

Abstract
The intestinal barrier becomes compromised during systemic inflammation, leading to entry of luminal bacteria into the host and gut origin sepsis. Pathogenesis and treatment of inflammatory gut barrier failure is an important problem in critical care. In this study we examined the role of cyclooxygenase-2 (COX-2), a key enzyme in the production of inflammatory prostanoids, in gut barrier failure during experimental peritonitis in mice. I.p. injection of LPS or cecal ligation and puncture (CLP) increased the levels of COX-2 and its product prostaglandin E2 (PGE2) in the ileal mucosa, caused pathologic sloughing of the intestinal epithelium, increased passage of FITC-dextran and bacterial translocation across the barrier, and increased internalization of the tight junction-associated proteins JAM-A and ZO-1. Luminal instillation of PGE2 in an isolated ileal loop increased transepithelial passage of FITC-dextran. Low doses (0.5–1 mg/kg), but not a higher dose (5 mg/kg) of the specific COX-2 inhibitor Celecoxib partially ameliorated the inflammatory gut barrier failure. These results demonstrate that high levels of COX-2-derived PGE2 seen in the mucosa during peritonitis contribute to gut barrier failure, presumably by compromising tight junctions. Low doses of specific COX-2 inhibitors may blunt this effect while preserving the homeostatic function of COX-2-derived prostanoids. Low doses of COX-2 inhibitors may find use as an adjunct barrier-protecting therapy in critically ill patients.
Keywords: peritonitis, gut barrier failure, gut origin sepsis, cyclooxygenase-2, prostaglandin E2, Celecoxib
The intestinal epithelium serves as a barrier that prevents systemic entry of bacteria, toxins, and antigens. Derangement of the gut barrier with its associated bacterial translocation may play an important role in the pathogenesis of sepsis in critically ill patients (1–4). Gut-origin sepsis may complicate necrotizing enterocolitis (5) shock (6–8), burns (9–11), trauma (12–14), and acute illness (15–17). Furthermore, inflammatory gut barrier failure is a part of multiple organ dysfunction syndrome (3, 18). Finding ways to prevent or ameliorate the inflammatory gut barrier failure is therefore an important area of ongoing basic and clinical research.
How inflammation leads to gut barrier failure is not well understood. Barrier function of the intestinal epithelium critically depends on tight junctions (TJ), the structures that seal enterocyte borders. TJ consist of integral proteins (occludin and claudins) that form a tight seal of the apical intercellular space, and associated cytosolic proteins (zonula occludens-1, ZO-1; and junction-associated molecule-A, JAM-A) that connect the TJ to intracellular actinomyosin complexes (19). TJ undergo continuous remodeling during epithelial renewal, as evidenced by internalization and re-deposition of the junctional proteins (20). Under conditions causing barrier breakdown, a dynamic balance between these two opposing processes is shifted towards the internalization. A variety of inflammatory factors including IL-1β (21, 22), IL-4 (23), IL-6 (24), TNF (25–31), IFN-γ (32–35), High Mobility Group B (36, 37), nitric oxide (38–41), and inflammatory prostanoids (42–45) each can increase gut barrier permeability in vitro and/or in vivo, indicating the multifactorial character of gut barrier failure.
Roles of prostanoids in gut barrier function under normal and pathologic conditions are fairly complex. Prostanoids are oxygenated fatty acids derived from membrane arachidonate via the rate-limiting oxygenation and peroxidation reactions catalyzed by COX-1 and COX-2 cyclooxygenases. Since COX-1 COX-2 double KO mice die early postpartum, the cyclooxygenase activity is essential (46). The main prostanoid in the intestine is PGE2 (47). Low levels of prostanoids produced under normal conditions by constitutive expression of COX-1 and basal expression of COX-2 regulate ion transport, intestinal secretion, cell migration, blood vessel and smooth muscle tone; they suppress production of inflammatory cytokines and are required for barrier maintenance and intestinal homeostasis (48). High levels of prostanoids resulting from transcriptional induction of COX-2 during inflammation orchestrate the innate and adaptive immune responses (49). They promote production of nitric oxide and vasodilation (48), angiogenesis (50), hyperalgesia (51, 52), smooth muscle contraction (53), and enterocyte proliferation (54). Increased expression of COX-2 is one of the key factors in the pathogenesis of gut barrier failure during inflammatory bowel disease (55–57) and necrotizing enterocolitis (58). Apparently, responses to low and high concentrations of prostanoids are quite different.
If high levels of prostanoids damage the gut barrier, inhibition of prostanoid production may seem an obvious treatment strategy. Under the assumption that protective effects of low-level prostanoids are provided by COX-1, COX-2 appears the preferred target. Indeed, under certain scenarios COX-2 inhibitors have been reported to protect the barrier (59–62). However, other studies have found that COX-2 inhibitors do not protect, but rather aggravate the barrier damage in experimental colitis and necrotizing enterocolitis (63–66). Moreover, specific COX-2 inhibitors have been found to exhibit gut toxicity, albeit not to the extent of nonspecific COX inhibitors (67, 68). This is not surprising, since both COX-1 and COX-2 are necessary for barrier protection under adverse conditions (69, 70), with COX-2 actually being more important than COX-1 in this role (71). Thus, it might seem that COX-2 inhibitors are not useful as a therapy for inflammatory gut barrier failure.
We hypothesized that low doses of a specific inhibitor that attenuate, rather than completely inhibit COX-2, would protect the barrier during inflammation by dampening the detrimental effects of high levels, yet preserving the beneficial effects of low levels of COX-2 activity. This hypothesis was tested using experimental peritonitis models in mice. LPS injection or cecal ligation and puncture (CLP) increased expression of COX-2 and levels of PGE2 in the ileal mucosa and caused barrier derangement. The barrier was also compromised by luminal instillation of PGE2. Low (0.5 mg/kg), but not high (5 mg/kg) dose of the COX-2-specific inhibitor Celecoxib significantly protected the barrier during peritonitis. Low doses of COX-2 inhibitors may thus find use as adjunct barrier protection therapy in critically ill patients.
Materials and Methods
Reagents and Antibodies
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. Commensal E. coli strain 35354T was obtained from ATCC (Manassas, VA) and transformed with the pUC18 plasmid to confer ampicillin resistance. Antibodies were from the following suppliers: COX-2 (cat. 16016), EP1 (cat.101740), EP2 (cat. 101750), EP3 (cat. 101760) and EP4 (cat. 101755) receptors, Cayman Chemical (Ann Arbor, MI); iNOS (cat. 610432), BD Transduction Laboratories (San Jose, CA); ZO-1 (cat. B2129), Lifespan Biosciences (Seattle, WA), JAM-A (cat. 361700), Invitrogen (Carlsbad, CA); β-actin (cat. A1978), Sigma-Aldrich. All primary antibodies were rabbit polyclonal except iNOS and β-actin, which were mouse monoclonal. Secondary antibodies for immunofluorescence (FITC or Texas Red-conjugated donkey anti-rabbit) or Western blot (horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse) were from Jackson ImmunoResearch (West Grove, PA). PGE2 (cat. 514531) and IL-6 (cat. 583371) ELISA kits were from Cayman Chemical.
Animals
All procedures were designed in adherence to the National Institutes of Health Guidelines on Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Children’s Hospital Los Angeles. C57Bl/6J mice were either bred in-house or purchased from Jackson Labs (Sacramento, CA). COX-2-deficient mice were purchased from Jackson Labs and bred heterozygous. cox-2−/− mice were derived from crosses involving cox-2+/− parents. Mice were genotyped as recommended by the supplier. Healthy animals of both sexes, 8–12 wk old and weighting 18–26 g were used in experiments. We did not observe significant differences between sexes or between younger and older animals in this study. Survival surgeries were performed under isoflurane anesthesia and followed by buprenorphine s.c. to minimize post-operative pain. Experimental peritonitis was induced by i.p. injection of 40 mg/kg LPS from E. coli 0127:B8, or by CLP (72). In the latter procedure, the cecum was externalized through abdominal incision, tied with a silk suture, and punctured two times with 21-gauge needle. The cecum was returned to the peritoneal cavity and incision closed with staples. Sham operation was performed similarly, but punctures were omitted.
Assessment of barrier function
30 min prior to the induction of peritonitis, mice were orally gavaged with 200 μl of test mixture, normal saline (NS) containing 22 mg/ml FITC-Dextran, MW=4 kDa, and 5×108 cfu/ml ampicillin-resistant E. coli 35354T. 16 h post LPS injection or CLP, blood samples (~400 μl) were collected by cardiac puncture. Serum was prepared by centrifugation at 10,000xg for 10 min. Concentrations of FITC-dextran in serum were determined by fluorometry and interpolation to standard curve. Mesenteric lymph nodes, spleens, and livers were aseptically excised, weighted, ground in a Dounce homogenizer, and serially diluted. Titers of E. coli were determined by plating dilutions onto LB agar supplemented with 100 mg/L ampicillin.
Effect of luminally-instilled PGE2
2 h before surgery, mice were gavaged with 200 μl of 22 mg/ml FITC-dextran in NS. Isoflurane-anesthetized animals were placed on a warm pad. A 3 cm loop of terminal ileum was externalized through abdominal incision and clamped. 200 μl of 50 μM PGE2 in NS was injected into the lumen of the isolated loop through a 27-gauge needle. The externalized intestinal loop was covered with moist gauze to prevent drying. 25 min after PGE2 treatment, blood was collected for measuring serum FITC-dextran.
Immunoflorescence microscopy
4 μm paraffin sections of terminal ileum were deparaffinized and re-hydrated by boiling in 10 mM sodium citrate. After blocking with 5% normal donkey serum, the sections were incubated with primary and secondary Cy3-conjugated antibodies as recommended by manufacturers. Slides were mounted with DAPI mounting medium. Images were acquired on an LSM 700 confocal system mounted on an AxioObserver Z1 microscope equipped with a 40×/1.3 Plan-NEOFLUAR oil-immersion lens and controlled by ZEN 2009 software (Carl Zeiss Microimaging, Thornwood, NY). Fluorescence excitation lasers and emission filters were 405 nm excitation with short-pass 490 nm emission for DAPI and 555 nm excitation with long-pass 560 nm emission for Cy3. The confocal pinhole was set to 1 Airy unit for Cy3. To increase the contiguous area that was imaged, the optical zoom factor was set to 0.5 and adjacent fields of view were tiled together with no overlap. For comparisons, sections were processed in parallel on the same slide, and images were acquired and adjusted identically.
Western blots
Mucosal scrapings from terminal ileum were extracted on ice with buffer (20 mM Tris pH 8.0, 100 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with 0.5 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mM benzamidine. Solubilized proteins (50 μg) were separated by SDS-PAGE. Gels were electroblotted onto nitrocellulose membranes, which were then blocked in PBS-Tween with 2% fish gelatin, incubated with primary antibody, washed, and incubated with horseradish peroxidase-conjugated secondary antibody. After washing and soaking in luminol-peroxide reagent, membranes were exposed to X-ray film.
ELISA
30 mg (wet wt) of ileal mucosal scrapings were homogenized with 1 ml PBS supplemented with 0.5 mM PMSF in a Dounce homogenizer. The homogenate was cleared by centrifugation. ELISA was performed using PGE2 and IL-6 kits as directed by their manufacturers.
Statistics
Quantitative data were compared using Wilcoxon’s test or Student’s t test of the statistical software package JMP≤ (Cary, NC). A p-value of < 0.05 was considered significant.
Results
Peritonitis is associated with COX-2 induction and high levels of prostaglandin E2 in ileal mucosa
To gain insight into the mechanisms of inflammatory gut barrier failure in vivo, we employed the two well-characterized mouse models of systemic inflammation/sepsis/peritonitis, i.p. injection of LPS and CLP. Each model has its advantages and disadvantages: the former is more controlled but less clinically relevant, whereas the opposite is true for the latter. We reasoned that using two different models would at least partially mitigate their disadvantages. The dose of LPS or the parameters of the CLP procedure (number of punctures and needle gauge) were adjusted to cause ~50% mortality in 36 h. Within hours after peritonitis-inducing treatments, mice developed outward signs of fever including lethargy, hunched posture, ruffled fur, and anorexia. By the time of sacrifice (16 h) over 96% of animals were alive.
Systemic inflammation is associated with high levels of prostanoids resulting from upregulation of COX-2. We hypothesized that high levels of COX-2-derived prostanoids, particularly PGE2, might be directly responsible for the increased gut barrier permeability. Since prostanoids are metabolically labile and act locally (73), such a mechanism would imply induction of the COX-2 gene and increased concentrations of PGE2 in the intestinal tissue. To test this, we assessed expression of the COX-2 protein and measured levels of PGE2 in the mucosa of the small intestine during experimental peritonitis. Since PGE2 is rapidly converted in vivo into inactive stable metabolites 13,14-dihydro-15-keto PGE2 and 13,14-dihydro-15-keto PGA2 (hereinafter PGE2 metabolites), actually the levels of these metabolites were measured by the assay.
COX-2 protein levels in the ileal mucosa were markedly elevated following LPS injection or CLP compared to NS injection or sham operation, as judged by Western blots (Figure 1a). Consistent with high levels of COX-2, there was also a significant increase in production of PGE2 in the mucosa (Figure 1b). Immunofluorescence microscopy revealed that the bulk of COX-2 was induced in the epithelium and in few cells within the lamina propria, presumably lymphocytes (Figure 1c). Immunostaining for COX-2 was specific because no signal was detected in the intestinal sections from cox2−/− mice (Figure 1c, Neg).
Figure 1. Expression of COX-2, PGE2, and PGE2 receptors during peritonitis. (a) COX-2 protein.

expression in the ileal mucosa 16 h after control treatments (normal saline, NS, and sham), LPS injection, or CLP. Pos and Neg, positive and negative controls, IEC-6 cells treated with and without LPS. β-actin reprobe is shown to demonstrate lane load. Positions of size markers and their molecular weight in kD are on the left. Bar graph shows COX-2 band densities normalized to actin signal. *, significant differences, p<0.01. Data are representative of 3 independent experiments with 1 animal in each group. (b) Levels of stable PGE2 metabolites in ileal mucosa 16 h after indicated treatments. Data are average ±SEM of 3 animals for each data point. Significant differences **(p=0.44), # (p=0.005). (c) Localization of COX-2 (red) in the ileum 16 h after indicated treatments. Neg, negative control, intestinal section from cox-2−/− animal stained for COX-2. (d) Localization of indicated PGE2 receptors (red) in the ileum 16 h after i.p. injection with normal saline. Neg, negative control, a section stained with normal serum substituting primary antibody. DAPI-stained nuclei appear in blue. Bar=100 μm. All immunofluorescence images are representative of at least 3 animals in each group.
Responses to PGE2 are mediated by the four PGE receptors, EP1–4. In order to test whether the intestinal epithelium is capable of sensing PGE2, we performed immunofluorescence analysis of EP receptor expression in small intestinal sections. For all four receptors, highest levels of expression were observed in the epithelium. For EP1, 2, and 4, the signal was strongest at the luminal interface and somewhat weaker within enterocyte bodies; EP3 was largely limited to crypt epithelium (Figure 1d). Intracellular EPs are likely due to the ongoing receptor biogenesis and/or internalization. Immunostaining for EP receptors was specific because no signal was detected if the primary antibody was substituted for normal serum (Figure 1d, Neg). We did not observe any consistent change in EP receptor expression or localization associated with experimental peritonitis (unpublished results). Taken together, these data indicate that COX-2 is induced, and PGE2 levels increase in the mucosa during experimental peritonitis. Moreover, enterocytes possess the full complement of PGE receptors to enable sensing of the inflammatory PGE2.
Experimental peritonitis is associated with gut barrier derangement
Morphology, passage of FITC-dextran from gut lumen to bloodstream, and bacterial translocation to lymphatic organs and liver were used to characterize the state of the gut barrier during peritonitis. The most prominent morphologic feature on the hematoxylin-eosin (H&E)-stained sections of the small intestine following LPS injection was profuse epithelial sloughing (Figure 2a). The sloughed enterocytes were mostly single cells with very few fragments of the epithelium. Although sometimes there were signs of villus tip destruction, generally the epithelial architecture appeared intact (Figure 2a).
Figure 2. Peritonitis is associated with gut barrier derangement.

(a) H&E-stained sections of ileum from mice 16 h after injection with NS, injection with 40 mg/kg LPS, sham operation, or CLP. Note abundant epithelial sloughing in animals injected with LPS or subjected to CLP. Data are representative of at least 9 animals in each group. (b) Transepithelial passage of FITC-dextran and bacteria during peritonitis. Mice were gavaged with FITC-dextran and E. coli 35354T prior to indicated treatments. Serum concentrations of FITC-dextran (top) and counts of viable E. coli in combined mesenteric lymph nodes and spleens (bottom) were determined 16 h post treatment. *, Significant differences (p ≤ 0.01, n ≥ 12 in each group). (c) Localization of JAM-A (red) in ileal epithelium 16 h after indicated treatments. Note increased re-distribution of JAM-A from borders to intracellular space of enterocytes in LPS and CLP samples. (d) Localization of ZO-1 (red) in ileal epithelium 16 h after indicated treatments. Note increased redistribution of ZO-1 from TJ to intracellular space of enterocytes in LPS and CLP samples. DAPI-stained nuclei appear in blue. Bar=50 μm. All images are representative of at least 3 animals in each group.
To assess barrier function, mice were gavaged with FITC-dextran and ampicillin-resistant commensal strain of E. coli. FITC-dextran is commonly used to study passage of high molecular weight substances across the epithelium. Commensal strains of E. coli were used to examine bacterial translocation across the barrier. Our attempts to use E. coli K12 for this purpose were unsuccessful, presumably due to poor survival of this strain in the gastrointestinal tract (unpublished data). The serum concentration of FITC-dextran and E. coli translocation to lymphatic organs (mesenteric lymph nodes + spleen) were determined 16 h after LPS injection or CLP. As expected, acute endotoxemia resulted in dramatically increased serum FITC-dextran, and bacterial translocation (Figure 2b). Even more dramatic barrier dysfunction was observed in the CLP model (Figure 2c).
To evaluate the state of TJ, we performed immunostaining of intestinal thin sections with antibodies against the TJ-associated proteins ZO-1 and JAM-A. Control epithelia displayed prominent immunoreactivity of both proteins at enterocyte borders, consistent with localization to TJ. Some of the signal was intracellular (Figures 2c and d, NS and sham), which is in agreement with equilibrium between internalization and re-deposition of junctional proteins in the process of dynamic remodeling of TJ (20). In animals subjected to LPS injection, localization of ZO-1 and JAM-A to enterocyte borders was less pronounced, whereas intracellular localization was more pronounced. In many enterocytes the signal was diffusely spread over the cell (Figures 2c and d, LPS), indicating increased internalization of junctional proteins and derangement of TJ. Similar pathologic changes were observed in the CLP model (Figures 2c and d, CLP). Because virtually no signal was observed when primary antibodies were substituted for normal rabbit serum, immunostaining for JAM-A and ZO-1 was specific. Thus, in two different models, peritonitis was associated with sloughing of the intestinal epithelium, increased gut barrier permeability to macromolecules and bacteria, and increased internalization of TJ proteins, all indicating derangement of the gut barrier.
Luminally applied PGE2 increases barrier permeability in vivo
Systemic inflammation is intricately associated with elevation of COX-2-derived prostanoids in multiple tissues (73). To dissect the role of inflammatory prostanoids in the gut barrier failure, it was necessary to separate the effects of prostanoids from the effects of other inflammatory factors known to participate in this process. Since prostanoids act in temporally- and spatially-restricted manner, and EP receptors tend to localize to the luminal aspect of the epithelium, gut-specific effects can be achieved by local, as opposed to systemic, application. Accordingly, we tested the ability of luminally instilled PGE2 to increase FITC-dextran transfer from the lumen into the bloodstream. Prior to experiment mice were gavaged with the test solution containing FITC-dextran. Following isoflurane anesthesia, a loop of distal ileum was externalized and a segment was clamped. A solution of PGE2 or equal amount of solvent was injected into the lumen of the isolated segment. Animals were kept under anesthesia for 25 min to allow solute translocation across the barrier, and serum FITC-dextran concentrations were then measured. Luminally instilled PGE2 caused dramatic elevation of serum FITC-dextran (Figure 3), indicating that locally applied PGE2 increases gut barrier permeability.
Figure 3. Luminal application of PGE2 increases transepithelial passage of FITC-dextran.
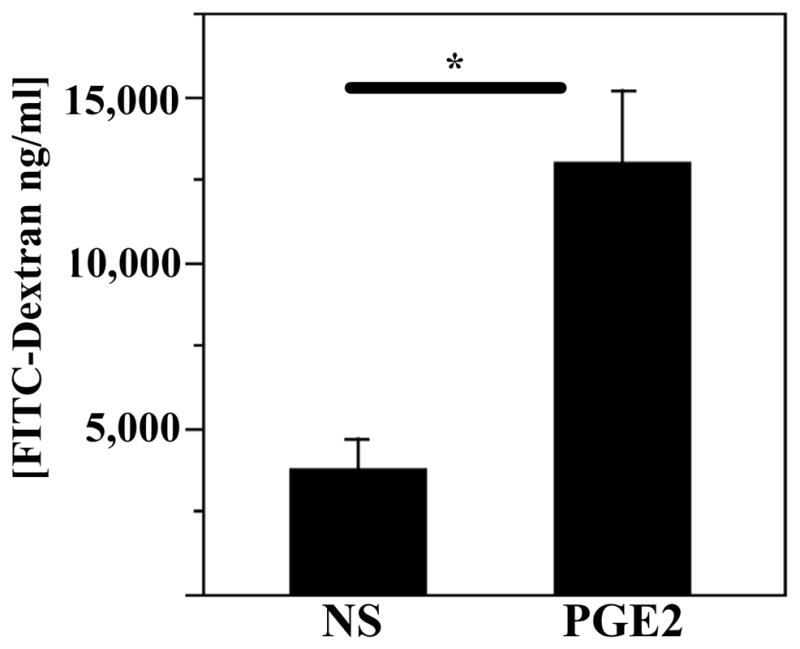
Mice were gavaged with FITC-dextran solution 2 h prior to experiments. Following anesthesia, 200 μl of normal saline (NS) or 50 mM PGE2 (PGE2) were injected into the lumen of isolated ileal loop. Serum levels of FITC-dextran were determined 20 min after treatment. Data are average ±SEM for 5 animals in each group. *, Significant difference at p=0.02.
Low dose of Celecoxib partially ameliorates barrier derangement during peritonitis
Our data indicated that levels of COX-2 and its product PGE2 in the intestine increase during experimental peritonitis. Furthermore, luminal application of PGE2 increased intestinal barrier permeability. If upregulation of COX-2 contributes to the inflammatory gut barrier failure, inhibition of this enzyme could help preserve the barrier by reversing this effect. To determine whether inhibition of COX-2 might limit barrier dysfunction, we assessed barrier integrity in LPS-injected or CLP-operated mice treated with various doses of the specific COX-2 inhibitor Celecoxib.
Mice were treated with Celecoxib at doses of 0 – 5 mg/kg prior to LPS injection. The drug was introduced by oral gavage together with the test mixture of FITC-dextran and ampicillin-resistant E. coli. Serum levels of FITC-dextran and bacterial translocation were measured 16 h after LPS injection. Levels of FITC-dextran and bacterial translocation were significantly reduced by 0.5 mg/kg Celecoxib. However, higher doses provided no protection (1 mg/kg), or even exacerbated gut barrier failure (5 mg/kg, Figures 4a and b). The 0.25 mg/kg dose did not have significant effect on barrier function during endotoxemia (unpublished results). When E. coli translocation to lymphoid organs and liver was used as readout for barrier integrity, there was similar dose-dependent effect of Celecoxib: significant protection at 0.5 and 1 mg/kg; and no protection at 5 mg/kg (Figure 4c, d, and e). At 5 mg/kg, Celecoxib caused relatively mild, yet significant barrier impairment by itself, in the absence of inflammatory stimuli (Figures 4a, b, and c).
Figure 4. Dose-dependent effects of Celecoxib on gut barrier permeability during peritonitis.

Mice were gavaged with the test mixture of FITC-dextran and E. coli prior to experiments. Celecoxib at indicated doses (mg/kg) or vehicle were added to the test mixture. (a) Serum FITC-dextran levels 16 h after injection with NS or LPS, with or without various doses of Celecoxib, as indicated. (b) Serum FITC-dextran levels 16 h after sham operation or CLP, with or without various doses of Celecoxib, as indicated. (c) E. coli counts in lymphatic organs 16 h after injection with NS or LPS, with or without various doses of Celecoxib, as indicated. (d) E. coli counts in lymphatic organs 16 h after sham operation or CLP, with or without Celecoxib, as indicated. (e) E. coli counts in liver 16 h after indicated treatments, with or without 0.5 mg/kg Celecoxib, as indicated. Data are average ± SEM, n≥12 in each group. Significant differences: *, p ≤0.01 vs. LPS or CLP alone; **, p≤0.02 vs. LPS or CLP alone; #, p≤0.02 vs. NS or sham.
Having established protective effect of low doses of Celecoxib on barrier function, we next sought to determine what effect this drug might have on other aspects of epithelial homeostasis. In the range of doses used, Celecoxib failed to significantly reduce epithelial sloughing during peritonitis (unpublished results). However, at 0.5 mg/kg Celecoxib reversed the increased internalization of the TJ-associated proteins JAM-A and ZO-1 following LPS injection or CLP (Figures 5 a–d), which is consistent with improved barrier function. Celecoxib somewhat reduced mucosal accumulation of IL-6, an inflammatory cytokine with a prominent role in intestinal inflammation, although the reduction was insignificant (unpublished data). Celecoxib significantly reduced mucosal levels of PGE2 during peritonitis (Figure 6a), and dramatically attenuated induction of the two hallmark inflammatory enzymes, COX-2 and inducible nitric oxide synthase (Figures 6b and c). In summary, Celecoxib, administered at lower, but not at higher doses, protected the epithelial barrier during experimental peritonitis and reduced expression of several key inflammatory factors.
Figure 5. Effects of Celecoxib on localization of TJ-associated proteins JAM-A and ZO-1.

(a) JAM-A (red) after injection with NS or LPS, with or without indicated doses (mg/kg) of Celecoxib. (b) JAM-A (red) after sham operation or CLP, with or without indicated doses of Celecoxib. (c) ZO-1 (red) after injection with NS or LPS, with or without indicated doses of Celecoxib. (d) ZO-1 (red) after sham operation or CLP, with or without indicated doses of Celecoxib. All images show sections of ileal samples taken 16 h after peritonitis-inducing treatments and are representative of at least 3 animals in each group. DAPI-stained nuclei appear in blue. Bar=100 μm.
Figure 6. Effects of Celecoxib on expression of inflammatory mediators.

(a) Levels of stable PGE2 metabolites in the ileal mucosa 16 h after LPS injection (left) or CLP (right) and treatment with Celecoxib at indicated doses (mg/kg). Data are average ± SEM for at least 3 animals in each data point. *, significant differences from LPS alone or CLP alone, p ≤ 0.05. (b) COX-2 and (c) iNOS protein levels in ileal mucosa 16 h after LPS injection or CLP, with and without 0.5 mg/kg Celecoxib. β-actin reprobes are shown to demonstrate lane load. Positions of size markers and their molecular weight in kD are on the left. Bar graphs show average band densities normalized to β-actin signal. Significance levels: *p=0.022, **p=0.044, ##p=0.047. Data are representative of 3 independent experiments.
Discussion
In this report we present evidence for the role of COX-2-derived prostanoids in gut barrier failure during peritonitis. Upon induction of experimental peritonitis by LPS injection or CLP there are multiple signs of gut barrier failure including epithelial sloughing, increased permeability to FITC-dextran, bacterial translocation, and internalization of TJ-associated proteins. At the same time, COX-2 is induced and levels of PGE2 increase in the mucosa. COX-2 induction is most prominent in the epithelium. Expression of COX-2 in the epithelial cells is consistent with our previous findings (63). The fact that epithelial cells express full complement of prostaglandin E receptors raises the possibility of autocrine response to inflammatory PGE2 in enterocytes. The causal relationship between COX-2-derived prostanoids and gut barrier failure is supported by the fact that luminally applied PGE2 increases barrier permeability in the absence of systemic inflammation. The role of COX-2 as a factor in gut barrier failure is further supported by the beneficial effect of COX-2 attenuation with the specific inhibitor Celecoxib.
Effects of Celecoxib in the inflammatory gut barrier failure are double-edged. We have found that lower doses (0.5–1 mg/kg), but not the higher dose (5 mg/kg) of this drug provide significant barrier protection in the endotoxemia model. In fact, the 5 mg/kg dose caused significant barrier derangement by itself, in the absence of an inflammatory stimulus, and it exacerbated barrier damage during endotoxemia. For comparison, the average daily dose of Celecoxib for treating rheumatoid arthritis and other inflammatory disorders in human patients is 5 mg/kg. A corresponding mouse dose should be 2–5 times higher due to faster metabolism in smaller animals (74). An optimal therapeutic dose of Celecoxib for gut barrier protection is therefore at least an order of magnitude lower than a typical clinical dose. The double-edged effect of Celecoxib on barrier function may be explained by a dual role of PGE2 in the pathogenesis of inflammatory gut barrier failure. Lower levels of PGE2 resulting from constitutive expression of COX-1 and basal expression of COX-2 might protect the intestine by regulating ion transport, secretion, blood supply, and smooth muscle tone via the high affinity PGE2 receptors. It is also possible that low concentrations of PGE2 promote barrier integrity by regulating turnover of TJ proteins. Thus, a certain level of COX-2 activity might be required for the intestinal homeostasis and protection, whereas higher levels typically associated with exuberant inflammation might be pathogenic. Such dual role of COX-2 has been suspected (75) but never demonstrated. A lower dose of a COX-2 inhibitor, as exemplified by 0.5 and 1 mg/kg in our mouse experiments, might blunt the pathogenic inflammatory burst of COX-2 activity while preserving the protective function of this enzyme.
The LPS dose of 40 mg/kg might seem excessive. However, our assumption was that gut origin sepsis would typically occur under conditions of severe systemic inflammation, which our intent was to mimic. Even at 40 mg/kg, LPS caused still lesser degree of inflammation than CLP. Various studies have found vastly different LD50 for LPS in mice, from as low as 1.2 mg/kg to as high as 50 mg/kg. These differences are likely due to different potencies of LPS from different sources, and different sensitivities to LPS of different strains of mice.
Although low doses of Celecoxib protect the intestinal barrier during peritonitis, this protection is only partial. Levels of pathologic FITC-dextran transfer and bacterial translocation during peritonitis decrease significantly upon treatment with 0.5–1 mg/kg Celecoxib, but they still remain above the background. At 0.25 – 5 mg/kg, Celecoxib does not noticeably reduce enterocyte sloughing or IL-6 expression during peritonitis. These partial effects support the idea of gut barrier failure as a multi-factorial phenomenon, to which COX-2-derived prostanoids contribute alongside a plethora of other pathogenic and protective factors (76). The fact that cox-2−/− mice are protected against endotoxemia (77), but have exacerbated barrier dysfunction during polymicrobial sepsis (78) also indicates that COX-2 is not the only actor in the drama of inflammatory gut barrier failure. The mechanisms by which PGE2 regulates the intestinal barrier remain largely unknown. In Caco-2 enterocyte monolayers, high concentrations of PGE2 increase TJ permeability via EP1 and EP4 receptors, Ca2+ release, and myosin light chain kinase (45). A similar mechanism might operate in vivo. Another possibility is that high levels of PGE2 contribute to barrier failure by enhancing other inflammatory responses. Our observation of decreased induction of iNOS and COX-2 during peritonitis in mice treated with low dose Celecoxib might be consistent with this mechanism. Furthermore, reduction of COX-2 protein expression by Celecoxib may indicate that this drug interrupts a putative positive feedback regulation of the COX-2 gene by its end product, PGE2 (79). Nevertheless, because luminal application of PGE2 rapidly increases barrier permeability in the absence of systemic inflammation, we believe that the effect of high levels of PGE2 on the barrier is direct. The putative mechanisms behind PGE2-induced gut barrier failure and protective effect of low dose Celecoxib are summarized in Figure 7.
Figure 7. A diagram of the proposed role of COX-2-derived PGE2 on the epithelial barrier and effects of Celecoxib.
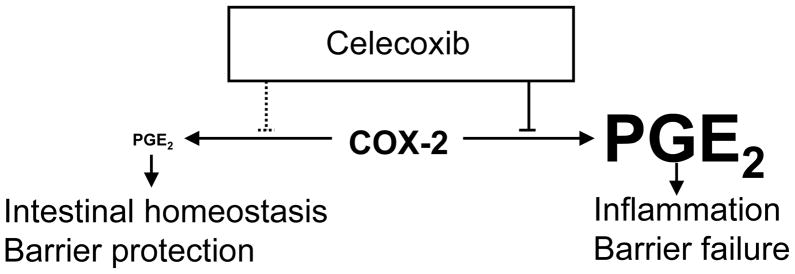
Low levels of COX-2-derived PGE2 are required for epithelial homeostasis and gut protection, whereas high levels of PGE2 promote gut barrier failure. High doses of Celecoxib are not beneficial because they obliterate the protective effect of PGE2. Low doses of Celecoxib blunt the inflammatory burst of PGE2 while preserving the protective effect.
Our findings suggest that low doses of selective COX-2 inhibitors may find use as adjunct gut barrier protection therapy in critically ill patients. The same approach may be useful in the broad array of intestinal inflammatory disorders whereby gut barrier failure plays a prominent role, e.g. necrotizing enterocolitis, Crohn’s disease, and inflammatory bowel disease. Randomized clinical trials are needed to establish the validity of this approach.
Acknowledgments
Grant support: National Institutes of Health Grant R01 AI 014032 to H.R.F.
The authors have no conflicting financial interests. We thank Larry Wang for help with microscopic pathology. This study was supported by National Institutes of Health Grant R01 AI 014032 to H.R.F.
Abbreviations
- CLP
cecal ligation and puncture
- H&E
hematoxylin and eosin
- NS
normal saline
- PGE
prostaglandin E
- TJ
tight junctions
References
- 1.Alverdy JC, Chang EB. The re-emerging role of the intestinal microflora in critical illness and inflammation: why the gut hypothesis of sepsis syndrome will not go away. J Leukoc Biol. 2008 Mar;83(3):461–6. doi: 10.1189/jlb.0607372. [DOI] [PubMed] [Google Scholar]
- 2.Deitch EA. Gut-origin sepsis: evolution of a concept. Surgeon. Dec;10(6):350–6. doi: 10.1016/j.surge.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fink MP, Delude RL. Epithelial barrier dysfunction: a unifying theme to explain the pathogenesis of multiple organ dysfunction at the cellular level. Crit Care Clin. 2005 Apr;21(2):177–96. doi: 10.1016/j.ccc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Gatt M, Reddy BS, MacFie J. Review article: bacterial translocation in the critically ill--evidence and methods of prevention. Aliment Pharmacol Ther. 2007 Apr 1;25(7):741–57. doi: 10.1111/j.1365-2036.2006.03174.x. [DOI] [PubMed] [Google Scholar]
- 5.Petrosyan M, Guner YS, Williams M, Grishin A, Ford HR. Current concepts regarding the pathogenesis of necrotizing enterocolitis. Pediatr Surg Int. 2009 Apr;25(4):309–18. doi: 10.1007/s00383-009-2344-8. [DOI] [PubMed] [Google Scholar]
- 6.Luyer MD, Jacobs JA, Vreugdenhil AC, Hadfoune M, Dejong CH, Buurman WA, et al. Enteral administration of high-fat nutrition before and directly after hemorrhagic shock reduces endotoxemia and bacterial translocation. Ann Surg. 2004 Feb;239(2):257–64. doi: 10.1097/01.sla.0000108695.60059.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saini MS, Liberati DM, Diebel LN. Sequential changes in mucosal immunity after hemorrhagic shock. Am Surg. 2001 Aug;67(8):797–801. [PubMed] [Google Scholar]
- 8.Sheth SU, Lu Q, Twelker K, Sharpe SM, Qin X, Reino DC, et al. Intestinal mucus layer preservation in female rats attenuates gut injury after trauma-hemorrhagic shock. J Trauma. Feb;68(2):279–88. doi: 10.1097/TA.0b013e3181caa6bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen LW, Hwang B, Wang JS, Chen JS, Hsu CM. Hypertonic saline-enhanced postburn gut barrier failure is reversed by inducible nitric oxide synthase inhibition. Crit Care Med. 2004 Dec;32(12):2476–84. doi: 10.1097/01.ccm.0000147831.07329.32. [DOI] [PubMed] [Google Scholar]
- 10.Olguin F, Araya M, Hirsch S, Brunser O, Ayala V, Rivera R, et al. Prebiotic ingestion does not improve gastrointestinal barrier function in burn patients. Burns. 2005 Jun;31(4):482–8. doi: 10.1016/j.burns.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 11.Reys LG, Ortiz-Pomales YT, Lopez N, Cheadle G, de Oliveira PG, Eliceiri B, et al. Uncovering the neuroenteric-pulmonary axis: Vagal nerve stimulation prevents acute lung injury following hemorrhagic shock. Life Sci. Feb 22; doi: 10.1016/j.lfs.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 12.Fishman JE, Levy G, Alli V, Sheth S, Lu Q, Deitch EA. Oxidative modification of the intestinal mucus layer is a critical but unrecognized component of trauma hemorrhagic shock-induced gut barrier failure. Am J Physiol Gastrointest Liver Physiol. Jan 1;304(1):G57–63. doi: 10.1152/ajpgi.00170.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lau LL, Halliday MI, Lee B, Hannon RJ, Gardiner KR, Soong CV. Intestinal manipulation during elective aortic aneurysm surgery leads to portal endotoxaemia and mucosal barrier dysfunction. Eur J Vasc Endovasc Surg. 2000 Jun;19(6):619–24. doi: 10.1053/ejvs.2000.1063. [DOI] [PubMed] [Google Scholar]
- 14.Li Z, Yang X, Lu L, Yu Y, Yao Y. Gut barrier function damage following multiple firearm injuries in a porcine model. Chin Med Sci J. 2001 Dec;16(4):209–13. [PubMed] [Google Scholar]
- 15.Peregudov SI, Khanevich MD. The small intestine as the origin of bacteremia in acute diffuse peritonitis. Nutr Hosp. 1996 Nov-Dec;11(6):317–20. [PubMed] [Google Scholar]
- 16.Rahman SH, Ammori BJ, Holmfield J, Larvin M, McMahon MJ. Intestinal hypoperfusion contributes to gut barrier failure in severe acute pancreatitis. J Gastrointest Surg. 2003 Jan;7(1):26–35. doi: 10.1016/S1091-255X(02)00090-2. discussion -6. [DOI] [PubMed] [Google Scholar]
- 17.Souza DG, Vieira AT, Soares AC, Pinho V, Nicoli JR, Vieira LQ, et al. The essential role of the intestinal microbiota in facilitating acute inflammatory responses. J Immunol. 2004 Sep 15;173(6):4137–46. doi: 10.4049/jimmunol.173.6.4137. [DOI] [PubMed] [Google Scholar]
- 18.Deitch EA. Role of the gut lymphatic system in multiple organ failure. Curr Opin Crit Care. 2001 Apr;7(2):92–8. doi: 10.1097/00075198-200104000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006 Dec;169(6):1901–9. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalmers AD, Whitley P. Continuous endocytic recycling of tight junction proteins: how and why? Essays Biochem. 53:41–54. doi: 10.1042/bse0530041. [DOI] [PubMed] [Google Scholar]
- 21.Al-Sadi R, Guo S, Dokladny K, Smith MA, Ye D, Kaza A, et al. Mechanism of interleukin-1beta induced-increase in mouse intestinal permeability in vivo. J Interferon Cytokine Res. Oct;32(10):474–84. doi: 10.1089/jir.2012.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al-Sadi R, Ye D, Said HM, Ma TY. Cellular and molecular mechanism of interleukin-1beta modulation of Caco-2 intestinal epithelial tight junction barrier. J Cell Mol Med. Apr;15(4):970–82. doi: 10.1111/j.1582-4934.2010.01065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colgan SP, Resnick MB, Parkos CA, Delp-Archer C, McGuirk D, Bacarra AE, et al. IL-4 directly modulates function of a model human intestinal epithelium. J Immunol. 1994 Sep 1;153(5):2122–9. [PubMed] [Google Scholar]
- 24.Yang R, Han X, Uchiyama T, Watkins SK, Yaguchi A, Delude RL, et al. IL-6 is essential for development of gut barrier dysfunction after hemorrhagic shock and resuscitation in mice. Am J Physiol Gastrointest Liver Physiol. 2003 Sep;285(3):G621–9. doi: 10.1152/ajpgi.00177.2003. [DOI] [PubMed] [Google Scholar]
- 25.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003 Dec 1;171(11):6164–72. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 26.Costantini TW, Deree J, Loomis W, Putnam JG, Choi S, Baird A, et al. Phosphodiesterase inhibition attenuates alterations to the tight junction proteins occludin and ZO-1 in immunostimulated Caco-2 intestinal monolayers. Life Sci. 2009 Jan 2;84(1–2):18–22. doi: 10.1016/j.lfs.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Hindryckx P, De Vos M, Jacques P, Ferdinande L, Peeters H, Olievier K, et al. Hydroxylase inhibition abrogates TNF-alpha-induced intestinal epithelial damage by hypoxia-inducible factor-1-dependent repression of FADD. J Immunol. Nov 15;185(10):6306–16. doi: 10.4049/jimmunol.1002541. [DOI] [PubMed] [Google Scholar]
- 28.Paiotti AP, Ribeiro DA, Silva RM, Marchi P, Oshima CT, Neto RA, et al. Effect of COX-2 inhibitor lumiracoxib and the TNF-alpha antagonist etanercept on TNBS-induced colitis in Wistar rats. J Mol Histol. Jun;43(3):307–17. doi: 10.1007/s10735-012-9400-8. [DOI] [PubMed] [Google Scholar]
- 29.Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G, et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am J Gastroenterol. 2002 Aug;97(8):2000–4. doi: 10.1111/j.1572-0241.2002.05914.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang F, Schwarz BT, Graham WV, Wang Y, Su L, Clayburgh DR, et al. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006 Oct;131(4):1153–63. doi: 10.1053/j.gastro.2006.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yajima S, Morisaki H, Serita R, Suzuki T, Katori N, Asahara T, et al. Tumor necrosis factor-alpha mediates hyperglycemia-augmented gut barrier dysfunction in endotoxemia. Crit Care Med. 2009 Mar;37(3):1024–30. doi: 10.1097/CCM.0b013e31819b53b6. [DOI] [PubMed] [Google Scholar]
- 32.Beaurepaire C, Smyth D, McKay DM. Interferon-gamma regulation of intestinal epithelial permeability. J Interferon Cytokine Res. 2009 Mar;29(3):133–44. doi: 10.1089/jir.2008.0057. [DOI] [PubMed] [Google Scholar]
- 33.Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005 Jun;19(8):923–33. doi: 10.1096/fj.04-3260com. [DOI] [PubMed] [Google Scholar]
- 34.Luyer MD, Buurman WA, Hadfoune M, Wolfs T, van’t Veer C, Jacobs JA, et al. Exposure to bacterial DNA before hemorrhagic shock strongly aggravates systemic inflammation and gut barrier loss via an IFN-gamma-dependent route. Ann Surg. 2007 May;245(5):795–802. doi: 10.1097/01.sla.0000251513.59983.3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, et al. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005 Oct;16(10):5040–52. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 2002 Sep;123(3):790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- 37.Yang R, Miki K, Oksala N, Nakao A, Lindgren L, Killeen ME, et al. Bile high-mobility group box 1 contributes to gut barrier dysfunction in experimental endotoxemia. Am J Physiol Regul Integr Comp Physiol. 2009 Aug;297(2):R362–9. doi: 10.1152/ajpregu.00184.2009. [DOI] [PubMed] [Google Scholar]
- 38.Dickinson E, Tuncer R, Nadler E, Boyle P, Alber S, Watkins S, et al. NOX, a novel nitric oxide scavenger, reduces bacterial translocation in rats after endotoxin challenge. Am J Physiol. 1999 Dec;277(6 Pt 1):G1281–7. doi: 10.1152/ajpgi.1999.277.6.G1281. [DOI] [PubMed] [Google Scholar]
- 39.Han X, Fink MP, Yang R, Delude RL. Increased iNOS activity is essential for intestinal epithelial tight junction dysfunction in endotoxemic mice. Shock. 2004 Mar;21(3):261–70. doi: 10.1097/01.shk.0000112346.38599.10. [DOI] [PubMed] [Google Scholar]
- 40.Suzuki Y, Deitch EA, Mishima S, Lu Q, Xu D. Inducible nitric oxide synthase gene knockout mice have increased resistance to gut injury and bacterial translocation after an intestinal ischemia-reperfusion injury. Crit Care Med. 2000 Nov;28(11):3692–6. doi: 10.1097/00003246-200011000-00026. [DOI] [PubMed] [Google Scholar]
- 41.Upperman JS, Potoka D, Grishin A, Hackam D, Zamora R, Ford HR. Mechanisms of nitric oxide-mediated intestinal barrier failure in necrotizing enterocolitis. Semin Pediatr Surg. 2005 Aug;14(3):159–66. doi: 10.1053/j.sempedsurg.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 42.Lejeune M, Leung P, Beck PL, Chadee K. Role of EP4 receptor and prostaglandin transporter in prostaglandin E2-induced alteration in colonic epithelial barrier integrity. Am J Physiol Gastrointest Liver Physiol. Nov;299(5):G1097–105. doi: 10.1152/ajpgi.00280.2010. [DOI] [PubMed] [Google Scholar]
- 43.Martin-Venegas R, Roig-Perez S, Ferrer R, Moreno JJ. Arachidonic acid cascade and epithelial barrier function during Caco-2 cell differentiation. J Lipid Res. 2006 Jul;47(7):1416–23. doi: 10.1194/jlr.M500564-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Rodriguez-Lagunas MJ, Ferrer R, Moreno JJ. Effect of eicosapentaenoic acid-derived prostaglandin E on intestinal epithelial barrier function. Prostaglandins Leukot Essent Fatty Acids. Feb 27; doi: 10.1016/j.plefa.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Lagunas MJ, Martin-Venegas R, Moreno JJ, Ferrer R. PGE2 promotes Ca2+-mediated epithelial barrier disruption through EP1 and EP4 receptors in Caco-2 cell monolayers. Am J Physiol Cell Physiol. Aug;299(2):C324–34. doi: 10.1152/ajpcell.00397.2009. [DOI] [PubMed] [Google Scholar]
- 46.Loftin CD, Tiano HF, Langenbach R. Phenotypes of the COX-deficient mice indicate physiological and pathophysiological roles for COX-1 and COX-2. Prostaglandins Other Lipid Mediat. 2002 Aug;68–69:177–85. doi: 10.1016/s0090-6980(02)00028-x. [DOI] [PubMed] [Google Scholar]
- 47.Hawkey CJ, Rampton DS. Prostaglandins and the gastrointestinal mucosa: are they important in its function, disease, or treatment? Gastroenterology. 1985 Nov;89(5):1162–88. doi: 10.1016/0016-5085(85)90225-2. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi K, Kato S, Amagase K. Prostaglandin EP receptors involved in modulating gastrointestinal mucosal integrity. J Pharmacol Sci. 114(3):248–61. doi: 10.1254/jphs.10r06cr. [DOI] [PubMed] [Google Scholar]
- 49.Hirata T, Narumiya S. Prostanoids as regulators of innate and adaptive immunity. Adv Immunol. 116:143–74. doi: 10.1016/B978-0-12-394300-2.00005-3. [DOI] [PubMed] [Google Scholar]
- 50.Perini RF, Ma L, Wallace JL. Mucosal repair and COX-2 inhibition. Curr Pharm Des. 2003;9(27):2207–11. doi: 10.2174/1381612033454027. [DOI] [PubMed] [Google Scholar]
- 51.Nakayama Y, Omote K, Namiki A. Role of prostaglandin receptor EP1 in the spinal dorsal horn in carrageenan-induced inflammatory pain. Anesthesiology. 2002 Nov;97(5):1254–62. doi: 10.1097/00000542-200211000-00032. [DOI] [PubMed] [Google Scholar]
- 52.Stock JL, Shinjo K, Burkhardt J, Roach M, Taniguchi K, Ishikawa T, et al. The prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J Clin Invest. 2001 Feb;107(3):325–31. doi: 10.1172/JCI6749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sametz W, Hennerbichler S, Glaser S, Wintersteiger R, Juan H. Characterization of prostanoid receptors mediating actions of the isoprostanes, 8-iso-PGE(2) and 8-iso-PGF(2alpha), in some isolated smooth muscle preparations. Br J Pharmacol. 2000 Aug;130(8):1903–10. doi: 10.1038/sj.bjp.0703522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology. 2006 Sep;131(3):862–77. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacDermott RP. Alterations in the mucosal immune system in ulcerative colitis and Crohn’s disease. Med Clin North Am. 1994 Nov;78(6):1207–31. doi: 10.1016/s0025-7125(16)30096-7. [DOI] [PubMed] [Google Scholar]
- 56.Sheibanie AF, Yen JH, Khayrullina T, Emig F, Zhang M, Tuma R, et al. The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23-->IL-17 axis. J Immunol. 2007 Jun 15;178(12):8138–47. doi: 10.4049/jimmunol.178.12.8138. [DOI] [PubMed] [Google Scholar]
- 57.Wang D, Dubois RN. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene. Feb 11;29(6):781–8. doi: 10.1038/onc.2009.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lugo B, Ford HR, Grishin A. Molecular signaling in necrotizing enterocolitis: regulation of intestinal COX-2 expression. J Pediatr Surg. 2007 Jul;42(7):1165–71. doi: 10.1016/j.jpedsurg.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 59.Cuzzocrea S, Mazzon E, Serraino I, Dugo L, Centorrino T, Ciccolo A, et al. Celecoxib, a selective cyclo-oxygenase-2 inhibitor reduces the severity of experimental colitis induced by dinitrobenzene sulfonic acid in rats. Eur J Pharmacol. 2001 Nov 9;431(1):91–102. doi: 10.1016/s0014-2999(01)01403-0. [DOI] [PubMed] [Google Scholar]
- 60.Martin AR, Villegas I, La Casa C, Alarcon de la Lastra C. The cyclo-oxygenase-2 inhibitor, rofecoxib, attenuates mucosal damage due to colitis induced by trinitrobenzene sulphonic acid in rats. Eur J Pharmacol. 2003 Nov 28;481(2–3):281–91. doi: 10.1016/j.ejphar.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 61.Moses T, Wagner L, Fleming SD. TLR4-mediated Cox-2 expression increases intestinal ischemia/reperfusion-induced damage. J Leukoc Biol. 2009 Oct;86(4):971–80. doi: 10.1189/jlb.0708396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zamuner SR, Warrier N, Buret AG, MacNaughton WK, Wallace JL. Cyclooxygenase 2 mediates post-inflammatory colonic secretory and barrier dysfunction. Gut. 2003 Dec;52(12):1714–20. doi: 10.1136/gut.52.12.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grishin AV, Wang J, Potoka DA, Hackam DJ, Upperman JS, Boyle P, et al. Lipopolysaccharide induces cyclooxygenase-2 in intestinal epithelium via a noncanonical p38 MAPK pathway. J Immunol. 2006 Jan 1;176(1):580–8. doi: 10.4049/jimmunol.176.1.580. [DOI] [PubMed] [Google Scholar]
- 64.Singh VP, Patil CS, Jain NK, Kulkarni SK. Aggravation of inflammatory bowel disease by cyclooxygenase-2 inhibitors in rats. Pharmacology. 2004 Oct;72(2):77–84. doi: 10.1159/000079135. [DOI] [PubMed] [Google Scholar]
- 65.Tsubouchi R, Hayashi S, Aoi Y, Nishio H, Terashima S, Kato S, et al. Healing impairment effect of cyclooxygenase inhibitors on dextran sulfate sodium-induced colitis in rats. Digestion. 2006;74(2):91–100. doi: 10.1159/000097657. [DOI] [PubMed] [Google Scholar]
- 66.Zhang L, Lu YM, Dong XY. Effects and mechanism of the selective COX-2 inhibitor, celecoxib, on rat colitis induced by trinitrobenzene sulfonic acid. Chin J Dig Dis. 2004;5(3):110–4. doi: 10.1111/j.1443-9573.2004.00164.x. [DOI] [PubMed] [Google Scholar]
- 67.Carothers AM, Davids JS, Damas BC, Bertagnolli MM. Persistent cyclooxygenase-2 inhibition downregulates NF-{kappa}B, resulting in chronic intestinal inflammation in the min/+ mouse model of colon tumorigenesis. Cancer Res. Jun 1;70(11):4433–42. doi: 10.1158/0008-5472.CAN-09-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Paiotti AP, Marchi P, Miszputen SJ, Oshima CT, Franco M, Ribeiro DA. The role of nonsteroidal antiinflammatory drugs and cyclooxygenase-2 inhibitors on experimental colitis. In Vivo. May-Jun;26(3):381–93. [PubMed] [Google Scholar]
- 69.Ishikawa TO, Oshima M, Herschman HR. Cox-2 deletion in myeloid and endothelial cells, but not in epithelial cells, exacerbates murine colitis. Carcinogenesis. Mar;32(3):417–26. doi: 10.1093/carcin/bgq268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morteau O, Morham SG, Sellon R, Dieleman LA, Langenbach R, Smithies O, et al. Impaired mucosal defense to acute colonic injury in mice lacking cyclooxygenase-1 or cyclooxygenase-2. J Clin Invest. 2000 Feb;105(4):469–78. doi: 10.1172/JCI6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hotz-Behofsits C, Simpson RJ, Walley M, Bjarnason IT. Role of COX-2 in nonsteroidal anti-inflammatory drug enteropathy in rodents. Scand J Gastroenterol. Aug;45(7–8):822–7. doi: 10.3109/00365521003797205. [DOI] [PubMed] [Google Scholar]
- 72.Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moldawer LL, Efron PA. Cecal ligation and puncture. Curr Protoc Immunol. Nov;Chapter 19(Unit 19):3. doi: 10.1002/0471142735.im1913s91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bos CL, Richel DJ, Ritsema T, Peppelenbosch MP, Versteeg HH. Prostanoids and prostanoid receptors in signal transduction. Int J Biochem Cell Biol. 2004 Jul;36(7):1187–205. doi: 10.1016/j.biocel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 74.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008 Mar;22(3):659–61. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 75.Coruzzi G, Venturi N, Spaggiari S. Gastrointestinal safety of novel nonsteroidal antiinflammatory drugs: selective COX-2 inhibitors and beyond. Acta Biomed. 2007 Aug;78(2):96–110. [PubMed] [Google Scholar]
- 76.Suzuki T. Regulation of intestinal epithelial permeability by tight junctions. Cell Mol Life Sci. Feb;70(4):631–59. doi: 10.1007/s00018-012-1070-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ejima K, Layne MD, Carvajal IM, Kritek PA, Baron RM, Chen YH, et al. Cyclooxygenase-2-deficient mice are resistant to endotoxin-induced inflammation and death. FASEB J. 2003 Jul;17(10):1325–7. doi: 10.1096/fj.02-1078fje. [DOI] [PubMed] [Google Scholar]
- 78.Fredenburgh LE, Velandia MM, Ma J, Olszak T, Cernadas M, Englert JA, et al. Cyclooxygenase-2 deficiency leads to intestinal barrier dysfunction and increased mortality during polymicrobial sepsis. J Immunol. Nov 15;187(10):5255–67. doi: 10.4049/jimmunol.1101186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ishikawa TO, Jain N, Herschman HR. Feedback regulation of cyclooxygenase-2 transcription ex vivo and in vivo. Biochem Biophys Res Commun. 2009 Jan 16;378(3):534–8. doi: 10.1016/j.bbrc.2008.11.099. [DOI] [PMC free article] [PubMed] [Google Scholar]