

Abstract
Background and Purpose
Pharmacological activation of glucokinase (GK) lowers blood glucose in animal models and humans, confirming proof of concept for this mechanism. However, recent clinical evidence from chronic studies suggests that the glucose-lowering effects mediated by glucokinase activators (GKAs) are not maintained in patients with type 2 diabetes (T2D). Existing preclinical data with GKAs do not explain this loss of sustained glucose-lowering efficacy in patients. Here, we have assessed the effects of chronic (up to 11 months) treatment with two different GKAs in two models of T2D.
Experimental Approach
Two validated animal models of T2D, insulin-resistant obese Zucker rats and hyperglycaemic gkwt/del mice, were treated with two different GKAs for 1 or 11 months respectively at exposures that translate to clinical exposures in humans. Blood glucose, cholesterol, triglycerides and insulin were measured. GKA pharmacokinetics were also determined.
Key Results
Treatment with either GKA provided sustained lowering of blood glucose for up to 1 month in the Zucker rat and up to 11 months in hyperglycaemic gkwt/del mice, with maintained compound exposures. This efficacy was achieved without increases in plasma or hepatic triglycerides, accumulation of hepatic glycogen or impairment of glucose-stimulated insulin secretion.
Conclusions and Implications
Chronic treatment with two GKAs in two animal models of diabetes provided sustained lowering of blood glucose, in marked contrast to clinical findings. Therefore, either these animal models of T2D are not good predictors of responses in human T2D or we need a better understanding of the consequences of GK activation in humans.
Keywords: type 2 diabetes, glucokinase activator, AZD1656, GKA71, glucose, translational model, efficacy, durability, rodent, pharmacokinetic/pharmacodynamic
Introduction
Glucokinase activators (GKAs) bind to an allosteric binding site of glucokinase (GK) and stabilize a conformation of the enzyme that promotes the binding of glucose and its phosphorylation to glucose 6-phosphate (Coghlan and Leighton, 2008; Matschinsky, 2009). The glucose-lowering efficacy of GKAs is mediated through GK activation in pancreas and liver. In the pancreas, GK acts as the ‘glucose sensor’ controlling glucose-stimulated insulin secretion (GSIS). Hepatic GK, together with the GK regulatory protein, is a key regulator of glucose uptake by the liver (Futamura et al., 2006; Agius, 2008; Bourbonais et al., 2012) and hence of net hepatic glucose balance.
Recent clinical data demonstrate that GKAs can acutely lower glucose levels in non-diabetic and type 2 diabetic subjects (Bonadonna et al., 2010; Ericsson et al., 2012; Morrow et al., 2012; Norjavaara et al., 2012; Sjöstrand et al., 2013). Repeat dose studies have shown glucose control can be maintained for 4 weeks (Morrow et al., 2012) and up to 14 weeks (Meininger et al., 2011). However, the glucose-lowering efficacy of GKAs over the longer term has been called into question (Meininger et al., 2011; Kiyosue et al., 2013; Wilding et al., 2013).
A number of acute (1 day; Grimsby et al., 2003; Efanov et al., 2005; Coope et al., 2006) and repeat dose (9–35 days; Fyfe et al., 2007; Ohyama et al., 2010; Eiki et al., 2011; Sörhede Winzell et al., 2011; Futamura et al., 2012) preclinical studies have demonstrated effective glucose lowering by GKAs in both non-diabetic and diabetic animal models. Two chronic studies report sustained glucose lowering with two different GKAs at 20 and 40 weeks (Nakamura et al., 2009; 2011). However there are no data describing exposure in these studies, making it difficult to relate these results to clinical observations. In addition to the issue of a sustained glucose response, questions have recently been raised about the effect of chronic GKA administration on hepatic and plasma triglyceride levels (De Ceuninck et al., 2013; Rees and Gloyn, 2013). It is also unclear what might be the long-term consequences of GKA activation on beta cell function, (Bebernitz et al., 2009) given that sulphonylureas gradually lose efficacy in humans (Kahn et al., 2006).
In order to better understand whether or not GKAs can deliver robust and sustained glucose lowering under diabetic conditions, we have investigated the chronic efficacy of two GKAs (AZD1656 and GKA71) in two animal models of type 2 diabetes (T2D) at clinically relevant exposures. Sustained glucose lowering was observed over a 4 week study in male obese Zucker fa/fa rats that are insulin-resistant, hyperinsulinaemic and hyperlipidaemic (but not markedly hyperglycaemic), and also over 11 months in high-fat (HF)-fed male gkwt/del mice (Gorman et al., 2008; Baker et al., 2014) that are insulin-resistant and hyperglycaemic but not hyperlipidaemic. Reductions in blood glucose were achieved without increases in plasma or hepatic triglycerides or hepatic glycogen content and without adverse effects on GSIS after 48 weeks of dosing in gkwt/del mice.
Methods
Animals
All animal care and experimental procedures were approved by the UK Home Office and carried out in accordance with the Animals (Scientific Procedures) Act 1986. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 75 animals were used in the experiments described here. Male obese Zucker fa/fa rats and male gkwt/del mice (Gorman et al., 2008) were supplied from the AstraZeneca Biological Animal Breeding Unit. Zucker fa/fa rats were received at 9–10 weeks of age, housed in groups of 3–4 at 19–21°C on a reverse 12 h : 12 h light : dark cycle (lights off 08:00 h) and maintained on RM3 diet (Special Diets Services, Witham, Essex, UK) for 3 weeks before the start of the study. Body weights at the start of the study were 440–535 g. Male gkwt/del mice or wild-type littermates were received at 8 weeks of age, housed in groups of three at 21–23°C on a reverse 12 h : 12 h light : dark cycle (lights off 09:00 h), and maintained on standard chow RM1 diet (Special Diets Services) for 3 weeks acclimatization. Animals had ad libitum access to food and tap water. Blood samples for in-study glucose (∼5 μL) and pharmacokinetic (PK) analyses (20 μL) were obtained at the same time from the tail vein, as previously described (Baker et al., 2014).
Experimental designs and measurements
Details of individual study designs are provided in the Supporting Information. Doses of GKAs were selected to provide translatable drug exposures based on known PK properties in the rodent species and known tolerated and therapeutically relevant exposures and PK in humans (Ericsson et al., 2012; Morrow et al., 2012). Correction was made for species differences in plasma protein binding and potency against species-specific GK isoforms to ensure comparable free plasma exposure.
Study 1: chronic efficacy of AZD1656 in male obese Zucker fa/fa rats
Rats received daily oral doses of vehicle [1% Pluronic F127 (Sigma-Aldrich, Gillingham, Dorset, UK) in water; n = 9] or corresponding solutions of AZD1656 at either 3 mg·kg−1 (n = 9) or 10 mg·kg−1 (n = 7) over 29 days. Free-feeding blood glucose and PK profiles were measured over 12 h post-dose on days 1 and 28, and an oral glucose tolerance test (OGTT) was performed on day 14. Terminal blood samples were taken on day 29 for clinical chemistry analysis.
Study 2: long-term efficacy of GKA71 in HF-fed male gkwt/del mice
Thirteen days prior to first administration of compound, 30 male gkwt/del mice and 10 male wild-type mice were transferred from RM1 chow diet to a 60% HF diet (RD12492i, Research Diets, New Brunswick, NJ, USA). Another group of 10 male wild-type mice remained on RM1 chow. Body weights at the start of the study were in the range 26–33 g; average 29.7 g (gkwt/del, HF diet), 30.2 g (wild-type, HF diet) and 30.6 g (wild-type, chow diet). gkwt/del mice on HF diet were randomized into three groups (n = 10 per group) based on body weight, and baseline 24 h free-feeding glucose profiles were measured at day 3. Three days later (day 0), two of these groups were switched to HF diet supplemented with GKA71 (formulated in RD12492i by Research Diets) to provide a daily dose of 2.5 or 5 mg·kg−1, based on historical records of food intake in these mice. The third group was maintained on unsupplemented HF diet. Food intake was checked at the start and midway through the study. Compound exposures were confirmed by PK measurements throughout the course of the study.
Beginning on day 3 and continuing weekly up to week 49, blood glucose measurements were taken on all mice in the study, 1 h before start of dark phase. On day 5, and thereafter monthly up to month 11 (week 41), 24 h free-feeding blood glucose profiles were measured on gkwt/del mice only, and on all mice at month 12 (week 45). PK profiles were measured in gkwt/del mice for months 1–11. The study was terminated at the start of week 50, and blood samples were taken for blood chemistry analysis. Tissue samples (eyes, sciatic nerves, kidneys, liver, heart, skeletal muscle and pancreas) were collected from each animal and fixed in 10% neutral buffered formalin prior to processing into paraffin wax. Sections (4 μm thick) were stained with haematoxylin and eosin and evaluated by light microscopy.
Three gkwt/del mice (all from 2.5 mg·kg−1·day−1 group) were withdrawn from this study on weeks 18, 19 and 39 due to injury sustained from fighting. The incidence of fighting is in line with our historical records for this animal model. Two wild-type mice from the HF diet control group were also withdrawn in weeks 32 and 43 for welfare reasons unrelated to treatment. In both cases, data from these mice were excluded from statistical analysis.
Blood glucose measurements and OGTT
Free-feeding blood glucose profiles from blood microsamples were measured using a hand-held Accu-chek® glucose monitor (Roche Diagnostics, Burgess Hill, West Sussex, UK). For OGTTs, food was withdrawn 4 h before the test. OGTTs were performed by administration of 2 g·kg−1 bolus of glucose (5 mL·kg−1) 2 h after compound dosing (Baker et al., 2014). Accu-chek blood glucose readings were taken at −120 min (baseline) and 5–90 min (after glucose bolus). Free-feeding blood glucose profile AUCs were calculated as total AUC, OGTT glucose and insulin. AUC values were corrected for differences in baseline at t = 0 min.
PK methods
To measure compound exposure, 20 μL blood samples were collected and mixed with 80 μL PBS pH 7.0. Protein precipitation with acetonitrile was used to extract compounds from plasma matrix. GKAs (including active metabolite) were quantified by HPLC-MS/MS. HPLC methods used a Phenomenex (Macclesfield, Cheshire UK) Synergi 4 μm max-RP column (50 × 2.0 mm), with a gradient of 5% methanol/95% 10 mM aqueous ammonium acetate (or formic acid), to 95% methanol/5% 10 mM aqueous ammonium acetate (or formic acid) at a flow rate of 0.7 mL·min−1 over 4 min. Detection used a Waters (Elstree, Hertfordshire, UK) tandem quadrupole mass spectrometer.
Previously, we have shown that a GKA will lower blood glucose by an amount proportional to free drug concentration relative to the GK EC50 (Coope et al., 2006). Values for compounds GKA71 and AZD1656 (and its active metabolite AZD5658) are detailed in Table 1.
Table 1.
GKA potencies in mouse, rat and human
| AZD1656 | GKA71 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Human | Rat | Human | Mouse | |||||||
| Dose (mg·kg−1·day−1)a | 0.6 | – | 2.3 | – | 3 | – | 10 | – | – | 2.5 | 5 |
| 1656 | Metb | 1656 | Met | 1656 | Met | 1656 | Met | ||||
| GKA EC50 (μM)c | 0.09 | 0.04 | 0.09 | 0.04 | 0.1 | 0.05 | 0.1 | 0.05 | 0.1 | 0.4 | 0.4 |
| fud | 0.056 | 0.136 | 0.056 | 0.136 | 0.041 | 0.041 | 0.041 | 0.041 | 0.013 | 0.0021 | 0.0021 |
| Cmax (μM) | 1.1 | 0.12 | 5 | 0.47 | 3.5 | 0.87 | 6.9 | 2.6 | – | 33.7 | 53.1 |
| Css,av (μM)e | 0.3 | 0.1 | 1.7 | 0.2 | 0.3 | 0.2 | 1.6 | 0.7 | – | 26.2 | 47.1 |
| Cmax,free (μM) | 0.062 | 0.016 | 0.28 | 0.064 | 0.14 | 0.036 | 0.28 | 0.11 | – | 0.071 | 0.11 |
| Css,av,free (μM) | 0.019 | 0.008 | 0.096 | 0.031 | 0.013 | 0.006 | 0.066 | 0.027 | – | 0.055 | 0.099 |
| Cmax,free/EC50 | 0.68 | 0.41 | 3.1 | 1.6 | 1.4 | 0.71 | 2.8 | 2.1 | – | 0.18 | 0.28 |
| Css,av,free/EC50 | 0.21 | 0.21 | 1.1 | 0.76 | 0.13 | 0.13 | 0.66 | 0.54 | – | 0.14 | 0.25 |
| Total Cmax,free/EC50f | 1.1 | 4.7 | 2.1 | 5.0 | – | 0.18 | 0.28 | ||||
Human dose based on 70 kg individual (Morrow et al., 2012), oral twice daily. Rat dose oral once daily. Mouse dosed with compound in diet.
Met = active metabolite AZD5658; (Morrow et al., 2012).
Equipotent against hepatic and pancreatic forms of GK.
fu = fraction unbound.
Css,av = mean drug concentration over 24 h at steady state.
Combined AZD1656 + active metabolite activity.
Blood and plasma chemistry
Insulin concentrations were measured using elisa [Insulin (Mouse) Ultrasensitive elisa, ALPCO Diagnostics, Salem, NH, USA]. Assays for total plasma cholesterol, lactate and triglycerides were performed as previously described (Gorman et al., 2008). HbA1c was measured on a Variant II analyser (Bio-Rad, Hercules, CA, USA).
Tissue analysis
Hepatic triglyceride and glycogen concentrations were determined as described previously (Poucher et al., 2007; Sörhede Winzell et al., 2011).
Data analysis
Data collected on free-feeding glucose AUC measurements in male gkwt/del mice demonstrate that a 10% difference at P < 0.05 with 79% power can be detected with a group size of n = 6. Animals were randomized to initial experimental groups based on body weight. All results are presented as mean values ± SEM for the indicated number of animals. Statistical analysis was performed using anova adjusting for treatment groups, followed by pairwise t-tests using pooled estimates of variance. One-sided tests were performed for reductions in blood glucose and OGTT AUC values, and for changes in metabolic parameters. A two-sided test was applied to changes in body weight. Differences were considered significant at P < 0.05. Studies were powered assuming no multiple comparison procedures would be used, and statistical significance was judged based on raw P-values. Multiple comparison adjustments such as Bonferroni did not materially change the conclusions.
Materials
AZD1656 and GKA71 have been described previously (Waring et al., 2011b; 2012) and were provided by AstraZeneca (Macclesfield, UK). EC50 values against recombinant forms of GK are shown in Table 1.
Results
Study 1: chronic efficacy of AZD1656 in male obese Zucker fa/fa rats
Effect of AZD1656 on body weight
Male obese Zucker rats dosed orally with AZD1656 at either 3 or 10 mg·kg−1·day−1 showed a slight (non-significant) increase in body weight compared with animals receiving vehicle (Figure 1) over the 28 days of the study.
Figure 1.
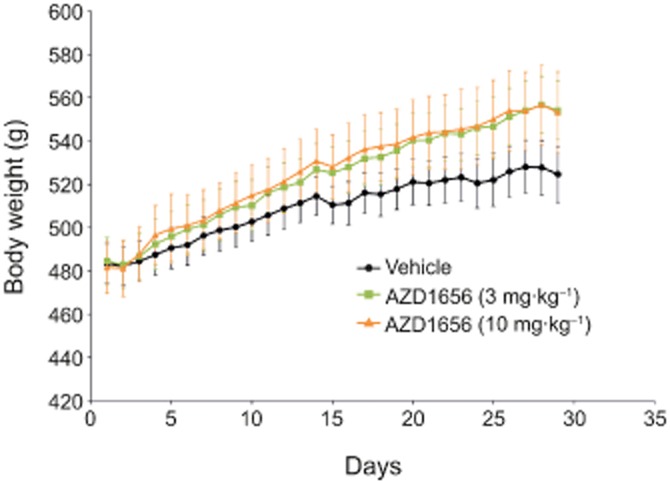
Effect of AZD1656 at 3 mg·kg−1·day−1 (n = 9) and 10 mg·kg−1·day−1 (n = 7) or vehicle (n = 9) on body weight gain over 29 days in male obese (fa/fa) Zucker rats. Data are mean ± SEM.
Effect on free-feeding glucose profiles
AZD1656 dose-dependently decreased glucose AUC(0–12 h) in free-feeding blood glucose profiles after 1 day (Figure 2A,B) and 28 days (Figure 2C,D) dosing, compared with vehicle. Rats dosed with AZD1656 at 10 mg·kg−1·day−1 for 28 days also showed a small but significant reduction in pre-dose glucose levels compared with vehicle (5.6 ± 0.3 vs. 6.3 ± 0.2 mM, P < 0.05). AZD1656 forms an active metabolite AZD5658 in vivo that has equivalent glucose-lowering efficacy (Morrow et al., 2012; Waring et al., 2012). At both days 1 and 28, total free combined levels of AZD1656 and active metabolite exceeded the efficacy threshold for 5–6 h at 3 mg·kg−1 and > 12 h at 10 mg·kg−1 doses.
Figure 2.
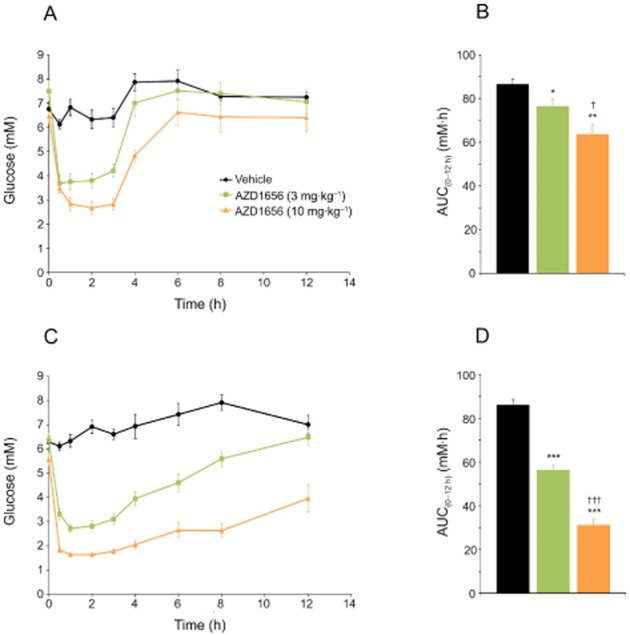
Blood glucose lowering in male obese (fa/fa) Zucker rats after daily dosing of AZD1656 at 3 mg·kg−1·day−1 (n = 9) and 10 mg·kg−1·day−1 (n = 7) or vehicle (n = 9). (A) 12 h free-feeding blood glucose profiles at day 1. Sample sizes: 3 mg·kg−1·day−1, n = 9; 10 mg·kg−1·day−1, n = 6; vehicle, n = 9. (B) Glucose AUC(0–12 h) values (mM·h) at day 1. (C) 12 h free-feeding blood glucose profiles at day 28. Sample sizes: 3 mg·kg−1·day−1, n = 9; 10 mg·kg−1·day−1, n = 7; vehicle, n = 9. (D) Glucose AUC(0–12 h) values (mM·h) at day 28. Data are mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle; †P < 0.05, †††P < 0.001 versus AZD1656 at 3 mg·kg−1·day−1.
Effect of AZD1656 on OGTT
AZD1656 gave a dose-dependent reduction in baseline-corrected glucose excursion (Figure 3A, B) in an OGTT performed at day 14. AZD1656 did not further stimulate insulin secretion at 20 min post-glucose bolus compared with vehicle (Figure 3C).
Figure 3.
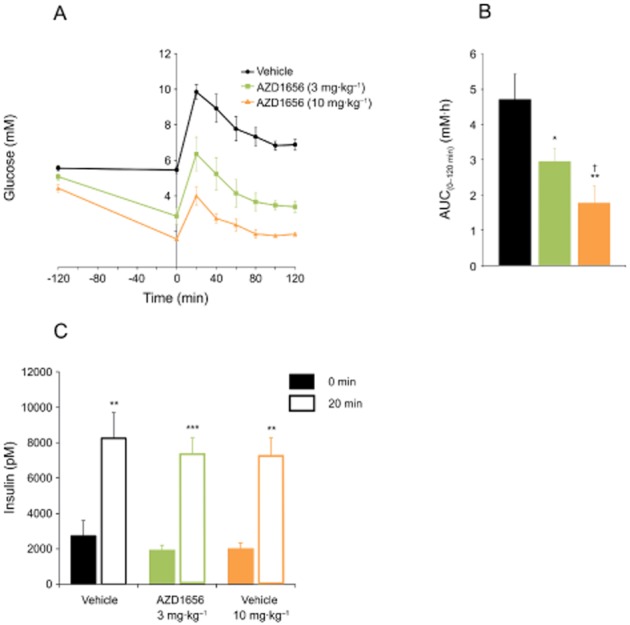
Blood glucose and insulin levels following OGTT in male obese (fa/fa) Zucker rats after 14 days dosing of AZD1656 at 3 mg·kg−1·day−1 (n = 9) and 10 mg·kg−1·day−1 (n = 7) or vehicle (n = 9). (A) Glucose levels during OGTT. Sample sizes: 3 mg·kg−1·day−1, n = 9; 10 mg·kg−1·day−1, n = 7; vehicle, n = 9. (B) Glucose AUC(0–12 h) values (mM·h). Data are mean ± SEM. *P < 0.05, **P < 0.01 versus vehicle, †P < 0.01, versus AZD1656 at 3 mg·kg−1·day−1. (C) Insulin levels at t = 0 and t = 20 min during OGTT. Sample sizes: 3 mg·kg−1·day−1, n = 8; 10 mg·kg−1·day−1, n = 8; vehicle: n = 9 (t = 0 min); 3 mg·kg−1·day−1, n = 8; 10 mg·kg−1·day−1, n = 6; vehicle, n = 9 (t = 20 min). Data are mean ± SEM. **P < 0.01, ***P < 0.001 versus t = 0 min.
Effect of AZD1656 on lipids
Analysis of terminal blood samples (day 29) showed that AZD1656 had no effect on plasma cholesterol, lactate or triglyceride concentrations (Table 2).
Table 2.
Study 1: terminal metabolic parameters in plasma samples from obese (fa/fa) and lean (fa/+) male Zucker rats
| Genotype | Treatment | Total cholesterol (mM) (n) | Lactate (mM) (n) | Triglycerides (mM) (n) |
|---|---|---|---|---|
| fa/fa | Vehicle | 18.76 ± 5.37 (9) | 4.69 ± 0.19 (8) | 4.92 ± 1.98 (9) |
| fa/fa | AZD1656 (3 mg·kg−1·day−1) | 13.91 ± 3.16 (8) | 5.09 ± 0.50 (8) | 5.93 ± 2.07 (8) |
| fa/fa | AZD1656 (10 mg·kg−1·day−1) | 15.64 ± 4.46 (7) | 4.91 ± 0.60 (7) | 4.75 ± 1.72 (7) |
| fa/+ | Vehicle | 2.41 ± 0.11a (10) | 4.58 ± 0.43 (10) | 1.00 ± 0.10b (10) |
Values are mean ± SEM.
P < 0.01 versus fa/fa vehicle.
P < 0.05 versus fa/fa vehicle.
Study 2: chronic efficacy of GKA71 in HF-fed male gkwt/del mice
Effect of GKA71 on body weight gain
The body weights of gkwt/del mice on HF diet increased in a biphasic manner consistent with established diet-induced obesity (Sörhede Winzell and Ahrén, 2004), with a slower rate of increase after about 17 weeks. GKA71 at 2.5 mg·kg−1·day−1 did not have a significant effect on body weight gain over the study period (Figure 4). At a dose of 5 mg·kg−1·day−1 GKA71, there was a downward trend in mean body weight for the treatment group after week 41 attributable to weight decrease in three animals, such that significance (P < 0.05) for the group as a whole was reached at week 49.
Figure 4.
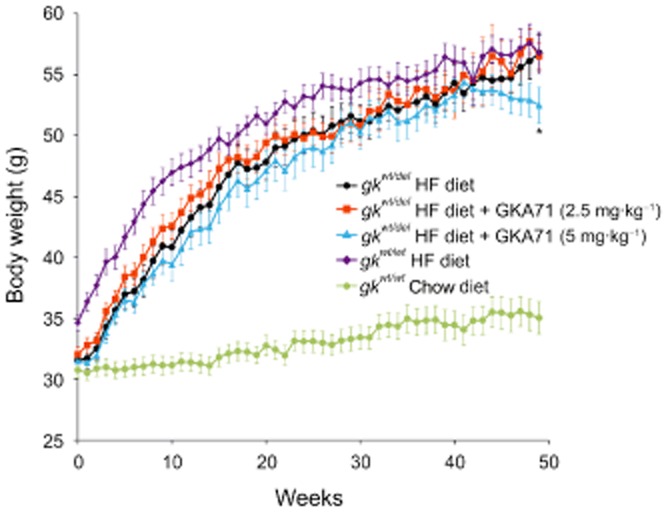
Body weight gain over 49 weeks in male gkwt/del mice on HF diet ± GKA71 and male wild-type mice on HF or chow diets: gkwt/del mice on HF diet alone (n = 10), gkwt/del mice on HF + GKA71 at 2.5 mg·kg−1·day−1 (n = 7–10), gkwt/del mice on HF + GKA71 at 5 mg·kg−1·day−1 (n = 10), wild-type mice on HF alone (n = 8–10), wild-type mice on chow diet alone (n = 10). Data are mean ± SEM. *P < 0.05 versus gkwt/del mice on HF diet alone.
Effect of GKA71 on weekly blood glucose levels
Baseline blood glucose values in gkwt/del mice on HF diet were 16.0 ± 0.2 mM, compared with 9.9 ± 0.5 mM for wild-type mice on HF diet and 8.2 ± 0.2 mM on chow diet. Over the 49 weeks of the study, mean blood glucose values of gkwt/del mice receiving HF diet alone remained within a range of 13.5–18.3 mM (Figure 5). The range for wild-type mice on HF diet was 7.1–9.9 mM, and on standard chow diet 7.0–10.5 mM.
Figure 5.

Weekly blood glucose values (1 h before start of dark phase) and compound exposures over 49 weeks in male gkwt/del mice on HF diet ± GKA71. (A) Blood glucose values for mice on HF diet alone (n = 10) or HF diet + GKA71 at 2.5 mg·kg−1·day−1 (n = 7–10), and compound exposure, expressed as a multiple of the mouse EC50. (B) Blood glucose values for mice on HF diet alone or HF diet + GKA71 at 5 mg·kg−1·day−1 (n = 10), and compound exposure. Blood glucose values are mean ± SEM. Compound exposure ratios are mean values over 24 h. Error bars indicate Cmax and Cmin. Week 0 values show mean baseline glucose in control and treatment groups at day −3 relative to diet switch. Three animals from the 2.5 mg·kg−1·day−1 GKA71 group were removed during the study as noted under Methods.
Glucose lowering stabilized within 2 weeks in animals receiving GKA71 in the diet (Figure 5). Over the remaining 47 weeks of the study, mean glucose values for gkwt/del mice receiving GKA71 in the diet at either dose were significantly lower (P < 0.001) than animals on HF diet alone (Table 3). GKA71 reduced mean blood glucose by 2.4 ± 0.3 mM at 2.5 mg·kg−1·day−1 and 3.2 ± 0.3 at 5 mg·kg−1·day−1, compared with control gkwt/del mice on HF diet. There was a difference (0.8 ± 0.3 mM, P < 0.01) in mean blood glucose levels between the two dose groups over the study. Blood glucose levels were lower compared with the HF diet control group for all weeks except weeks 5, 28, 29, 34, 36, 44 and 46–49 for the 2.5 mg·kg−1·day−1 GKA71 group, and weeks 29, 34, 36 and 47 for the 5 mg·kg−1·day−1 GKA71 group.
Table 3.
Study 2: mean blood glucose levels in male gkwt/del and wild-type mice over weeks 2–49
| Genotype | Diet | Mean blood glucose (mM) (n) | Mean total GKA71 over 24 h interval (μM) | Free drug/EC50 |
|---|---|---|---|---|
| gkwt/del | HF | 15.1 ± 0.2 (10) | – | – |
| gkwt/del | HF + GKA71 (2.5 mg·kg−1·day−1) | 12.7 ± 0.1a, b (7) | 21.64 ± 1.20 | 0.21 ± 0.01 |
| gkwt/del | HF + GKA71 (5 mg·kg−1·day−1) | 11.9 ± 0.7a (10) | 39.70 ± 2.35 | 0.11 ± 0.006 |
| gkwt/wt | HF | 8.4 ± 0.1 (8) | – | – |
| gkwt/wt | Chow | 8.8 ± 0.1 (10) | – | – |
Values are mean ± SEM.
P < 0.001 versus gkwt/del on HF diet.
P < 0.01 versus gkwt/del on HF diet + GKA71 (5 mg·kg−1·day−1).
Mean daily systemic exposure of GKA71 obtained for weeks 1–45 is shown in Table 3. Exposure was proportional to dose. Drug concentration in animals receiving 5 mg·kg−1·day−1 was above the efficacy threshold at all time points (Figure 5B), while for the group dosed at 2.5 mg·kg−1·day−1, 24 h drug concentration was significantly lower over the entire experiment, and only reached the efficacy threshold at weeks 17, 21 and 25 (Figure 5A).
Effect of GKA71 on monthly 24 h free-feeding blood glucose profiles
Once a month, 24 h free-feeding blood glucose profiles (−1 to 23 h) were measured. gkwt/del mice randomized to groups receiving HF diet alone, or HF + 2.5 or 5 mg·kg−1·day−1 GKA71 showed no significant difference in pre-study baseline glucose profiles (Figure 6A). Free-feeding glucose profiles at months 1, 3, 6, 9 and 12 are shown in Figure 6B–F. For animals receiving 2.5 mg·kg−1·day−1 GKA71, significant glucose lowering was achieved at all time points of the monthly glucose profile except for months 1 and 12 (see Figure 6B,F). For mice dosed at 5 mg·kg−1·day−1, significant glucose lowering was achieved at all time points at all months. Glucose AUC(–1 to 23 h) values for all monthly glucose profiles are shown in Figure 6G. The mean glucose AUC(–1 to 23 h) value averaged over the 12 month treatment period for mice receiving HF diet + 5 mg·kg−1·day−1 GKA71 (260.2 ± 3.9 mM·h) was lower than that of mice on HF diet + 2.5 mg·kg−1·day−1 (283.2 ± 2.9, P < 0.001), and both values were lower compared with mice on HF diet alone (328.5 ± 2.6, P < 0.001). It should be noted that the glucose AUC(–1 to 23 h) value for mice receiving HF diet alone at month 12 was significantly lower than the mean AUC(–1 to 23 h) value for this group over the whole study (P < 0.01).
Figure 6.
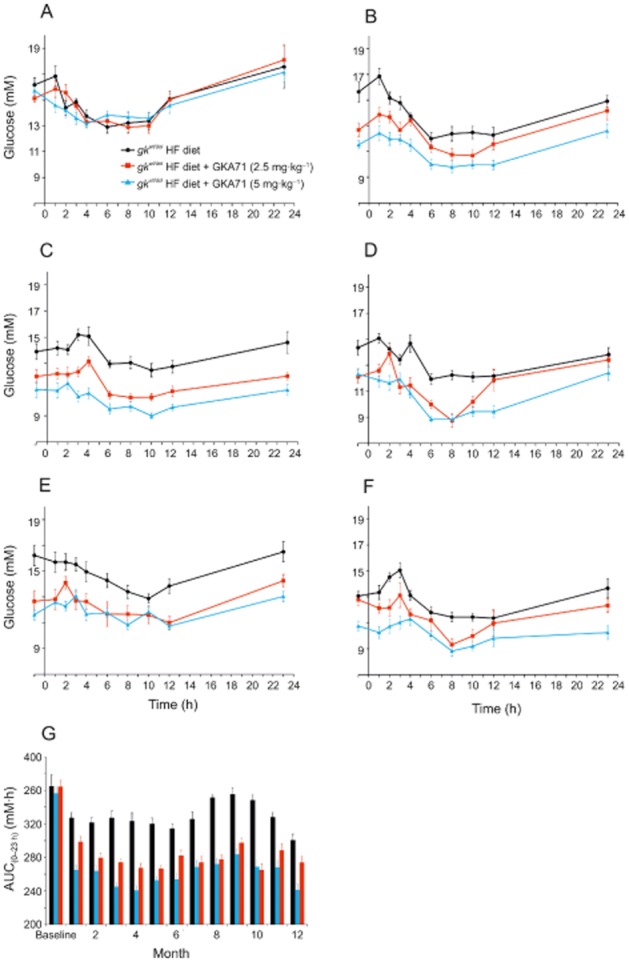
Twenty-four hour free-feeding blood glucose profiles for male gkwt/del mice on HF diet ± GKA71. (A) Baseline, (B) 1 month, (C) 3 months, (D) 6 months, (E) 9 months, (F) 12 months. Mice on HF diet alone (n = 10), HF diet + GKA71 at 2.5 mg·kg−1·day−1 (n = 7–10), HF diet + GKA71 at 5 mg·kg−1·day−1 (n = 10). Data are mean ± SEM. (G) Glucose AUC(–1 to 23 h) values (mM·h) for all monthly free-feeding glucose profiles over 12 months. Data are mean ± SEM.
Effect of GKA71 on glucose tolerance
At week 49, an OGTT was given to all mice in the study. Four hours fasted blood glucose levels were lower for mice dosed at 5 mg·kg−1·day−1 GKA 71 (14.4 ± 0.5 mM) versus controls (16.9 ± 1.0, P < 0.05), but not at 2.5 mg·kg−1·day−1 (15.7 ± 0.6 mM). Glucose clearance was slower in both gkwt/del and wild-type mice on HF diet compared with chow diet, consistent with diet-induced insulin resistance in this model (Gorman et al., 2008), and similar with untreated and treated gkwt/del mice (Figure 7A). Glucose AUC(0–90 min) values for gkwt/del mice on HF diet + GKA71 were not significantly lower than mice on HF diet alone. As expected, the AUC(0–90 min) value for wild-type littermates on HF diet was significantly lower than HF-fed gkwt/del mice (P < 0.001) and also lower than wild-type mice on chow diet (Figure 7B).
Figure 7.
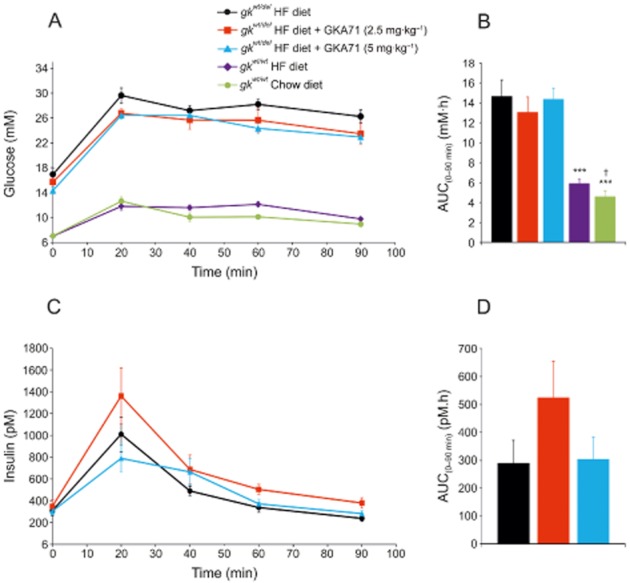
Blood glucose and insulin levels following OGTT in male gkwt/del mice on HF diet ± GKA71 and wild-type littermates on HF or chow diet after 49 weeks. (A) Glucose levels during OGTT. gkwt/del mice on HF diet alone (n = 10), HF diet + GKA71 at 2.5 mg·kg−1·day−1 (n = 7), HF diet + GKA71 at 5 mg·kg−1·day−1 (n = 10). Wild-type littermates on HF diet alone (n = 8), chow diet alone (n = 10). (B) Glucose AUC(0–90 min) values (mM·h). (C) Insulin levels during OGTT. (D) Insulin AUC(0–90 min) values (pM.h). Data are mean ± SEM. ***P < 0.001 versus gkwt/del mice on HF diet alone, †P < 0.05, versus wild-type on HF diet.
Insulin levels during the OGTT are shown in Figure 7C. The insulin excursions in animals on HF + 2.5 or 5 mg·kg−1·day−1 GKA71 were not significantly different than mice on HF diet alone (Figure 7D).
Effect of GKA71 on lipids and glycated haemoglobin (HbA1c)
GKA71 at 2.5 and 5 mg·kg−1·day−1 did not affect terminal concentrations of plasma cholesterol, plasma triglyceride, hepatic triglyceride or hepatic glycogen content compared with control gkwt/del mice on HF diet alone (Table 4). At the end of the study, GKA71 at both doses had significantly reduced blood % HbA1c levels compared with controls (Table 4).
Table 4.
Study 2: terminal metabolic parameters from male gkwt/del mice and gkwt/wt littermates
| Genotype | Diet | Plasma total cholesterol (mM) (n) | Plasma triglycerides (mM) (n) | Hepatic triglycerides (μmol·g−1) (n) | Hepatic glycogen (μmol·g−1) (n) | HbA1c (%) (n) |
|---|---|---|---|---|---|---|
| gkwt/del | HF | 6.40 ± 0.63 (7) | 1.02 ± 0.06 (7) | 0.19 ± 0.04 (8) | 26.34 ± 7.46 (9) | 7.97 ± 0.16 (9) |
| gkwt/del | HF + GKA71 (2.5 mg·kg−1·day−1) | 5.96 ± 0.40 (6) | 1.03 ± 0.08 (6) | 0.29 ± 0.06 (6) | 23.84 ± 3.35 (7) | 7.37 ± 0.22a (7) |
| gkwt/del | HF + GKA71 (5 mg·kg−1·day−1) | 5.29 ± 0.72 (8) | 1.00 ± 0.14 (8) | 0.25 ± 0.04 (10) | 25.95 ± 6.00 (10) | 7.09 ± 0.29a (7) |
| gkwt/wt | HF | 8.41 ± 0.41a (6) | 1.02 ± 0.12 (6) | 0.34 ± 0.06a (8) | 19.59 ± 5.27 (7) | 5.73 ± 0.09b (7) |
| gkwt/wt | Chow | 2.88 ± 0.10b (9) | 1.26 ± 0.07 (9) | 0.06 ± 0.01b (9) | 7.70 ± 3.62a (8) | 6.22 ± 0.07b (8) |
Values are mean ± SEM.
P < 0.05 versus gkwt/del on HF diet.
P < 0.001 versus gkwt/del on HF diet.
Histopathology
A slight reduction in hepatocyte vacuolation from ‘marked’ in the gkwt/del mice given HF diet to ‘moderate’ in HF-fed gkwt/del animals on 5 mg·kg−1·day−1 was seen histologically. No significant differences were observed in other tissues examined. HF feeding of gkwt/wt or gkwt/del mice resulted in substantially elevated hepatocellular cytoplasmic lipid vacuolation in comparison with the former strain-fed chow diet, which is reflected in elevated hepatic triglycerides (Table 4).
Discussion
Recent dose-ranging clinical studies with GKAs in differing type 2 diabetic cohorts (Meininger et al., 2011; Kiyosue et al., 2013; Wilding et al., 2013) have raised questions about whether this class of agents can elicit chronic glucose control. We have explored the ability of GKAs (AZD1656 and GKA71) to deliver sustained glucose control in two different animal models of insulin resistance and T2D. The male obese Zucker (fa/fa) rat was chosen to explore the effects of a GKA on long-term glucose control and lipid metabolism in a hyperlipidaemic environment (Kasiske et al., 1992; King, 2012). However, Zucker rats are not hyperglycaemic. Therefore, HF-fed male gkwt/del mouse was selected to investigate the effects of chronic GKA administration in the context of stable hyperglycaemia (Baker et al., 2014).
AZD1656 and GKA71 have very similar physico-chemical properties and are close analogues (Waring et al., 2011b; 2012), AZD1656 being slightly more potent. Acute studies in rodents have demonstrated that these compounds have similar PK/pharmacodynamic (PD) relationships (unpublished results). AZD1656 has been characterized in both acute and repeat-dose studies in humans (Ericsson et al., 2012; Morrow et al., 2012; Norjavaara et al., 2012; Kiyosue et al., 2013; Wilding et al., 2013).
In the obese Zucker fa/fa rat, AZD1656 gave a sustained reduction in free-feeding glucose profiles over 28 days, but only delivered a modest reduction in pre-dose glucose levels at 10 mg·kg−1·day−1, where compound exposure is sufficient to drive glucose-lowering efficacy for >12 h. At this dose, the free drug level of AZD1656 is comparable with the highest dose used in the multiple ascending dose study reported by Morrow et al., (2012), and is at the higher end of the dose range used in dose-finding studies (Kiyosue et al., 2013; Wilding et al., 2013). In normal rats and mice, AZD1656 and other GKAs give sustained reductions in blood glucose (Ohyama et al., 2010; unpublished results), but obese Zucker rats are extremely insulin-resistant (Kasiske et al., 1992; King, 2012) and GKAs have a smaller effect on pre-dose blood glucose.
To explore the long-term efficacy of a GKA at human translatable exposures in a rodent model with stable hyperglycaemia, we selected the HF-fed male gkwt/del mouse model (Baker et al., 2014). GKA71 has previously been shown to reduce basal glucose over 4 weeks of dietary dosing in the DIO mouse (Sörhede Winzell et al., 2011). However, the mice in this study were not markedly hyperglycaemic (8.8 ± 0.4 mM) and the glucose phenotype in the DIO model changes following prolonged HF feeding (Sörhede Winzell and Ahrén, 2004), making it unsuitable for an extended study. gkwt/del mice in study 2 remained consistently hyperglycaemic with an average blood glucose concentration in the HF diet control group over the study period of 15.1 ± 0.2 mM. GKA71 at both 2.5 and 5 mg·kg−1·day−1 delivered a sustained and significant reduction in plasma glucose for at least 45 weeks, together with a significant reduction in AUC values for monthly free-feeding blood glucose profiles up to week 45 and a significant reduction (P < 0.05) in glycated haemoglobin of ∼0.6 to 0.9% respectively. Free exposure of GKA71, as a multiple of the EC50, at 5 mg·kg−1·day−1, was consistently at levels associated with glucose lowering based on the defined relationship between exposure and in vitro GK enzyme potency (Coope et al., 2006), and slightly lower than the multiple obtained with AZD1656 in the clinic (Table 1). Exposure at 2.5 mg·kg−1·day−1 was significantly lower (P < 0.05; approximately 50% in magnitude), thus accounting for the correspondingly lower glucose-lowering efficacy observed at this dose.
Between weeks 45 and study termination, there is a suggestion in Figure 5A that efficacy may be diminishing in the mice receiving 2.5 mg·kg−1·day−1 GKA71, although this may also be interpreted as random fluctuation in glucose levels, as similar transient reductions in efficacy are seen over the course of the study. Unfortunately we did not measure drug exposure or food intake after week 45, but there was no decline in body weight at this dose during the last 4 weeks of the study, suggestive of no decrease in food intake and therefore drug dosage. However, we cannot be sure that there was no change in drug exposure between weeks 45 and 49. Efficacy is still present in the group receiving 5 mg·kg−1·day−1, although in this group there is a downward trend in mean body weight for the final few weeks of the study. The free-feeding glucose profile at week 45 gave AUC(–1 to 23 h) values for both doses of GKA71 in line with corresponding values from previous months (Figure 7). However, the AUC(–1 to 23 h) value at week 45 for mice on HF diet alone was significantly lower than the study mean for this group. Some strains of mice (including C57Bl/6) maintained for a long period on HF diet (>> 4 weeks) are known to adapt and show improvements in glucose homeostasis (Sörhede Winzell and Ahrén, 2004; Park et al., 2005; Ko et al., 2011; Sakurai et al., 2012). It is possible that we are seeing a similar effect beginning to appear in this study, albeit after a much longer period of stability.
Study 2 clearly demonstrated that GKA71 delivered sustained exposure-dependent efficacy in the male gkwt/del mouse model for at least 45 weeks. This finding is consistent with other chronic studies with GKAs reported in rodent models. Nakamura et al. (2009, 2011) showed that GKAs gave sustained glucose lowering up to 40 weeks in hyperglycaemic (∼11 mM) beta cell-specific GK knockout mice on HF diet. A different GKA gave sustained glucose lowering over 5 weeks at a dose of 10 mg·kg−1·day−1 but not at 3 mg·kg−1·day−1 in the progressively diabetic male Zucker diabetic fatty (ZDF) model (Futamura et al., 2012).
After 4 weeks treatment with AZD1656 in obese Zucker rats, we found neither increase in plasma total cholesterol and plasma triglycerides, nor any increase in plasma lactate levels. After 50 weeks dosing with GK71 in HF-fed gkwt/del mice, we also found neither increase in plasma total cholesterol and triglycerides, nor did we find any evidence for increased hepatic steatosis compared with that due to HF diet alone. We also found no evidence for elevation of hepatic glycogen above that associated with the HF diet alone. These findings are consistent with previous studies (Nakamura et al., 2009; 2011; Eiki et al., 2011; Sörhede Winzell et al., 2011; Futamura et al., 2012). Recently, De Ceuninck et al. (2013) have reported that a number of GKAs related to the benzoyl amino nicotinic acid GKA50 (McKerrecher et al., 2006) increase hepatic and plasma triglycerides. We have also observed elevated plasma triglycerides after dosing GKA50 (3 and 10 mg·kg−1·day−1) in obese Zucker rats (8 days) and ZDF rats (35 days; unpublished results). However, GKA50 and some other compounds from the benzoyl amino nicotinic acid series were found to have secondary pharmacology and a glucose-lowering efficacy that did not follow the established PK/PD relationship (Coope et al., 2006). GKA50, for example, is both a retinoic acid receptor antagonist (Waring et al., 2011a) and agonist. These findings led to the discovery and development of the neutral benzamide GKAs (Waring et al., 2011b; 2012), including AZD1656 and GKA71. Both GKAs are free of this secondary pharmacology with no effect on plasma lipids or hepatic triglyceride levels in efficacy studies such as those reported here, or at higher doses in safety studies performed in rats and dogs (unpublished results). In the clinic, AZD1656 induced a small transient rise in triglycerides in a diverse European and Latin American population (Wilding et al., 2013), but had no effect on triglycerides as monotherapy in a Japanese cohort (Kiyosue et al., 2013). The issue of triglyceride accumulation with GKAs therefore appears inconclusive, and is likely to benefit from further investigation.
AZD1656 gave a significant improvement in the glucose excursion profile after an OGTT at 2 weeks in the obese Zucker rat, as assessed by total glucose AUC and AUC after adjusting for differences in baseline glucose at t = 0 min. In contrast, although GKA71 lowered the total glucose AUC in an OGTT in HF-fed gkwt/del mice at 49 weeks there was neither significant reduction in the AUC after correcting for differences in baseline glucose, nor was there any change in the glucose elimination constant (KG; Sörhede Winzell and Ahrén, 2004). In study 1, AZD1656 was administered orally to fasted rats 120 min before the glucose bolus, and the free drug concentration over the period of the OGTT was significantly above the efficacy threshold. In study 2, drug was administered in the diet, which was withdrawn 4 h before the OGTT, so free drug levels during the glucose challenge are predicted to be below the efficacy threshold for both doses. This would in part explain the lack of marked efficacy in the OGTT. However, the lack of any effect of GKA71 on glucose tolerance over the 49 weeks treatment period, despite the sustained reduction in pre-feeding blood glucose, indicated that this GKA did not improve overall insulin sensitivity. Nakamura et al. (2009) have reported that a different GKA did improve glucose tolerance after 20 weeks treatment in the beta cell haploinsufficient gkwt/del mouse, although exposure was not reported. Neither AZD1656 nor GKA71 gave any improvement in insulin secretion in response to the oral glucose challenge, suggesting that these GKAs may be controlling blood glucose concentration primarily by a reduction in net hepatic glucose output. Histopathological examination of pancreatata from mice in study 2 showed no detrimental findings associated with treatment with GKA71. Hence, we conclude that long-term treatment with GKAs has no significant adverse functional effects on the aging pancreas. In support of this conclusion, it has recently been reported that a GKA can still increase beta cell proliferation rates in 1-to 2-year-old mice (Stolovich-Rain et al., 2012).
There is a growing view that GKA-mediated glucose-lowering efficacy diminishes over time in diabetic patients. However, almost all preclinical animal model data, including current studies, support sustained efficacy for GKA-mediated glucose-lowering efficacy. Compelling evidence on human target validation from GK-activating mutations in humans also indicates that GK activation delivers sustained glucose lowering (Osbak et al., 2009). Therefore, these contradictory data question either the value of animal models of diabetes as predictors of long-term responses in patients or the extent of our understanding of this mechanism in humans. GKAs mediate short-term perturbations in fluxes in and out of the hepatic triglyceride and glycogen pools (Nissim et al., 2012), and it is certainly possible that these will differ between rodents and humans. For example, triglyceride synthesis in man relies less on de novo lipogenesis than in rodents (Murphy, 2006) and also varies significantly between lean and obese human subjects (Donnelly et al., 2005). Further studies on hepatic metabolic flux in vivo may be the key to a better understanding of how GKAs achieve glycaemic control in rodents and humans, and their potential for safe and sustained glucose lowering in patients with T2D.
Acknowledgments
We gratefully acknowledge the assistance of AstraZeneca Animal Sciences and Welfare for technical assistance, Dr Julie Cook for analytical support, Dr Mike Waring and Dr Darren McKerrecher for discussions, and Dr Elizabeth Pilling and Dr Peter Ceuppens for statistical analyses.
Glossary
- GK
glucokinase
- GKA
glucokinase activator
- GSIS
glucose-stimulated insulin secretion
- HF
high fat
- OGTT
oral glucose tolerance test
- PD
pharmacodynamics
- PK
pharmacokinetics
- T2D
type 2 diabetes
- ZDF rats
Zucker diabetic fatty rats
Conflicts of interest
Authors are either past or present employees of AstraZeneca and hold stock in the company.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12504
Figure S1 Study design: Study 1.
Figure S2 Study design: Study 2.
References
- Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414:1–18. doi: 10.1042/BJ20080595. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Atkinson AM, Wilkinson GP, Coope GJ, Charles AD, Leighton B. Characterisation of the heterozygous glucokinase knockout mouse as a translational disease model for glucose control in type 2 diabetes. Br J Pharmacol. 2014;171:1629–1641. doi: 10.1111/bph.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebernitz GR, Beaulieu V, Dale BA, Deacon R, Duttaroy A, Gao JP, et al. Investigation of functionally liver selective glucokinase activators for the treatment of type 2 diabetes. J Med Chem. 2009;52:6142–6152. doi: 10.1021/jm900839k. [DOI] [PubMed] [Google Scholar]
- Bonadonna RC, Heise T, Arbet-Engels C, Kapitza C, Avogaro A, Grimsby J, et al. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol. 2010;95:5028–5036. doi: 10.1210/jc.2010-1041. [DOI] [PubMed] [Google Scholar]
- Bourbonais FJ, Chen J, Huang C, Zhang Y, Pfefferkorn JA, Landro JA. Modulation of glucokinase by glucose, small-molecule activator and glucokinase regulatory protein: steady-state kinetic and cell-based analysis. Biochem J. 2012;441:881–887. doi: 10.1042/BJ20110721. [DOI] [PubMed] [Google Scholar]
- Coghlan M, Leighton B. Glucokinase activators in diabetes management. Expert Opin Investig Drugs. 2008;17:145–167. doi: 10.1517/13543784.17.2.145. [DOI] [PubMed] [Google Scholar]
- Coope GJ, Atkinson AM, Allott C, McKerrecher D, Johnstone C, Pike KG, et al. Predictive blood glucose lowering efficacy by glucokinase activators in high fat fed female Zucker rats. Br J Pharmacol. 2006;149:328–335. doi: 10.1038/sj.bjp.0706848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ceuninck F, Kargar C, Ilic C, Caliez A, Rolin J-O, Umbdenstock T, et al. Small molecule glucokinase activators disturb lipid homeostasis and induce fatty liver in rodents: a warning for therapeutic applications in humans. Br J Pharmacol. 2013;168:339–353. doi: 10.1111/j.1476-5381.2012.02184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efanov AM, Barrett DG, Brenner MB, Briggs SL, Delaunois A, Durbin JD, et al. A novel glucokinase activator modulates pancreatic islet and hepatocyte function. Endocrinology. 2005;146:3696–3701. doi: 10.1210/en.2005-0377. [DOI] [PubMed] [Google Scholar]
- Eiki J-I, Nagata Y, Futamura M, Sasaki-Yamamoto K, Iino T, Nishimura T, et al. Pharmacokinetic and pharmacodynamic properties of the glucokinase activator MK-0941 in rodent models of type 2 diabetes and healthy dogs. Mol Pharmacol. 2011;80:1156–1165. doi: 10.1124/mol.111.074401. [DOI] [PubMed] [Google Scholar]
- Ericsson H, Roshammar D, Wollbratt M, Heijer M, Persson M, Ueda S, et al. Tolerability, pharmacokinetics, and pharmacodynamics of the glucokinase activator AZD1656, after single ascending doses in healthy subjects during euglycemic clamp. Int J Clin Pharm Ther. 2012;50:765–777. doi: 10.5414/CP201747. [DOI] [PubMed] [Google Scholar]
- Futamura M, Hosaka H, Kadotani A, Shimazaki H, Sasaki K, Ohyama S, et al. An allosteric activator of glucokinase impairs the interaction of glucokinase and glucokinase regulatory protein and regulates glucose metabolism. J Biol Chem. 2006;281:37668–37674. doi: 10.1074/jbc.M605186200. [DOI] [PubMed] [Google Scholar]
- Futamura M, Yao J, Li X, Bergeron R, Tran J-L, Zycband E, et al. Chronic treatment with a glucokinase activator delays the onset of hyperglycaemia and preserves beta cell mass in the Zucker diabetic fatty rat. Diabetologia. 2012;55:1071–1080. doi: 10.1007/s00125-011-2439-3. [DOI] [PubMed] [Google Scholar]
- Fyfe MC, White JR, Taylor A, Chatfield R, Wargent E, Printz RL, et al. Glucokinase activator PSN-GK1 displays enhanced antihyperglycaemic and insulinotropic actions. Diabetologia. 2007;50:1277–1287. doi: 10.1007/s00125-007-0646-8. [DOI] [PubMed] [Google Scholar]
- Gorman T, Hope DCD, Brownlie R, Yu A, Gill D, Löfvenmark J, et al. Effect of high-fat diet on glucose homeostasis and gene expression in glucokinase knockout mice. Diabetes Obes Metab. 2008;10:885–897. doi: 10.1111/j.1463-1326.2007.00819.x. [DOI] [PubMed] [Google Scholar]
- Grimsby J, Sarabu R, Corbett WL, Haynes N-E, Bizzarro FT, Coffey JW, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301:370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- Kasiske BL, O'Donnell MP, Keane WF. The Zucker rat model of obesity, insulin resistance, hyperlipidemia, and renal injury. Hypertension. 1992;19:I110–I115. doi: 10.1161/01.hyp.19.1_suppl.i110. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King AJF. The use of animal models in diabetes research. Br J Pharmacol. 2012;166:877–894. doi: 10.1111/j.1476-5381.2012.01911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyosue A, Hayashi N, Komori H, Leonsson-Zachrisson M, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 as monotherapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2013;15:923–930. doi: 10.1111/dom.12100. [DOI] [PubMed] [Google Scholar]
- Ko HJ, Lee Y, Lee JH, Azuma Y, Friedline RH, Kinicki S, et al. Metabolic aging affects energy balance and insulin sensitivity response to chronic high-fat feeding. Diabetes. 2011;60(Suppl. 1):A423. [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerrecher D, Allen JV, Caulkett PW, Donald CS, Fenwick ML, Grange E, et al. Design of a potent, soluble glucokinase activator with excellent in vivo efficacy. Bioorg Med Chem Lett. 2006;16:2705–2709. doi: 10.1016/j.bmcl.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nature Rev Drug Discov. 2009;8:399–416. doi: 10.1038/nrd2850. [DOI] [PubMed] [Google Scholar]
- Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2560–2566. doi: 10.2337/dc11-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow LA, Leonsson-Zachrisson M, Ericsson H, Wollbratt M, Knutsson M, Hompesch M, et al. Safety, pharmacokinetics and pharmacodynamics of multiple-ascending doses of the novel glucokinase activator AZD1656 in patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14:1114–1122. doi: 10.1111/j.1463-1326.2012.01661.x. [DOI] [PubMed] [Google Scholar]
- Murphy EJ. Stable isotope methods for the in vivo measurement of lipogenesis and triglyceride metabolism. J Anim Sci. 2006;84(E.Suppl):E94–E104. doi: 10.2527/2006.8413_supple94x. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Terauchi Y, Ohyama S, Kubota J, Shimazaki H, Nambu T, et al. Impact of small-molecule glucokinase activator on glucose metabolism and beta-cell mass. Endocrinology. 2009;150:1147–1154. doi: 10.1210/en.2008-1183. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Shimazaki H, Ohyama S, Eiki J, Terauchi Y. Effect of long-term treatment with a small-molecule glucokinase activator on glucose metabolism, lipid profiles and hepatic function. J Diabetes Invest. 2011;2:276–279. doi: 10.1111/j.2040-1124.2011.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissim I, Horyn O, Nissim I, Daikhin Y, Wehrli SL, Yudkoff M, et al. Effects of a glucokinase activator on hepatic intermediary metabolism: study with 13C-isotopomer-based metabolomics. Biochem J. 2012;444:537–551. doi: 10.1042/BJ20120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norjavaara E, Ericsson H, Sjoberg F, Leonsson-Zachrisson M, Sjostrand M, Morrow LA, et al. Glucokinase activators AZD6370 and AZD1656 do not affect the central counterregulatory response to hypoglycemia in healthy males. J Clin Endocrinol. 2012;97:3319–3325. doi: 10.1210/jc.2012-1496. [DOI] [PubMed] [Google Scholar]
- Ohyama S, Takano H, Iino T, Nishimura T, Zhou Y-P, Langdon RB, et al. A small-molecule glucokinase activator lowers blood glucose in the sulfonylurea-desensitized rat. Eur J Pharmacol. 2010;640:250–256. doi: 10.1016/j.ejphar.2010.04.054. [DOI] [PubMed] [Google Scholar]
- Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanne-Chantelot C, Ellard S, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–1526. doi: 10.1002/humu.21110. [DOI] [PubMed] [Google Scholar]
- Park S-Y, Cho Y-R, Kim H-J, Higashimori T, Danton C, Lee M-K, et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. 2005;54:3530–3540. doi: 10.2337/diabetes.54.12.3530. [DOI] [PubMed] [Google Scholar]
- Poucher SM, Freeman S, Loxham SJG, Convey G, Bartlett JB, De Schoolmeester J, et al. An assessment of the in vivo efficacy of the glycogen phosphorylase inhibitor GPi688 in rat models of hyperglycaemia. Br J Pharmacol. 2007;152:1239–1247. doi: 10.1038/sj.bjp.0707502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees MG, Gloyn AL. Small molecular glucokinase activators: has another new anti-diabetic therapeutic lost favour? Br J Pharmacol. 2013;168:335–338. doi: 10.1111/j.1476-5381.2012.02201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai Y, Inoue H, Shintani N, Arimori A, Hamagami K-I, Hayata-Takano A, et al. Compensatory recovery of blood glucose levels in KKA(y) mice fed a high-fat diet: insulin-sparing effects of PACAP overexpression in cells. J Mol Neurosci. 2012;48:647–653. doi: 10.1007/s12031-012-9758-9. [DOI] [PubMed] [Google Scholar]
- Sjöstrand M, Ericsson H, Hartford M, Norjavaara E, Eriksson JW. Pharmacodynamic effects of the oral glucokinase activator AZD6370 after single doses in healthy volunteers assessed with euglycaemic clamp. Diabetes Obes Metab. 2013;15:35–41. doi: 10.1111/j.1463-1326.2012.01672.x. [DOI] [PubMed] [Google Scholar]
- Sörhede Winzell M, Ahrén B. The high-fat diet-fed mouse. A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes. 2004;53:S215–S219. doi: 10.2337/diabetes.53.suppl_3.s215. [DOI] [PubMed] [Google Scholar]
- Sörhede Winzell M, Coghlan M, Leighton B, Frangioudakis G, Smith DM, Storlien LH, et al. Chronic glucokinase activation reduces glycaemia and improves glucose tolerance in high-fat diet fed mice. Eur J Pharmacol. 2011;663:80–86. doi: 10.1016/j.ejphar.2011.05.009. [DOI] [PubMed] [Google Scholar]
- Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y. Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem. 2012;287:27407–27414. doi: 10.1074/jbc.M112.350736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waring MJ, Brogan IJ, Coghlan M, Johnstone C, Jones HB, Leighton B, et al. Overcoming retinoic acid receptor-alpha based testicular toxicity in the optimisation of glucokinase activators. Med Chem Comm. 2011a;2:771–774. [Google Scholar]
- Waring MJ, Johnstone C, McKerrecher D, Pike KG, Robb G. Matrix-based multiparameter optimisation of glucokinase activators: the discovery of AZD1092. Med Chem Comm. 2011b;2:775–779. [Google Scholar]
- Waring MJ, Clarke DS, Fenwick MD, Godfrey L, Groombridge SD, Johnstone C, et al. Property based optimisation of glucokinase activators – discovery of the phase IIb clinical candidate AZD1656. Med Chem Comm. 2012;3:1077–1081. [Google Scholar]
- Wilding J, Leonsson-Zachrisson M, Wessman C, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes Metab. 2013;15:750–759. doi: 10.1111/dom.12088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study design: Study 1.
Figure S2 Study design: Study 2.