

Abstract
Age-related hearing loss (ARHL), a degenerative disorder characterized by age-dependent progressive increase in the threshold of auditory sensitivity, affects 40% of people over the age of 65, and it has emerged as an important social and public health problem. Various factors, including genetic and environmental components, are known to affect both the onset and severity of ARHL. In particular, age-dependent changes in cellular oxidative stress and inflammatory responses accompanied by altered cellular signaling and gene expression progressively affect the function of the auditory system and eventually lead to hearing impairment. Recent findings suggest that a disturbance of intracellular NAD+ levels is clinically related to the progression of age-associated disorders. Therefore, maintenance of optimal intracellular NAD+ levels may be a critical factor for cellular senescence, and thus, understanding its molecular signaling pathways would provide critical insights into the prevention and treatment of ARHL as well as other age-related diseases. In this review, we describe the role of NAD+ metabolism in aging and age-related diseases, including ARHL, and discuss a potential strategy for prevention or treatment of ARHL with a particular interest in NAD+-dependent cellular pathways.
Keywords: Age-related hearing loss, NAD+, degenerative disorder, metabolism
Age-related hearing loss (ARHL), known as presbycusis, is a degenerative disorder characterized by progressive worsening of auditory sensitivity with age. Although ARHL is not life-threatening, it affects communication and functional ability, and is strongly associated with decreased quality of life, cognitive decline, and increased incidence of clinical depression and social isolation [1]. ARHL affects around 40% of people over the age of 65. The percentage continues to increase with age, reaching a maximum of 60–80% of people over the age of 85 [2]. Owing to the high prevalence of ARHL in aged people, especially, with a steady but significant increase in human lifespan, ARHL is emerging as a major social and public health problem. Therefore, early prevention, detection/diagnosis, treatment, and improvement of the quality of hearing health care are crucial for hearing impaired patients.
ARHL is critically influenced by a host of environmental factors as well as genetic factors [3]. For example, lifetime insults to the auditory systems by noise, smoking, and ototoxic medication are mainly considered as critical predisposing factors that accelerate the hearing loss with age. Inherited and acquired mutations in the chromosomal and mitochondrial DNAs also enhance the susceptibility to ARHL. ARHL is generally accompanied by various types of auditory dysfunction that progresses with age. The functional integrity of three major components of the cochlea, organ of Corti, stria vascularis/spiral ligament, and spiral ganglion neurons are compromised in different types of ARHL. A high-frequency hearing loss may reflect a sensory hearing loss due to outer hair cell damage in the organ of Corti [4]. An early non-specific progressive hearing loss may indicate a metabolic hearing loss caused by the damage to stria vascularis and spiral ligament prior to significant hair cell degeneration [5]. Because the stria vascularis is heavily vascularized and has an extremely high metabolic rate, the hearing loss caused by the damage to stria vascularis is called as a metabolic hearing loss. Neural hearing loss, which is thought to affect speech comprehension, usually refers to the damage to the spiral ganglion neurons. Many histological studies support the loss of outer hair cells as well as cellular components in stria vascularis and spiral ligament in ARHL.
Although several hypotheses have been postulated to explain the molecular mechanism of age-associated chronic diseases, including ARHL, many of them are yet to be proved. However, oxidative stress, mitochondrial dysfunction, inflammatory response, and altered cell signaling and gene expressions have been reported to facilitate tissue damages that play a central role in age-related disease that are characterized by morphological and ultrastructural degeneration. Recently, the intracellular nicotinamide adenine dinucleotide (NAD+)/NADH ratios have been reported to be decreased in various organs, including liver, heart, kidney and lung, in aged animals and human skin tissue [6, 7]. The decrease of NAD+/NADH ratio is attributed to the NAD+-consuming Poly(ADP-ribose) polymerase 1 (PARP1) hyper-activation induced by the accumulation of age-associated oxidative damage due to altered redox mechanisms and consequent DNA damage [6]. Since silent mating type information regulation 2 homolog 1 (sirtuin1, SIRT1) activity is influenced by the NAD+/NADH ratio [8], a significant reduction in the NAD+/NADH ratio in the aging process causes a decrease in SIRT1 activity with age. In addition, the α-ketoglutarate dehydrogenase (α-KGDH) complex, an enzyme complex of Krebs cycle in the mitochondria, facilitates the generation of reactive oxygen species (ROS) after the reduction in NAD+/NADH [9]. Also, decreased NAD+/NADH favors ROS generation in the respiratory chain complex I [10]. Therefore, maintaining adequate NAD+ levels may be a critical factor for cellular senescence and could emerge as a useful strategy for treating many diseases, including age-related diseases. In this review, we describe the role of NAD+ metabolism in aging and age-related diseases, including ARHL, and describe a potential strategy for prevention or treatment of ARHL, by targeting the NAD+-dependent cellular pathways.
Aging, ARHL, and Calorie Restriction (CR)
Influence of ROS and Inflammation in Aging
Aging refers to a series of time-dependent changes at molecular and cellular levels leading to characteristic phenotypic alterations that negatively affect the function of various organisms. Recently, increased attention is being focused on the involvement of ROS in age-related cellular degeneration throughout the organism. More than 50 years ago, Harman proposed that aging is the result of damage to tissues by free radicals that ultimately causes functional deficits of organs [11]. Free radicals or ROS, including superoxide anion, hydrogen peroxide, and hydroxyl radicals, are unavoidable byproducts of cellular respiration. They are very unstable, highly reactive, and thereby damage proteins, lipids, and DNA in the target organs. Mitochondria has been reported to play a key role in aging, both as a major source of ROS and a target for their damaging effects, and, therefore, mitochondrial oxidative stress appears to be a cause, rather than a consequence [12, 13]. This oxidative stress leads to the damage of mitochondrial molecules such as mitochondrial DNA (mtDNA), lipids, or proteins. A recent study on transgenic mice with a high rate of accumulated mtDNA mutations demonstrated that increased mtDNA mutations leads to signs of accelerated aging, including early onset of hearing loss [14]. Interestingly, ototoxic insults, such as noise, ototoxic drugs, as well as aging, increase the oxidative stress and ROS production, that inflicts oxidative damage on mtDNA and mutations of mtDNA leading to defective electron transport [15]. ROS create a vicious cycle in which the mtDNA mutation increases the ROS-induced cellular damage, activating the apoptotic cascade and thereby inducing cell death [16, 17].
The increased inflammation is considered as one of the hallmarks of aging and is closely associated with chronic diseases. While acute inflammation is normally tightly controlled and is a part of the healing process, chronic low-grade inflammation with increases in circulating pro-inflammatory markers, such as TNF-α, IL-6, and C-reactive protein (CRP) have been associated with a number of age-associated chronic disorders, including cardiovascular disease, diabetes, physical disability, and cognitive decline [18]. Several potential mechanisms contribute to age-related inflammation. Decline in immune function with aging promotes inflammation. Although it is somewhat difficult to decipher if age-related chronic diseases are a cause or consequence of the excessive inflammation, these age-related chronic disorders, such as obesity, physical inactivity, cardiovascular disease, diabetes, chronic kidney disease, osteoarthritis, and Alzheimer’s disease, are closely associated with increased inflammation [19]. Interestingly, chronic inflammation results in the generation of free radicals that facilitate the damage and deterioration of target cells and organs, which further leads to chronic disease. Furthermore, it is well known that oxidative stress induced ROS increases in tissues and circulating sera, while antioxidant capacity declines with age [20]. Though it is not easy to determine which is a cause or consequence, literary evidences indicate a complex relationship between inflammation and oxidative stress in age-associated diseases.
Influence of ROS and Inflammation in ARHL
Although the biological mechanisms underlying ARHL remain to be elucidated, one possible mechanism causing ARHL is the effect of inflammatory processes on the cochlea. It has been reported that there is a relationship between ARHL and chronic inflammatory diseases such as diabetes [21] and cardiovascular disease [22]. A link between immune function and experimental ARHL has also been identified in an animal model study using a senescence-accelerated mouse (SAM), the SAMP1 [23]. Furthermore, pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, play a critical role in cochlear tissue damage in noise- or cisplatin-induced hearing loss [24, 25]. Immunohistochemical localization of pro-inflammatory cytokines in the cochlea following noise or cisplatin exposure indicated that these cytokines are predominantly expressed in the stria vascularis, spiral ganglion neuron, and organ of Corti. These studies suggest the possibility that a chronic age-related inflammation could induce, or accelerate, long-term damage to the cochlear tissue with age, thereby eventually facilitating ARHL.
Influence of CR and SIRT in Aging and ARHL
Caloric restriction (CR) extends the lifespan of most mammalian species and appears to consistently decrease the biological rate of aging in a variety of organisms as well as delay the onset of age-associated diseases, including chronic inflammatory disorders such as hypertension, cardiovascular disease, diabetes, nephropathy, and stroke [26–28]. CR also delays the onset of ARHL and reduces the age-related cochlear pathology in rodents [29–31]. The mechanisms underlying the beneficial effects of CR in aging and age-associated diseases are attributed to the reduced oxidative damage to protein, lipid, and DNA and reduced levels of mtDNA mutations by decreasing the metabolism and the associated production of damaging ROS [26, 32]. However, recent findings indicate that CR in fact increases the metabolism, which suggests the possibility that there are other alternative mechanisms that mediate the beneficial effects of CR in aging and age-associated diseases [33, 34]. In order to identify these alternative pathways, recent genetic and molecular studies in model organisms began to elucidate the involvement of sirtuins, a mammalian ortholog of Saccharomyces cerevisiae silent information regulator 2 (SIR2) gene, on the beneficial effect of CR. Interestingly, SIRTs require NAD+ as a cofactor for enzymatic histone deacetylase activity [35]. The deacetylation catalyzed by SIRTs is coordinated with the cleavage of NAD+ into nicotinamide (NAM) and 1-O-acetyl-ADP-ribose [36]. Thus, the NAD+ requirement for SIRT function indicates a potential link between aging and metabolism.
Enzymes in Aging-related Hearing Loss
Role of sirtuin enzymes in aging and ARHL
NAD+ acts as a metabolic cofactor and a rate-limiting co-substrate for many enzymes, including the sirtuins. The mammalian family of sirtuins consists of 7 enzymes, named SIRT1-7 [37]. They are ubiquitously expressed and they show a specific cellular localization and function. SIRT1, SIRT6, and SIRT7 are generally localized in the nuclei of cells [38], whereas SIRT3, SIRT4, and SIRT5 are localized in the mitochondria [38–40]. SIRT1 and SIRT5 act exclusively as deacetylases [41, 42], whereas SIRT2, SIRT3, SIRT4, and SIRT6 may also have a mono-ADP-ribosyl transferase activity [41, 43–45]. SIRT1, the most widely studied sirtuin, has a Km for NAD+ that lies within the range of the physiological changes in intracellular NAD+ content. This suggests that sirtuin activity could be critically influenced by the physiological changes in intracellular NAD+ levels [46]. Considering that the intracellular NAD+/NADH ratios are decreased in various tissues of aged animals and humans [6, 7], this decreased NAD+/NADH ratio reduces the SIRT1 activity with age. Furthermore, it has been reported that CR extends yeast life span by increasing the cellular NAD+/NADH ratio [47, 48]. SIRT1 also plays a key role in energy homeostasis and extension of rodent life span after CR [49, 50]. Quintas et al. reported the age-associated decrease in SIRT1 expression in rat hippocampus [50], whereas CR increased the expression of SIRT1 in the brain, liver, adipose tissue, and kidney [51]. Therefore, these findings suggest that the simultaneous decreases of NAD+/NADH ratio and SIRT1 expression with age may play an important role in the aging process and the pathogenesis of age-related disorders.
SIRT1 regulates diverse biological functions through direct interaction and subsequent deacetylation of its targets such as FOXOs, PGC-1α, p53, and NF-κB, which closely relate to its function in the aging process and CR-induced effect [49]. NF-κB transcription factor is one of the key regulators of inflammation. NF-κB activation is achieved by either an IκB-dependent pathway through IκB phosphorylation and subsequent degradation or an IκB-independent pathway through post-translational modifications of Rels, including acetylation of the NF-κB p65 subunit. NF-κB p65 can be acetylated at 5 specific lysine residues (Lys-122, -123, -218, -221, and -310). In particular, acetylation of Lys-310 is required for the transcriptional activity of NF-κB, whereas the other acetylation sites are involved in DNA binding [52]. A large body of recent evidence indicates that SIRT1 regulates the inflammatory responses through NF-κB p65 deacetylation. SIRT1 knockdown leads to inflammatory pathway activation with increased inflammatory gene expression, whereas SIRT1 activation produces anti-inflammatory effects [53]. SIRT1 physically interacts with the nuclear translocated NF-κB p65 and deacetylates NF-κB p65 at Lys-310, thereby inhibiting the transcriptional activity of NF-κB [54].
The tumor suppressor p53 is a key transcription factor in the cellular stress response [55]. A number of post-translational modifications can occur in p53 that have critical effects on its stability and function, including phosphorylation, acetylation, sumoylation, neddylation, and methylation [56]. Cytosolic p53 is bound to Mdm2, a RING finger E3 ubiquitin ligase that facilitates degradation under normal conditions. Cellular stress, including DNA damage, hypoxia, or oxidative stress, induces rapid mitochondrial translocation and post-translational modification, such as acetylation of p53 by p300/CBP or PCAF acetyltransferase [57]. The p53 is acetylated at lysine residues, including Lys 370, 372, 382, and 386 in the carboxy-terminal region. Because acetylated p53 cannot bind to Mdm2, increased p53 acetylation levels strongly correlates with protein stabilization and activation in response to cellular stress [58]. Interestingly, nuclear SIRT1 and mitochondrial SIRT3 regulate p53 function through direct interaction and subsequent deacetylation of p53 [59]. In the nucleus, acetylation of p53 stimulates its sequence-specific DNA-binding and subsequent recruitment of other transcription cofactors to promoter regions, and thereby enhances transcription of target genes [60], such as the p53-upregulated modulator of apoptosis (PUMA), NADPH activator A (NOXA), and p53-induced gene 3 (PIG3) that are involved in ROS production through mitochondrial dysfunction or apoptosis. Deacetylation of p53 by nuclear-localized SIRT1 inactivates this sequence-specific transcriptional activity of the protein and represses p53-mediated cell growth arrest and apoptosis in response to DNA damage and oxidative stress [58].
Mitochondria-localized SIRT3 deacetylates and activates several enzymes that are critical in maintaining cellular ROS levels and apoptosis. Though it is not well known whether acetylated p53 in mitochondria has other functions, mitochondrial p53 interacts with anti- and proapoptotic Bcl-2 family members to either inhibit or activate them, and thereby promotes apoptosis through robust mitochondrial outer membrane permeabilization and subsequent cytochrome c release [61, 62]. Deacetylation of p53 by mitochondrial-localized SIRT3 represses p53-mediated cell growth arrest and apoptosis in response to DNA damage and oxidative stress [59]. SIRT3 also deacetylates SOD2 and thereby enhances the catalytic activity of SOD2 to attenuate oxidative stress [63]. SIRT3 knockout mice could not decrease the levels of lipid peroxidation that are typically observed during CR indicating that SIRT3 is necessary during CR to mitigate oxidative stress. In addition, a recent report by Someya et al. demonstrated that SIRT3 is critical for the prevention of ARHL by CR as it enhances the deacetylation of mitochondrial isocitrate dehydrogenase, IDH2 [64]. SIRT3 enhances the enzymatic activity of IDH2 by deacetylation during CR [64]. Activated IDH2 stimulates the reduction of NADP+ to NADPH, which in turn facilitates the reduction of oxidized glutathione and thereby reduces oxidative stress. Furthermore, CR reduced the loss of hair cells and spiral ganglion neurons in the cochlea of WT mice, but not in SIRT3 KO mice. Therefore, adequate SIRT3 levels and activity are critical for the maintenance of normal hearing function.
Role of non-sirtuin NAD+-consuming enzyme PARPs in aging and ARHL
Excessive oxidative stress may also cause DNA damage, and accumulation of DNA damage can lead to cell cycle arrest or genomic instability, which are common features in the aging process. The removal of oxidative DNA damage through repair of DNA single strand breaks is facilitated by poly(ADP-ribose) polymerases (PARPs). PARP-1 is the most critical protein modifying nuclear enzyme involved in DNA repair. PARP-1 is a major NAD+ consumer in the cellular processes, in which the ADP-ribose moiety is not transferred to an acetyl group, as it happens with sirtuins, but to acceptor proteins in order to build ADP-ribosyl polymers[65]. PARP-1 is strongly activated by DNA damage and oxidative stress. Under physiological conditions, mild activation of PARP-1 can regulate several cellular processes, including DNA repair, cell cycle progression, cell survival, chromatin remodeling, and genomic stability. However, hyperactivation of PARP-1 upon severe oxidative damage causes rapid depletion of intracellular NAD+ levels because PARP-1 uses NAD+ as the endogenous substrate for poly-ADP-ribosylation [60, 66]. Therefore, SIRT1 activity is down-regulated during PARP-1 hyperactivation [67]. These observations indicate that PARP-1 and SIRT1 activity are inter-dependent as they compete for a limited pool of cellular NAD+. In addition to this competitive utilization of NAD+, SIRTs and PARPs are linked by another functional interaction. SIRT1 directly interacts and deacetylates PARP-1, and thereby reduces PARP-1 activity [68]. SIRT1 also negatively regulates the transcriptional activity of PARP-1 gene promoter, leading to decreased PARP-1 protein synthesis [68]. Furthermore, depletion of NAD+ following PARP-1 hyperactivation has been shown to deplete intracellular ATP stores leading to the release of apoptosis-inducing factors (AIF) and consequent cell death due to energy restriction. PARP-1 activation has been implicated in the pathogenesis of many diseases such as hypertension, atherosclerosis, lung injury, hemorrhagic shock, and diabetic, cardiovascular, and kidney complications. In these diseases, the oxidant-mediated cell injury is dependent on PARP activation, and can be attenuated by pharmacological inhibitors of PARP. Therefore, precise regulation of PARP activity through the regulation of NAD+ and SIRTs may be also crucial to prevent the development of several age-related pathological disorders.
Therapeutic Considerations of NAD+ Regulation
Although the exact mechanism responsible for age-associated cellular damages are not fully understood yet, numerous studies indicate that ROS and increased inflammation are important factors. The role of these two factors, ROS and inflammation, seems to be closely related each other and thus influences the cellular processes when they are abnormally regulated. Accordingly, pharmacological interventions that can reduce systemic inflammation and/or oxidative stress may prevent or alleviate the development and progression of age-associated diseases. However, their application is severely restricted by the side effects associated with drug usage [19]. Alternatively, maintaining a proper level of intracellular NAD+ and NADH seems to be a better option, since recent studies suggest it as an indicator of the metabolic state of a given cell [69]. Pathological conditions such as diabetes, oxidative stress, and neurodegeneration are well correlated with decreased cellular NAD+ levels [70–72]. Interestingly, a recent report indicates that cellular NAD+ level declines with age [6, 7], which implies the importance of maintaining optimal intracellular NAD+ levels to prevent age-associated cellular dysfunction. Since NAD+ modulates SIRTs involved in various cellular processes, including energy metabolism, the beneficial effects observed with enhanced SIRT activity could be attributed to increased intracellular NAD+ levels. This was clearly reflected by the beneficial effects of CR on the lifespan as well as the rate of the aging process mediated by NAD+-dependent SIRT activation, which leads to enhanced mitochondrial function [50, 73]. Consistent with this notion, the therapeutic potential of NAD+ has recently been reported, in which increase in SIRT activity by treatment with resveratrol or synthetic small SIRT-activating molecules enhanced the muscle strength, motor coordination, and anti-aging effects [74–76]. However, additional studies are required in the future to address target specificity of resveratrol or small molecules and also the efficacy on human subject [77].
Since both PARPs and SIRTs are NAD+-consuming enzymes and thus compete for NAD+, the effect of NAD+ treatment may also result in altered SIRT activity through the crosstalk between PARPs and SIRTs. Supplementation of nicotinamide riboside has been reported to improve the mitochondrial function and alleviate phenotypic manifestation in high fat diet-induced obesity, which correlates well with the increased activities of SIRT1 and SIRT3 [73]. Moreover, selective blockage of NAD+-consuming enzymes other than SIRTs may also be a potentially good strategy to increase NAD+ levels. Consistent with this notion, targeted PARP inactivation increased NAD+ levels and increased SIRT1 activity [67], suggesting that the modulation of PARP activity could be a therapeutic strategy for the treatment of metabolic diseases associated with SIRT activity.
Approaches aimed at increasing the NAD+ levels by supplementing NAD+ precursors through the activation of de novo and salvage pathways for NAD+ biosynthesis have demonstrated cytoprotective effects against cellular damages. In fact, this specific strategy increased NAD+ levels in vitro and in vivo. For examples, administration of nicotinamide, a NAD+ precursor, showed a protective effect against oxidative stress and glucose deprivation in vitro, and also alleviated tissue damages from animal models of ischemia [78, 79], spinal cord injury [80], and multiple sclerosis [81]. Similarly, nicotinic acid, another NAD+ precursor, has also been used to treat hyperlipidemia [82], and nicotinamide riboside showed extended lifespan in yeast [83], indicating the therapeutic potential of NAD+ precursors. Although NAD+ treatment has not been tested extensively for its cytoprotective effects, a recent report suggests that it may reduce brain damage by protecting against PARP-1-induced cell death [84, 85]. In addition to de novo and salvage NAD+ biosynthesis pathways for regulating cellular NAD+ levels, intracellular NADH:quinone oxidoreductase 1 (NQO1) enzyme, a cytosolic flavoprotein, normally participates in reduction of quinone compounds in exchange for NADH oxidation to NAD+ as shown in Fig. 1 [86, 87]. Moreover, NQO1 expression was recently reported not only in the cytoplasm but also in the mitochondria [88], suggesting that endogenous NQO1 may act in close relationship with cellular metabolic pathways. Therefore, it is plausible that endogenous factors or chemical agents that potentially activate NQO1 enzymatic activity or act as strong substrates of NQO1 may be beneficial by increasing intracellular NAD+ levels. Indeed, several reports indicate that pharmacological activation of NQO1 ameliorates phenotypic manifestations associated with pathological conditions in rodent models. In particular, metabolic diseases such as obesity and spontaneous hypertension were shown to be reversed upon NQO1 activation by β-lapachone, a strong NQO1 substrate [89, 90], and pathological conditions such as arterial restenosis due to tissue injury and cisplatin-associated nephrotoxicity were also ameliorated by NQO1 activation [91, 92]. In these experiments, activated NQO1 facilitated cellular metabolism and mitochondrial function because of increased intracellular NAD+ levels through NADH oxidation, which in turn facilitated phosphorylation of AMPK and activation of SIRT1 protein. Therefore, pharmacological modulation of NQO1 activity in a short-term seems to be an effective way to treat several pathological conditions, including metabolic dysfunctions, such as diabetes. Although the long-term treatment of a NQO1 activator has not been adequately studied to validate its therapeutic potential in the aging process or more specifically in ARHL, a recent report indicated that NQO1 activation leading to increased cellular NAD+ levels prevented motor and cognitive declines associated with aging by mimicking CR effects [93].
Figure 1.
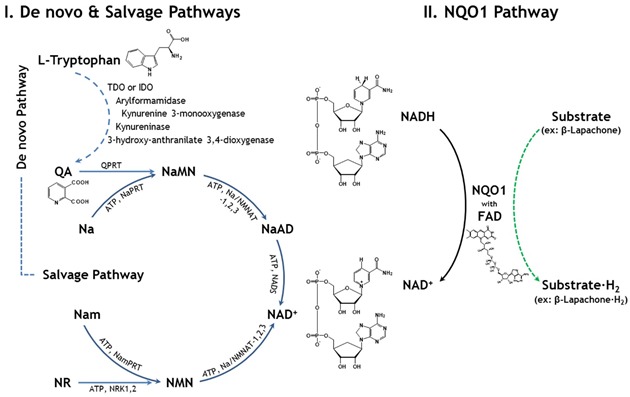
Mammalian NAD+ synthetic pathways. The biosynthesis of NAD+ through de novo, salvage, and NQO1 pathways. ATP: adenoshine triphosphate, FAD: flavin adenine dinucleotide, IDO: indoleamine 2,3-dioxygenase, Na: nicotinic acid, NaAD: nicotinic acid adenine dinucleotide, NAD: nicotinamide adenine dinucleotide, NADS: NAD snythetase, Nam: nicotinamide, NaMN: nicotininc acid mononucleotide, NaPRT: nicotinic acid phosphoribosyl transferase, NMN: nicotinamide mononucleotide, NMNAT: nicotinamide mononucleotide adenylyltransferase, NQO1: NAD(P)H:quinone oxydoreductase 1, NR: nicotinamide riboside, NRK1,2: nicotinamide reboside kinase1, 2, NamPRT: nicotinamide phosphoribosyltransferase, NMNAT: nicotinamide mononucleotide adenyltransferase, QA: quinolinic acid, QPRT: quinolinate phosphoribosyltransferase, and TDO: tryptophan 2,3-dioxygenase
Taken together, these reports strongly suggest that modulating NAD+ levels may be an effective therapeutic target in the treatment of ARHL as well as other aging-associated metabolic diseases, and could be considered as a prophylactic target. However, several questions are yet to be answered to prove the long-term effect of modulating NAD+ levels and to exclude any potential adverse effects that may be associated with prolonged increase in NAD+.
Acknowledgments
This work was supported by National Research Foundation of Korea [NRF] grants funded by the Korean government [MSIP]: [No. 2011-0028866] and [No. 2011-0030715].
Reference
- [1].Cacciatore F, Napoli C, Abete P, Marciano E, Triassi M, Rengo F. Quality of life determinants and hearing function in an elderly population: Osservatorio Geriatrico Campano Study Group. Gerontology. 1999;45:323–328. doi: 10.1159/000022113. [DOI] [PubMed] [Google Scholar]
- [2].Desai M, Pratt LA, Lentzner H, Robinson KN. Trends in vision and hearing among older Americans. Aging Trends. 2001;2:1–8. doi: 10.1037/e620682007-001. [DOI] [PubMed] [Google Scholar]
- [3].Van Eyken E, Van Camp G, Van Laer L. The complexity of age-related hearing impairment: contributing environmental and genetic factors. Audiol Neurootol. 2007;12:345–358. doi: 10.1159/000106478. [DOI] [PubMed] [Google Scholar]
- [4].Spongr VP, Flood DG, Frisina RD, Salvi RJ. Quantitative measures of hair cell loss in CBA and C57BL/6 mice throughout their life spans. J Acoust Soc Am. 1997;101:3546–3553. doi: 10.1121/1.418315. [DOI] [PubMed] [Google Scholar]
- [5].Hequembourg S, Liberman MC. Spiral ligament pathology: a major aspect of age-related cochlear degeneration in C57BL/6 mice. J Assoc Res Otolaryngol. 2001;2:118–129. doi: 10.1007/s101620010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS One. 2011;6:e19194. doi: 10.1371/journal.pone.0019194. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [7].Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. 2012;7:e42357. doi: 10.1371/journal.pone.0042357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, et al. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Adam-Vizi V, Tretter L. The role of mitochondrial dehydrogenases in the generation of oxidative stress. Neurochem Int. 2013;62:757–763. doi: 10.1016/j.neuint.2013.01.012. [DOI] [PubMed] [Google Scholar]
- [10].Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- [12].Miquel J, Economos AC, Fleming J, Johnson JE., Jr Mitochondrial role in cell aging. Exp Gerontol. 1980;15:575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- [13].Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. 1994;91:10771–10778. doi: 10.1073/pnas.91.23.10771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- [15].Fetoni AR, Picciotti PM, Paludetti G, Troiani D. Pathogenesis of presbycusis in animal models: a review. Exp Gerontol. 2011;46:413–425. doi: 10.1016/j.exger.2010.12.003. [DOI] [PubMed] [Google Scholar]
- [16].Liu XZ, Yan D. Ageing and hearing loss. J Pathol. 2007;211:188–197. doi: 10.1002/path.2102. [DOI] [PubMed] [Google Scholar]
- [17].Pickles JO. Mutation in mitochondrial DNA as a cause of presbyacusis. Audiol Neurootol. 2004;9:23–33. doi: 10.1159/000074184. [DOI] [PubMed] [Google Scholar]
- [18].Singh T, Newman AB. Inflammatory markers in population studies of aging. Ageing Res Rev. 2011;10:319–329. doi: 10.1016/j.arr.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Woods JA, Wilund KR, Martin SA, Kistler BM. Exercise, inflammation and aging. Aging Dis. 2012;3:130–140. [PMC free article] [PubMed] [Google Scholar]
- [20].Kregel KC, Zhang HJ. An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations. Am J Physiol Regul Integr Comp Physiol. 2007;292:R18–36. doi: 10.1152/ajpregu.00327.2006. [DOI] [PubMed] [Google Scholar]
- [21].Mitchell P, Gopinath B, McMahon CM, Rochtchina E, Wang JJ, Boyages SC, et al. Relationship of Type 2 diabetes to the prevalence, incidence and progression of age-related hearing loss. Diabet Med. 2009;26:483–488. doi: 10.1111/j.1464-5491.2009.02710.x. [DOI] [PubMed] [Google Scholar]
- [22].Gates GA, Cobb JL, D’Agostino RB, Wolf PA. The relation of hearing in the elderly to the presence of cardiovascular disease and cardiovascular risk factors. Arch Otolaryngol Head Neck Surg. 1993;119:156–161. doi: 10.1001/archotol.1993.01880140038006. [DOI] [PubMed] [Google Scholar]
- [23].Iwai H, Lee S, Inaba M, Sugiura K, Tomoda K, Yamashita T, et al. Prevention of accelerated presbycusis by bone marrow transplantation in senescence-accelerated mice. Bone Marrow Transplant. 2001;28:323–328. doi: 10.1038/sj.bmt.1703152. [DOI] [PubMed] [Google Scholar]
- [24].Fujioka M, Kanzaki S, Okano HJ, Masuda M, Ogawa K, Okano H. Proinflammatory cytokines expression in noise-induced damaged cochlea. J Neurosci Res. 2006;83:575–583. doi: 10.1002/jnr.20764. [DOI] [PubMed] [Google Scholar]
- [25].So H, Kim H, Lee JH, Park C, Kim Y, Kim E, et al. Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J Assoc Res Otolaryngol. 2007;8:338–355. doi: 10.1007/s10162-007-0084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Weindruch R, Sohal RS. Seminars in medicine of the Beth Israel Deaconess Medical Center. Caloric intake and aging. N Engl J Med. 1997;337:986–994. doi: 10.1056/NEJM199710023371407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Marzetti E, Wohlgemuth SE, Anton SD, Bernabei R, Carter CS, Leeuwenburgh C. Cellular mechanisms of cardioprotection by calorie restriction: state of the science and future perspectives. Clin Geriatr Med. 2009;25:715–732. doi: 10.1016/j.cger.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Someya S, Yamasoba T, Weindruch R, Prolla TA, Tanokura M. Caloric restriction suppresses apoptotic cell death in the mammalian cochlea and leads to prevention of presbycusis. Neurobiol Aging. 2007;28:1613–1622. doi: 10.1016/j.neurobiolaging.2006.06.024. [DOI] [PubMed] [Google Scholar]
- [30].Henry KR. Effects of dietary restriction on presbyacusis in the mouse. Audiology. 1986;25:329–337. doi: 10.3109/00206098609078397. [DOI] [PubMed] [Google Scholar]
- [31].Sweet RJ, Price JM, Henry KR. Dietary restriction and presbyacusis: periods of restriction and auditory threshold losses in the CBA/J mouse. Audiology. 1988;27:305–312. doi: 10.3109/00206098809081601. [DOI] [PubMed] [Google Scholar]
- [32].Masoro EJ. Caloric restriction and aging: an update. Exp Gerontol. 2000;35:299–305. doi: 10.1016/s0531-5565(00)00084-x. [DOI] [PubMed] [Google Scholar]
- [33].Lin SJ, Kaeberlein M, Andalis AA, Sturtz LA, Defossez PA, Culotta VC, et al. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature. 2002;418:344–348. doi: 10.1038/nature00829. [DOI] [PubMed] [Google Scholar]
- [34].Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447:545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- [35].Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- [36].Tanner KG, Landry J, Sternglanz R, Denu JM. Silent information regulator 2 family of NAD-dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc Natl Acad Sci U S A. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J. Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med. 2007;39:335–345. doi: 10.1080/07853890701408194. [DOI] [PubMed] [Google Scholar]
- [38].Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A. 2002;99:13653–13658. doi: 10.1073/pnas.222538099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schwer B, North BJ, Frye RA, Ott M, Verdin E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J Cell Biol. 2002;158:647–657. doi: 10.1083/jcb.200205057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- [42].Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- [43].Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941–954. doi: 10.1016/j.cell.2006.06.057. [DOI] [PubMed] [Google Scholar]
- [44].Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem. 2005;280:21313–21320. doi: 10.1074/jbc.M413296200. [DOI] [PubMed] [Google Scholar]
- [45].Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem. 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- [46].Imai S, Johnson FB, Marciniak RA, McVey M, Park PU, Guarente L. Sir2: an NAD-dependent histone deacetylase that connects chromatin silencing, metabolism, and aging. Cold Spring Harb Symp Quant Biol. 2000;65:297–302. doi: 10.1101/sqb.2000.65.297. [DOI] [PubMed] [Google Scholar]
- [47].Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Easlon E, Tsang F, Skinner C, Wang C, Lin SJ. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 2008;22:931–944. doi: 10.1101/gad.1648308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Canto C, Auwerx J. Caloric restriction, SIRT1 and longevity. Trends Endocrinol Metab. 2009;20:325–331. doi: 10.1016/j.tem.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Quintas A, de Solis AJ, Diez-Guerra FJ, Carrascosa JM, Bogonez E. Age-associated decrease of SIRT1 expression in rat hippocampus: prevention by late onset caloric restriction. Exp Gerontol. 2012;47:198–201. doi: 10.1016/j.exger.2011.11.010. [DOI] [PubMed] [Google Scholar]
- [51].Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- [52].Neumann M, Naumann M. Beyond IkappaBs: alternative regulation of NF-kappaB activity. Faseb J. 2007;21:2642–2654. doi: 10.1096/fj.06-7615rev. [DOI] [PubMed] [Google Scholar]
- [53].Yoshizaki T, Milne JC, Imamura T, Schenk S, Sonoda N, Babendure JL, et al. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol Cell Biol. 2009;29:1363–1374. doi: 10.1128/MCB.00705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. doi: 10.1016/j.cell.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Brooks CL, Gu W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell. 2011;2:456–462. doi: 10.1007/s13238-011-1063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Li S, Banck M, Mujtaba S, Zhou MM, Sugrue MM, Walsh MJ. p53-induced growth arrest is regulated by the mitochondrial SirT3 deacetylase. PLoS One. 2010;5:e10486. doi: 10.1371/journal.pone.0010486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Goodwin PM, Lewis PJ, Davies MI, Skidmore CJ, Shall S. The effect of gamma radiation and neocarzinostatin on NAD and ATP levels in mouse leukaemia cells. Biochim Biophys Acta. 1978;543:576–582. doi: 10.1016/0304-4165(78)90312-4. [DOI] [PubMed] [Google Scholar]
- [61].Amaral JD, Xavier JM, Steer CJ, Rodrigues CM. Targeting the p53 pathway of apoptosis. Curr Pharm Des. 2010;16:2493–2503. doi: 10.2174/138161210791959818. [DOI] [PubMed] [Google Scholar]
- [62].Vaseva AV, Moll UM. The mitochondrial p53 pathway. Biochim Biophys Acta. 2009;1787:414–420. doi: 10.1016/j.bbabio.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- [64].Someya S, Yu W, Hallows WC, Xu J, Vann JM, Leeuwenburgh C, et al. Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell. 2010;143:802–812. doi: 10.1016/j.cell.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Krishnakumar R, Kraus WL. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Skidmore CJ, Davies MI, Goodwin PM, Halldorsson H, Lewis PJ, Shall S, et al. The involvement of poly(ADP-ribose) polymerase in the degradation of NAD caused by gamma-radiation and N-methyl-N-nitrosourea. Eur J Biochem. 1979;101:135–142. doi: 10.1111/j.1432-1033.1979.tb04225.x. [DOI] [PubMed] [Google Scholar]
- [67].Bai P, Canto C, Oudart H, Brunyanszki A, Cen Y, Thomas C, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S, et al. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol. 2009;29:4116–4129. doi: 10.1128/MCB.00121-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Berger F, Ramirez-Hernandez MH, Ziegler M. The new life of a centenarian: signalling functions of NAD(P) Trends Biochem Sci. 2004;29:111–118. doi: 10.1016/j.tibs.2004.01.007. [DOI] [PubMed] [Google Scholar]
- [70].Ido Y. Pyridine nucleotide redox abnormalities in diabetes. Antioxid Redox Signal. 2007;9:931–942. doi: 10.1089/ars.2007.1630. [DOI] [PubMed] [Google Scholar]
- [71].Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- [72].Pearson KJ, Lewis KN, Price NL, Chang JW, Perez E, Cascajo MV, et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proc Natl Acad Sci U S A. 2008;105:2325–2330. doi: 10.1073/pnas.0712162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, et al. Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab. 2008;8:157–168. doi: 10.1016/j.cmet.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, et al. SRT1720 improves survival and healthspan of obese mice. Sci Rep. 2011;1:70. doi: 10.1038/srep00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Mouchiroud L, Houtkooper RH, Auwerx J. NAD(+) metabolism: a therapeutic target for age-related metabolic disease. Crit Rev Biochem Mol Biol. 2013;48:397–408. doi: 10.3109/10409238.2013.789479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ayoub IA, Lee EJ, Ogilvy CS, Beal MF, Maynard KI. Nicotinamide reduces infarction up to two hours after the onset of permanent focal cerebral ischemia in Wistar rats. Neurosci Lett. 1999;259:21–24. doi: 10.1016/s0304-3940(98)00881-7. [DOI] [PubMed] [Google Scholar]
- [79].Ayoub IA, Maynard KI. Therapeutic window for nicotinamide following transient focal cerebral ischemia. Neuroreport. 2002;13:213–216. doi: 10.1097/00001756-200202110-00008. [DOI] [PubMed] [Google Scholar]
- [80].Brewer KL, Hardin JS. Neuroprotective effects of nicotinamide after experimental spinal cord injury. Acad Emerg Med. 2004;11:125–130. doi: 10.1111/j.1553-2712.2004.tb01421.x. [DOI] [PubMed] [Google Scholar]
- [81].Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, et al. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci. 2006;26:9794–9804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Sauve AA. NAD+ and vitamin B3: from metabolism to therapies. J Pharmacol Exp Ther. 2008;324:883–893. doi: 10.1124/jpet.107.120758. [DOI] [PubMed] [Google Scholar]
- [83].Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell. 2007;129:473–484. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- [84].Ying W, Garnier P, Swanson RA. NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem Biophys Res Commun. 2003;308:809–813. doi: 10.1016/s0006-291x(03)01483-9. [DOI] [PubMed] [Google Scholar]
- [85].Ying W, Alano CC, Garnier P, Swanson RA. NAD+ as a metabolic link between DNA damage and cell death. J Neurosci Res. 2005;79:216–223. doi: 10.1002/jnr.20289. [DOI] [PubMed] [Google Scholar]
- [86].Gaikwad A, Long DJ, 2nd, Stringer JL, Jaiswal AK. In vivo role of NAD(P)H:quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J Biol Chem. 2001;276:22559–22564. doi: 10.1074/jbc.M101053200. [DOI] [PubMed] [Google Scholar]
- [87].Ross D, Kepa JK, Winski SL, Beall HD, Anwar A, Siegel D. NAD(P)H:quinone oxidoreductase 1 (NQO1): chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem Biol Interact. 2000;129:77–97. doi: 10.1016/s0009-2797(00)00199-x. [DOI] [PubMed] [Google Scholar]
- [88].Dong H, Shertzer HG, Genter MB, Gonzalez FJ, Vasiliou V, Jefcoate C, et al. Mitochondrial targeting of mouse NQO1 and CYP1B1 proteins. Biochem Biophys Res Commun. 2013;435:727–732. doi: 10.1016/j.bbrc.2013.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Hwang JH, Kim DW, Jo EJ, Kim YK, Jo YS, Park JH, et al. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes. 2009;58:965–974. doi: 10.2337/db08-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kim YH, Hwang JH, Noh JR, Gang GT, Kim do H, Son HY, et al. Activation of NAD(P)H:quinone oxidoreductase ameliorates spontaneous hypertension in an animal model via modulation of eNOS activity. Cardiovasc Res. 2011;91:519–527. doi: 10.1093/cvr/cvr110. [DOI] [PubMed] [Google Scholar]
- [91].Kim SY, Jeoung NH, Oh CJ, Choi YK, Lee HJ, Kim HJ, et al. Activation of NAD(P)H:quinone oxidoreductase 1 prevents arterial restenosis by suppressing vascular smooth muscle cell proliferation. Circ Res. 2009;104:842–850. doi: 10.1161/CIRCRESAHA.108.189837. [DOI] [PubMed] [Google Scholar]
- [92].Oh GS, Kim HJ, Choi JH, Shen A, Choe SK, Karna A, et al. Pharmacological activation of NQO1 increases NAD levels and attenuates cisplatin-mediated acute kidney injury in mice. Kidney Int. 2013;85(3):547–60. doi: 10.1038/ki.2013.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lee JS, Park AH, Lee SH, Lee SH, Kim JH, Yang SJ, et al. Beta-lapachone, a modulator of NAD metabolism, prevents health declines in aged mice. PLoS One. 2012;7:e47122. doi: 10.1371/journal.pone.0047122. [DOI] [PMC free article] [PubMed] [Google Scholar]