

Abstract
The chemokine receptor CXCR2 is vital for inflammation, wound healing, angiogenesis, cancer progression, and metastasis. Adaptor protein 2 (AP2), a clathrin binding heterotetrameric protein comprised of α, β2, μ2, and σ2 subunits, facilitates clathrin-mediated endocytosis. Mutation of the LLKIL motif in the CXCR2 carboxyl-terminal domain (CTD) results in loss of AP2 binding to the receptor and loss of ligand mediated receptor internalization and chemotaxis. AP2 knockdown also results in diminished ligand-mediated CXCR2 internalization, polarization and chemotaxis. Using knockdown/rescue approaches with AP2-μ2 mutants, the binding domains were characterized in reference to CXCR2 internalization and chemotaxis. When in an open conformation, μ2 Patch 1 and Patch 2 domains bind tightly to membrane PIP2 phospholipids. When AP2-μ2 is replaced with μ2 mutated in Patch 1 and/or Patch 2 domains, ligand-mediated receptor binding and internalization are not lost. However chemotaxis requires AP2-μ2 Patch 1, but not Patch 2. AP2-σ2 has been demonstrated to bind dileucine motifs to facilitate internalization. Expression of AP2-σ2 V88D and V98S dominant negative mutants resulted in loss of CXCR2 mediated chemotaxis. Thus, AP2 binding to both membrane phosphatidylinositol phospholipids and dileucine motifs is crucial for directional migration or chemotaxis. Moreover, AP2-mediated receptor internalization can be dissociated from AP2-mediated chemotaxis.
Keywords: CXCR2, AP2, AP2-μ2, AP2-σ2, PIP2 patches, chemotaxis, internalization
Introduction
The chemokine receptor CXCR2 is a Giα-coupled receptor that mediates migration of many cell types including leukocytes and endothelial cells (1). CXCR2 mediates numerous biological processes such as wound healing, inflammation, angiogenesis and cancer metastasis (2–6). The adaptor protein 2 complex (AP2) is involved in the internalization of CXCR2 upon ligand binding by coupling it to clathrin-mediated endocytosis (7, 8). AP2 is also critical for CXCR2-mediated directed cell migration or chemotaxis (7). We have previously shown that the LLKIL motif present in the carboxyl-terminal domain (CTD) of CXCR2 is important for binding to AP2 and for the CXCL8-mediated polarization of early signals during chemotaxis (7, 9). However, it has not been established whether it is the lack of AP2 binding or failure to internalize the receptor that results in diminished CXCR2-mediated chemotaxis when the LLKIL motif is mutated.
The AP2 complex is a hetero-tetramer comprised of subunits, α and β2 (~100 kDa each), μ2 (~50 kDa), and σ2 (~17kDa) (10). The membrane phosphoinositides play a key role in the docking of the AP2 to the plasma membrane (11–13). According to the current model, when AP2 encounters phosphatidylinositol (4,5) bisphosphate (PIP2)-enriched regions at the inner leaflet of the plasma membrane, the α and β subunits initiate a weak association with the PIP2 enriched region (14, 15). Impairment of the association of α and β subunits with a PIP2-enriched region prevents this initial recruitment and abolishes the binding of the AP2 complex to PIP2-containing model membranes (11, 14, 16). Reduction of the PIP2 level at the plasma membrane drastically affects the association of AP2 to the membrane without significantly affecting clathrin assembly (17), indicating the importance of PIP2. The AP2-μ2 subunit has two patches (patch 1 and 2) of electropositive surface that presumably strengthen the binding of AP2 to the PIP2 or PIP3 enriched inner leaflet of the plasma membrane (14). Lysine residues K341, K343 and K345 are key components of Patch 1 and these amino acids have been shown to be critical for AP2 binding to PIP2 (11, 18). Others have shown that in addition to K345 (from Patch 1), K354 and K356 amino acid residues were also important for binding to PIP2-containing model membranes (18). The recent crystal structure of AP2 revealed that lysine K319 and K356 are also part of Patch 1(14). Amino acid residues K167, R169, R170, K421 form Patch 2 of AP2-μ2, but the functional contribution of Patch 2 to PIP2 binding is yet to be determined (14). These 2 patches are spatially well separated and may serve as the PIP2 anchors for AP2-μ2. Adaptor associated kinase (AAK1) bound to the ear domain of the α subunit, phosphorylates T156 on AP2-μ2. This further strengthens the recruitment of the entire AP2 complex as the avidity of AP2-μ2 increases several fold to the PIP2 present in the plasma membrane. The conversion of inactive and closed to active and open conformation of AP2 initiates a significant conformational change (10 Ǻ) that brings both Patch 1 and Patch 2 of AP2-μ2 co-planar to the inner leaflet of lipid bilayer along with σ2 subunit so that both AP2-μ2 and AP2-σ2 subunits can bind to their respective cargoes for internalization (14). AP2-μ2 binds to the Yxxϕ motif (ϕ - bulky hydrophobic) and AP2-σ2 binds to the dileucine based [DE]XXXL[LI] motif presented by the transmembrane cargo proteins (19). Furthermore, the recruited AP2 complex acting through the AP2-μ2 subunit stimulates the enzyme phosphatidylinositol 4-phosphate 5-kinase (PIP5K type I) to produce more PIP2 (20). This positive feedback loop produces more localized accumulation of PIP2 thus enhancing the activity of phosphoinositides.
CXCR2 lacks the Yxxϕ motif at the CTD, so the μ2 subunit cannot directly bind to CXCR2. However, the major conformational rearrangement of the AP2 complex occurs upon binding to the plasma membrane. Initially, the AP2 complex binds to the plasma membrane through interaction with the α2 and β2 subunits. Through the electrostatic attraction and subsequent binding of the carboxyl domain of μ2 to high local concentration of PIP2, AP2 adopts an open conformation (major conformation change) (15). This reorientation might be necessary for the engagement of the σ2 subunit with the LLKIL motif present in the CTD of CXCR2. The disordered μ2-linker becomes a four-turn helix and the phosphorylation of the μ2 subunit on T156 may stabilize the helical form of the μ2-linker and a high affinity binding to the sorting motifs (41). The CTD domain of CXCR2 involved in AP2 binding lacks the classical acidic residues preceding the dileucine motif. Variations in the motif have been previously observed (21). The AP2-σ2 subunit and part of the α-subunit are involved in binding to the dileucine motif of the cargo (22). Mutation of the LLKIL motif ablates the interaction of CXCR2 with AP2 and importantly, the CXCR2-mediated chemotaxis (7, 9, 23). We sought here to determine whether binding of AP2 to the PIP2 phospholipid in the plasma membrane is required for the proper orientation and binding of the σ2 subunit of AP2 to the LLKIL motif of CXCR2 to mediate receptor internalization and/or chemotaxis. We also sought to determine whether AP2 mediated CXCR2 internalization is critical for CXCR2-mediated chemotaxis. Here, we clearly show that AP2 binding to CXCR2 is vital for ligand-induced receptor internalization, cell polarization toward a chemokine gradient, and chemotaxis. Our data also show that AP2-mediated receptor internalization can be dissociated from AP2-mediated chemotaxis.
Results
Co-localization of CXCR2 with the AP2 complex in neutrophil-like differentiated HL60 cells expressing CXCR2 (dHL-60-CXCR2)
To evaluate the distribution of the AP2 complex in polarized differentiated neutrophil-like HL-60 cells (dHL-60) cells responding to a gradient of CXCL8, dHL-60 cells were loaded onto fibronectin-coated coverslips and stimulated with a gradient of CXCL8 in the Zigmond chamber. Results show that dHL-60 cells stably expressing CXCR2 were highly polarized in the direction of the CXCL8 gradient with cell bodies carrying distinctive tails (Fig. 1A). To determine whether AP2-μ2 co-localizes with CXCR2, polarized cells were stained for AP2-μ2, CXCR2 and filamentous actin (F-actin) where F-actin staining was employed to mark the active leading edge. F-actin was distributed in a polarized manner towards the CXCL8 gradient, with concentrated accumulation at the anterior leading edge. F-actin, CXCR2 and AP2-μ2 appear to co-localize to the leading edge (Fig. 1A). Line scan analysis of representative polarized cells clearly indicates that the distribution profile of CXCR2 mirrors that of the AP-2 complex (Fig. 1B).
Figure 1. CXCR2 co-localizes with AP2-μ2 and clathrin fails to polarize in neutrophil-like dHL-60 CXCR2 cells upon stimulation with CXCL +8.
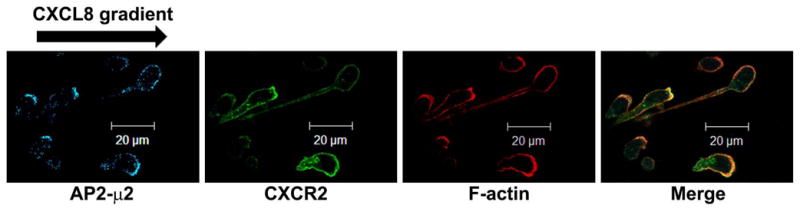
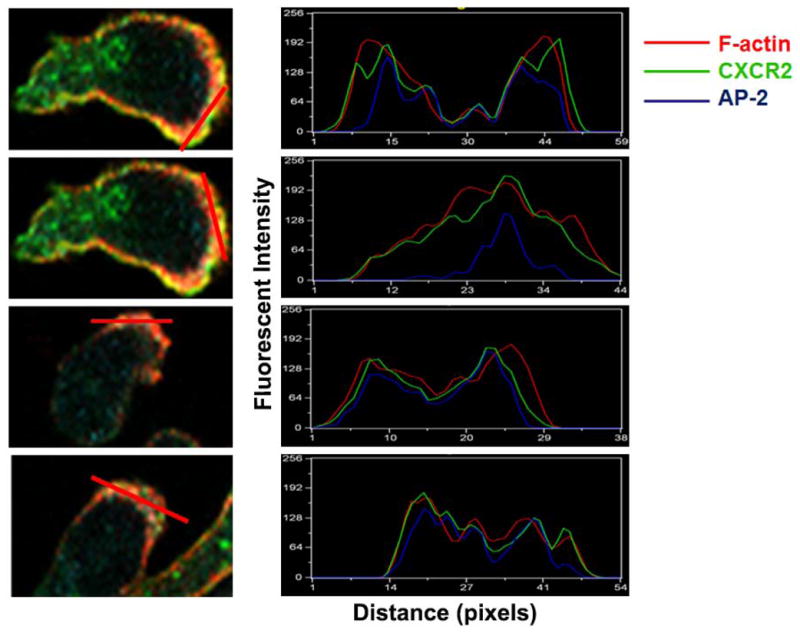
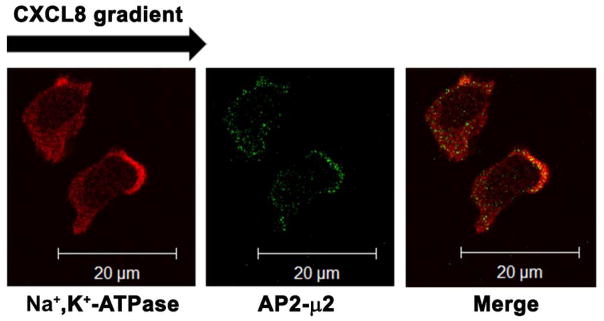

A) Confocal images of dHL-60-CXCR2 cells that were directionally stimulated with 50 ng/ml of CXCL8 for 10 min. CXCR2, AP2-μ2 and F-actin were pseudo-colored green, cyan and red respectively. Images represent single z-stack sections of 0.5 μm. Scale bar – 20 μm. B) Line scan analysis of representative dHL-60-CXCR2 cells from fig.1A. The enlarged confocal image of the individual cells is shown on the left and the corresponding line scan distribution profile of fluorescence intensities of F-actin, CXCR2 and AP-2 obtained by Metamorph analysis is shown on the right. Red trace – F-actin; Green trace – CXCR2; Blue trace – AP-2. C) Confocal images of neutrophil-like dHL-60-CXCR2 cells were directionally stimulated with 50 ng / ml of CXCL8 for 10 min. Na+, K+-ATPase and AP2-μ2 were pseudo-colored red and green respectively. Images represent single z-stack sections of 0.5 μm. Scale bar – 20 μm. D) Representative confocal images of dPLB-985-CXCR2 cells that were directionally stimulated with 50 ng / ml of CXCL8 for 10 min are shown. AP2 and clathrin were pseudo-colored red and green respectively (left panel). Enlarged areas of the leading edges of the polarized cells were depicted on the right top and right bottom panels.
In the polarized cells, Na+, K+-ATPase was employed to mark the plasma membrane of migrating cells. The AP2 complex clearly co-localized at the leading edge along with Na+, K+-ATPase. The Na+, K+-ATPase also had a polarized distribution, instead of uniformly marking the thin plasma membrane (Fig. 1C). The polarized appearance of Na+, K+-ATPase may be due to a thickening of the membrane at the leading edge. Alternatively, Na+, K+-ATPase may be directed to the polarized leading edge to inhibit cell motility as suggested by Barve et al (24).
Next, we determined whether clathrin is polarized at the leading edge in a manner similar to the AP-2 complex. With high resolution confocal microscopy, polarized PLB-985-CXCR2 cells showed clathrin as distinct dots, as well as converged dots at the leading edge of the polarized cells after 10 min of gradient stimulation with CXCL8 (Fig. 1D left panel). The magnified view of the leading edge of the polarized cells is shown for better clarity (right top and bottom panels). The images were quantitatively analyzed for co-localization of AP2 with clathrin by Metamorph analysis. Twenty six CXCL8-polarized cells were subjected to same threshold settings for both AP2 and clathrin and the % co-localization was measured. Nearly, 70% of AP2 present at the leading edge co-localized with clathrin (69.3±4.5% - S.D.).
Functional consequences of the association of CXCR2 with the AP2 complex and β-arrestin1 in terms CXCR2-mediated chemotaxis and receptor internalization
CXCR2 binds to two major adaptor proteins, namely AP2 and β-arrestin. AP2 also binds to β-arrestin and both proteins can bind to clathrin to facilitate endocytosis (7). The binding of AP2 to CXCR2 is through the LLKIL motif present in the CTD of CXCR2 (7). Earlier, we demonstrated that CXCR2 lacking the ‘LLKIL’ motif in the CTD is incapable of binding to AP2 but still can bind to β-arrestin1 efficiently (7). Sequence alignment and analysis of the sequences between CXCR2, CXCR1, CXCR3 and CXCR4 CTDs with a web-based CLUSTAL W program indicated that the LLKIL motif or a highly similar motif in the CTD of these chemokine receptors is conserved among CXC chemokine receptors. The IL residues important for AP-2 binding and the phosphorylation sites that are important for β-arrestin binding are highlighted in the top panel (Fig. 2A). The bottom panel illustrates positions of the mutations in 4A-CXCR2 and 4A/IL-CXCR2.
Figure 2. AP2 is essential for CXCR2-mediated chemotaxis, but β-arrestin1 is dispensable.



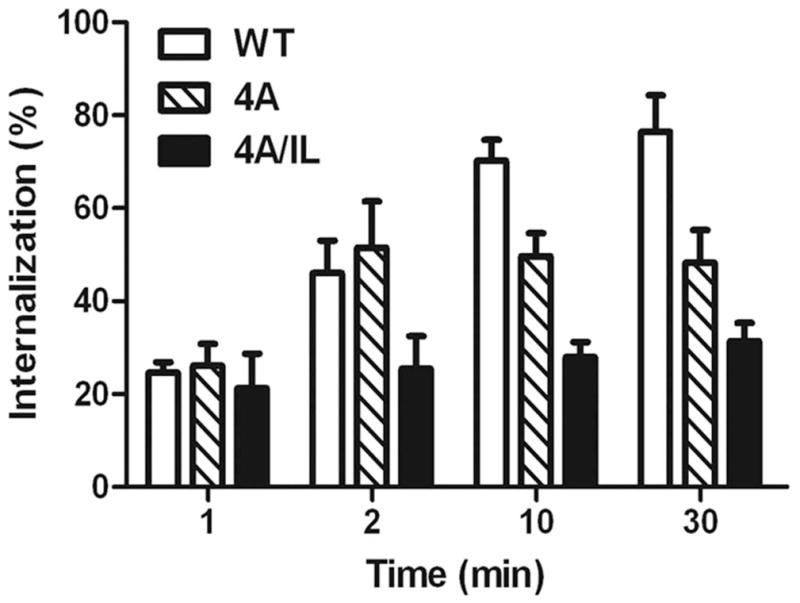
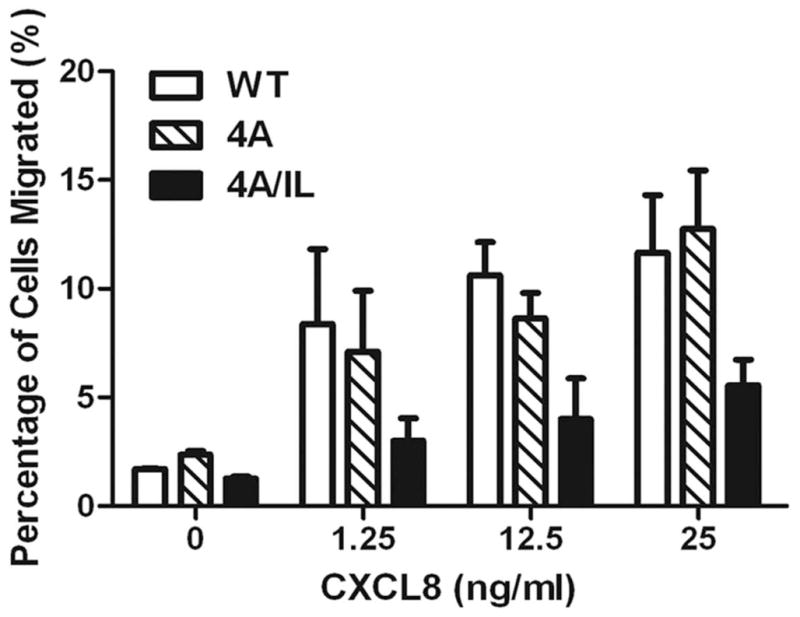
A) Top panel: LLKIL motif in CTDs of human CXC chemokine receptors is conserved. The CTDs of CXCR2 (45 residues), CXCR1 (44 residues), CXCR3 (49 residues) and CXCR4 (47 residues) were aligned with CLUSTALW (1.83) multiple sequence alignment program. The LLKIL functional motif of CXCR2 and similar putative motifs in other CXC receptor CTDs are in bold. Also, the serine residues known to be phosphorylated in CXCR2 CTD in response to CXCL8 stimulation are in bold. Bottom panel: The mutations in CXCR2 important for binding of AP2 and β-arrestin are illustrated. B) Decreased association of CXCR2 mutants with AP2 and/or β-arrestin1 after stimulation with CXCL8. dHL-60 cells stably expressing CXCR2-WT, 4A or CXCR2-4A/IL mutants were stimulated with or without CXCL8. CXCR2 was immunoprecipitated with anti-CXCR2 antibody, and blotted for AP2-β2 subunit or β-arrestin1. The blot was stripped and re-blotted for CXCR2. The relative values of fold increase in response to CXCL8 stimulation for each cell line calculated from 3 independent experiments is shown under the western blots (fold ± S.E.M.). One tenth of the total lysate input for co-immunoprecipitation was used for Western analysis of the total lysates. C) CXCL8-mediated internalization of CXCR2 is abolished in 4A/IL mutant of CXCR2, but only partially attenuated in 4A-CXCR2 mutant. The internalization of CXCR2 was performed by following the internalization of 125I-CXCL8 in dHL60-CXCR2 cells stably expressing CXCR2-WT, 4A or 4A/IL mutant. Error bars are S.E.M and the experiments were repeated 3 times with duplicates for each treatment. ANOVA: 2 min – 4A vs. 4A/IL, p<0.05; 10 min - WT vs. 4A/IL, p<0.001; 30 min – WT vs. 4A, p<0.05 and WT vs. 4A/IL, p<0.001. D) Impairment of CXCR2-mediated chemotaxis when association of AP-2 with CXCR2 is ablated by site-directed mutagenesis0. Chemotaxis assays were performed in a modified Boyden chamber with dHL-60 cells stably expressing CXCR2 WT, 4A, or 4A/IL mutants. Error bars are S.E.M and experiments were repeated 3 times with 5 replicates each treatment. ANOVA: CXCL8 12.5 ng/mL - WT vs. 4A/IL, p<0.01; 4A vs. 4A/IL, p<0.05; CXCL8 25 ng/mL - WT vs. 4A/IL, p<0.05; 4A vs. 4A/IL, p<0.01; WT vs. 4A at all of the time points – n.s.
The binding of β-arrestin to CXCR2 is greatly enhanced when ligand binds to receptor and activates the phosphorylation of serine residues in the CTD (342, 346,347,348). Mutation of these four serine residues to alanine, or truncation of the receptor at S331 reduces the binding of β-arrestin to the receptor without inhibiting chemotaxis and resulted in markedly delayed desensitization (25, 26). A site-directed mutagenesis approach was employed 1) to evaluate whether binding events of AP2 and β-arrestin1 to CXCR2 are interdependent or mutually exclusive events and 2) to dissect the role of AP2 and β-arrestin1 on CXCR2-internalization in CXCR2-mediated chemotaxis.
The AP2 complex may stabilize the binding of β-arrestin1 to CXCR2 when major phosphorylation sites on CXCR2 are mutated
The dHL-60 CXCR2 cells stably expressing WT-CXCR2, 4A-CXCR2 and 4A/IL-CXCR2 mutants were stimulated with CXCL8, the CXCR2-associated proteins were cross-linked, CXCR2 was immunoprecipitated with anti-CXCR2 antibody and analyzed for the binding of AP2 and β-arrestin1 to CXCR2. Considering the relative low expression level of mutant receptors in 4A/IL cells, we increased the loading of this type of cells to match the level of receptors with other cell types in co-immunoprecipitation assays. β-arrestin1 is known to exhibit increased affinity for GPCRs when the carboxyl-terminal domain is phosphorylated (27). When the key phosphorylation sites of CXCR2 that are involved with its desensitization were mutated to alanine residues (4A mutant), the receptor mutant bound weakly to β-arrestin1 (41% reduction), but exhibited normal binding to AP2 in comparison with WT-CXCR2 (Fig. 2B). The CXCR2 receptor with both IL/AA (mutant deficient in AP2 binding) and 4A (mutant with reduced binding to β-arrestin) mutations, designated ‘4A/IL’ mutant, displayed reduced binding to AP2 (64% reduction) as expected and even further reduced binding of β-arrestin1 (90% reduction), in comparison to 4A mutant (Fig. 2B) even though β-arrestin is known to bind with low affinity to GPCRs through the intracellular loops of the receptor (28–30). The deficiency of binding of AP-2 complex to IL/AA-CXCR2 mutant has been shown previously (24).
Ablation of the binding of the AP2 complex to CXCR2 profoundly affects CXCR2 internalization; however reducing β-arrestin1 binding has little effect on CXCR2 internalization
We examined whether binding or loss of binding of AP2 and / or β-arrestin to CXCR2 would modulate the receptor internalization. The internalization of CXCR2 was measured by the [125I]-CXCL8 binding assay. One min following CXCL8 stimulation (100 ng / ml of CXCL8), there was no significant difference in the internalization between WT-CXCR2-WT, 4A-CXCR2 and 4A/IL-CXCR2 mutants. Between 1 min to 30 min, the CXCR2-WT receptor exhibited an increasing percentage of internalization, reaching a maximum of 80% internalization by 30 min. On the other hand, the 4A-CXCR2 mutant showed an initial increase of internalization (50%) at 2 min, with no additional increase in internalization up to 30 min, and remained lower than the WT control (p=0.04). Interestingly, the internalization of the 4A/IL mutant remained significantly low (around 30%) for all of the time points compared to WT and 4A mutant (p<0.001) (Fig. 2C).
Binding of the AP2 complex but not β-arrestin1 to CXCR2 is absolutely essential for CXCL8-mediated chemotaxis
We tested whether a compromise in binding of AP2 or β-arrestin1 to CXCR2 with concomitant effects on CXCR2 internalization would have any functional effect on CXCR2-mediated chemotaxis. Even though there was reduced binding of β-arrestin1 and marginal reduction in CXCR2 internalization (20% at 10 and 30 min of CXCL8 stimulation) with the 4A-CXCR2 mutant (Fig. 2C), the chemotaxis was not compromised, either because there was sufficient receptor internalization, or because the 4A mutant is fully capable of binding to the AP2 complex. On the other hand, when IL/AA substitution was added to the 4A-CXCR2 mutant, the chemotaxis of dHL60 cells was greatly impaired (66% loss), implying that formation of the ‘chemosynapse’ between CXCR2 and the AP2 complex is critical for the CXCR2-directed migration (p<0.01 for WT vs. 4A/IL and p<0.05 for 4A vs. 4A/IL at 12.5 ng/mL of CXCL8; p<0.05 for WT vs. 4A/IL and p<0.01 for 4A vs. 4A/IL at 25 ng/mL of CXCL8; WT vs. 4A at all of the time points – n.s.) (Fig. 2D).
Functional role of AP2-μ2/PIP2 interaction in CXCR2-mediated chemotaxis and internalization
To evaluate how AP2 facilitates CXCR2-mediated chemotaxis, we sought to determine whether binding of AP2 to the PIP2 phospholipid at the inner leaflet of the plasma membrane is an absolute requirement for CXCR2-mediated chemotaxis and / or CXCR2 internalization.
Analysis of the effects of knocking down of AP2-μ2
The functional roles of AP2-μ2/PIP2 and AP2-σ2/CXCR2 interactions in CXCR2-mediated chemotaxis and CXCL8 induced internalization of CXCR2 were evaluated by knocking down AP2-μ2. AP2-μ2 was stably silenced using a lentiviral based shRNA construct. Of the two short hairpin constructs AP2-μ2-sh1 and AP2-μ2-sh5, sh5 was highly efficient in knocking down endogenous AP2-μ2 in HEK-293-CXCR2 cells (93%) (Fig. 3A). The NIH Image J program (31) was employed to quantify the level of knock down of the endogenous μ2 subunit of AP2. Silencing of the μ2 subunit of the AP2 complex also resulted in a concurrent loss of the β2 and σ2 subunits (Fig. 3A). This is not surprising since others have shown that the α2 subunit is also lost when the μ2 subunit of the AP2 complex is silenced (32). In all subsequent experiments, the AP2-μ2-sh5 (denoted as AP2-μ2-KD or simply KD) construct was used for stable knockdown cell lines. AP2-μ2 was also stably knocked down in PLB-985-CXCR2 cells (70%) which were subsequently differentiated into neutrophil-like cells and used for some of the experiments.
Figure 3. Stable silencing of AP2-μ2 impairs CXCR2-mediated chemotaxis.
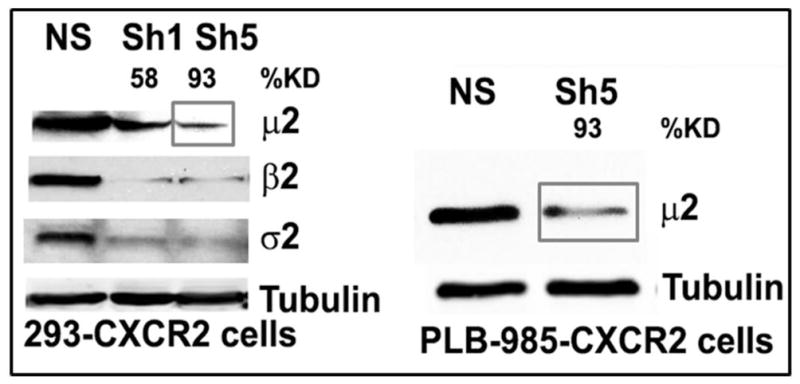
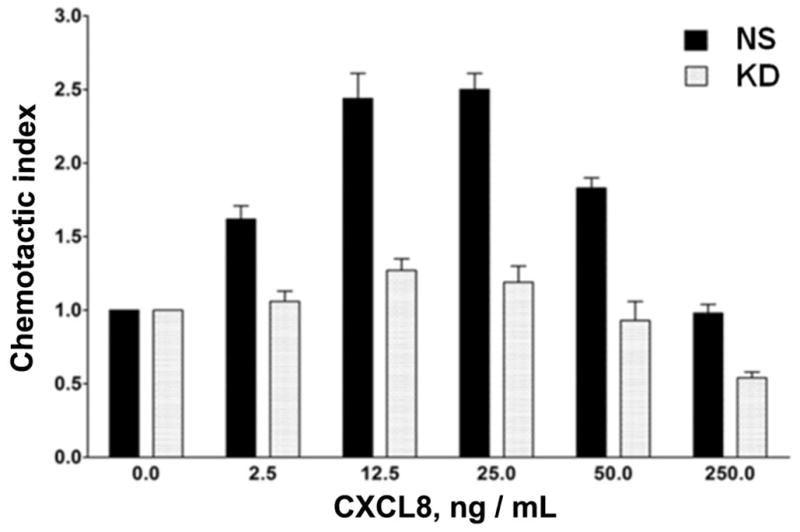
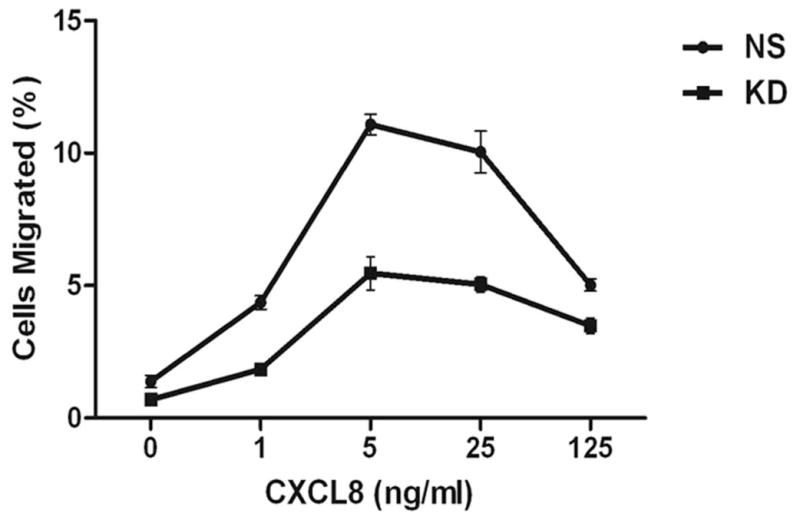
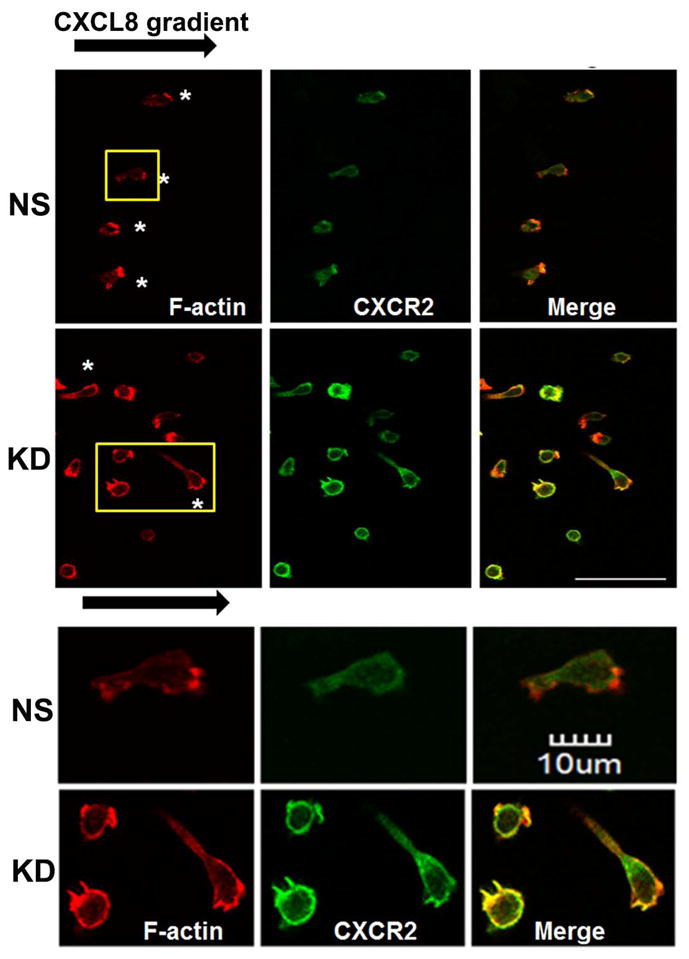
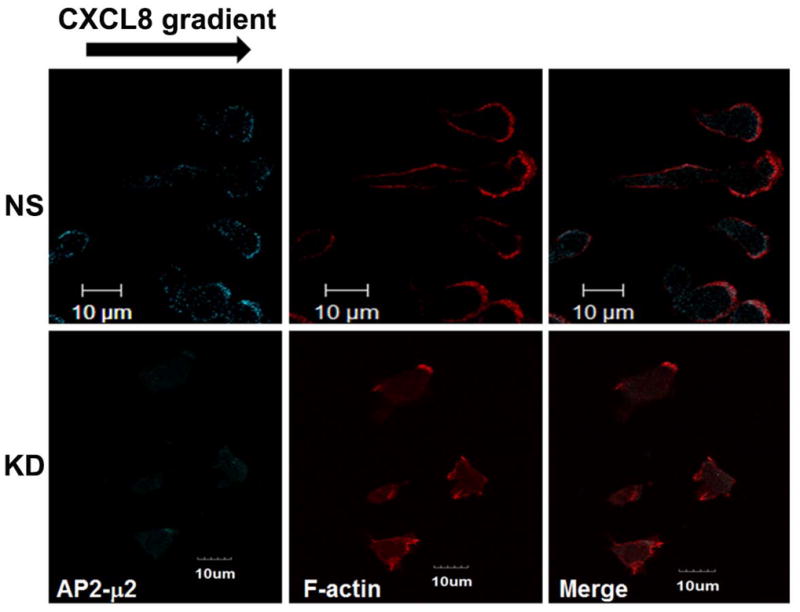
A) Stable knock down of the AP2-μ2 subunit in HEK-293-CXCR2 and PLB-985-CXCR2 cells. Two shRNA constructs sh5 and sh1 were employed to knock down AP2-μ2 in HEK-293-CXCR2 cells and one shRNA construct sh5 was utilized in PLB-985-CXCR2 cells. NS - Non-silencing; % KD – Level of AP2-μ2 knock down. β-tubulin serves as the loading control. B) Stable silencing of the AP2-μ2 subunit impairs CXCR2-mediated chemotaxis in HEK-293-CXCR2 cells. Chemotaxis assays were performed in a modified Boyden chamber. Data were plotted from two independent experiments with 3 replicates in each experiment. NS - Non-silencing; KD-5 - AP2-μ2 knock down; Error bar = S.E.M.; ANOVA: NS vs. KD – for 12.5, 25, and 50 ng / ml CXCL8, p < 0.001; for 2.5 ng / ml CXCL8, p < 0.01; for 250 ng / mL CXCL8, p < 0.05. C) Impairment of CXCR2-mediated chemotaxis of dPLB-985-CXCR2 cells with stable knock down of the AP2-μ2 subunit. Chemotaxis assays were performed in a modified Boyden chamber. Data were plotted from two independent experiments with 4–6 replicates in each experiment. NS - Non-silencing; KD - AP2-μ2 knock down; Error bars - S.E.M. ANOVA: NS vs. KD, p < 0.001 for 1, 5 and 25 ng / mL of CXCL8 and p<0.05 for 125 ng / mL of CXCL8. D) Impairment of polarization of dPLB-985-CXCR2 cells toward the CXCL8 gradient when AP2-μ2 subunit was stably knocked down. Confocal images of dPLB-985-CXCR2 cells were directionally stimulated with 50 ng / ml of CXCL8 in the Zigmond chamber for 10 min. F-actin and CXCR2 were pseudo-colored red and green respectively. Images represent single z-stack sections of 0.5 μm. NS – Non-silenced; KD - AP2-μ2 knock down; Scale bar – 50 μm. An enlarged view of some cells is shown below. E) Comparison of AP-μ2 at the leading edge in polarized dPLB-985-CXCR2 cells with and without AP-μ2 knock down. Confocal images of dPLB-985-CXCR2 cells were directionally stimulated with 50 ng / ml of CXCL8 in the Zigmond chamber for 10 min. F-actin and CXCR2 were pseudo-colored red and green respectively. Images represent single z-stack sections of 0.5 μm. NS – Non-silenced; KD - AP2-μ2 knock down. F) CXCL8-mediated internalization of CXCR2 is attenuated after stable silencing of AP2-μ2 subunit in HEK-293-CXCR2 cells. Non-silencing (NS) and AP2-μ2 knock down (KD) HEK-293-CXCR2 cells (2–5 × 105 cells) were stimulated with 100 ng / ml CXCL8 for different time points, the non-internalized cell surface CXCR2 was quantified by FACS analysis as described in ‘Methods’. The geometric mean of the fluorescence intensity of cell surface CXCR2 was plotted. Error bars - S.E.M. ANOVA: 15 min and 30 min - NS vs. KD – p<0.01.
Impairment of the chemotaxis and internalization upon knockdown of AP2-μ2
We tested the efficiency of the CXCR2-mediated chemotaxis when AP2-μ2 was knocked down. CXCR2-mediated chemotaxis in response to CXCL8 was abolished when the AP2-μ2 was knocked down in HEK-293-CXCR2 cells (p<0.001 for NS vs. KD – at 12.5, 25, and 50 ng / mL CXCL8; p<0.01 for 2.5 ng / mL CXCL8; p<0.05 for 250 ng / mL CXCL8) (Fig. 3B). A similar trend in impairment of CXCR2-mediated chemotaxis was observed in neutrophil-like PLB-985-CXCR2 cells with stable knock down of AP2-μ2 (p<0.001 for 1, 5 and 25 ng / mL CXCL8; p<0.05 for 125 ng / mL CXCL8) (Fig. 3C). The neutrophil-like PLB-985-CXCR2 cells were evaluated in the Zigmond chamber to determine whether if the impairment in chemotaxis was due to a defective polarization. Using confocal analysis of cells, F-actin was employed to mark the leading edge of polarized cells. In the non-silenced control (NS), the migrating cells clearly had F-actin at the tip or the leading edge aligning with the direction of the CXCL8 gradient (marked by asterisks). CXCR2 remained fairly evenly distributed around the cells (Fig. 3D). The CXCL8-mediated polarization of F-actin was impaired in PLB-985-CXCR2 cells with the AP2-μ2 knock down. Interestingly, many AP2-μ2 knock down PLB-985-CXCR2 cells had relatively more CXCR2 on their plasma membrane, presumably due to the reduced internalization due to the knock down of AP2. An enlarged view of both NS and KD cells is shown in the bottom panel depicting mostly round, non-polarized AP2-μ2 knock down PLB-985-CXCR2 cells (Fig. 3D). Similarly, when the non-silenced PLB-985-CXCR2 cells are stained for AP2-μ2, AP2-μ2 had a polarized distribution similar to F-actin in 85% of the cells (46 out of 54 cells), whereas the AP2-μ2 knock down cells exhibited a dim staining for AP2-μ2 and were mostly not polarized towards the CXCL8 gradient (only 4 of 31 cells analyzed (11%) were polarized) (Fig. 3E). The CXCL8-mediated internalization was also impaired in the AP2-μ2 knock down HEK-293-CXCR2 cells (Fig. 3F) (p<0.01 for NS vs. KD at 15 and 30 min.).
Dissecting the role of AP2-μ2/PIP2 interaction in CXCR2-mediated chemotaxis and internalization
In order to determine the role of the two PIP2 binding patches on AP2-μ2 in CXCR2-mediated chemotaxis and internalization, the Patch 1 (residues K341, K343 and K345) (12), Patch 2 (K167, R169 and R170) (15) and the Threonine-156 (T156) sites of μ2 were mutated. A schematic view of the mutations on AP2-μ2 is shown (Fig. 4A). All mutants of AP2-μ2, namely Patch 1(K341E/K343E/K345E), Patch 2 (K167E/R169E/R170E), T156A, and the combination mutant (P1P2T) were expressed in AP2-μ2-sh5 cells (Fig. 4B).
Figure 4. Patch 1 electropositive residues of AP2-μ2 that bind to PIP2 are critical for CXCR2-mediated chemotaxis.
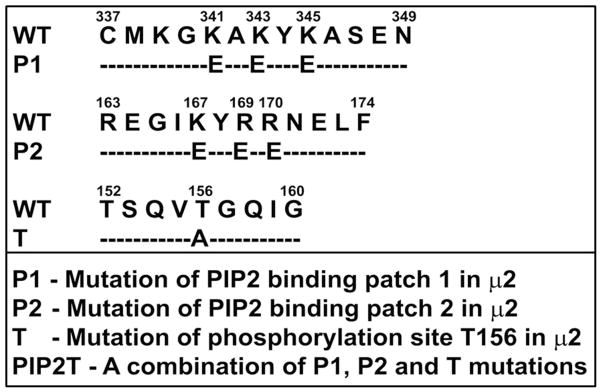
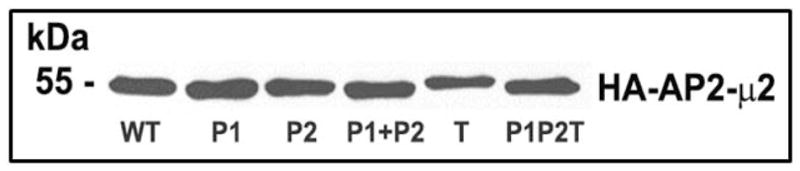

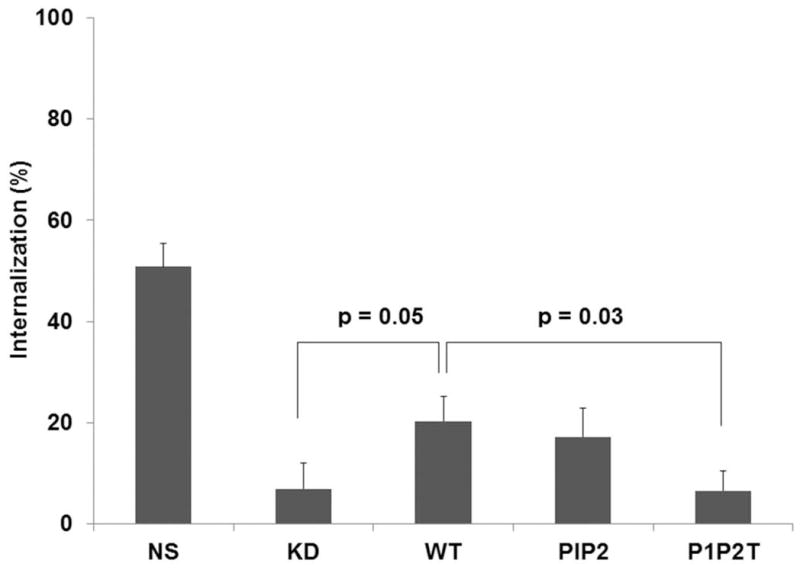
A) Schematic diagram of the different site-directed mutants of AP2-μ2 subunit. The PIP2 binding elements (Patch 1 and 2 residues of AP2-μ2) and the phosphorylation site (T156) were differentially mutated and assessed for their role in CXCR2-mediated chemotaxis. B) Transient expression profile of AP2-μ2 mutants in HEK-293-CXCR2 cells. HA-tagged shRNA-resistant WT, patch 1 (P1), patch 2 (P2), patch 1 and 2 (P1+P2), phosphorylation site (T) and a combination (P1P2T) mutants were transiently overexpressed in HEK-293-CXCR2 cells where AP2-μ2 had been stably knocked down. The proteins from the lysate were separated by 10%SDS-PAGE and analyzed by Western blotting. C) Patch 1 and phosphorylation site T156 but not Patch 2 residues of the AP2-μ2 subunit were vital for CXCR2-mediated chemotaxis. Patch 1 (P1), Patch 2 (P2), phosphorylation site (T156) and the combination (P1P2T) AP2-μ2 mutants were evaluated for their ability to rescue the chemotaxis from the effects of μ2 knock down. The chemotactic index was plotted from three independent experiments with duplicates in each experiment. KD = AP2-μ2-sh5; Error bars - S.E.M. ANOVA: KD vs. WT and WT vs P2 – 2.5, 12.5, 25 and 250 ng / ml CXCL8, p<0.001; KD vs. P1, T and P1P2T – At all concentrations of CXCL8 – p= n.s. D) CXCL8-mediated internalization of CXCR2 is partially rescued by AP2-μ2-WT after stably knocking down the AP2-μ2 subunit in HEK-293-CXCR2 cells. Non-silencing HEK-293 (NS), HEK-293-μ2-sh5 cells and HEK-293-μ2-sh5 cells with transient over-expression of AP2-μ2-WT, AP2-μ2-P1P2 and AP2-μ2 mutant (P1P2T) were stimulated with 100 ng / ml CXCL8 for 5 min and the non-internalized plasma membrane-bound CXCR2 was quantified by FACS analysis as described in ‘Methods’. The geometric mean of the fluorescence intensity was used to calculate the percentage of cell surface CXCR2. KD = AP2-μ2-sh5; ANOVA: KD vs. WT, p= 0.05; KD+WT vs. KD+P1P2T, p=0.03; KD+WT vs. KD+P1P2, p=not significant (n.s.).
In order to test the effects of charge-reversal mutants of Patch 1 and Patch 2 and the phospho-mutant (T156A) of AP2-μ2 on CXCR2-mediated chemotaxis, these mutants and the WT AP2-μ2 were transiently over-expressed in AP2-μ2-KD cells. WT AP2-μ2 was able to rescue CXCL8-stimulated chemotaxis in AP2-μ2-KD cells (p<0.001 for KD vs. WT – at 2.5, 12.5, 25, and 250 ng / mL CXCL8); however, the Patch 1 mutant was totally impaired in CXCL8-stimulated chemotaxis similar to AP2-μ2-KD cells (p=n.s. for KD vs. P1 – at all concentrations of CXCL8). The Patch 2 mutant behaved like the WT AP2-μ2 (p<0.001 for KD vs. P2 – at 2.5, 12.5, 25, and 250 ng / mL CXCL8). The phospho-null mutant (T156A) and the combination mutant comprising Patch 1, 2 and T156 mutations (P1P2T) were also deficient in CXCR2-mediated chemotaxis (p=n.s. for KD vs. T and KD vs. P1P2T – at all concentrations of CXCL8) (Fig. 4C). These results suggest that Patch 1 and T156 of the AP2 complex are critical in CXCR2-mediated chemotaxis. We next determined whether CXCR2 internalization can be rescued by any of the AP2-μ2 mutants after μ2 knock down. Expression of the shRNA resistant WT AP2-μ2 can rescue approximately 40% of the CXCR2 internalization. Overexpression of the PIP2T mutant of AP2-μ2 could not rescue internalization, leaving it similar to the μ2 knockdown level, but the P1P2 mutant (with intact T156) was able to rescue internalization to the level of WT AP2 rescue (Fig. 4D). These data imply that T156 is critical for CXCR2 mediated internalization.
PIP2 interaction patches on AP2-μ2 are dispensable for its interaction with CXCR2 through the σ2 subunit
Next, we examined whether the AP2 complex with Patch 1 and/or Patch 2 charge reversal mutants, the T156A mutant, or the combination (P1, P2 and T156) mutations were capable of interacting with CXCR2. AP2-μ2 WT and its mutants were transiently overexpressed in AP2-μ2-sh5 knock down cells. Following stimulation with 100 ng /ml CXCL8, CXCR2 and its interacting proteins were cross-linked with DSP and the cells were lysed. CXCR2 was immunoprecipitated and any associated AP2 complex was analyzed by blotting with anti-β2 antibody. The AP2 complex with the AP2-μ2 subunit as either WT or Patch 1 mutant or a combination of patch 1 and 2 mutants continued to exhibit binding to CXCR2 (KD). However, the combination mutant, P1P2T (Patch 1, 2 and T156A), exhibited diminished binding to CXCR2 and the levels were very similar to the AP2-μ2 knock down condition (lane 2 under Co-IP panel) (Fig. 5A).
Figure 5. Patch 1 and Patch 2 residues of the AP2-μ2 subunit that bind to PIP2 lipid are not critical for the binding of the AP2 complex to CXCR2.
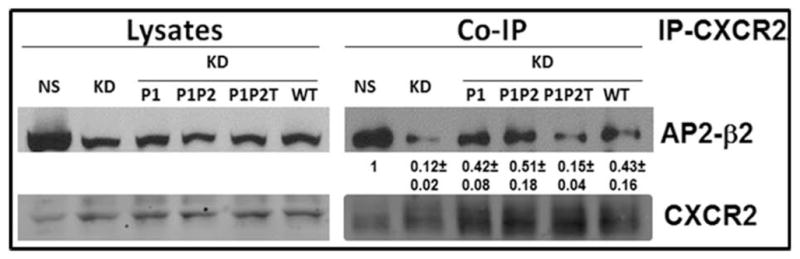
A) Non-silencing (NS), HEK-293-μ2-KD cells and HEK-293-μ2-KD cells with transiently over-expressed AP2-μ2 mutants (P1, P1+P2, P1P2T and WT) were serum starved, stimulated with 100 ng /ml CXCL8 and cross-linked with DSP. The cells were lysed and a CXCR2 co-immunoprecipitation assay was performed. The CXCR2 associated proteins were eluted with 50 mM DTT and separated by 10%SDS-PAGE. The CXCR2 associated AP2 complex was probed with an anti-β2 antibody. Experiments were repeated 3 times and the mean band densities as normalized to co-immunoprecipitation with CXCR2 ±S. D. are shown.
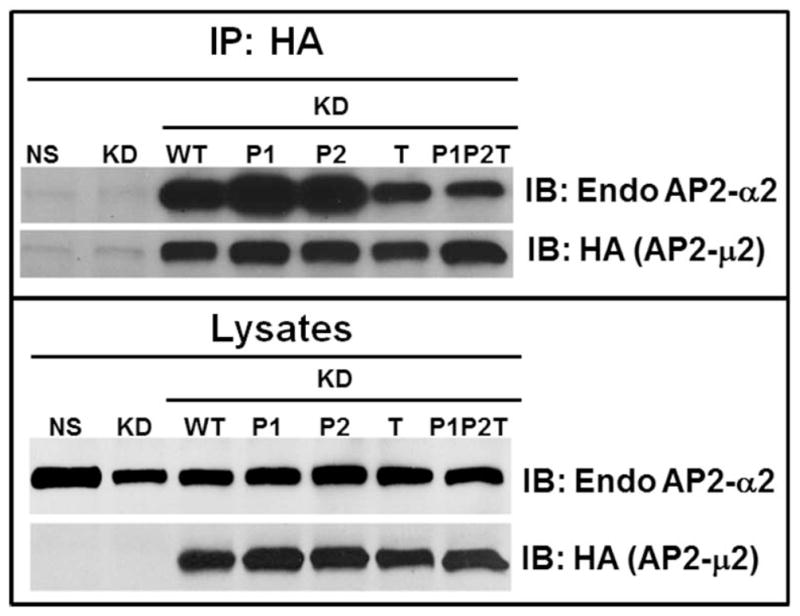
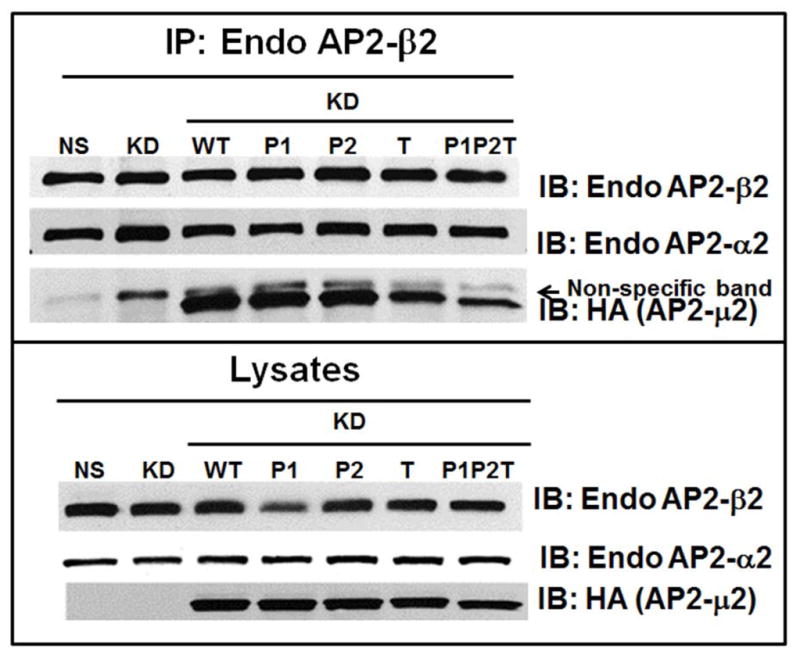
B) HA-AP2-μ2 associates with endogenous α2 subunit of AP-2. HEK-293-μ2-KD cells with transiently over-expressed HA-AP2-μ2 mutants (P1, P1+P2, T, P1P2T and WT), were lysed subjected to Western blot analysis for α2 and HA-μ2 subunits (upper panel). Each HA-tagged μ2 subunit was immunoprecipitated with anti-HA-agarose and blotted for endogenous α2 and for HA-μ2 (upper panel). A representative blot from 3 individual experiments is shown. C) Functional AP-2 complexes successfully incorporate transiently expressed HA-AP2-μ2 as shown by a reciprocal co-immunoprecipitation. A reciprocal co-immunoprecipitation of the endogenous AP2-β2 from 1.5 mg of total lysate shows that the functional AP-2 complexes contain both endogenous AP2-α2 and overexpressed HA-AP2-μ2. One thirtieth (1/30) of the total lysate input for co-immunoprecipitation was used for Western analysis of the total lysates. The top panel shows co-immunoprecipitation and the bottom panel shows the lysates. A representative blot from 2 individual experiments is shown.
To evaluate whether the exogenously expressed HA-AP2-μ2 WT or its mutants can be incorporated with other endogenous subunits of the AP-2 complex to form a functional AP-2 complex, we performed Western analysis of HEK293-CXCR2 cells without or with μ2 knock down/rescue with Sh-RNA resistant HA-AP2-μ2 and determined the levels of expression of the AP2-α2 subunit. Even though the level of the α2 subunit decreased after knock down of the μ2 subunit, a considerable level of α2 was still present and this level increased when exogenous HA-tagged μ2 subunits were transiently overexpressed (Fig 5 B, upper frame). Additionally, we performed co-immunoprecipitation experiments wherein overexpressed μ2 subunits were immunoprecipitated from cell lysates with HA-antibody. Immunoprecipitated proteins were electrophoresed, Western blotted, and blots were probed for associated endogenous α2 subunits. It is clear that HA-tagged μ2 subunits formed robust association with endogenous α2 subunits, indicating a functional AP-2 complex formed with the HA-tagged μ2 (Figure 5B upper frame). The level of association of μ2 with α2 subunits was somewhat reduced with the T156A mutant and the PIP2T mutant as compared to the WT or Patch1 and Patch2 mutants of the μ2 subunit of AP2. In addition, a reciprocal co-immunoprecipitation was also performed in which the endogenous AP2-β2 was immunoprecipitated and blotted for endogenous AP2-α2 and overexpressed AP2-HA-μ2 present in the functionally assembled AP2 complexes. Clearly, overexpressed HA-tagged AP2-μ2 was successfully incorporated into the AP2 holocomplexes for WT, Patch1, Patch 2 and T156A mutants. There was a comparatively small reduction in incorporation of the AP2-μ2-P1P2T mutant into the AP2 holocomplex (Fig. 5C). Others have also shown that HA-μ2 can be successfully incorporated with other endogenous subunits into the AP-2 complex (19, 46).
Hydrophobic residues on AP2-σ2 are indispensable for CXCR2-mediated chemotaxis
The crystal structure of the AP2 complex with the acidic dileucine motif containing peptides revealed a hydrophobic binding pocket formed by amino acid residues V88 and V98 on the σ2 subunit and part of the α-subunit (33). V88D and V98S site-directed mutants of AP2-σ2 were over-expressed in HEK-293-CXCR2 cells and the effects of these mutations on CXCR2-mediated chemotaxis were analyzed. Substitution of Valine88 with aspartate (V88D) produced a modest reduction in CXCR2-mediated chemotaxis (p=0.03) while the V98S mutant had a severe impairment on CXCR2-mediated chemotaxis in HEK-293-CXCR2 cells (p<0.001) (Fig. 6A). Both V88D and V98S mutants of AP2-σ2 displayed small but significant effects on the internalization of CXCR2 at 30 min only following CXCL8 stimulation (p=0.03 for WT vs. V88D; p=0.03 for WT vs. V98S; Bonferroni – p<0.05 for WT vs. V98S at 30 min) (Fig. 6B). These data imply that the V98S mutant of σ2 subunit is a dominant negative mutant in terms of chemotaxis, while both V88D and V98S can have small effects on CXCR2 internalization.
Figure 6. Over-expression of the dominant negative AP2-σ2 inhibits CXCR2-mediated chemotaxis without significantly affecting the internalization of CXCR2.
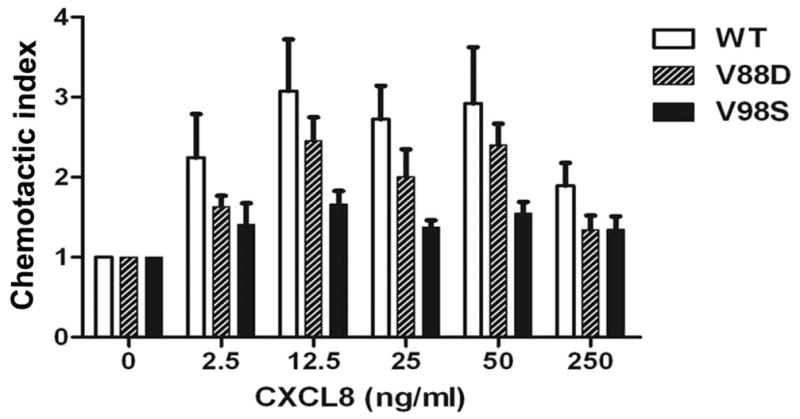
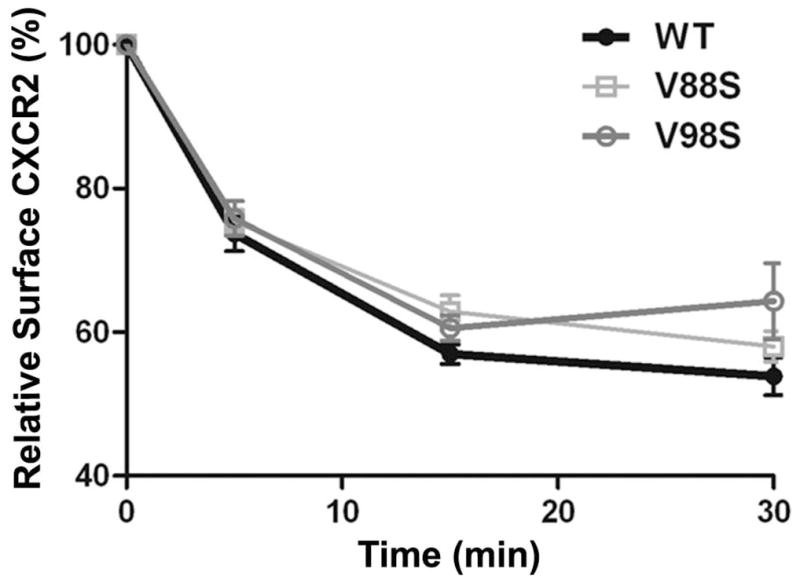
A) WT and dominant negative V88D and V98S AP2-σ2 constructs were transiently over-expressed in HEK-293-CXCR2 cells and chemotaxis assays were performed in a modified Boyden chamber as described in ‘Methods’. Experiments were repeated 3 times with triplicates in each treatment. ANOVA: WT vs. V88D, p = 0.03; WT vs. V98S, p < 0.001. B) CXCR2 internalization assay was performed with FACS analysis as described in ‘Methods’. The geometric mean of the fluorescence intensity was used to calculate the percentage of cell surface CXCR2. ANOVA: WT vs. V88D, p= 0.0262; WT vs. V98S, p = 0.0279. Bonferroni post-hoc tests – WT vs. V98S at 30 min (p < 0.05).
Clathrin does not appear to be required for CXCR2 endocytosis
To evaluate a role for clathrin in the endocytosis of CXCR2, clathrin heavy chain was transiently knocked down in CXCR2-HEK293 cells (Fig. 7A). A small but significant loss (10–20%) of CXCR2 internalization was observed at the 15 min and 30 min time points, presumably due to other clathrin- and caveolae-independent pathways [p<0.0001 for NS vs. KD-clathrin; Bonferroni test – p<0.05 at 5 and 15 min and p<0.01 at 30 min] (Fig. 7B) (51, 52). The effect of loss of clathrin on CXCR2-mediated chemotaxis was not significant from the control (p=0.5 for NS vs. KD-clathrin). (Fig. 7C).
Figure 7. Transient knock down of clathrin heavy chain impedes CXCR2 internalization but has little effect on CXCR2-mediated chemotaxis.
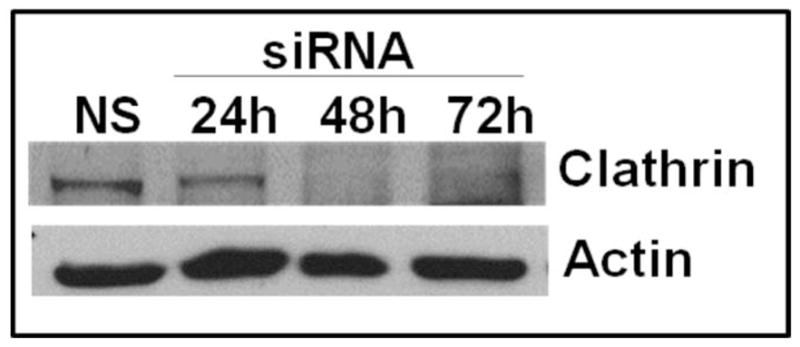
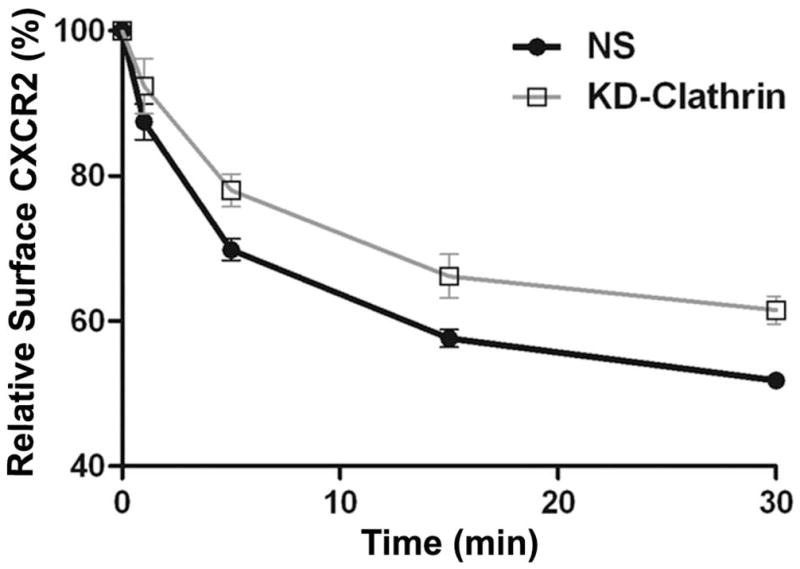
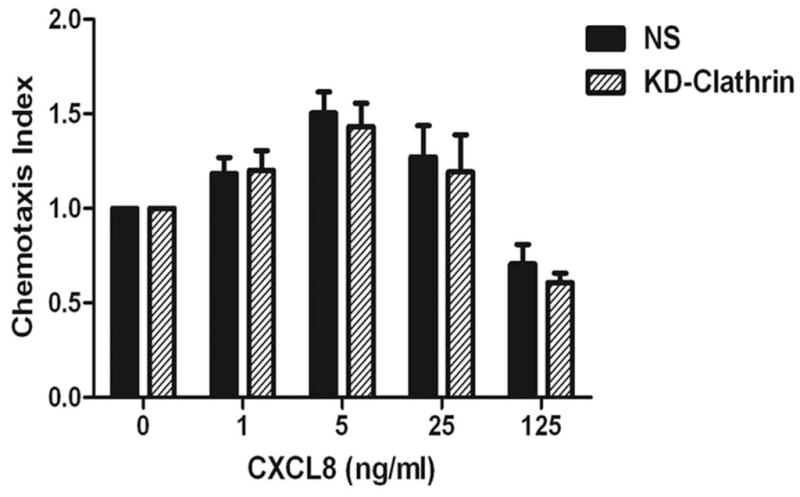
A) siRNA targeting the heavy chain of clathrin was transfected into HEK-293 cells stably expressing CXCR2 and the efficiency of the knock down was assessed by Western blotting with actin serving as the loading control. B) CXCR2 internalization assay was performed with FACS analysis as described in “Methods”. Error bars are S.E.M and experiments were repeated 3 times with triplicates in each treatment. ANOVA: NS vs. KD-clathrin, p < 0.0001. Bonferroni post-hoc tests show significant differences at 5 and 15 min (p < 0.05) and highly significant difference at 30 min (p < 0.01). C) Chemotaxis assays were performed in a modified Boyden chamber as described in ‘Methods’. Error bars are S.E.M and experiments were repeated 3 times with 2–3 replicates in each treatment. ANOVA: NS vs. KD-clathrin, p = 0.512. Bonferroni post-hoc test - All data points – N.S.
Discussion
AP2 is involved in the endocytosis of ligand-activated GPCRs and tyrosine kinase receptors such as the EGF receptor. It also plays a key role in directed cell migration or chemotaxis (7, 23), especially in CXCR2-mediated chemotaxis (9). The LLKIL motif in the CTD of CXCR2 is absolutely essential, as mutation of the dileucine or isoleucine-leucine motif on the CXCR2 CTD ablates ligand-mediated chemotaxis (7, 23). In this study, we initially evaluated whether AP2 and β-arrestin1 play a role in CXCR2-mediated chemotaxis and CXCR2 internalization. Based on results from CXCR2/4A mutant, CXCR2-mediated directed migration and CXCR2 internalization are mainly attributable to AP2 binding. A similar trend is observed in the case of the viral chemokine receptor US28 and the protease activated receptor-1 receptor where AP2 is required for endocytosis but β-arrestins are dispensable (34, 35). Moreover, AP2 binding to clathrin displaces β-arrestin and other accessory proteins from AP2 (36, 37). Also, data from the CXCR2 4A/IL mutant nicely demonstrate that AP2 may play a role in stabilizing the β-arrestin1 binding in the absence of phosphorylation of key serine residues in the CTD of CXCR2. Clearly, the requirement of β-arrestins may depend on the GPCR and also the cell type.
PIP2 generation and signaling at the inner leaflet of the plasma membrane are considered to be integrators of cell polarity in directionally migrating cells (38). CXCR2-mediated polarization is compromised in cells where AP2-μ2 is stably knocked down (Fig. 3D and 3E). This supports the concept that AP2 is a component of the CXCR2 ‘chemosynapse’ formed at the leading edge of a cell in response to a ligand gradient (39), orchestrating the events for initiating and sustaining cell polarization, ultimately leading to directed migration. Interestingly, AP2 directly interacts with the enzyme α-tubulin acetyl transferase at the clathrin coated pits and this interaction is required for microtubule acetylation. This ensures that the stable acetylated microtubules that are oriented towards the leading edge can effectively deliver the cargo necessary for promoting directional migration (40). The presence of Na+, K+-ATPase at the leading edge is probably due to its regulatory roles on phosphatidylinositol-3-kinase and c-Src activity (24, 41) which are involved in CXCR2-mediated chemotaxis (42). AP2 also accumulated at the cell’s leading edge (where the CXCR2-triggered localized activation of PI3K with resultant focal accumulation of PIP2 and PIP3). This is in contrast to the uniform distribution of punctae all over the cell observed in epithelial cells (43–45). This could also be due to the membrane thickening because of the membrane ruffling at the leading edge. However, since CXCR2 remains uniformly distributed along the cell membrane, our data argue for a polarization of AP-2 at the leading edge of the cell.
This is the first report that PIP2 binding residues on Patch 1 (K341, K343 and K345) but not Patch 2 (K167, R169 and R170) of the μ2 subunit of AP2 are critical for CXCR2-mediated chemotaxis. Interestingly, both Patch 1 and Patch 2 residues of AP2-μ2 are not necessary for the interaction of the σ2 subunit of the AP2 with the dileucine motif in the CXCR2 CTD. This iterates the functional independence of the PIP2 binding patches of AP2-μ2 and the cargo binding (LLKIL motif binding to AP2-σ2) (11). Thus it is very intriguing that the Patch 1 domain of AP2-μ2 is necessary for CXCR2-mediated chemotaxis, but Patches 1 and 2 of AP2-μ2 are dispensable for the binding of AP2 to CXCR2. However, the phosphorylation of T156 on AP2-μ2 is essential for CXCR2 binding to AP2. When AP2-μ2 binds the PIP2/PIP3 enriched membrane, this enables the phosphorylation of T156, which results in a 25-fold increase in the affinity for the sorting motifs (46). This makes AP2 a coincidence detector, simultaneously detecting PIP2, Yxxϕ and / or LL/IL-bearing cargoes (47). This also explains the observation that the σ2/α-hemi-appendage continues to bind to the CXCR2 CTD when both Patch 1 and 2 sites on AP2-μ2 are ablated by site-directed mutagenesis.
We were unable to fully restore the CXCR2 internalization by exogenous expression of WT AP2 μ2 in the μ2 KD cells. The only partial rescue of the internalization by the transient over-expression of WTμ2 might be due to a lack of restoration of sufficient levels of other subunits of the AP2 complex (α2, β2, σ2) in the 48h window after transfection to accommodate the overexpressed WTμ2 (Fig. 4D). For example, the level of the AP2-β2 subunit did not show a compensatory increase to accommodate enhanced expression of WTμ2. However, AP2-β1 might potentially substitute for AP2-β2 to accommodate the elevated expression of μ2 (48, 49). We show here that in contrast to β2, α2 is expressed at sufficiently high levels to accommodate either WT AP2-μ2 or mutant forms of μ2 expressed in μ2 KD cells and form the AP2 complex (Fig. 5B). It is not clear whether the AP2 complex is co-translationally assembled with all of the subunits as they are made. If this is the case, then exogenously expressed WT AP2-μ2 may not be thermodynamically stable if it is not co-translationally assembled. Arguing against this possibility, our confocal images of HEK-293-CXCR2 cells overexpressing AP2-μ2 did not show extensive μ2 accumulation around the Golgi area (data not shown). Another reason why exogenously expressed WT μ2 might not allow complete restoration of receptor internalization is that over-expressed AP2-μ2 might sequester synaptotagmin (a protein known to interact with AP2-μ2) that normally is involved in the nucleation of the clathrin-coated pit (50). A similar situation is observed when over-expression of AP180 sequestered clathrin and inhibited clathrin-dependent endocytosis (51). Still another alternative is that other AP2-μ2 interacting proteins could sequester a fraction of the exogenously expressed AP2-μ2 and thus prevent it from forming the functional holocomplex of AP2. Finally, the lack of complete re-expression of the endogenous α2, β2 and σ2 to the level present in non-silenced cells might result in less functional AP2 holocomplexes and therefore result in the lower internalization levels of CXCR2 in μ2KD cells with transient expression of a knock in of AP2- μ2.
It is interesting that hydrophobic amino acidsVal98 and Val 88) of AP2-σ2 are vital for CXCR2-mediated chemotaxis but not absolutely required for the internalization of CXCR2. This might be due to the existence of alternative endocytic pathways like AP2-Arf6 mediated internalization but this remains controversial (52, 53). The CXCR2 receptor is probably capable of invoking alternative endocytic pathways like RhoA, Cdc42 and cortactin-dynamin mediated pathways (54, 55) when the default AP2-σ2-clathrin pathway is compromised. CXCR2 is known to activate Cdc42 and RhoA signaling molecules (9, 56, 57). The knockdown of clathrin did not ablate CXCR2 internalization but had only moderate effects. Importantly, clathrin knockdown had little effect on CXCR2-mediated chemotaxis.
In conclusion, we show here that AP2 is critical for CXCR2-mediated polarization and chemotaxis of cells. Patch1 of AP2-μ2 and the hydrophobic residues Val98 and Val 88 of AP2-σ2 are vital for CXCR2-mediated chemotaxis with Val 88 playing a minor role. Lastly, AP2 mediated CXCR2 receptor internalization can be dissociated from AP2-orchestrated, CXCR2-mediated chemotaxis.
Materials and Methods
Cell Culture
i) HL-60 CXCR2 cells
Human promyelocytic leukemia (HL-60) cells stably expressing human CXCR2 WT and the mutants – CXCR2-4A (S342A/S346A/S347A/S348A) and CXCR2-4A/IL (S342A/S346A/S347A/S348A and I323A/L324A) were grown in Roswell Park Memorial Institute-1640 (RPMI-1640) (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Norcross, GA), 25 mM HEPES, 3 mM L-glutamine and Penicillin (50 units / ml) / Streptomycin (50 μg / ml) (Mediatech, Inc., Herndon, VA). Differentiated HL-60 cells were prepared as previously described (23). These will be denoted as ‘dHL-60- CXCR2’ cells.
ii) HEK-293-CXCR2 cells
Human embryonic kidney HEK-293 cells (HEK-HEK-293) (American Type Culture Collection, Manassas, VA) that were transfected and selected to stably expresses human CXCR2 were cultured in Dulbecco-modified minimum essential medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Norcross, GA), 3 mM L-glutamine and Penicillin (50 units / ml) / Streptomycin (50 μg/ml) (Mediatech, Inc., Herndon, VA). These cells will be denoted as ‘HEK-293-CXCR2’cells.
iii) PLB-985-CXCR2 cells
PLB-985 cells, a variant of HL-60 cells, were kindly provided by Dr. Carole Parent at the National Institute of Health (NIH, Bethesda, MD). These cells were retrovirally transfected and selected to stably express human CXCR2. These will be denoted as PLB-985-CXCR2 cells. These were differentiated into neutrophil-like cells as described previously (23).
Co-localization of CXCR2 and AP2-μ2 in dHL60 cells and PLB-985-CXCR2 cells
The co-localization of CXCR2 with AP2-μ2 in dHL-60 CXCR2 cells was examined by stimulating the cells with CXCL8 by a chemokine gradient generated in the Zigmond chamber (Neuroprobe, Gaithersbug, MD) as described (42). The paraformaldehyde fixed cover slips were processed for staining as described. CXCR2 and AP2-μ2 were probed with mouse anti-CXCR2 (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and rabbit anti-AP2-μ2 antibodies respectively (Abcam, Cambridge, MA). After washing thrice with PBST, the cover slips were incubated with either donkey anti-mouse or donkey anti-rabbit antibodies or goat anti-rabbit AlexaFluor643 that were conjugated to the flourophores cy3 or cy5 (Jackson Immunoresearch Inc., West Grove, PA). F-actin was stained with Rhodamine-phalloidin (Invitrogen, Carlsbad, CA). Washed cover slips were mounted with ProLong Gold antifade reagent (Invitrogen, Carlsbad, CA). Confocal images of the cells were acquired using a Olympus FV-1000 inverted laser scanning confocal microscope (Olympus NDT Inc., Waltham, MA) with a 60X / 1.45 Plan-APOCHROMAT oil immersion lens. The images were processed and assembled with Photoshop computer program (Adobe Systems, San Jose, CA).
Stable knockdown of μ2 subunit of the AP2 complex by RNA interference (RNAi)
HEK-293-CXCR2 cells
To determine the functional significance of the μ2 subunit of the AP2 complex in CXCR2-mediated chemotaxis and internalization, AP2-μ2 was stably knocked down in HEK-293-CXCR2 cells by transfecting shRNA miR plasmids for AP2-μ2 (clone ID – V2LHS_172835 (sh5) and V2LHS_172831 (sh1)). These short hairpin RNAi clones were selected from the GIPZ lentiviral shRNA miR library (Open Biosystems, Huntsville, AL). A non-silencing construct (NS) in the same vector served as the control. These constructs are bicistronic for GFP and shRNA expression. Briefly, 4 μg of the shRNA miR plasmid-Fugene-6 mixture (1:3) were transfected into HEK-293-CXCR2 cells. Puromycin selection pressure was applied 48h later and continued for 4 weeks. The level of knock down in the polyclonal stable cell line was determined by Western analysis of the stable lines and selected cells with at least 70% knock down were functionally analyzed. The AP2-μ2 knock down HEK-293-CXCR2 cells were denoted as ‘HEK-293-μ2-KD’ cells.
PLB-985-CXCR2 cells
To determine the functional significance of the μ2 subunit of the AP2 complex in CXCR2-mediated chemotaxis and internalization, AP2-μ2 was stably knocked down in PLB-985-CXCR2 cells. Briefly, 6 μg of the shRNA miR plasmid (clone ID – V2LHS_172835) (Open Biosystems, Huntsville, AL) for AP2-μ2 in pGIPZ lentiviral vector, 4 μg psPAX2 and 2 μg pGDM.2 of viral envelope plasmids were transfected as a 1:3 Fugene mixture into HEK-293-FT cells, the lenti-virus packaging cells. A non-silencing construct (NS) in the same vector served as the control. Viral supernatants were collected at 48h and 72 h intervals post-transfection, concentrated by ultrafiltration (50K cut off) using Amicon filters (EMD Millipore, Billerica, MA). The viral concentrate was added onto PLB-985-CXCR2 cells plated onto 6-well plates. Puromycin selection pressure (0.5 μg / ml) was applied after 48h and continued for 4 weeks. The level of knock down in the polyclonal stable cell line was determined by Western analysis of the stable lines and selected stable cells were functionally analyzed.
Engineering of CXCR2-4A and CXCR2-4A/IL retroviral constructs
The CXCR2 WT construct in the pMSCV vector served as the template for the site-directed mutagenesis. Mutations in CXCR2 – CXCR2-4A (S342A/S346A/S347A/S348A), and CXCR2-4A/IL (S342A/S346A/S347A/S348A and I323A/L324A) were carried out with the QuikChange site-directed mutagenesis kit following the manufacturer’s protocol.
Engineering of shRNA resistant AP2-μ2 construct
Oligonucleotides were synthesized commercially (Sigma/Aldrich, St. Louis, MO). WT AP2-μ2 was HA-tagged as described previously and this was shown to be functional in terms of association with other subunits as well as endocytosis (58). Mutations in AP2-μ2 were carried out with the QuikChange site-directed mutagenesis kit following the manufacturer’s protocol. Site-directed mutagenesis of the region on AP2-μ2 that binds to the short hairpin RNAi construct (shRNA5) was designed to include three rounds of silent mutations (9 silent mutations) that would ablate the ability of the shRNA5 to destroy the rescue DNA constructs. This was termed ‘shRNA resistant WT AP2-μ2’. The first round of silent mutagenesis involved N310 (AAC to AAT) and F311 (TTT to TTC). For the second round, the mutant DNA from the first round of silent mutagenesis served as the template and silent mutations were performed on K308 (AAG to AAA) and Ser309 (TCC to AGT). For the third round, the mutant DNA from the first two rounds of silent mutagenesis was used as the template to add in degenerate codons for V305 (GTG to GTA), V306 (GTC to GTT) and I307 (ATC to ATT). The primers used for all three rounds of mutagenesis were as follows: first round – forward -5′-GTGGTCATCAAGTCCAA TTTCAAACCCTCACTGCTG-3′ and reverse -5′-CAGCAGT GAGGGTTTGAAATTGGACTTGATGACCAC-3′; second round – forward -5′-CAAGGTGGTCATCAAAAGTAATTT CAAACCCTCAC-3′ and reverse – 5′-GTGAGGGTTT GAAATTACTTTTGATGACCACCTTG-3′; third round - forward 5′-CTGGAGGTCAAGGTAGTTATTAAAAGTAA TTTCAAAC and reverse 5′-GTTTGAAATTACTTTT AATAACTACCTTGACCTCCAG-3′.
Site-directed mutagenesis of AP2-μ2
Patch 1 mutant
For mutation of patch 1, shRNA resistant WT AP2-μ2 DNA served as the template. Mutation of patch-1 involved conversion of K341, K343 and K345 into E341, E343 and E345 respectively. The mutagenic primers employed were as follows: forward – 5′-CTGCATGAAGGGGGAGG CCGAGTACGAGGCCAGCGAG-3′ and reverse–5′-CT CGCTGGCCTCGTACTCGGCCTCCCCCTTCATGCAG-3′.
Patch 2 mutant
For mutation of patch 2, shRNA resistant WT AP2-μ2 DNA was employed as the template. Mutation of patch-2 involved mutation of K167, R169, and R170 to E167, E169 and E170 respectively. The mutagenic primers employed were as follows: forward – 5′-CGGCGAGAGGGTATC GAGTATGAAGAGAATGAGCTCTTCCTGG-3′ and reverse – 5′-CCAGGAAGAGCTCATTCTCTTCATACTCGATACCC TCTCGCCG-3′.
Patch 1 and 2 combination mutant
For combining patch 1 and 2 mutations, patch 2 mutant DNA (K167E/R169E/R170E) was utilized as the template and Patch 1 mutagenic oligonucleotides were employed to generate patch 1 mutations (K341E/K343E/K345E).
T156A phosphorylation site mutant
Using shRNA resistant WT AP2-μ2 DNA as the template, T156 was mutated to A156. The mutagenic primers employed were as follows: forward–5′-CAC CAGCCAGGTAGCTGGGCAGATTGGC-3′ and reverse – 5′-GCCAATCTGCCCAGCTACCTGGCT GGTG-3′.
Patch 1, patch 2 and T156A combination mutant
For combining patch 1(K341E/K343E/K345E), patch 2 (K167E/R169E/R170E) and T156A mutations, patch 1 and patch 2 combination mutant DNA was utilized as the template and T156A mutagenic oligonucleotides were employed to generate patch 1, patch 2 and T156A combination mutant.
Shuttling of AP2-μ2 mutants into a hygromycin-resistant vector
Since the knockdown shRNA plasmids for AP2-μ2 conferred puromycin resistance to HEK-293-CXCR2 cells, the mutants of AP2-μ2 were shuttled into pcDNA3-hygromycin vector. The final AP2-μ2 mutant constructs in pcDNA3 with hygromycin resistance were employed for all transient expression of WT AP2-μ2 and its mutants into HEK-293-CXCR2 cells with the stable knock down of AP2-μ2.
Co-immunoprecipitation of HA-AP2-μ2 with the endogenous α 2 and β2 subunit of the AP2-complex
HEK-293-CXCR2 cells (NS, μ2-KD and μ2-KD cells with transient overexpression of WT or mutant subunits of AP2) were harvested 72h post transfection. The cells were lysed in ice-cold co-immunoprecipitation buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1% Nonidet P-40, 5 mM EDTA) containing proteinase inhibitor cocktail I and phosphatase inhibitor cocktails 3 and 2 (Sigma/Aldrich, St. Louis, MO). HA-tagged AP2-μ2 subunit was immunoprecipitated from 1.5 mg of total lysate by adding monoclonal anti-HA antibody conjugated to Sepharose beads (containing the equivalent of 2 μg of the antibody) (Thermo Scientific, Waltham, MA) and nutated for 2 h at 4°C. Immunoprecipited AP2-μ2 subunit in complex association with the other AP2 subunits were washed thrice with co-immunoprecipitation buffer and eluted from beads by incubating with Laemmli sample buffer under reducing conditions with 50 mM DTT for 10 min at 70°C. Eluted proteins were separated by 10% SDS-PAGE and analyzed by Western blotting.
For reciprocal co-immunoprecipitation of endogenous AP2-β2 subunit with transiently overexpressed HA-AP2-μ2, mouse monoclonal anti-β2 antibodies were cross-linked to Protein G-Dynabeads using Bis[sulfosuccinimidyl] suberate (BS3). Immunoprecipitation of endogenous β2-subunits were performed as described above and blotted for HA-AP2-μ2. NS and μ2-KD cell lysates served as the control as they were not transfected with HA-AP2-μ2 construct.
Transient knockdown of clathrin heavy chain by siRNA
The target sequence for knockdown of clathrin heavy chain was adopted from Motley et al. (16). The siRNAs for clathrin knock down (UAAUCCAAUUCGAAGACCAAU) and the control scramble sequence (AACAGUCGCGUUUGCGACUGG) were commercially synthesized (Thermo Scientific, Waltham, MA). These siRNAs were transfected into HEK-293 cells that stably express human CXCR2 cells using Lipofectamine/RNAimax (Grand Island, NY) following the manufacturer’s protocol. Briefly, 1×106 cells were seeded overnight onto 6 cm dishes. The next day, 15 μl of Lipofectamine/RNAimax was mixed with 200 μl OptiMEM medium (Life Technologies,) and 4 μl 100 μM RNAi into another 200 μl aliquot of OptiMEM medium. These two solutions were mixed and incubated for 10 – 20 min at R.T. The Lipofectamine/RNAimax-siRNA complex was added drop wise into the dish. The final concentration of siRNA was 100 nM in 4 ml of media. Cells were harvested at 24h, 48h and 72h time points and examined for the efficiency of the knock down. The 48h time point post siRNA was chosen for CXCR2-meditated chemotaxis and internalization of CXCR2.
Chemotaxis assays in modified Boyden chamber
The HEK-293-CXCR2 cells with AP2-μ2 knocked down were transiently transfected with shRNA resistant WT AP2-μ2 or various mutants and the chemotaxis was performed in a modified Boyden chamber and were analyzed for their ability to undergo CXCL8-mediated chemotaxis as described previously (7, 23, 39, 59). The chemotactic index was calculated by normalizing to the number of cells that migrated under basal conditions, which was set as 1. Chemotaxis of differentiated neutrophil-like HL60-CXCR2 cells (dHL-60-CXCR2 cells) and differentiated neutrophil-like PLB-985-CXCR2 cells (dPBL-985-CXCR2 cells) were performed in a modified Boyden chamber as described previously (42).
CXCR2 internalization assay
In dHL60-CXCR2 cells, the internalization of CXCR2 was evaluated by following the internalization of [125I]-CXCL8 at different points as described previously (23). CXCR2 internalization in HEK-293-CXCR2 cells was measured by flow cytometry assay. Subconfluent HEK-293-CXCR2 cells were trypsinized, washed and resuspended in DMEM containing 10% FBS. Cells were allowed to recover for at least 2 h at 37°C. The cells were then washed thrice with serum-free DMEM containing 0.1% bovine serum albumin (BSA). An aliquot of these cells (2–5 × 105 cells) were stimulated with 100 ng / ml CXCL8 for different time points and incubated on ice right after stimulation. After washing with ice-cold Ca2+ and Mg2+-free phosphate buffered saline (PBS), pH 7.5, containing 0.5% BSA, Phycoerythrin (PE)-conjugated anti-CXCR2 antibody (1:100) (BD Biosciences Pharmingen, San Diego, CA) was added to bind to non-internalized cell surface CXCR2 and incubated in dark for 15 min. Cells were then washed thrice with PBS containing 0.1% BSA and fixed with 0.5% paraformaldehyde in PBS. Flow cytometry was performed at the Vanderbilt core facilities. Geometric mean of the fluorescence intensity was used to calculate the percentage of cell surface CXCR2.
Co-immunoprecipitation of CXCR2 with AP-2 complex and β-arrestin1
Rabbit anti-CXCR2 antibody was conjugated to paramagnetic Dynabeads (Invitrogen, Carlsbad, CA) as described previously (60). Co-immunoprecipitation was performed after cell lysates were cross-linked. Briefly, HEK-293-CXCR2 cells (NS, μ2-KD and μ2-KD cells with transient over-expression of mutant subunits of AP2 were serum starved for 2h and stimulated for 2 min with CXCL8 at 100 ng / ml. Stimulated cells were washed with ice-cold Ca2+ and Mg2+-free phosphate-buffered saline (PBS), pH 7.5 and cross-linked with 2 mM Dithio-bis [succinimidylpropionate] (DSP) (Sigma/Aldrich, St. Louis, MO) in PBS for 30 min at R.T. Cross-linking was terminated by adding 10 mM Tris-HCl (PH 7.5) for 15 min at R.T. The cells were then lysed in radioimmunoprecipitation assay (RIPA) buffer containing proteinase inhibitor cocktail I and phosphatase inhibitor cocktails 3 and 2 (Sigma/Aldrich, St. Louis, MO). CXCR2 was immunoprecipitated by adding anti-CXCR2-conjugated Dynabeads (containing the equivalent of 1 μg of the antibody) to the cell lysates (300 μg) and nutated for 2 h at 4°C. Immunoprecipited CXCR2 in complex with the associated proteins were washed thrice with PBS containing 0.01% Tween-20 and eluted by incubating with lysis buffer containing 50 mM dithiothreitol (DTT) for 30 min at 37°C. Eluted CXCR2 and its associated proteins were separated by 10% SDS-PAGE and analyzed by Western blotting.
Analysis of co-localization of AP2 with Clathrin
Before acquiring the confocal images of polarized PLB-985 neutrophil-like cells that stably express CXCR2, the microscope was tested to see if the fluorescent images acquired have similar centroids i.e., no misalignment of centroids of the images acquired in different channels from the same fluorescent microbeads (FocalCheck fluorescent microspheres) (Invitrogen). The acquired confocal images were viewed in FV10-ASW-3.0 viewer and the micrographs were exported out as TIFF files. The TIFF files were imported into the ‘Metamorph’ program (Molecular Devices, Sunnyvale, CA) and same threshold was applied for the images of both AP2 and clathrin and analyzed for % co-localization of AP2 with clathrin at the leading edge of CXCL8-polarized PLB-985-CXCR2 cells.
DNA sequencing
The cDNA constructs and mutations were verified by DNA sequencing using Big Dye Terminator chemistry at the (VANderbilt Technologies for Advanced GEnomics) VANTAGE-DNA sequencing core facility at Vanderbilt University.
Statistical analysis
Data analysis was performed using GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA). Statistical significance between groups was determined by analysis of variance (ANOVA) followed by either a post-hoc Bonferroni’s test or Tukey’s multiple comparison test with statistical significance set at p<0.05.
Synopsis.
The dileucine motif of CXCR2 interacts with adaptor protein 2 (AP2) to facilitate ligand-mediated receptor internalization and chemotaxis. Using knockdown/rescue approaches involving mutations in AP2-μ2 and AP2-σ2 subunits involved in PIP2 binding versus receptor, respectively, we evaluated ligand-mediated receptor internalization, cell polarization and chemotaxis. Both electropositive patch1 amino acid residues on AP2-μ2 and the hydrophobic residues on AP2-σ2 are vital for directional migration and chemotaxis, while clathrin is dispensable. Importantly, AP2-mediated receptor internalization can be dissociated from AP2-mediated chemotaxis.
Acknowledgments
We thank Dr. Paige Baugher for her help with CXCR2-4A and CXCR2-4A/IL constructs, Ayub Karwandyar for cross-linking of AP2-β2 antibodies to Dynabeads and Shuai Yuan for his valuable help in engineering of most of the AP2-μ2 mutant constructs. We thank Dr. Sam Wells for his assistance with confocal microscopy. This work was supported by NIH grants CA-34590 (to A.R.), T32 119925 and F32 CA171895 (to O.E.H), VA Merit Award (to A. R.), Career Scientist Award (to A.R.) from the Tennessee Valley Healthcare System and the Department of Veterans Affairs, Vanderbilt Ingram Cancer Center Support grant CA68485, and Ingram Professorship (to A.R.).
References
- 1.Arai H, Charo IF. Differential regulation of G-protein-mediated signaling by chemokine receptors. The Journal of biological chemistry. 1996;271(36):21814–21819. doi: 10.1074/jbc.271.36.21814. [DOI] [PubMed] [Google Scholar]
- 2.Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A, Strieter RM. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol. 2000;165(9):5269–5277. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- 3.Devalaraja RM, Nanney LB, Du J, Qian Q, Yu Y, Devalaraja MN, Richmond A. Delayed wound healing in CXCR2 knockout mice. J Invest Dermatol. 2000;115(2):234–244. doi: 10.1046/j.1523-1747.2000.00034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matsuo Y, Raimondo M, Woodward TA, Wallace MB, Gill KR, Tong Z, Burdick MD, Yang Z, Strieter RM, Hoffman RM, Guha S. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int J Cancer. 2009;125(5):1027–1037. doi: 10.1002/ijc.24383. [DOI] [PubMed] [Google Scholar]
- 5.Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256(2):137–165. doi: 10.1016/j.canlet.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strieter RM, Burdick MD, Mestas J, Gomperts B, Keane MP, Belperio JA. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer. 2006;42(6):768–778. doi: 10.1016/j.ejca.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Fan GH, Yang W, Wang XJ, Qian Q, Richmond A. Identification of a motif in the carboxyl terminus of CXCR2 that is involved in adaptin 2 binding and receptor internalization. Biochemistry. 2001;40(3):791–800. doi: 10.1021/bi001661b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Traub LM. Tickets to ride: selecting cargo for clathrin-regulated internalization. Nat Rev Mol Cell Biol. 2009;10(9):583–596. doi: 10.1038/nrm2751. [DOI] [PubMed] [Google Scholar]
- 9.Sai J, Fan GH, Wang D, Richmond A. The C-terminal domain LLKIL motif of CXCR2 is required for ligand-mediated polarization of early signals during chemotaxis. J Cell Sci. 2004;117(Pt 23):5489–5496. doi: 10.1242/jcs.01398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ. Molecular architecture and functional model of the endocytic AP2 complex. Cell. 2002;109(4):523–535. doi: 10.1016/s0092-8674(02)00735-3. [DOI] [PubMed] [Google Scholar]
- 11.Honing S, Ricotta D, Krauss M, Spate K, Spolaore B, Motley A, Robinson M, Robinson C, Haucke V, Owen DJ. Phosphatidylinositol-(4,5)-bisphosphate regulates sorting signal recognition by the clathrin-associated adaptor complex AP2. Mol Cell. 2005;18(5):519–531. doi: 10.1016/j.molcel.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 12.Gaidarov I, Keen JH. Membrane targeting of endocytic adaptors: cargo and lipid do it together. Dev Cell. 2005;8(6):801–802. doi: 10.1016/j.devcel.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 13.Kwiatkowska K. One lipid, multiple functions: how various pools of PI(4,5)P(2) are created in the plasma membrane. Cellular and molecular life sciences : CMLS. 2010;67(23):3927–3946. doi: 10.1007/s00018-010-0432-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jackson LP, Kelly BT, McCoy AJ, Gaffry T, James LC, Collins BM, Honing S, Evans PR, Owen DJ. A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell. 2010;141(7):1220–1229. doi: 10.1016/j.cell.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaidarov I, Chen Q, Falck JR, Reddy KK, Keen JH. A functional phosphatidylinositol 3,4,5-trisphosphate/phosphoinositide binding domain in the clathrin adaptor AP-2 alpha subunit. Implications for the endocytic pathway. J Biol Chem. 1996;271(34):20922–20929. doi: 10.1074/jbc.271.34.20922. [DOI] [PubMed] [Google Scholar]
- 16.Gaidarov I, Keen JH. Phosphoinositide-AP-2 interactions required for targeting to plasma membrane clathrin-coated pits. The Journal of cell biology. 1999;146(4):755–764. doi: 10.1083/jcb.146.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abe N, Inoue T, Galvez T, Klein L, Meyer T. Dissecting the role of PtdIns(4,5)P2 in endocytosis and recycling of the transferrin receptor. J Cell Sci. 2008;121(Pt 9):1488–1494. doi: 10.1242/jcs.020792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohde G, Wenzel D, Haucke V. A phosphatidylinositol (4,5)-bisphosphate binding site within mu2-adaptin regulates clathrin-mediated endocytosis. J Cell Biol. 2002;158(2):209–214. doi: 10.1083/jcb.200203103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- 20.Krauss M, Kukhtina V, Pechstein A, Haucke V. Stimulation of phosphatidylinositol kinase type I-mediated phosphatidylinositol (4,5)-bisphosphate synthesis by AP-2mu-cargo complexes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(32):11934–11939. doi: 10.1073/pnas.0510306103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozik P, Francis RW, Seaman MN, Robinson MS. A screen for endocytic motifs. Traffic. 2010;11(6):843–855. doi: 10.1111/j.1600-0854.2010.01056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doray B, Lee I, Knisely J, Bu G, Kornfeld S. The gamma/sigma1 and alpha/sigma2 hemicomplexes of clathrin adaptors AP-1 and AP-2 harbor the dileucine recognition site. Molecular biology of the cell. 2007;18(5):1887–1896. doi: 10.1091/mbc.E07-01-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sai J, Walker G, Wikswo J, Richmond A. The IL sequence in the LLKIL motif in CXCR2 is required for full ligand-induced activation of Erk, Akt, and chemotaxis in HL60 cells. J Biol Chem. 2006;281(47):35931–35941. doi: 10.1074/jbc.M605883200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barwe SP, Anilkumar G, Moon SY, Zheng Y, Whitelegge JP, Rajasekaran SA, Rajasekaran AK. Novel role for Na, K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol Biol Cell. 2005;16(3):1082–1094. doi: 10.1091/mbc.E04-05-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mueller SG, White JR, Schraw WP, Lam V, Richmond A. Ligand-induced desensitization of the human CXC chemokine receptor-2 is modulated by multiple serine residues in the carboxyl-terminal domain of the receptor. The Journal of biological chemistry. 1997;272(13):8207–8214. doi: 10.1074/jbc.272.13.8207. [DOI] [PubMed] [Google Scholar]
- 26.Ben-Baruch A, Grimm M, Bengali K, Evans GA, Chertov O, Wang JM, Howard OM, Mukaida N, Matsushima K, Oppenheim JJ. The differential ability of IL-8 and neutrophil-activating peptide-2 to induce attenuation of chemotaxis is mediated by their divergent capabilities to phosphorylate CXCR2 (IL-8 receptor B) J Immunol. 1997;158(12):5927–5933. [PubMed] [Google Scholar]
- 27.Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. The Journal of biological chemistry. 1993;268(23):16879–16882. [PubMed] [Google Scholar]
- 28.Cheng ZJ, Zhao J, Sun Y, Hu W, Wu YL, Cen B, Wu GX, Pei G. beta-arrestin differentially regulates the chemokine receptor CXCR4-mediated signaling and receptor internalization, and this implicates multiple interaction sites between beta-arrestin and CXCR4. The Journal of biological chemistry. 2000;275(4):2479–2485. doi: 10.1074/jbc.275.4.2479. [DOI] [PubMed] [Google Scholar]
- 29.Wu G, Krupnick JG, Benovic JL, Lanier SM. Interaction of arrestins with intracellular domains of muscarinic and alpha2-adrenergic receptors. The Journal of biological chemistry. 1997;272(28):17836–17842. doi: 10.1074/jbc.272.28.17836. [DOI] [PubMed] [Google Scholar]
- 30.Wu N, Macion-Dazard R, Nithianantham S, Xu Z, Hanson SM, Vishnivetskiy SA, Gurevich VV, Thibonnier M, Shoham M. Soluble mimics of the cytoplasmic face of the human V1-vascular vasopressin receptor bind arrestin2 and calmodulin. Molecular pharmacology. 2006;70(1):249–258. doi: 10.1124/mol.105.018804. [DOI] [PubMed] [Google Scholar]
- 31.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motley AM, Berg N, Taylor MJ, Sahlender DA, Hirst J, Owen DJ, Robinson MS. Functional analysis of AP-2 alpha and mu2 subunits. Mol Biol Cell. 2006;17(12):5298–5308. doi: 10.1091/mbc.E06-05-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kelly BT, McCoy AJ, Spate K, Miller SE, Evans PR, Honing S, Owen DJ. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature. 2008;456(7224):976–979. doi: 10.1038/nature07422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fraile-Ramos A, Kohout TA, Waldhoer M, Marsh M. Endocytosis of the viral chemokine receptor US28 does not require beta-arrestins but is dependent on the clathrin-mediated pathway. Traffic. 2003;4(4):243–253. doi: 10.1034/j.1600-0854.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 35.Chen B, Dores MR, Grimsey N, Canto I, Barker BL, Trejo J. Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. J Biol Chem. 2011;286(47):40760–40770. doi: 10.1074/jbc.M111.299776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owen DJ, Vallis Y, Pearse BM, McMahon HT, Evans PR. The structure and function of the beta 2-adaptin appendage domain. EMBO J. 2000;19(16):4216–4227. doi: 10.1093/emboj/19.16.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schmid EM, Ford MG, Burtey A, Praefcke GJ, Peak-Chew SY, Mills IG, Benmerah A, McMahon HT. Role of the AP2 beta-appendage hub in recruiting partners for clathrin-coated vesicle assembly. PLoS biology. 2006;4(9):e262. doi: 10.1371/journal.pbio.0040262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thapa N, Anderson RA. PIP2 signaling, an integrator of cell polarity and vesicle trafficking in directionally migrating cells. Cell adhesion & migration. 2012;6(5):409–412. doi: 10.4161/cam.21192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raman D, Neel NF, Sai J, Mernaugh RL, Ham AJ, Richmond AJ. Characterization of chemokine receptor CXCR2 interacting proteins using a proteomics approach to define the CXCR2 “chemosynapse”. Methods Enzymol. 2009;460:315–330. doi: 10.1016/S0076-6879(09)05215-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montagnac G, Meas-Yedid V, Irondelle M, Castro-Castro A, Franco M, Shida T, Nachury MV, Benmerah A, Olivo-Marin JC, Chavrier P. alphaTAT1 catalyses microtubule acetylation at clathrin-coated pits. Nature. 2013;502(7472):567–570. doi: 10.1038/nature12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem. 2000;275(36):27832–27837. doi: 10.1074/jbc.M002951200. [DOI] [PubMed] [Google Scholar]
- 42.Sai J, Raman D, Liu Y, Wikswo J, Richmond A. Parallel phosphatidylinositol 3-kinase (PI3K)-dependent and Src-dependent pathways lead to CXCL8-mediated Rac2 activation and chemotaxis. J Biol Chem. 2008;283(39):26538–26547. doi: 10.1074/jbc.M805611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong K, Pertz O, Hahn K, Bourne H. Neutrophil polarization: spatiotemporal dynamics of RhoA activity support a self-organizing mechanism. Proc Natl Acad Sci U S A. 2006;103(10):3639–3644. doi: 10.1073/pnas.0600092103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Keymeulen A, Wong K, Knight ZA, Govaerts C, Hahn KM, Shokat KM, Bourne HR. To stabilize neutrophil polarity, PIP3 and Cdc42 augment RhoA activity at the back as well as signals at the front. J Cell Biol. 2006;174(3):437–445. doi: 10.1083/jcb.200604113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Onsum MD, Wong K, Herzmark P, Bourne HR, Arkin AP. Morphology matters in immune cell chemotaxis: membrane asymmetry affects amplification. Physical biology. 2006;3(3):190–199. doi: 10.1088/1478-3975/3/3/004. [DOI] [PubMed] [Google Scholar]
- 46.Ricotta D, Conner SD, Schmid SL, von Figura K, Honing S. Phosphorylation of the AP2 mu subunit by AAK1 mediates high affinity binding to membrane protein sorting signals. J Cell Biol. 2002;156(5):791–795. doi: 10.1083/jcb.200111068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carlton JG, Cullen PJ. Coincidence detection in phosphoinositide signaling. Trends in cell biology. 2005;15(10):540–547. doi: 10.1016/j.tcb.2005.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahle S, Mann A, Eichelsbacher U, Ungewickell E. Structural relationships between clathrin assembly proteins from the Golgi and the plasma membrane. EMBO J. 1988;7(4):919–929. doi: 10.1002/j.1460-2075.1988.tb02897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keen JH. Clathrin and associated assembly and disassembly proteins. Annu Rev Biochem. 1990;59:415–438. doi: 10.1146/annurev.bi.59.070190.002215. [DOI] [PubMed] [Google Scholar]
- 50.Haucke V, Wenk MR, Chapman ER, Farsad K, De Camilli P. Dual interaction of synaptotagmin with mu2- and alpha-adaptin facilitates clathrin-coated pit nucleation. Embo J. 2000;19(22):6011–6019. doi: 10.1093/emboj/19.22.6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao X, Greener T, Al-Hasani H, Cushman SW, Eisenberg E, Greene LE. Expression of auxilin or AP180 inhibits endocytosis by mislocalizing clathrin: evidence for formation of nascent pits containing AP1 or AP2 but not clathrin. Journal of cell science. 2001;114(Pt 2):353–365. doi: 10.1242/jcs.114.2.353. [DOI] [PubMed] [Google Scholar]
- 52.Lau AW, Chou MM. The adaptor complex AP-2 regulates post-endocytic trafficking through the non-clathrin Arf6-dependent endocytic pathway. J Cell Sci. 2008;121(Pt 24):4008–4017. doi: 10.1242/jcs.033522. [DOI] [PubMed] [Google Scholar]
- 53.Montagnac G, de Forges H, Smythe E, Gueudry C, Romao M, Salamero J, Chavrier P. Decoupling of activation and effector binding underlies ARF6 priming of fast endocytic recycling. Curr Biol. 2011;21(7):574–579. doi: 10.1016/j.cub.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 54.Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8(8):603–612. doi: 10.1038/nrm2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sauvonnet N, Dujeancourt A, Dautry-Varsat A. Cortactin and dynamin are required for the clathrin-independent endocytosis of gammac cytokine receptor. J Cell Biol. 2005;168(1):155–163. doi: 10.1083/jcb.200406174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neel NF, Sai J, Ham AJ, Sobolik-Delmaire T, Mernaugh RL, Richmond A. IQGAP1 is a novel CXCR2-interacting protein and essential component of the “chemosynapse”. PLoS One. 2011;6(8):e23813. doi: 10.1371/journal.pone.0023813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. American journal of physiology Lung cellular and molecular physiology. 2001;280(6):L1094–1103. doi: 10.1152/ajplung.2001.280.6.L1094. [DOI] [PubMed] [Google Scholar]
- 58.Nesterov A, Carter RE, Sorkina T, Gill GN, Sorkin A. Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. Embo J. 1999;18(9):2489–2499. doi: 10.1093/emboj/18.9.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raman D, Sai J, Neel NF, Chew CS, Richmond A. LIM and SH3 protein-1 modulates CXCR2-mediated cell migration. PLoS One. 2010;5(4):e10050. doi: 10.1371/journal.pone.0010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith AL, Friedman DB, Yu H, Carnahan RH, Reynolds AB. ReCLIP (reversible cross-link immuno-precipitation): an efficient method for interrogation of labile protein complexes. PloS one. 2011;6(1):e16206. doi: 10.1371/journal.pone.0016206. [DOI] [PMC free article] [PubMed] [Google Scholar]