

Abstract
A common genetic polymorphism that results in increased activity of the dopamine regulating enzyme COMT (the COMT Val158 allele) has been found to associate with poorer cognitive performance and increased susceptibility to develop psychiatric disorders. It is generally assumed that this increase in COMT activity influences cognitive function and psychiatric disease risk by increasing dopamine turnover in cortical synapses, though this cannot be directly measured in humans. Here we explore a novel transgenic mouse model of increased COMT activity, equivalent to the relative increase in activity observed with the human COMT Val158 allele. By performing an extensive battery of behavioral tests, we found that COMT overexpressing mice (COMT-OE mice) exhibit cognitive deficits selectively in the domains that are affected by the COMT Val158 allele, stimulus–response learning and working memory, functionally validating our model of increased COMT activity. Although we detected no changes in the level of markers for dopamine synthesis and dopamine transport, we found that COMT-OE mice display an increase in dopamine release capacity in the striatum. This result suggests that increased COMT activity may not only affect dopamine signaling by enhancing synaptic clearance in the cortex, but may also cause changes in presynaptic dopamine function in the striatum. These changes may underlie the behavioral deficits observed in the mice and might also play a role in the cognitive deficits and increased psychiatric disease risk associated with genetic variation in COMT activity in humans.
The catechol-O-methyl transferase (COMT) enzyme degrades catecholamines, including dopamine. A single nucleotide polymorphism in the human COMT gene (Val158) increases the thermostability of the enzyme—thereby increasing its level of activity compared to the 158Met allele (Chen et al. 2004). In healthy subjects, the Val158 allele has been found to associate with poorer cognitive performance, specifically on tasks involving executive function (Bruder et al. 2005). Also in healthy subjects, Val158 has been found to associate with increased binding of a D1 receptor PET ligand in the cortex, which is suggested to reflect a compensation for reduced dopamine tone (Slifstein et al. 2008). In clinical studies, the Val158 allele has been linked with increased risk for schizophrenia in some populations (Tunbridge et al. 2006), as well as a poorer response to antipsychotic treatment in affected individuals (Bertolino et al. 2007). The same polymorphism has also been found to influence vulnerability to environmental risk factors. For example, cannabis use in adolescence increases the risk of developing psychosis in adulthood selectively in Val158 homozygotes (Caspi et al. 2005).
These many associations have driven a great deal of interest into the function of the COMT protein, yet how increased COMT activity affects brain functions is still not fully understood. In the striatum, the dopamine transporter (DAT) is highly abundant and responsible for rapid dopamine uptake into dopaminergic terminals (Cass et al. 1993) where it is either packaged into storage vesicles or metabolized by monoamine oxidase (MAO) (Eisenhofer et al. 2004). In contrast, dopamine transporters are both less abundant and located farther from synaptic sites in prefrontal cortex (PFC) neurons (Sesack et al. 1998), where uptake by the norepinephrine transporter (NET) and subsequent metabolism by COMT predominates. It has therefore been proposed that the increase in COMT activity conferred by the Val158 allele selectively influences dopamine signaling in the PFC and not the striatum. However, as yet, no markers are available for measuring synaptic dopamine turnover in living human brain tissue. Studies with Comt gene knockout mice using microdialysis and HPLC to quantify extracellular levels of dopamine and dopamine metabolites have found that Comt deletion has differential effects on PFC and striatum (Yavich et al. 2007; Kaenmaki et al. 2010), but it is unclear how this may relate to the situation of increased COMT activity, relative to normal levels.
Genetic studies have determined that the human ancestral allele for COMT 158 encodes Val, and that Met is a derived allele (Palmatier et al. 1999). No such polymorphism exists in mice. Wild-type mice encode Leu at this position (Chen et al. 2004), which results in higher activity relative to either of the human alleles. Because in humans it is the allele with higher relative activity (Val) that is associated with poorer cognitive function and increased risk for psychiatric disorders, we generated a transgenic mouse model of increased COMT activity relative to the level in wild-type mice.
In humans and mice, endogenous COMT protein is present in several different cell types. Neuronal cells expressing COMT include pyramidal neurons, cerebellar Purkinje and granular cells, and striatal spiny neurons. Nonneuronal cells expressing COMT include microglial cells, intestinal macrophages, and astroglia (Myohanen et al. 2010). A transgenic mouse model of increased COMT activity selectively in neurons was previously reported (Papaleo et al. 2008). In that model, Comt overexpression occurred in all neurons, without any regional restriction. In order to identify the contribution of COMT activity level specifically in neurons restricted primarily to the forebrain, we utilized a promoter from the CamKIIα gene (Mayford et al. 1996).
Here we report the characterization of our novel mouse model of Comt overexpression (COMT-OE mice). We have quantified the relative increase in COMT activity and found it to be similar to the increase in relative activity measured in human brain tissue from COMT Val158 homozygotes compared to 158Met homozygotes (Chen et al. 2004). We also present the results from an extensive battery of behavioral tests, which identified specific cognitive deficits similar to those associated with the Val158 allele in humans. Furthermore, we investigated the effects of increased COMT activity on components of the dopamine system and, using cyclic voltammetry, we have identified an increase in the capacity to release dopamine in the striatum, suggesting a novel mechanism by which increased COMT activity may impact behavioral functions.
Results
Generation of a transgenic mouse model of increased COMT activity
To investigate the relationship among COMT activity level, dopamine regulation, and behavior, we developed a transgenic mouse model of a relative increase of wild-type Comt expression in neurons (COMT- OE mice). Because genetic variation in COMT activity is known to affect male and female subjects differently (Harrison and Tunbridge 2008), we used exclusively male mice for all experiments in this study.
To create this model, we generated mice in which wild-type Comt is under the control of the tetracycline response element, tet-operator (tet-O), and crossed these mice to a line in which the tetracycline transactivator tTA is driven by the CamKIIα promoter (Mayford et al. 1996). Bi-transgenic mice carrying both the CamKIIα-tTA and tet-O-Comt transgenes express the Comt transgene predominantly in forebrain neurons, with some staining also apparent in the midbrain (Fig. 1A). By using an in situ hybridization with cellular resolution, we observed transgene expression in the ventral tegmental area of the midbrain (Fig. 1B). Although CamKIIα expression is normally restricted to the forebrain, this unusual transgenic expression pattern is fortuitous because COMT has previously been localized to the cell bodies of melanized midbrain neurons (presumed to be dopamine neurons) in human tissue (Kastner et al. 1994). To determine whether transgenic Comt was expressed in dopamine neurons, we combined in situ hybridization using a transgene specific probe with immunostaining using an anti-TH antibody. Figure 1C shows co-expression of Comt transgenic mRNA and TH protein within the same VTA cells, demonstrating that transgene expression occurred in identified dopamine neurons.
Figure 1.

Analysis of Comt mRNA expression in COMT-OE Transgenic mice. (A) Radiolabeled oligonucleotide probe specific for the Comt transgene gene hybridized to midsagittal sections from a COMT-OE (top) and control mouse (bottom) shows expression of the transgene in the forebrain as well as in the midbrain. Staining in the cerebellum represents nonspecific binding as it is apparent in control as well as COMT-OE mice. (B) Coronal sections of COMT-OE mouse (left hemisphere) and control (right hemisphere) hybridized with a digoxigenin-labeled RNA riboprobe specific for the transgene reveals transgene expression in the midbrain. (C) VTA neurons from COMT-OE mice labeled for TH protein (green) and Comt transgenic mRNA (red) reveals coexpression in the cytoplasm (DAPI nuclear stain in blue). (D) A radiolabeled oligonucleotide probe specific for Comt mRNA reveals the total level of Comt expression (endogenous plus transgenic) in COMT-OE mice. (E) The same probe hybridized to sections from a control mouse shows endogenous Comt expression level. (F) A probe specific for the transgene hybridized to sections from a control mouse shows background signal. The approximate locations of the sections in D–F, relative to bregma (anterior–posterior [AP]), are provided beneath the sections.
To determine the extent of Comt mRNA overexpression in transgenic mice, we performed in situ hybridization using a Comt antisense probe that will hybridize to both the endogenous as well as transgenic Comt mRNA. Figure 1D demonstrates the level of expression of Comt mRNA in a series of coronal sections from a COMT-OE mouse, compared to the level of Comt mRNA in a control littermate (Fig. 1E).
Transgenic overexpression of COMT protein leads to a 30% increase in COMT enzyme activity in the forebrain
This increase in Comt mRNA expression resulted in an increase in COMT protein levels in the cortex, striatum, and hippocampus (Fig. 2A). As expected, COMT protein level in the cerebellum was unaffected because the transgene was not expressed in the cerebellum. We used an ex vivo COMT enzyme activity assay to confirm that transgenic Comt mRNA expression resulted in an increase in COMT enzymatic activity in the forebrain (Fig. 2B). A two-way ANOVA revealed a highly significant effect of genotype across the three forebrain structures tested, F(1,31) = 55.97, P < 0.0001. As expected, activity level in the cerebellum was unaffected by genotype (Control = 2253 ± 87.75, COMT-OE = 2202 ± 130.9, P = 0.750). The 20%–40% increase in activity observed across the forebrain of COMT-OE mice is equivalent to the increase in relative activity measured in human brain tissue from COMT Val158 homozygotes compared to 158Met homozygotes (Chen et al. 2004).
Figure 2.

Transgenic overexpression of COMT protein leads to a 30% increase in COMT enzyme activity in the forebrain. (A) Western blot confirmed an increase in COMT protein in the striatum. No such increase in COMT protein level was seen in the cerebellum, as expected. Each blot contains protein extract from three control and three COMT-OE mice. The blot containing cerebellum (right) also includes an extract from a COMT KO mouse to determine antibody specificity, and no signal was observed in this lane. (B) COMT-OE mice show a 20%–40% increase in COMT enzyme activity in the forebrain, while activity in the cerebellum is unaffected. (F.ctx) Frontal cortex: COMT-OE n = 3, Control = 3. (Str) Striatum: COMT-OE n = 8, Control = 7. (Hip) Hippocampus: COMT-OE n = 8, Control = 8. (Cb) Cerebellum: COMT-OE n = 8, Control = 8. (***) P < 0.0001.
Increased COMT activity does not result in generalized motor or cognitive deficits
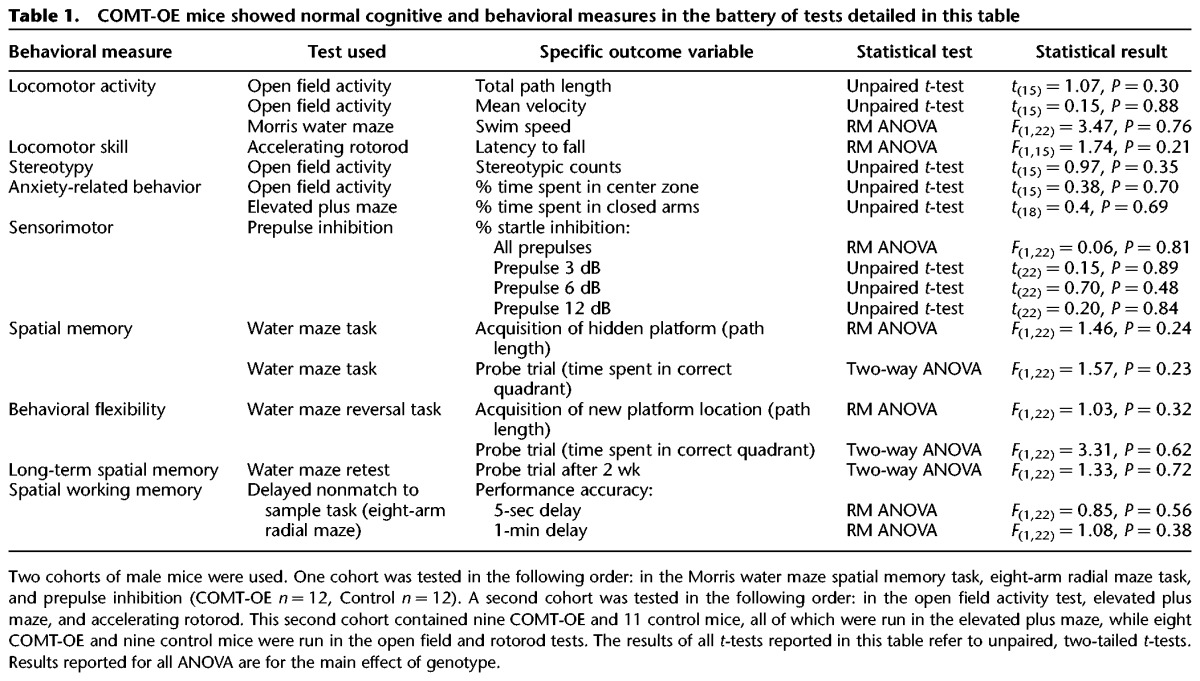
We examined COMT-OE and control littermate mice in an extensive battery of tests and found that COMT-OE mice do not present any generalized motor or cognitive deficits. These tests included measures of locomotor activity, stereotypy, motor coordination on a rotarod, anxiety-related behavior, sensorimotor gating, spatial memory, behavioral flexibility, long-term spatial memory, and spatial working memory. The tests we implemented, the specific outcome variables we measured, and the results of statistical analysis we used to compare COMT-OE and control mice are presented in Table 1. Two separate cohorts of mice were used; the tests administered to each group and the order of testing are presented in the legend for Table 1.
Table 1.
COMT-OE mice showed normal cognitive and behavioral measures in the battery of tests detailed in this table
Increased COMT activity results in specific cognitive deficits in stimulus–response learning and working memory
In contrast to the normal performance we saw in the tests listed in Table 1, like human subjects homozygous for the COMT Val158 allele, the COMT-OE mice show poorer performance in some cognitive domains linked to dopamine signaling in the striatum and prefrontal cortex (Diaz-Asper et al. 2008). Mice were tested on a conditional associative learning (CAL) paradigm (Bach et al. 2008) in which reinforcement was dependent upon selecting an action associated with one of two specific auditory cues (Fig. 3A). COMT-OE mice showed slower learning on this task (Fig. 3B), documented by a repeated measures ANOVA test for % correct over all 45 sessions. There was a significant main effect of session, F(44,1364) = 63.46, P < 0.0001, a significant effect of genotype, F(1,31) = 7.24, P = 0.0114, and a significant interaction between session and genotype, F(44,1364) = 1.73, P = 0.0025. There was no effect of genotype on performance in the first 4 d of the testing, during which the performance of all mice was not significantly different from chance as none of the subjects had yet begun acquiring the rule. This is evidenced by the fact that in this period of first exposure to the task there was not yet an effect of session, F(3,93) = 0.15, P = 0.927. There was also no effect of genotype, F(2,31) = 1.68, P = 0.202, and no interaction between session and genotype, F(6,93) = 0.33, P = 0.918. While displaying a specific learning deficit, COMT-OE mice ultimately reached the same level of performance at asymptote. A repeated measures ANOVA test for percent correct over the last 8 d of training revealed no significant main effect of session, F(7,217) = 0.41, P = 0.89, demonstrating that the mice had reached asymptote. During these 8 d of stable performance there was no effect of genotype, F(1,31) = 1.81, P = 0.19, and no significant interaction between genotype and session, F(7,217) = 1.30, P = 0.25. This equal performance at asymptote was also supported by an unpaired, two-tailed t-test for mean % correct for the last 8 d of training, t(31) = 1.35, P = 0.188 (Fig. 3C). After 45 sessions on the CAL task, we then tested nonspatial working memory by imposing a delay between the offset of the stimulus cue and the availability of the response lever and found that the COMT-OE mice performed more poorly than control mice (Fig. 3D). A repeated measures ANOVA test for % correct revealed a significant main effect of delay length, F(4,60) = 59.36, P < 0.0001, indicating that the increasing delay made the task more difficult. There was a significant main effect of genotype, F(1,15) = 5.15, P = 0.038, and no significant interaction between genotype and delay length, F(4,60) = 0.85, P = 0.49.
Figure 3.

COMT-OE mice show slower stimulus–response learning and a deficit in working memory. (A) Stimulus–response learning was tested in an operant task in which auditory cue 1 was rewarded if a lever press was made, while auditory cue 2 was rewarded if the mouse withheld from lever pressing. (B) COMT-OE mice showed slower learning on this task, though they reached the same performance level at plateau. (C) A comparison of performance on the last 8 d of training revealed no effect of genotype. COMT n = 15, Control n = 18. (D) Working memory was tested in the same operant schedule by imposing a delay between the offset of the auditory cue and the availability of the response lever. COMT-OE mice performed worse than control mice across the delay sessions. Control n = 10, COMT n = 7. (*) P < 0.05.
COMT-OE mice show signs of impulsive and compulsive behavior in an operant assay of attention
Because COMT-OE mice showed slower learning on the CAL task we explored what specific aspects of their behavior might underlie this learning defect. Learning the CAL task requires attending to the cues and choosing a go or no-go response. We therefore tested the mice in the 5-Choice Serial Reaction Time Task (5-CSRTT), an established assay for selective attention that also provides a measure of the subject’s ability to selectively respond to cues at the appropriate time. We trained the mice to attend to five cue lights and reinforced nose poke responses made in the panel directly beneath the single light that was randomly selected to be activated for each trial. The duration of the stimulus (light) presentation was gradually decreased over training sessions. Figure 4A shows that COMT-OE mice required more sessions to reach training criterion (a session with <20% trial omission and >80% accuracy) for almost all stimulus durations. A repeated measures ANOVA test for sessions to criteria revealed a significant main effect of genotype, F(1,12) = 5.27, P = 0.0405. After mice reached criteria on the 1-sec stimulus duration version of the task they were tested on a range of shorter stimulus durations (1 sec, 0.5 sec, 0.25 sec, and 0.125 sec), one session for each stimulus duration condition. Figure 4B shows that % correct during these short stimulus duration effects was not affected by genotype, confirmed by the results of a repeated measures ANOVA test, F(1,10) = 1.83, P = 0.206. Therefore, as in the CAL task, COMT-OE mice showed slower acquisition of the 5-CSRRT, but once acquired were able to perform as well as controls.
Figure 4.

COMT-OE mice require more training on the 5-CSRTT and show signs of impulsive and compulsive behavior. (A) COMT-OE mice required significantly more training sessions to reach criterion compared to control mice. (B) Once COMT-OE mice had acquired the 1-sec stimulus duration version of the 5-CSRTT, they were equally accurate at the shorter stimulus durations. (C) During testing on the shorter stimulus durations, COMT-OE mice made more premature responses. (D) During testing on the shorter stimulus durations, COMT-OE mice made more perseverative responses. Control n = 6, COMT-OE n = 6. (*) P < 0.05.
In addition to measuring attention, the 5-CSRTT can also be used to measure impulsive and compulsive behaviors (Robbins 2002). An increase in responding prematurely, before the stimulus has been presented, is considered to indicate impulsive behavior, while perseveratively continuing to respond to any stimulus after the trial has been completed indicates compulsive behavior. We found that during testing on the short stimulus durations, COMT-OE mice made significantly more premature and perseverative responses (Fig. 4C,D, respectively). Average number of premature responses made per session: Control = 3.66 ± 0.37, COMT-OE = 7.61 ± 1.42, unpaired t-test, t(10) = 2.69, P = 0.0226. Average number of perseverative responses made per session, Control = 5.47 ± 1.048, COMT-OE = 11.66 ± 1.736, t(10) = 3.05, P = 0.0122. This result shows that COMT-OE mice display an increase in inappropriate lever pressing activity which may explain why they are slower to learn the go no-go CAL task.
Increased COMT activity does not alter the level of TH or DAT in the striatum
An increase in unspecific or inappropriate responding in operant conditioning paradigms has been reported in multiple perturbations that result in increased levels of extracellular dopamine such as following treatment with amphetamine (Robbins 2002) or in a genetic knockdown of the dopamine transporter DAT (Yin et al. 2006). We therefore investigated whether increased COMT activity results in compensatory changes in markers of dopamine synthesis or transport. Figure 5A shows no difference in the level of tyrosine hydroxylase (TH), the enzyme which catalyzes a rate-limiting step in the synthesis of catecholamines in the striatum of COMT-OE and in control mice. Figure 5B shows that the dopamine transporter protein DAT is similarly unaffected and quantification of these immunoblots confirmed no significant effect of genoptype on the level of these two proteins. For TH levels, Control = 1.16 ± 0.061, COMT-OE = 1.093 ± 0.044, unpaired t-test, t = 0.865(4), P = 0.436. For DAT levels, Control = 1.99 ± 0.274, COMT-OE = 2.38 ± 0.106, t = 1.32(4), P = 0.257.
Figure 5.

COMT hyperactivity does not alter the level of TH or DAT in the striatum. (A) Striatum (STR) extracts from COMT-OE and control mice probed with anti-TH antibody shows no difference in the level of TH protein. Cerebellum (CER) extracts were used as a negative control for the TH and DAT antibodies. An anti-tubulin antibody was used as a loading control and to normalize band density. (B) No difference in the level of DAT protein was detected between genotypes. (C) Quantification of the TH bands. (D) Quantification of the DAT bands. Control n = 3 mice, COMT-OE n = 3 mice.
Increased COMT activity results in increased dopamine release capacity in the striatum
Changes in the level of TH and DAT protein would represent a major compensatory shift in the dopamine system. It is possible that more subtle compensatory changes occur in COMT-OE mice. Because dopamine signaling in the cortex has been found to be important for working memory (Goldman-Rakic et al. 2000) the working memory deficit observed in COMT-OE mice could be due to perturbed dopamine signaling in the PFC; however, this is difficult to measure in mice, due to the low levels of dopamine present. On the other hand, the deficit in stimulus–response learning may come from suboptimal dopamine functioning in the dorsomedial striatum (Yin et al. 2008), which can more readily be assayed in vivo. We therefore investigated the consequences of COMT overexpression on dopamine activity in the dorsomedial striatum using fast-scan cyclic voltammetry (FSCV), a technique that allows striatal dopamine to be measured in vivo on a subsecond time scale (Millar et al. 1985). We electrically stimulated the medial forebrain bundle (MFB), which comprises the mesostriatal, mesolimbic, and mesocortical projection fibers, in urethane-anesthetized mice and recorded dopamine release within the dorsomedial striatum (Fig. 6A). We found a significant increase in dopamine release in COMT-OE mice compared to littermate controls. Figure 6B shows a representative example of the dopamine peaks obtained for each genotype in this experiment. Figure 6C shows that the average peak dopamine current obtained was significantly affected by genotype, Control = 13.8 ± 1.4 nA, COMT-OE = 24.83 ± 4.0 nA, f(24) = 2.81, P = 0.009. Figure 6D shows there was also a significant difference in the amount of dopamine released (area under the curve), Control = 32.85 ± 2.95 nA s, COMT-OE = 61.09 ± 9.66 nA s, f(24) = 3.17, P = 0.004. In contrast, the rate of dopamine decay after release was unchanged, Control = 1.071 ± 0.176 sec, COMT-OE = 0.918 ± 0.229 sec, f(24) = 0.668, P = 0.51 (Fig. 6E). Together these results suggest that increased COMT activity impacts dopamine function in the striatum and this impact is primarily on the capacity for dopamine release, rather than on the rate of clearance of extracellular dopamine.
Figure 6.

Dopamine release capacity is increased in the dorsomedial striatum of COMT-OE Mice. (A) Schematic of the placement of the recording and stimulating electrodes for FSCV experiments in anesthetized mice (image from Paxinos and Franklin [2001]). (B) Example peaks from two animals showing dopamine release in the dorsomedial striatum in response to a 1-sec, 60-Hz, 150-µA stimulation of the medial forebrain bundle (represented by the gray line below the x-axis, 5 sec after the start of recording). (C) Average peak size was significantly higher in COMT-OE mice. (D) Average area under the curve, representing total release volume, was also significantly increased in COMT-OE mice. (E) Average rate of decay was not altered. Control n = 15, COMT-OE n = 11. (*) P < 0.05, (**) P < 0.005.
Discussion
Genetic variation leading to increased COMT activity is associated with decreases in cognitive function and increased risk for developing psychiatric disorders (Tunbridge et al. 2006). Here we have modeled increased COMT activity in the forebrain and in midbrain dopaminergic neurons in mice. The relative increase in COMT activity is quantitatively similar to the relative increase resulting from the human COMT Val158 allele compared to COMT 158Met (Chen et al. 2004). This level of increase in activity resulted in deficits in stimulus response learning and working memory, which is negatively affected by the Comt Val158 allele in humans (Dickinson and Elvevag 2009). Interestingly, COMT-OE mice displayed normal spatial working memory in a T-maze, but a deficit in nonspatial working memory. This may be due to the nature of the information which must be maintained (spatial information, versus the choice between “go” and “no-go” action). Alternatively, it may be due to the sensitivity of each task. The T-maze task involves only four trials per day and therefore is not suitable for picking up subtle deficits. In contrast, the CAL memory task involves 55 trials per day and is therefore more sensitive to differences in performance that are relatively mild.
Further behavioral analysis suggests that COMT-OE mice are impaired in stimulus response learning because they have poorer control over their response actions, displaying more premature and perseverative responses in an operant test of attention. The distinct behavioral phenotype of the COMT-OE mice validates our COMT hyperactivity model and also suggests that increased COMT activity restricted to forebrain and midbrain neurons is sufficient to result in cognitive impairment. It has previously been reported that mice with increased COMT activity in all neurons display cognitive deficits (Papaleo et al. 2008). To our knowledge, we provide the first evidence of a cognitive impairment resulting from an increase in COMT activity selectively in forebrain and midbrain neurons. Interestingly there are both similarities and differences between our COMT-OE mice and the pan neuronal Comt overexpression model described by Papaleo and colleagues. While both models display cognitive deficits, only the pan neuronal model exhibited anxiety-related behavior. This may be due to Comt overexpression in either a neuronal cell-type or brain region not targeted by the CAMKIIα-tTA construct that we employed.
The cognitive deficits observed in COMT-OE mice were accompanied by an increase in dopamine release capacity in the striatum, with no change in the rate of decay of extracellular dopamine. This finding is somewhat unexpected given the current predominant hypothesis about the mechanism by which COMT activity influences cognitive function. Because the dopamine transporter (DAT) is present at low levels in the cortex and is localized somewhat distant from synapses (Sesack et al. 1998), it is assumed that COMT activity plays a dominant role in the synaptic turnover of dopamine, and increased activity results in more rapid clearance of dopamine from the synapse. In contrast, because of DAT’s relative abundance and localization proximal to synapses in the striatum (Cass et al. 1993), COMT activity has been demonstrated to have little influence on the clearance of dopamine from the synapse in the striatum (Kaenmaki et al. 2010). Therefore it has been proposed that the increase in COMT activity conferred by the VAL158 allele impacts cognitive function by enhancing the inactivation of extracellular dopamine directly at synapses, selectively in the cortex. Our result suggests that genetic variation in COMT activity may also influence dopamine release, and that this modulation occurs subcortically, in the striatum. While our finding is novel, other data have recently been published that may question the above assumption about how COMT regulates dopamine and suggest that mechanisms other than synaptic turnover are involved. COMT exists in both soluble (S-COMT) and membrane bound (MB-COMT) isoforms. MB-COMT has been presumed to be responsible for the breakdown of extracellular dopamine at cortical synapses. However, recent studies suggest that MB-COMT may be intracellularly oriented (Schott et al. 2010). Further, MB-COMT may not, in fact, reside on plasma membranes, but instead is present either in the cytoplasm or on cytosolic membrane (Myohanen et al. 2010). Therefore dopamine may first have to be transported inside the cell for inactivation by COMT, rather than this action occurring directly in the synaptic space.
The observation that increased COMT activity resulted in an increase in striatal dopamine release and not clearance prompts the question whether altered COMT activity may affect presynaptic dopamine function. In the human striatum COMT immunoreactivity has been detected in both the cell bodies and dendritic spines of medium spiny neurons (MSNs), but not in the presynaptic terminals which contact MSN dendritic spines (Kastner et al. 1994; Karhunen et al. 1995). However, COMT has been localized to the cell bodies of melanized midbrain neurons (Kastner et al. 1994). To determine whether the Comt transgene expression in the midbrain occurred in dopamine neurons, we performed a double fluorescence staining procedure using a riboprobe specific for the Comt transgene and an antibody selective for the dopamine cell marker tyrosine hydroxlyase (TH). We found double positive cells in both the ventral tegmental area (VTA) and substantia nigra (SN) of the midbrain, areas with projections to striatal, cortical, and limbic structures, confirming the dopamine identity of neurons expressing the transgene.
Our data indicate that COMT activity levels may influence dopamine regulation presynaptically and this would be consistent with the observation that COMT Val158 carriers show increased F-dopa uptake in vivo (Meyer-Lindenberg et al. 2005) compared to carriers of the COMT 158Met allele. The authors of this study suggest that increased F-dopa uptake may be a secondary compensation to the postulated decrease in cortical extracellular dopamine levels caused by COMT hyperactivity (Akil et al. 2003; Meyer-Lindenberg et al. 2005). However, given the evidence that COMT may be expressed in presynaptic dopamine neurons, increased F-dopa uptake may, in fact, also result from a cell autonomous developmental compensation for the increase in COMT activity within dopamine midbrain neurons.
It is well established that dopamine signaling in the dorsal striatum strongly influences motor control. While a reduction in dopamine tone occurs in Parkinson’s disease, drug-induced increase in dopamine release can result in hyperlocomotor activity. It has recently been shown that selective activation of striatal medium spiny neurons (MSNs) in the indirect pathway elicits a parkinsonian state with decreased locomotor initiations, while activation of direct-pathway MSNs reduces freezing and increases locomotion (Kravitz et al. 2010). The increase in dopamine release capacity observed in COMT-OE mice should simulate the direct pathway via D1 receptors and inhibit the indirect pathway via D2 receptors. This may elicit the question of whether the deficits we observed in cognitive tasks are perhaps driven by an underlying alteration in motor behavior. However, we saw no deficits in motor performance on an accelerating rotorod, or in locomotor activity in either an open field or during maze tasks (Table 1). This is consistent with the finding that mice with increased COMT activity in all neurons of the brain including the dorsal striatum showed normal locomotor activity (Papaleo et al. 2008). Further evidence that the poor performance of COMT-OE mice in cognitive tasks is not driven by a defect in motor control is provided by the selective nature of the cognitive deficits we observed. In the case of the CAL task, the mice eventually perform at an accuracy level equal to control mice, it is only during training that they differ. If poor task performance was due to a motor incapacity then poor accuracy on the task would presumably persist. The observed deficits in the 5-CSRTT are also unlikely to be due to an underlying motor defect because the animals do not perform poorly in all conditions of the task. COMT-OE mice making more premature and perseverative responses occurred only during the trials with a higher attentional demand (short cue duration trials). Again, if this is driven by impairment in motor performance, it would be expected that the increase in perseverative and premature responses would occur in all sessions, regardless of the duration of cue presentation.
The changes we observed in dopamine release in the striatum of COMT-OE mice could be responsible for the deficits observed in the learning of stimulus–response associations in the mice because similar behavioral phenotypes have been observed in models of increased extracellular dopamine (Robbins 2002; Yin et al. 2008). However, further experiments would be required to determine if these phenotypes are causally related. For example, recording DA release in the striatum in response to conditioned cues in awake animals would provide information about behaviorally evoked DA release. Blockade of DA receptors during such recordings might establish causal involvement. While our data raise the possibility of a presynaptic role for increased COMT activity in modulating dopamine release capacity at mesostriatal synapses, our study does not address the relative importance of increased COMT activity at pre- or postsynaptic sites, as both sites are targeted in our model. Thus, the observed deficits in stimulus–response learning and working memory could result from increased COMT activity in caudate-putamen, frontal cortex, SN/VTA, or a combination of these sites. Future studies are required to make this important distinction. Nevertheless, our findings suggest that further investigations into the mechanisms by which a relative increase in COMT activity alters cognition and affects psychiatric disease risk should include consideration of both sides of the synapse, and to do so in both subcortical as well as cortical structures. Understanding the mechanisms by which genetic modulation of COMT enzyme activity affects dopamine function is important because it will significantly influence our understanding, and capacity for treating, the cognitive deficits associated with psychiatric diseases.
Materials and Methods
Generation of COMT-OE transgenic mice
Mice expressing wild-type Comt under control of the tet-operator (tet-O-Comt mice) were crossed to mice expressing the tetracycline transactivator (tTA) transgene under the calcium/calmodulin-dependent kinase IIα promoter (CamKIIα-tTA mice (Mayford et al. 1996), and offspring were used for behavioral and molecular analysis. To control for genetic background, we followed the recommendations made by the Banbury conference on genetic background in mutant mice (Silva 1997). Namely, that mutations be maintained in congenic lines, and that mutants be analyzed in a defined hybrid (and preferably F1) genetic background. Therefore tet-O-Comt mice were maintained on a congenic C57BL/6J background and CamKIIα-tTA mice were maintained on a congenic 129S6/SvEvTac background. Crossing these lines resulted in F1 offspring including double transgenic mice which carry both the tet-O-Comt and CamKIIα-tTA transgenes and which express the Comt transgene in forebrain neurons (COMT-OE mice). Mice were genotyped by triplex polymerase chain reaction (PCR) using primers specific for tTA, tet-O, and a fragment of the endogenous D1 receptor gene (to provide a positive control for the PCR). To specifically test the effect of transgenic Comt overexpression we compared double transgenic mice to control mice that included single-transgenic and wild-type littermates. Behavior testing began when the mice were a minimum of 3 mo of age. To motivate mice to earn rewards in the operant tasks, mice were single housed, weighed every day, and provided enough regular maintenance chow to maintain their bodyweight at 85% of their normal bodyweight on an ad lib diet. Water was available ad libitum. Mice were housed, bred, and tested according to the local IACUC guidelines. They were maintained on a 12-h light–dark cycle and tested during the light phase.
Immunoblotting (COMT)
The striatum and cerebellum were dissected from COMT-OE and control littermate mice. Tissue was homogenized in lysis buffer (25 mM Tris pH 8.0, 125 mM NaCl, 1% Triton, with complete protease inhibitor cocktail [Roche]). Samples were centrifuged at 16 kg for 30 min at 4°C and the supernatant collected. A Biorad protein assay kit (Biorad) was used to determine protein concentration and 10-µg total protein was loaded in each lane of a 15% SDS Page gel. After transfer to Immobilon-FL PDVF membrane (Millipore) the blots were incubated with a purified mouse monoclonal anti-mouse catechol-O-methyltransferase antibody (BD Transduction Laboratories) 1:5000, then an HRP conjugated goat anti-mouse Ig antibody (1:5000). Amersham ECL Plus reagents (GE Healthcare Life Sciences) were used for detection. Following detection, blots were stripped and reprobed for tubulin as a loading control.
Immunoblotting (TH and DAT)
Tissue was homogenized in radioimmunoprecipitation buffer containing 25 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, and a protease inhibitor cocktail (Roche). Homogenates were rotated at 4°C overnight, centrifuged at 12 kg for 20 min at 4°C, and the supernatant collected. Protein concentration was determined as described above and samples were resolved on a 15% SDS Page gel. Fifteen micrograms and 30 µg of total protein were loaded per well for the TH and DAT blots, respectively, and gels were electrotransferred onto PVDF membranes. TH was detected using a mouse-monoclonal anti-TH antibody (Millipore MAB318) at 1:2500 (4°C overnight) followed by 1:2500 anti-mouse HRP antibody incubation for 2 h at RT. DAT was detected using a rat-generated anti-DAT antibody (Millipore MAB369) and an anti-rat HRP antibody at the same dilutions and incubation conditions. HRP on both blots was detected by chemiluminescent reaction to ECL-Plus (Amersham). Following detection, blots were stripped and reprobed for tubulin as a loading control.
COMT enzyme activity assay
COMT enzyme activity was assayed by measuring the incorporation of a radioactive methyl group into a catechol substrate in tissue homogenates as first described by Zurcher and Da Prada (1982) and adapted by Chen et al. (2004) and scaled down by us for these experiments. Briefly, brain regions were dissected on ice and homogenized in 10 × W/V buffer (50% glycerol, 1 mM Tris pH 7.4 with 1 × Complete-Mini EDTA-free protease inhibitor cocktail [Roche]) and stored at −80°C until used. For each reaction 2-μg tissue (20-μL extract) was transferred to a glass scintillation minivial, to each vial 250 μL of susbtrate mix (10 mM Tris pH 7.4, 1 mM MgCl2, 10-μL catechol [Sigma 135011], 1 μM DTT, 3.0 μCi/mL of 3H-SAM [Amersham], TRK614 Spec. activity 15 ci/mmol) was added. After 60-min incubation at 37°C the reactions were stopped by the addition of 1 M HCl. Monoflow I organic scintillation fluid (5 mL [National Diagnostics]) was then added and the mixture vigorously shaken. The 3H-labeled product of the reaction in the organic phase was then measured by scintillation counting, while the 3H-SAM remained in the aqueous phase. Reactions were run in duplicate for frontal cortex, striatum, and cerebellum, triplicates were run for hippocampal samples. Independent single reactions containing the 10 mg/mL of the COMT inhibitor Tropolone (FLUKA 93555) were also carried out for each sample to determine nonspecific activity, which was subtracted from the average of the uninhibited values obtained for each sample. Enzyme activity was calculated as counts per minute (Cpm) of incubation per microgram tissue.
Behavioral testing
Behavioral testing began when mice were 3 mo of age. The behavioral analysis listed in Table 1 included open field activity, elevated plus maze, spatial Morris water maze task, and prepulse inhibition. We have previously described the detailed methods used for each of these tests (Kellendonk et al. 2006). Table 1 also contains the analysis of an accelerating rotarod test in which the mice were tested for 2 d, four trials per day with a 1-h intertrial interval (ITI). The rotarod accelerated ∼10–37 rpm and each trial was terminated when the mouse fell off, or after a maximum of 5 min.
Conditional associative learning
Stimulus–response learning was measured using a conditional associative learning task (Bach et al. 2008), and nonspatial working memory was tested by imposing delays in this task.
Mice were tested daily, 5 d per week in standard mouse operant chambers equipped with ultrasensitive, retractable mouse levers, a tone generator, house light, and a pellet dispenser. The chambers were enclosed in sound-attenuating boxes (MED Associates). Correct responses were reinforced with a 20-mg Dustless Precision Pellet (Bio-serve).
Pretraining
Mice progressed through three different phases on a continuous reinforcement schedule. For each trial, it was randomly determined which lever, left or right, would be presented. On phase 1, the lever was presented for 60 sec and reinforcement occurred at the end of each trial regardless of whether or not a response occurred. On phase 2, the lever was present only for 30 sec and reinforcement was contingent on a lever press. On phase 3, the lever was present for 10 sec, and again, reinforcement was contingent on a response. Across all three phases, the intertrial interval (ITI) was 10 sec and the session ended when 55 reinforcers were earned or when 2 h elapsed. The mice advanced to the next phase when two or fewer trials occurred when no response was made.
Acquisition of the CAL task began immediately on completion of phase 3. For each trial, a random number generator determined which auditory stimulus (tone or pulsating tone) and which lever (left or right) would be presented. Reinforcement depended on learning the two stimulus–response associations, which were as follows: If stimulus 1 is present, then lever press is reinforced, whereas if stimulus 2 is present, withholding from responding is reinforced (the two stimulus–response associations were counterbalanced across all groups). Acquisition took place through trial and error. Each trial began with the onset of the auditory stimulus, and after 10 sec, one lever was inserted into the chamber. If a correct lever press response was emitted, a reinforcer was delivered immediately, the lever was retracted, and the auditory stimulus was terminated, ending the trial. If an incorrect lever press response was emitted (i.e., lever press during the presentation of stimulus 2), the lever was retracted and the trial was terminated. If no response was emitted 5 sec after the lever was presented (15 sec from the onset of the cue), the lever was retracted, the auditory stimulus ended, and reinforcement occurred if stimulus 2 had been presented. The ITI was 10 sec, and the session ended when 55 reinforcers were earned. The mice advanced to the delayed version of the task after five consecutive days of performing with an accuracy of 78% or better.
Delayed version of the CAL Task involved the insertion of a delay between the offset of the auditory stimulus and the insertion of the lever. The following four delays were used: 1, 2.5, 5, and 10 sec. Only one delay was used throughout a session, and each mouse was tested on each delay three times. Performance was assessed on the delayed version two of five sessions per week.
5-Choice Serial Reaction Time Task (5-CSRTT)
The 5-Choice Serial Reaction Time Task was performed essentially as described for the rat attention task in Chudasama and Robbins (2004), with some modifications for mice.
Apparatus
Testing was conducted in a standard mouse operant chamber equipped with a five hole nose poke wall (Med Associates). Each nose poke hole is equipped with an LED for illumination as well as an infrared photocell beam to record nose poke responses. On the chamber wall opposite the five nose poke wall was a receptacle providing access to a liquid dipper which was used to deliver evaporated milk as a reinforcer. Mice were tested twice daily (once in the morning and once in the afternoon) with 30-min training or testing sessions.
Training
Each trial began with the house light on and random illumination of one of the five nose poke holes for 1 min. If the mouse poked its nose into a hole while the hole was illuminated, or within 1 min of illumination offset (the limited hold [LH] period), this was recorded as a correct response and the mouse was reinforced with the dipper raised for 2 sec to provide access to the milk. A nose poke in other nonilluminated holes was recorded as an incorrect response. Incorrect responses and failure to nose poke into any hole within the LH period (omission) resulted in termination of the trial followed by a timeout or 10-sec intertrial interval (ITI) during which the house light is turned off. Timeouts were also imposed when the mouse nose pokes during the ITI (anticipatory responses). Multiple nose pokes into the correct hole (perseverative responses) were recorded but had no scheduled consequences. The cue duration and LH period were reduced on subsequent sessions according to the animal’s performance. Mice were trained on multiple sessions with cue duration and LH period of 1 min, 30 sec, 15 sec, 8 sec, 4 sec, 2 sec, 1 sec in that order. Mice started with the 1-min condition and progressed to the next condition after achieving >80% correct responses accompanied by <20% omissions over five consecutive sessions. The number of trials required to reach criteria on each condition were analyzed and presented.
Testing was conducted the same as training, except the duration of cue presentation continued to be decreased, while the LH period remained fixed at 1 sec. Rather than the mice being tested on multiple sessions until they reached criterion level performance, all mice were tested for five sessions for each cue duration, 1 sec, 0.5 sec, 0.25 sec, 0.125 sec.
Fast-scan cyclic voltammetry in anesthetized animals
Fast-scan cyclic voltammetry was performed as previously described (Zweifel et al. 2009). Briefly, animals were anesthetized (urethane, 450 mg/kg, i.p.), a 7-µm thick carbon-fiber microelectrode was positioned in the dorsomedial striatum (coordinates: anterior posterior [AP] 0.5 mm, medial lateral [ML] −2.2 mm, dorsal ventral [DV] −2.4 mm), and an Ag/AgCl reference, implanted under the skull bone (Wightman et al. 1986). The potential applied to the carbon-fiber microelectrode was held at –0.4 V (versus Ag/AgCl) and then linearly scanned at 400 V/s to +1.3 V and back every 100 msec. Dopamine release was evoked by stimulating the medial forebrain bundle (MFB). For this purpose a bipolar stimulating electrode was positioned in the MFB (AP −2.0 mm, ML medial wire −0.7 mm and lateral wire −1.5 mm, initial DV was −3.8 mm. The electrode was then lowered in 0.1 steps to obtain maximal dopamine release. The DV range for all subjects was between −3.8 mm and −4.2 mm. The stimulating electrode was connected to a battery-operated constant current unit (A-M Systems), which was used to generate biphasic current pulses of 150-µA intensity (2 msec/phase), delivered at 60 Hz for 1-sec duration. Dopamine was resolved using TarHeel CV analysis software (Heien et al. 2004) and by matching the voltammogram generated with a known reference voltammogram, based on peak sizes, area under the curve (AUC), and decay rates (Yavich et al. 2005).
After the recordings, brains were extracted and were cut in half. The striatum was sectioned in the coronal plane and stained with Nissl to determine the location of the recording electrode; the midbrain was sectioned in the sagittal plane and stained with an anti-tyrosine hydroxylase antibody (described below) to determine the location of the stimulating electrode.
Histology
DNA oligo in situ hybridization
Mice were killed by cervical dislocation, and the brains were dissected and rapidly frozen. Cryostat sections (20 μm) were post-fixed in 4% paraformaldehyde in PBS (pH 7.4), dehydrated, and stored in 100% ethanol at 4°C until use. Sections were hybridized to a 44-base antisense oligonucleotide specific to the transgenic mRNA (5′ GGA TTT TAG GGG CGC TTA CCT GTA GCC ATT GCA GCT AGG TGA GC 3′) which is specific to the human growth hormone polyA sequence that was cloned into the transgenic construct. Oligonucleotide (50 ng) was labeled with 50 μCi of [α33P]dATP (Perkin Elmer) using recombinant terminal transferase (Roche). Sections were hybridized with 100 cpm/mL oligonucleotide in hyb buffer (50% formamide [Fluka], 10% dextran sulfate, 4 × SSC, 50 mM sodium phosphate pH 7, 10 mM sodium pyrophosphate, 5 × Denhardt’s, 200 μg/mL denaturated salmon sperm DNA, 200 μg/mL poly[dA]) at 42°C overnight. Slides were washed with 1 × SSC first at 60°C for 30 min then 1 × SSC and 0.1 SSC at room temperature for 5 min. Slides were dehydrated with ethanol and exposed to autoradiographic film for 2 wk.
RNA riboprobe in situ hybridization
To achieve cellular resolution, we also performed in situ hybridization analysis using a digoxigenin-labeled cRNA probe against the human growth hormone polyA sequence. DIG in situ hybridization was carried out exactly as described by Schaeren-Wiemers and Gerfin-Moser (1993).
Combined RNA riboprobe in situ hybridization and immunohistochemistry
A combination of in situ hybridization and immunohistochemistry was used to detect both transgenic mRNA and TH protein in single brain slices. Combined RNA riboprobe in situ hybridization and immunohistochemistry: fresh frozen 20-µm cryosections were thaw mounted onto Superfrost Plus slides (VWR). To determine specificity of RNA riboprobe staining, sections from both COMT-OE and control mice were included on each slide. Sections were post-fixed 15 min, RT, w/4% PFA/PBS, then washed with PBS. Endogenous peroxidase activity was blocked by incubating at RT for 30 min in 3% H2O2/PBS, followed by PBS washes. Slides where then incubated in 0.1 M triethanolamine-HCl at RT for 10 min followed by PBS washes. Prehybridization for 4 h at RT was carried out in hyb buffer (50% deionized formamide, 10 mM Tris-HCl pH 8, 200 µg/mL yeast tRNA, 10% dextran sulfate, 1 × Denhardt’s solution, 600 mM NaCl, 0.25% SDS, 1 mM EDTA pH 8). For each slide 1 μL of digoxigenin-labeled cRNA probe against the transgene specific human growth hormone polyA sequence (the same probes used for the standard in situ above) was mixed with 200 μL of hyb buffer, denatured at 85°C, and applied to each slide. A glass cover slip was applied and then slides were incubated ON in a chamber humidified with 50% formamide in DEPC H2O at 68°C. Slides were extensively washed: 5 × SSC at 68°C for 10 min, 2 × SSC/50% formamide at 68°C for 30 min, 2 × SSC at 68°C for 20 min, 0.2 × SSC at 68°C for 20 min, 0.2 × SSC at 68°C for 20 min. Slides were then incubated for 10 min at RT with TN buffer (100 mM Tris-HCl pH7.5, 150 mM NaCl), then blocked for 2 h in TNB (10% FBS in TN buffer) before overnight incubation at 4°C with the following antibodies diluted in TNB: anti-DIG-POD (Roche) 1:5000, mouse monoclonal anti-tyrosine hydroxylase antibody 1:1000 (MAB318, Millipore). After washing with TNT (TN buffer with 0.05% Tween), the cRNA Dig probe was detected using a Tyramide Signal Amplification (TSA) Plus- Cy3 kit following the manufacturer’s instructions (PerkinElmer), then washed with TNT. For TH antibody detection the slides were then incubated for 2 h at RT with goat anti-mouse Alexa Fluor 488 at 1:1000 (Invitrogen) in TNB. After washing with TNT, slides were cover slipped with Vectashield hard set mounting medium with DAPI (Vector Labs).
Fluorescent imaging
Fluorescently stained sections were viewed using a laser scanning confocal microscope (Fluoview FV1000, Olympus) and images were captured using Fluoview software. Sections were scanned sequentially for each fluorophore.
Acknowledgments
This work was supported by The Lieber Institute for Brain Development (E.R.K. and V.W.) and the Howard Hughes Medical Institute (E.R.K.). We thank Heidi K. Smith, Tessa Hirschfeld-Stoler, Iram Haq, and Svetlana Vronskaya for technical assistance. We also thank J.A. Gogos for the COMT KO mouse used for Western blot analysis.
References
- Akil M, Kolachana BS, Rothmond DA, Hyde TM, Weinberger DR, Kleinman JE 2003. Catechol-O-methyltransferase genotype and dopamine regulation in the human brain. J Neurosci 23: 2008–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach ME, Simpson EH, Kahn L, Marshall JJ, Kandel ER, Kellendonk C 2008. Transient and selective overexpression of D2 receptors in the striatum causes persistent deficits in conditional associative learning. Proc Natl Acad Sci 105: 16027–16032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolino A, Caforio G, Blasi G, Rampino A, Nardini M, Weinberger DR, Dallapiccola B, Sinibaldi L, Douzgou S 2007. COMT Val158Met polymorphism predicts negative symptoms response to treatment with olanzapine in schizophrenia. Schizophr Res 95: 253–255 [DOI] [PubMed] [Google Scholar]
- Bruder GE, Keilp JG, Xu H, Shikhman M, Schori E, Gorman JM, Gilliam TC 2005. Catechol-O-methyltransferase (COMT) genotypes and working memory: associations with differing cognitive operations. Biol Psychiatry 58: 901–907 [DOI] [PubMed] [Google Scholar]
- Caspi A, Moffitt TE, Cannon M, McClay J, Murray R, Harrington H, Taylor A, Arseneault L, Williams B, Braithwaite A, et al. 2005. Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol Psychiatry 57: 1117–1127 [DOI] [PubMed] [Google Scholar]
- Cass WA, Zahniser NR, Flach KA, Gerhardt GA 1993. Clearance of exogenous dopamine in rat dorsal striatum and nucleus accumbens: role of metabolism and effects of locally applied uptake inhibitors. J Neurochem 61: 2269–2278 [DOI] [PubMed] [Google Scholar]
- Chen J, Lipska BK, Halim N, Ma QD, Matsumoto M, Melhem S, Kolachana BS, Hyde TM, Herman MM, Apud J, et al. 2004. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet 75: 807–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW 2004. Dopaminergic modulation of visual attention and working memory in the rodent prefrontal cortex. Neuropsychopharmacology 29: 1628–1636 [DOI] [PubMed] [Google Scholar]
- Diaz-Asper CM, Goldberg TE, Kolachana BS, Straub RE, Egan MF, Weinberger DR 2008. Genetic variation in catechol-O-methyltransferase: effects on working memory in schizophrenic patients, their siblings, and healthy controls. Biol Psychiatry 63: 72–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson D, Elvevag B 2009. Genes, cognition and brain through a COMT lens. Neuroscience 164: 72–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G, Kopin IJ, Goldstein DS 2004. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 56: 331–349 [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Muly EC III, Williams GV 2000. D(1) receptors in prefrontal cells and circuits. Brain Res 31: 295–301 [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Tunbridge EM 2008. Catechol-O-methyltransferase (COMT): a gene contributing to sex differences in brain function, and to sexual dimorphism in the predisposition to psychiatric disorders. Neuropsychopharmacology 33: 3037–3045 [DOI] [PubMed] [Google Scholar]
- Heien ML, Johnson MA, Wightman RM 2004. Resolving neurotransmitters detected by fast-scan cyclic voltammetry. Anal Chem 76: 5697–5704 [DOI] [PubMed] [Google Scholar]
- Kaenmaki M, Tammimaki A, Myohanen T, Pakarinen K, Amberg C, Karayiorgou M, Gogos JA, Mannisto PT 2010. Quantitative role of COMT in dopamine clearance in the prefrontal cortex of freely moving mice. J Neurochem 114: 1745–1755 [DOI] [PubMed] [Google Scholar]
- Karhunen T, Tilgmann C, Ulmanen I, Panula P 1995. Catechol-O-methyltransferase (COMT) in rat brain: immunoelectron microscopic study with an antiserum against rat recombinant COMT protein. Neurosci Lett 187: 57–60 [DOI] [PubMed] [Google Scholar]
- Kastner A, Anglade P, Bounaix C, Damier P, Javoy-Agid F, Bromet N, Agid Y, Hirsch EC 1994. Immunohistochemical study of catechol-O-methyltransferase in the human mesostriatal system. Neuroscience 62: 449–457 [DOI] [PubMed] [Google Scholar]
- Kellendonk C, Simpson EH, Polan HJ, Malleret G, Vronskaya S, Winiger V, Moore H, Kandel ER 2006. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron 49: 603–615 [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Freeze BS, Parker PR, Kay K, Thwin MT, Deisseroth K, Kreitzer AC 2010. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466: 622–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER 1996. Control of memory formation through regulated expression of a CaMKII transgene. Science 274: 1678–1683 [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Kohn PD, Kolachana B, Kippenhan S, McInerney-Leo A, Nussbaum R, Weinberger DR, Berman KF 2005. Midbrain dopamine and prefrontal function in humans: interaction and modulation by COMT genotype. Nat Neurosci 8: 594–596 [DOI] [PubMed] [Google Scholar]
- Millar J, Stamford JA, Kruk ZL, Wightman RM 1985. Electrochemical, pharmacological and electrophysiological evidence of rapid dopamine release and removal in the rat caudate nucleus following electrical stimulation of the median forebrain bundle. Eur J Pharmacol 109: 341–348 [DOI] [PubMed] [Google Scholar]
- Myohanen TT, Schendzielorz N, Mannisto PT 2010. Distribution of catechol-O-methyltransferase (COMT) proteins and enzymatic activities in wild-type and soluble COMT deficient mice. J Neurochem 113: 1632–1643 [DOI] [PubMed] [Google Scholar]
- Palmatier MA, Kang AM, Kidd KK 1999. Global variation in the frequencies of functionally different catechol-O-methyltransferase alleles. Biol Psychiatry 46: 557–567 [DOI] [PubMed] [Google Scholar]
- Papaleo F, Crawley JN, Song J, Lipska BK, Pickel J, Weinberger DR, Chen J 2008. Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J Neurosci 28: 8709–8723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ 2001. The mouse brain in stereotaxic coordinates, 2nd ed Academic Press, San Diego, CA [Google Scholar]
- Robbins TW 2002. The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology (Berl) 163: 362–380 [DOI] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A 1993. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry 100: 431–440 [DOI] [PubMed] [Google Scholar]
- Schott BH, Frischknecht R, Debska-Vielhaber G, John N, Behnisch G, Duzel E, Gundelfinger ED, Seidenbecher CI 2010. Membrane-bound catechol-O-methyl transferase in cortical neurons and glial cells is intracellularly oriented. Front Psychiatry 1: 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Hawrylak VA, Matus C, Guido MA, Levey AI 1998. Dopamine axon varicosities in the prelimbic division of the rat prefrontal cortex exhibit sparse immunoreactivity for the dopamine transporter. J Neurosci 18: 2697–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ 1997. Mutant mice and neuroscience: recommendations concerning genetic background. Banbury Conference on genetic background in mice. Neuron 19: 755–759 [DOI] [PubMed] [Google Scholar]
- Slifstein M, Kolachana B, Simpson EH, Tabares P, Cheng B, Duvall M, Frankle WG, Weinberger DR, Laruelle M, Abi-Dargham A 2008. COMT genotype predicts cortical-limbic D1 receptor availability measured with [11C]NNC112 and PET. Mol Psychiatry 13: 821–827 [DOI] [PubMed] [Google Scholar]
- Tunbridge EM, Harrison PJ, Weinberger DR 2006. Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry 60: 141–151 [DOI] [PubMed] [Google Scholar]
- Wightman RM, Kuhr WG, Ewing AG 1986. Voltammetric detection of dopamine release in the rat corpus striatum. Ann N Y Acad Sci 473: 92–105 [DOI] [PubMed] [Google Scholar]
- Yavich L, Jakala P, Tanila H 2005. Noradrenaline overflow in mouse dentate gyrus following locus coeruleus and natural stimulation: real-time monitoring by in vivo voltammetry. J Neurochem 95: 641–650 [DOI] [PubMed] [Google Scholar]
- Yavich L, Forsberg MM, Karayiorgou M, Gogos JA, Mannisto PT 2007. Site-specific role of catechol-O-methyltransferase in dopamine overflow within prefrontal cortex and dorsal striatum. J Neurosci 27: 10196–10209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Zhuang X, Balleine BW 2006. Instrumental learning in hyperdopaminergic mice. Neurobiol Learn Mem 85: 283–288 [DOI] [PubMed] [Google Scholar]
- Yin HH, Ostlund SB, Balleine BW 2008. Reward-guided learning beyond dopamine in the nucleus accumbens: the integrative functions of cortico-basal ganglia networks. Eur J Neurosci 28: 1437–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurcher G, Da Prada M 1982. Rapid and sensitive single-step radiochemical assay for catechol-O-methyltransferase. J Neurochem 38: 191–195 [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, et al. 2009. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci 106: 7281–7288 [DOI] [PMC free article] [PubMed] [Google Scholar]