assays and half-life measurements
To compare the binding characteristics of mAb mutants to HA, ELISA plates (Thermo Fisher Scientific, Waltham, MA) were coated with PR8 HA (BEI Resources, Manassas, VA) overnight at 4 °C, washed with PBS with 0.1% Tween 20, blocked with 1% BSA (Sigma-Aldrich, St. Louis, MO) in PBS and incubated with mAb diluted in PBS plus 1% BSA. Plates were incubated with the appropriate secondary antibody conjugated to alkaline phosphatase and developed with p-nitrophenyl phosphate (PNPP, Thermo Fisher Scientific).
To determine in vivo half-life of mAbs, wild-type mice were given 200 μg of mAb via intraperitoneal injection (i.p.) with serum harvested and HA-specific mAb concentrations determined by ELISA using plates coated with PR8 HA, as described above.
Plaque-reduction neutralization assay
Various dilutions of mAb were preincubated with 60 to 80 PFU of virus for 1 h at room temperature on a shaker. The virus and mAb mixture was then used to infect a monolayer of MDCK cells in duplicate in a six-well plate that was incubated at 37 °C for 1 h with intermittent rocking every 5–10 min. Without washing off the inoculum, an agar overlay that was supplemented with corresponding mAb dilutions was added to each well. Two days later, the monolayer was fixed with 4% paraformaldehyde in PBS for 30 min and then permeabilized with 0.5% Triton X-100 for 20 min. Cells were blocked with 5% nonfat milk in PBS for 30 min before incubation with an anti-HA mAb for 1 h at room temperature. An anti-mouse secondary antibody conjugated to horseradish peroxidase was used at a 1:1,000 dilution before plaques were visualized using TrueBlue peroxidase substrate (KPL, Gaithersburg, MD). Plaques were counted for each antibody, and the percentage inhibition was calculated versus PBS-treated wells. Alternatively, some plaques were counted using crystal violet staining.
In vivo viral challenge experiments
Mice (6–8 weeks old) received various doses of mAb via i.p. injection (in 200 μl) 2 h before being anesthetized with a ketamine (75 mg/kg)/xylazine (15 mg/kg) mixture and receiving an intranasal infection with 5 mLD50 of the PR8 or Neth09 virus in 30 μl. All mice were monitored daily, and their weights were recorded for 14 d. Death was determined by a 20% body weight loss threshold that was authorized by the Rockefeller University Institutional Animal Care and Use Committee.
To determine viral lung titers in mice treated with bNAb, mice received 4 mg/kg of the various 6F12 bNAb Fc constructs or PBS before intranasal infection with 5 mLD50 of PR8 virus. Three days later, the mice were killed and their lungs were harvested. The lungs were mechanically homogenized in 2 ml of PBS and centrifuged to pellet tissue debris before the supernatants were collected. Supernatant samples were stored at −80°C until titers were determined by plaque assay, as described previously53. To analyze NP expression in lung tissues, lungs from mice infected with 4 × 106 PFU of PR8 virus that was preincubated with various concentrations of mAb were digested with type II collagenase (Worthington Biochemicals, Lakewood, NJ) before intracellular immunofluorescence staining with flow cytometry analysis.
In vitro antibody-dependent cellular cytotoxicity natural killer cell activation assay
A549 lung epithelial cells were infected with an MOI of PR8 virus such that >95% of cells expressed surface HA. The infected A549 cells were labeled with various concentrations of various huIgG1 antibodies for 20 min, were washed twice and were cultured 1:1 with PBMCs purified from donor leukocytes (New York Blood Center, New York, NY) for 3 h in 96-well U-bottom plates. Surface CD107a expression by CD56+CD3− NK cells was assessed by flow cytometry analysis. IRB approval is granted under Rockefeller IRB registration number IRB00000385.
Immune complex binding assay
Immune complex generation and binding to FcγRs has been described in detail30. Briefly, immune complexes were generated by mixing antibody and antigen (HA) in a 1:1 molar ratio followed by overnight incubation at 4 °C. Immune complexes were then diluted and added to ELISA plates coated with 10 μg/ml soluble huFcγRIIIa Phe158. The plates were washed and incubated with alkaline phosphatase–conjugated goat Fab anti-huIgG (SouthernBiotech) and developed with p-nitrophenyl phosphate substrate. To determine the concentration of IgG in the immune complexes, anti–HA head and anti–HA stalk immune complexes were captured on ELISA plates coated with anti–HA stalk or anti–HA head mAb, respectively, and detected with goat Fab anti-huIgG. Concentrations of captured IgG were determined using a standard curve.
Surface plasmon resonance
All experiments were performed with a Biacore T100 SPR system (Biacore, GE Healthcare) at 25 °C in HBS-EP + buffer (10 mM Hepes (pH 7.4), 150 mM NaCl, 3.4 mM EDTA and 0.005% (vol/vol) surfactant P20). For the measurement of antibody affinity for PR8 HA, soluble protein G (diluted at 25 μg/mL in 10 mM sodium acetate, pH 4.5) was immobilized on a Series S CM5 chip by amine coupling at a density of 1,000 RU (response units). Anti–HA huIgG1 mAbs at 12.5 nM were injected through flowcells at a flow rate of 30 μL/min, with a contact time of 60 s. Next, recombinant PR8 HA was injected through flowcells at a flow rate of 20 μL/min at concentrations ranging from 200 nM to 0.390625 nM (1:2 successive dilutions). The association time was 120 s, followed by 300 s dissociation. At the end of each cycle, the sensor surface was regenerated with 100 mM glycine-HCL, pH 1.5 (50 μL/min; 40 s). Background binding to blank immobilized flow cells was subtracted, and affinity constants were calculated using Biacore T100 Evaluation software using the 1:1 Langmuir binding model.
Statistical analyses
Statistical differences between survival rates were analyzed by comparing Kaplan-Meier curves using the log-rank test and GraphPad Prism software. All other statistical differences were compared using the Student’s t-test analysis.
Supplementary Material
ACKNOWLEDGMENTS
We thank P. Wilson (University of Chicago), J. Wrammert (Emory University) and R. Ahmed (Emory University) for providing mAb constructs, F. Krammer (Mt. Sinai Medical Center) for providing recombinant soluble HA protein, S. Bournazos for assistance with surface plasmon resonance studies and J. Carroll and P. Smith for technical assistance. We also thank the Biodefense and Emerging Infections Research Resources Repository for supplying recombinant soluble HA protein. Research reported in this publication was partially supported by the National Institute of Allergy and Infectious Disease of the US National Institutes of Health (NIH) under award numbers P01AI081677, R01AI035875 and U54AI057158 to J.V.R. and AI097092 to P.P. Research support was also provided by the Bill & Melinda Gates Foundation grant OPP1033115 to J.V.R. This work was also supported in part by the US Army Research Laboratory and the US Army Research Office under contract number W911NF-13-2-0036 (to J.V.R.). Partial support for P.P. was provided by the Program for Appropriate Technology in Health and by the NIH-funded Center for Research on Influenza Pathogenesis (HHSN266200700010C). D.J.D. received fellowship support from the Leukemia and Lymphoma Society and received funding support from the New York Community Trust.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
Reprints and permissions information is available online at http://www.nature.com/reprints/index.html.
AUTHOR CONTRIBUTIONS D.J.D., G.S.T., P.P. and J.V.R. designed experiments, D.J.D. and G.S.T. performed the experiments, D.J.D., G.S.T., P.P. and J.V.R. analyzed data and D.J.D. and J.V.R. wrote the paper.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.World Health Organization Influenza (Seasonal) Fact sheet. 2009:211. http://www.who.int/mediacentre/factsheets/fs211/en/
- 2.Ahmed R, Oldstone MB, Palese P. Protective immunity and susceptibility to infectious diseases: lessons from the 1918 influenza pandemic. Nat. Immunol. 2007;8:1188–1193. doi: 10.1038/ni1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez O, Tsibane T, Basler CF. Neutralizing anti-influenza virus monoclonal antibodies: therapeutics and tools for discovery. Int. Rev. Immunol. 2009;28:69–92. doi: 10.1080/08830180802593540. [DOI] [PubMed] [Google Scholar]
- 4.Couch RB, Kasel JA. Immunity to influenza in man. Annu. Rev. Microbiol. 1983;37:529–549. doi: 10.1146/annurev.mi.37.100183.002525. [DOI] [PubMed] [Google Scholar]
- 5.Wang TT, Palese P. Biochemistry. Catching a moving target. Science. 2011;333:834–835. doi: 10.1126/science.1210724. [DOI] [PubMed] [Google Scholar]
- 6.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 7.Knossow M, et al. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology. 2002;302:294–298. doi: 10.1006/viro.2002.1625. [DOI] [PubMed] [Google Scholar]
- 8.Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289:373–378. doi: 10.1038/289373a0. [DOI] [PubMed] [Google Scholar]
- 9.Dreyfus C, et al. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barbey-Martin C, et al. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002;294:70–74. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- 11.Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sui J, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 14.Abboud N, et al. A requirement for FcgγR in antibody-mediated bacterial toxin neutralization. J. Exp. Med. 2010;207:2395–2405. doi: 10.1084/jem.20100995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bournazos S, Chow SK, Abboud N, Casadevall A, Ravetch JV. Human IgG Fc domain engineering enhances anti-toxin neutralizing antibody activity. J. Clin. Invest. doi: 10.1172/JCI72676. (in the press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schmitz N, et al. Universal vaccine against influenza virus: linking TLR signaling to anti-viral protection. Eur. J. Immunol. 2012;42:863–869. doi: 10.1002/eji.201041225. [DOI] [PubMed] [Google Scholar]
- 17.Huber VC, Lynch JM, Bucher DJ, Le J, Metzger DW. Fc receptor–mediated phagocytosis makes a significant contribution to clearance of influenza virus infections. J. Immunol. 2001;166:7381–7388. doi: 10.4049/jimmunol.166.12.7381. [DOI] [PubMed] [Google Scholar]
- 18.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 19.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 20.Tan GS, et al. A pan-H1 anti-hemagglutinin monoclonal antibody with potent broad-spectrum efficacy in vivo. J. Virol. 2012;86:6179–6188. doi: 10.1128/JVI.00469-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li GM, et al. Pandemic H1N1 influenza vaccine induces a recall response in humans that favors broadly cross-reactive memory B cells. Proc. Natl. Acad. Sci. USA. 2012;109:9047–9052. doi: 10.1073/pnas.1118979109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reale MA, et al. Characterization of monoclonal antibodies specific for sequential influenza A/PR/8/34 virus variants. J. Immunol. 1986;137:1352–1358. [PubMed] [Google Scholar]
- 23.Wrammert J, et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 2011;208:181–193. doi: 10.1084/jem.20101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc. Natl. Acad. Sci. USA. 2012;109:6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manicassamy B, et al. Protection of mice against lethal challenge with 2009 H1N1 influenza A virus by 1918-like and classical swine H1N1 based vaccines. PLoS Pathog. 2010;6:e1000745. doi: 10.1371/journal.ppat.1000745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jegaskanda S, et al. Cross-reactive influenza-specific antibody-dependent cellular cytotoxicity antibodies in the absence of neutralizing antibodies. J. Immunol. 2013;190:1837–1848. doi: 10.4049/jimmunol.1201574. [DOI] [PubMed] [Google Scholar]
- 27.Jegaskanda S, Weinfurter JT, Friedrich TC, Kent SJ. Antibody-dependent cellular cytotoxicity (ADCC) is associated with control of pandemic H1N1 influenza virus infection of macaques. J. Virol. 2013;87:5512–5522. doi: 10.1128/JVI.03030-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava V, et al. Identification of dominant ADCC epitopes on hemagglutinin antigen of pandemic H1N1 influenza virus. J. Virol. 2013;87:5831–5840. doi: 10.1128/JVI.00273-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 30.Nimmerjahn F, Ravetch JV. Analyzing antibody–Fc-receptor interactions. Methods Mol. Biol. 2008;415:151–162. doi: 10.1007/978-1-59745-570-1_9. [DOI] [PubMed] [Google Scholar]
- 31.El Bakkouri K, et al. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J. Immunol. 2011;186:1022–1031. doi: 10.4049/jimmunol.0902147. [DOI] [PubMed] [Google Scholar]
- 32.Jegerlehner A, Schmitz N, Storni T, Bachmann MF. Influenza A vaccine based on the extracellular domain of M2: weak protection mediated via antibody-dependent NK cell activity. J. Immunol. 2004;172:5598–5605. doi: 10.4049/jimmunol.172.9.5598. [DOI] [PubMed] [Google Scholar]
- 33.Tedder TF, Baras A, Xiu Y. Fcγ receptor–dependent effector mechanisms regulate CD19 and CD20 antibody immunotherapies for B lymphocyte malignancies and autoimmunity. Springer Semin. Immunopathol. 2006;28:351–364. doi: 10.1007/s00281-006-0057-9. [DOI] [PubMed] [Google Scholar]
- 34.Nordstrom JL, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 2011;13:R123. doi: 10.1186/bcr3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000;6:443–446. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 36.Yang X, et al. Cetuximab-mediated tumor regression depends on innate and adaptive immune responses. Mol. Ther. 2013;21:91–100. doi: 10.1038/mt.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Belser JA, Katz JM, Tumpey TM. The ferret as a model organism to study influenza A virus infection. Dis. Model. Mech. 2011;4:575–579. doi: 10.1242/dmm.007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 2007;19:239–245. doi: 10.1016/j.coi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 39.Nimmerjahn F, Ravetch JV. FcγRs in health and disease. Curr. Top. Microbiol. Immunol. 2011;350:105–125. doi: 10.1007/82_2010_86. [DOI] [PubMed] [Google Scholar]
- 40.Barkas T, Watson CM. Induction of an Fc conformational change by binding of antigen: the generation of protein A–reactive sites in chicken immunoglobulin. Immunology. 1979;36:557–561. [PMC free article] [PubMed] [Google Scholar]
- 41.Kilàr F, Zavodszky P. Non-covalent interactions between Fab and Fc regions in immunoglobulin G molecules. Hydrogen-deuterium exchange studies. Eur. J. Biochem. 1987;162:57–61. doi: 10.1111/j.1432-1033.1987.tb10541.x. [DOI] [PubMed] [Google Scholar]
- 42.Torres M, Fernandez-Fuentes N, Fiser A, Casadevall A. The immunoglobulin heavy chain constant region affects kinetic and thermodynamic parameters of antibody variable region interactions with antigen. J. Biol. Chem. 2007;282:13917–13927. doi: 10.1074/jbc.M700661200. [DOI] [PubMed] [Google Scholar]
- 43.Oda M, Kozono H, Morii H, Azuma T. Evidence of allosteric conformational changes in the antibody constant region upon antigen binding. Int. Immunol. 2003;15:417–426. doi: 10.1093/intimm/dxg036. [DOI] [PubMed] [Google Scholar]
- 44.Schlessinger J, Steinberg IZ, Givol D, Hochman J, Pecht I. Antigen-induced conformational changes in antibodies and their Fab fragments studied by circular polarization of fluorescence. Proc. Natl. Acad. Sci. USA. 1975;72:2775–2779. doi: 10.1073/pnas.72.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong H, et al. Peptide-conjugation induced conformational changes in human IgG1 observed by optimized negative-staining and individual-particle electron tomography. Sci. Rep. 2013;3:1089. doi: 10.1038/srep01089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takai T, Li M, Sylvestre D, Clynes R, Ravetch JV. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 47.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 48.Ioan-Facsinay A, et al. FcγRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 2002;16:391–402. doi: 10.1016/s1074-7613(02)00294-7. [DOI] [PubMed] [Google Scholar]
- 49.Hazenbos WL, et al. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc γ RIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 50.Nimmerjahn F, et al. FcγRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc. Natl. Acad. Sci. USA. 2010;107:19396–19401. doi: 10.1073/pnas.1014515107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li F, Ravetch JV. Inhibitory Fcγ receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030–1034. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcγRIV: a novel FcR with distinct IgG subclass specificity. Immunity. 2005;23:41–51. doi: 10.1016/j.immuni.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 53.Wang TT, et al. Broadly protective monoclonal antibodies against H3 influenza viruses following sequential immunization with different hemagglutinins. PLoS Pathog. 2010;6:e1000796. doi: 10.1371/journal.ppat.1000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.