

Abstract
Background and Purpose
The prevalence of concurrent use of two or more drugs that block human ether-a-go-go-related gene product (hERG) K+ channels is not uncommon, but is not well characterized. This study defined the effects of concurrent exposure of two hERG-blocking drugs on hERG current amplitude. Experiments were conducted to determine if concomitant exposure to two potent pore hERG blockers, thioridazine and terfenadine and a weak hERG blocker, erythromycin, would result in an additive, synergistic or inhibitory effect.
Experimental Approach
hERG currents from stably transfected HEK cells were measured using the whole-cell variant of the patch-clamp method at physiological temperatures. Concentration–response relationships for thioridazine or terfenadine were obtained with cells pre-exposed to erythromycin.
Key Results
Pre-exposure of cells to erythromycin resulted in an approximately 14–22-fold rightward shift in the hERG concentration–response curve for thioridazine and terfenadine respectively. This reduction in affinity was not the result of a change in the voltage-dependent characteristics of the channel. Results suggest an external binding site for erythromycin.
Conclusions and Implications
Pretreatment with erythromycin induced an approximately 14–22-fold reduction in hERG affinity for pore-binding drugs at concentrations of erythromycin, which by themselves only block hERG by 10% or less. These results suggest distinct, allosterically linked binding sites on opposite sides of the hERG channel. Occupancy of the external site by erythromycin reduces the affinity of the pore binding site. Furthermore, these results suggest that co-administration of erythromycin may provide some reduction in cardiac liability of potent hERG-blocking drugs.
Keywords: thioridazine, erythromycin, patch-clamp, allosteric, hERG
Introduction
Many different drug classes and structures are associated with cardiac QT prolongation. The most common underlying mechanism for this QT prolongation is block of the rapid component of the delayed rectifier current IKR, which in humans is carried by the protein product of the human ether-a-go-go-related (hERG) gene (the Kv11.x channels; channel nomenclature follows Alexander et al., 2013). For many drugs, the amino acids important in binding to this channel protein have been determined and the binding site is believed to be within the pore towards the cytoplasmic side of the channel (Milnes et al., 2006). Interestingly, the binding of the macrolide antibiotic, erythromycin is not dramatically changed by mutation of the same amino acids that have profound effects on pore channel blockers, such as thioridazine and terfenadine (Duncan et al., 2006). This suggests that the binding site for erythromycin is distinct from that of pore channel blockers. In support of this, large toxins, such as BeKm-1, which selectively and potently inhibit hERG current bind to an external binding site on the channel (Zhang et al., 2003; Yi et al., 2007). However, the location of the binding site for relatively large, non-peptide molecules like erythromycin has not been well described.
The prevalence of concurrent use of two or more QT prolonging drugs is not uncommon (Curtis et al., 2003; LaPointe, 2006). While the hERG channel blocking activity of many QT prolonging drugs has been characterized, the effects of concurrent exposure of two hERG-blocking drugs on hERG current is not well characterized. In light of these deficiencies, experiments were conducted to further characterize the location of the erythromycin binding site on the hERG channel and to characterize the hERG channel blocking properties of potent pore channel blockers, thioridazine and terfenadine, in combination with the weaker hERG blocker, erythromycin.
Methods
Transfection and cell culture
HEK-293 cells were stably transfected with the hERG clone as previously described (Ekins et al., 2002). Cells were maintained in minimum essential medium with Earle's salts supplemented with non-essential amino acids (1%), sodium pyruvate (1%), penicillin, streptomycin (1%) and FBS (10%; all from Gibco/Life Technologies, Grand Island, NY, USA).
Solutions
Drugs were dissolved in either DMSO or deionized water to make stock solutions on the day of use. Dilutions of stock solutions were made immediately before the experiment to prepare the desired concentrations. The external solution (solution bathing the cell) had an ionic composition of (in mM): 137 NaCl, 4 KCl, 1.8 CaCl2, 1.2 MgCl2, 11 dextrose, 10 HEPES, adjusted to a pH of 7.4 with NaOH. The internal (pipette) solution had an ionic composition of (in mM): 130 KCl; 1 MgCl2, 5 NaATP, 7 NaCl, 5 EGTA, 5 HEPES, pH = 7.2 using KOH. Experiments were performed at 36 ± 1°C.
Data acquisition and analysis
Currents were measured using the whole-cell variant of the patch-clamp method (Crumb and Cavero, 2002). Pipette tip resistance was approximately 1–2 MΩ when filled with internal solutions. Analogue capacity compensation and 40–60% series resistance compensation were used to yield voltage drops across uncompensated series resistance of less than 3 mV. Bath temperature was measured by a thermistor placed near the cell under study and was maintained by a thermoelectric device. An Axopatch 1-B amplifier (Axon Instruments, Union City, CA, USA) was used for whole-cell voltage clamping. Creation of voltage-clamp pulses and data acquisition were controlled by a personal computer running pClamp software (Molecular Devices, Sunnyvale, CA, USA).
After rupture of the cell membrane (entering whole-cell mode), current amplitude and kinetics were allowed to stabilize (3–5 min) before experiments were begun. Unless otherwise indicated, hERG currents were elicited by a voltage pulse to +10 mV (500 ms) from a holding potential of −75 mV. hERG tail currents were measured upon repolarization to −40 mV (500 ms). Drug effects on tail current amplitude were measured after a steady-state level of block had been achieved. Unless otherwise stated, drug was applied in the bath solution. The pacing rate was 0.1 Hz.
Data are given as percentage of reduction of current amplitude, which was measured as current reduction after a steady-state effect had been reached in the presence of drug, relative to current amplitude before drug was introduced (control). Each cell served as its own control. Log–linear plots were created of the mean percentage of blockade (± SEM) at the concentrations that were tested. A nonlinear curve fitting routine was used to fit a three-parameter Hill equation to the results using MicroCal Origin, version 6.0 software (MicroCal Software, Northhampton, MA, USA). The equation is of the following form:
where B (%) is the percentage of current block at drug concentration D, IC50 is the concentration of drug that causes 50% block and n is the Hill coefficient.
To determine whether drug combinations may exert additive, synergistic or antagonistic effects, the combination index (CI) was calculated (Chou, 2006; Friemel and Zunkler, 2010). The CI was calculated from
where the denominators stand for the concentrations of test compounds A and B with each individual test compound inhibiting hERG by x% and the numerators A1 and B1 in combination inhibit hERG by the same x%. A CI that equals 1 is additive, a CI smaller than 1 indicates synergism, and a CI greater than 1 indicates antagonism.
Data analysis
Data are shown as means ± SEM from four to six experiments. Differences between means were analysed by one way ANOVA with Tukey's post hoc test. P < 0.05 was taken to show significant differences between means.
Materials
Thioridazine, terfenadine, erythromycin and clotrimazole were obtained from Sigma-Aldrich. (St. Louis, MO, USA).
Results
Thioridazine and erythromycin block of hERG current
Experiments were performed to characterize the effects of drug combinations on hERG channel block. For this characterization a potent hERG channel blocker, thioridazine and a weak hERG channel blocker, erythromycin, were examined. The concentration–response relationships for thioridazine or erythromycin block of hERG currents are shown in Figure 1. Experiments were conducted at 36 ± 1°C. Fits of the mean concentration–response data with a Hill equation yielded IC50 values of 33.0 nM for thioridazine and 59.3 μM for erythromycin, similar to previous reports (Stanat et al., 2003; Crumb et al., 2006).
Figure 1.
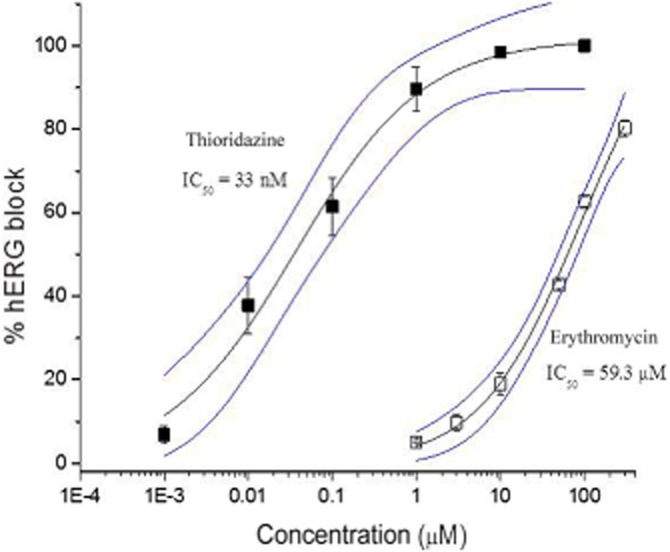
Dose–response relationships for thioridazine and erythromycin block of hERG current (n = 5–6). The IC50 values and 95% confidence intervals (blue lines) are also shown.
We next sought to determine the effects on hERG current amplitude of a combination of thioridazine and erythromycin. The effects on thioridazine block of hERG current after pretreating the cells with varying concentrations of erythromycin are illustrated in Figure 2. As indicated in panel A, in the absence of erythromycin pretreatment, 100 nM thioridazine substantially reduced hERG current amplitude. Panel B shows the effects of various concentrations of thioridazine on cells pretreated with 3 μM erythromycin for approximately 10–15 min. In the presence of erythromycin, much higher concentrations of thioridazine are required to block the hERG current. Panel C more clearly shows the effect erythromycin pretreatment has on thioridazine block of hERG. As the concentration of erythromycin used to pretreat cells is increased, the concentration–response curve for thioridazine block of hERG current is shifted rightward, with IC50 values increasing 16-fold as erythromycin concentrations increase from 0 to 3 μM (0 μM = 29.3 ± 3.9 nM, 0.3 μM = 177 ± 26.7 nM, 3 μM = 477 ± 76.0 nM, P < 0.005). No further shift was observed at an erythromycin pretreatment concentration of 30 μM compared with 3 μM (IC50 = 402 ± 58.9 nM for 30 μM).
Figure 2.
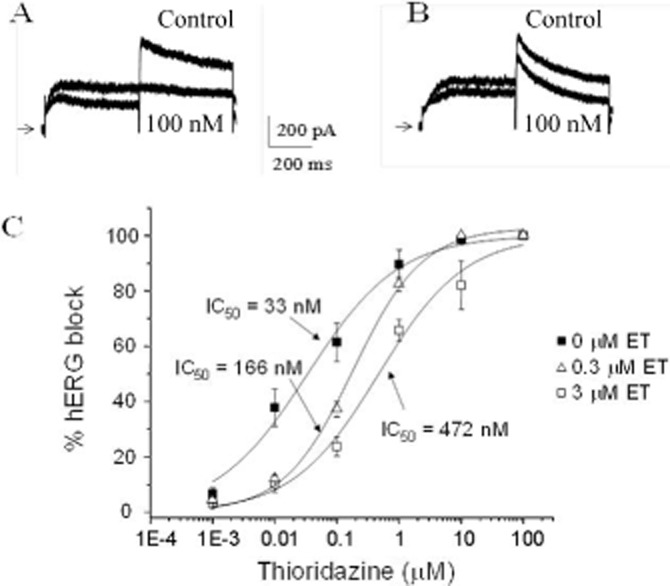
Effects of erythromycin (ET) pretreatment on thioridazine block of hERG. (A) Effect of 100 nM thioridazine on hERG current in the absence of erythromycin. (B) Effects of 100 nM thioridazine on hERG current in the presence of 3 μM erythromycin. Zero current levels are indicated by the arrow. (C) Concentration–response relationships for thioridazine block of hERG in the presence of varying concentrations of erythromycin (n = 4–6). Relationships have been fit with the equation given in the Methods to obtain IC50 values.
In order to better define the relationship between thioridazine additions to erythromycin-treated cells, the CI values were calculated at the various erythromycin concentrations. A CI value of 5.0 was calculated for an erythromycin concentration of 0.3 μM, and a CI value of 14.4 was calculated for an erythromycin concentration of 3 μM. According to Chou (2006), this indicates a moderate to strong antagonism.
To examine whether the effects of erythromycin pretreatment extend to drugs besides thioridazine, a series of experiments were performed to determine the effects of pretreating cells with 3 μM erythromycin on the terfenadine block of hERG. Figure 3 indicates a similar rightward shift in terfenadine affinity for hERG in the presence of erythromycin, with mean IC50 values increasing from 0.05 to 1.10 μM (n = 4). This represents a 22-fold increase in IC50, more pronounced than for thioridazine. A CI value of 22.1 was calculated for an erythromycin concentration of 3 μM, indicating strong antagonism.
Figure 3.
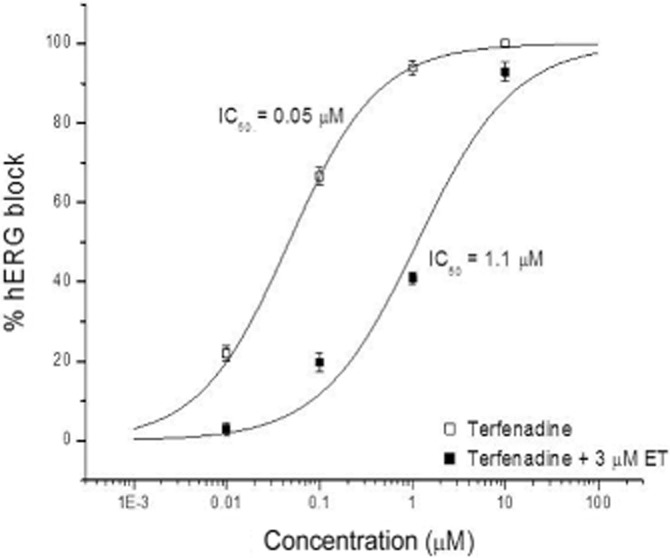
Effects of erythromycin (ET) pretreatment on terfenadine block of hERG current (n = 4). Cells were pretreated with either no (0) or 3 μM erythromycin.
Voltage-dependence of thioridazine and erythromycin block of hERG current
To further explore the effect of erythromycin pretreatment on thioridazine block of hERG, the voltage-dependent nature of hERG block for thioridazine, erythromycin, and a combination of the two were characterized. As indicated in Figure 4B, there was no difference in the current–voltage relationship observed for thioridazine, erythromycin or when cells were pretreated with 3 μM erythromycin and exposed to 100 nM thioridazine. The same was true for the voltage-dependence of hERG current activation (Figure 4D). Similarly, there was no difference in the kinetics of tail current decay at voltages ranging from −100 to −40 mV or in the timecourse of block development during a voltage pulse to +10 mV(data not shown).
Figure 4.
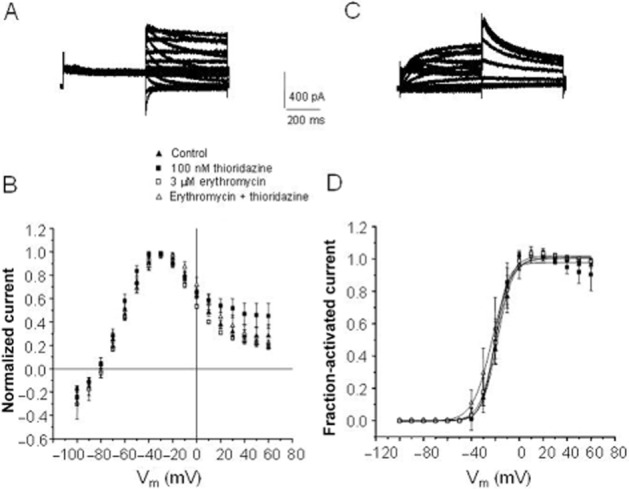
Voltage-dependent effects of thioridazine and erythromycin alone and in combination. (A) Example of a family of hERG current traces recorded at various voltages (see panel B) in the absence of any drug. Current was elicited by a prepulse to +10 mV followed by a voltage pulse to the voltages indicated in panel B. (B) Current–voltage relationship in control, 100 nM thioridazine, 3 μM erythromycin or 100 nM thioridazine in the presence of pretreatment with 3 μM erythromycin (n = 5). Current amplitude is normalized to maximal current amplitude. (C) family of hERG currents elicited by prepulses as indicated in panel D. Tail current was elicited by a pulse to −40 mV. (D) Voltage-dependence of hERG current activation under conditions the same as in panel B (n = 5). Current is normalized to maximal current amplitude.
Sidedness of thioridazine and erythromycin block
To gain some insight into where in the channel thioridazine and erythromycin might be exerting their effects on hERG, the voltage-dependence of hERG current block was determined at voltages where channel availability was maximal (Figure 5). At a voltage of +10 mV, hERG channel availability was virtually 100%. Over the voltage range of +10 mV to +60 mV, the voltage-dependence of block by either 100 nM thioridazine or 30 μM erythromycin was very flat. This voltage range was chosen to eliminate confusion between voltage-dependent block and the voltage-dependence of channel availability. For comparison, the block produced by 1 mM Ba++ shows considerably more voltage-dependence (Figure 5 inset). A similar lack of voltage-dependence has been shown for thioridazine block of hERG and suggests a binding site outside the electrical field of the hERG channel (Milnes et al., 2006). For thioridazine, the specific site on the inner mouth of the channel has been located by means of molecular biology (Milnes et al., 2006). Although the location of the binding site for erythromycin has not been determined, the same mutations that reduce or eliminate thioridazine and terfenadine binding, have little effect on erythromycin binding (Mitcheson, 2003; Duncan et al., 2006).
Figure 5.
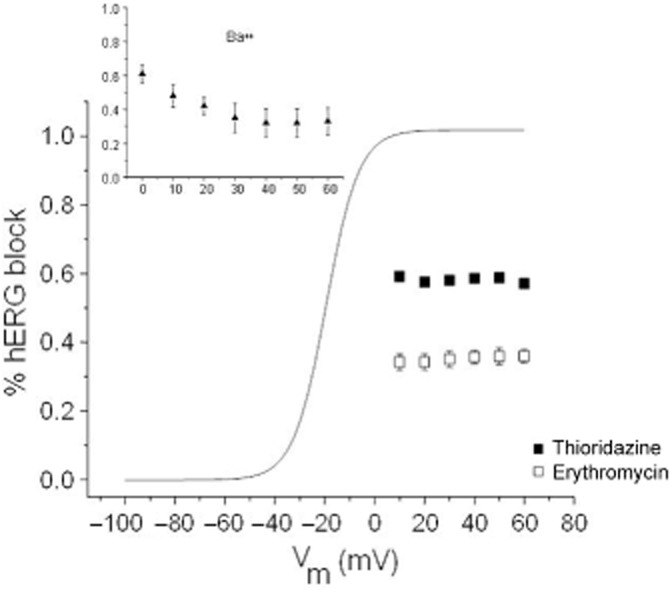
Voltage-dependence of hERG block by 100 nM thioridazine and 30 μM erythromycin (n = 5). For comparison, the voltage-dependence of hERG current activation is shown (solid line taken from Figure 4D). (Inset) Voltage-dependence of 1 mM Ba++ block of hERG current (n = 3).
To further characterize the location of the erythromycin binding site, a series of experiments in which the pH of either the external (bath) or internal (pipette) solution was changed were undertaken, similar to that used to characterize the interaction of verapamil with the hERG channel (Zhang et al., 1999). Erythromycin has a titratable nitrogen and a pKa of 8.8. In Figure 6A, B, the effects of acidifying the external pH from 7.4 to 6.5 are shown. As reported by others, lowering the external pH increases the rate of hERG tail current decay, an effect observed in the present report (Figure 6A). After the addition of 50 μM erythromycin, the hERG current was reduced by approximately the same amount whether the external pH was 7.4 or 6.5 (Figure 6B). Given the size of the erythromycin molecule and the fraction of molecules positively charged (greater than 90% at pH = 7.4 and greater than 99% at pH = 6.5) at the external pH values tested, the fact that the degree of block was not altered suggests an external erythromycin binding site. The same type of experiment was performed with acidifying the internal pH and applying erythromycin in the pipette solution (Figure 6C, D). If erythromycin does indeed block hERG channels by binding to a site on the extracellular side of the channel, then trapping the molecules inside the cell by acidifying the internal solution and protonating the erythromycin molecules should virtually eliminate hERG channel block. As illustrated in Figure 6C, pipette application of erythromycin at a pH of 5.0 where greater than 99.9% of the molecules are charged, dramatically reduces block. At concentrations ranging from 10 to 50 μM, only an approximately 10–15% reduction in current amplitude was observed (Figure 6D). In contrast, upon the addition of 50 μM erythromycin to the external solution, in the same cell, an approximately 50% reduction was observed (Figure 6C). Unfortunately, it is difficult to discern erythromycin block when applied internally under these conditions from normal time-and dialysis-dependent changes in hERG current amplitude once the whole-cell configuration is achieved. However, the fact that there is no distinguishable concentration-dependence to the pipette applied erythromycin effect suggests that what is occurring is in large part normal changes in current amplitude observed upon entering the whole-cell mode. To verify that the addition of drug to the pipette solution can indeed produce block, 50 nM thioridazine was added to the pipette solution. hERG current was rapidly reduced by 59.± 8.2% (n = 3), confirming current block by drug added to the pipette solution (data not shown).
Figure 6.
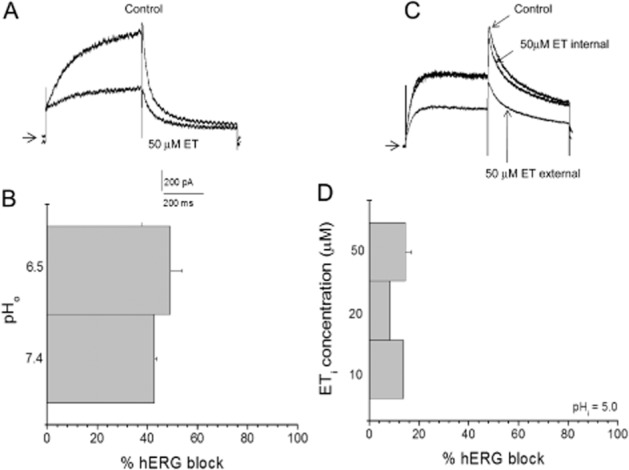
Sidedness of erythromycin block of hERG current. (A) hERG current traces in the absence and presence of 50 μM erythromycin (ET). Arrow indicates zero current level. (B) Effects of changing external pH on hERG block by 50 μM erythromycin applied to the bath solution (i.e. outside the cell) (n = 5). (C) hERG current traces in the absence and presence of 50 μM erythromycin (ET) added to the pipette solution (internal) or after washing in ET in the bath solution to the same cell (external). Arrow indicates zero current level. (D) Effects of various concentrations of erythromycin (ETi; 10, 20, 50 μM) added to the pipette solution at a pH of 5.0 (n = 5).
Effect of erythromycin pretreatment on clotrimazole block
In order to examine whether the effects of erythromycin pretreatment extend to drugs not known to block by binding to the channel pore, the effects of erythromycin pretreatment on clotrimazole block of hERG were examined. In contrast to thioridazine, erythromycin pretreatment had no effect on the clotrimazole block of hERG current (Figure 7A–C). At an erythromycin concentration (3 μM) that produced a maximal reduction in thioridazine affinity, the clotrimazole IC50 value (1.11 ± 0.19 μM, n = 5) remained virtually identical to that observed in the absence of erythromycin (1.08 ± 0.36 μM, n = 5). Like thioridazine and erythromycin, clotrimazole voltage-dependent block was flat, suggesting a binding site outside the electric field (Figure 7D). Furthermore, addition of clotrimazole to the pipette solution at a pH of 4.4 (clotromazole pKa = 6.12), which should protonate and trap the majority of clotrimazole molecules inside the cell was associated with relatively little reduction in hERG current. In contrast, addition of 1 μM clotrimazole to the bath solution produced a marked reduction in current amplitude (Figure 7E). The CI value calculated for the clotrimazole and erythromycin combination was 0.8, suggesting an additive or slightly synergistic effect.
Figure 7.
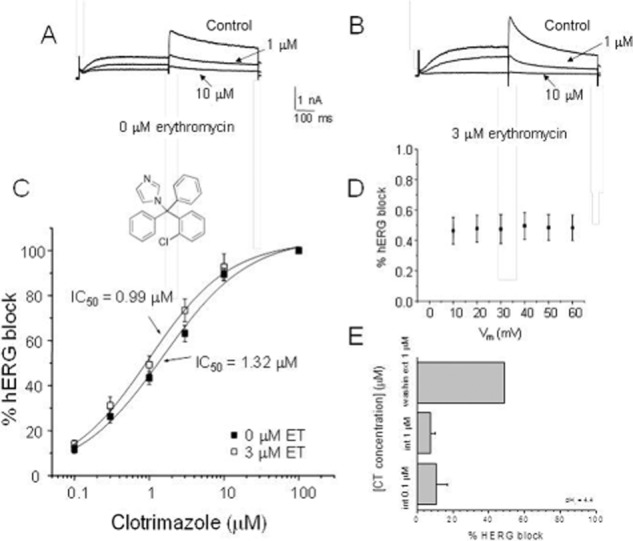
Effects of erythromycin pretreatment on clotrimazole block of hERG current. (A) hERG current reduction caused by the indicated concentrations of clotrimazole in the absence of erythromycin. (B) hERG current reduction caused by the indicated concentrations of clotrimazole in the presence of 3 μM erythromycin. (C) Mean concentration–response relationship for clotromazole block of hERG current in the absence and presence of 3 μM erythromycin (ET) (n = 5). (Inset) Structure of clotrimazole. (D) Voltage-dependence of clotrimazole block of hERG current (n = 5). (E) Effects of internal application of clotrimazole (i.e. in the pipette solution) and external application of clotrimazole (addition to the bath solution) at a pipette solution pH of 4.4 (n = 5). Clotrimazole (CT) was added either to the pipette solution at a concentration of 0.1 or 1.0 μM or to the bath solution at a concentration of 1 μM (washing ext 1 μM).
Discussion
The present study describes the ability of pretreatment with erythromycin to reduce the hERG-blocking affinity of the pore channel blockers, thioridazine and terfenadine, but not for clotrimazole, which was shown to have an external binding site. What is particularly interesting is how a 14-fold reduction in thioridazine affinity and a 22-fold reduction in terfenadine affinity can be produced by concentrations of erythromycin, which by themselves only block hERG by approximately 10% or less (see Figures 1, 2). This interaction occurred in the absence of any changes in the voltage-dependence or kinetics of the hERG current block produced by the combination of the two drugs. It has previously been shown that pretreatment of cells with complete blocking concentrations of verapamil can antagonize the complete block of hERG channels by dofetilide (Zhang et al., 1999). This result was assumed to reflect either a competition for a common binding site or some form of allosteric interaction. Similarly, the combination of ondansetron and an undisclosed drug were shown to not produce synergestic or additive effects on QT prolongation in dogs, but the addition of the undisclosed drug actually interfered with the ability of ondansetron to prolong QT (Valentin et al., 2010). Hreiche et al. (2011) showed that pretreatment of cells with ketoconazole inhibited the ability of domperidone to block hERG current. All of these drug–drug interactions were assumed to reflect competition at a common binding site.
Evidence for an allosteric interaction between thioridazine and erythromycin
The results in the present study can most easily be explained by an allosteric interaction between distinct binding sites for erythromycin and thioridazine on opposite sides of the channel and not competition at a common site. Firstly, it has been shown that the S6 helix point mutation F656A almost completely eliminates thioridazine block of hERG channels (Milnes et al., 2006). The site of this amino acid is believed to be on the cytoplasmic side of the channel protein within the pore of the channel. Mutations of the same amino acid, as well as others, have also been shown to reduce terfenadine block of hERG channels (Kamiya et al., 2008), suggesting a common location for binding. Secondly, in contrast, the point mutation F656A had little effect on erythromycin block of hERG channels (Duncan et al., 2006), suggesting that erythromycin does not share the common pore binding site. Thirdly, reducing external pH to externally applied erythromycin has no effect on the degree of hERG block while reducing internal pH to internally applied erythromycin dramatically reduces block. These results are most easily explained by an extracellular binding site for erythromycin, a speculation supported by Mitcheson (2003). It is therefore proposed that erythromycin binds to a site on the extracellular surface of the hERG channel while thioridazine and terfenadine bind to a site within the pore towards the intracellular surface. Occupancy of the external binding site reduces the affinity for binding to the cytoplasmic pore site. These sites are likely not deep within the pore as the voltage-dependence of block for both thioridazine and erythromycin is very shallow (Figure 5), suggesting a location outside the membrane electric field (Duncan et al., 2006; Milnes et al., 2006). It is interesting to note, that the antagonism of thioridazine and terfenadine block of hERG by erythromycin has a very different concentration-dependence from that of direct block of the channel. For instance, the erythromycin block of hERG is very weak at concentrations 3 μM or less (10% or less) and saturates at concentrations above 100 μM (Figure 1). In contrast, the antagonism of thioridazine block of hERG was obvious at 0.3 μM erythromycin and was maximal at 3–30 μM (Figure 2). Further experiments are necessary to define these relationships and possible mechanisms underlying them.
The ability of erythromycin to influence drug binding affinity did not extend to clotrimazole (see Figure 7). Pretreatment with the same concentrations of erythromycin that maximally reduced thioridazine blockade of hERG current had no effect on clotrimazole–induced hERG block. One possible explanation for this difference is that clotrimazole has a binding site that is not allosterically coupled to the erythromycin binding site. A more likely possibility is that clotrimazole and erythromycin compete for the same external binding site and the concentration of erythromycin (3 μM) is simply too low to have any meaningful effect. This contention is supported in the present study by pH experiments and the voltage-dependence of clotrimazole block, which suggest an external binding site for clotrimazole, like observed for erythromycin. Furthermore, the results of the clotrimazole-erythromycin combination were very different from those of the thioridazine-erythromycin combination. For the combination of thioridazine or terfenadine and erythromycin (3 μM), the CI value calculated (14.4 for thioridazine and 22.1 for terfenadine) suggests a strong antagonism for the binding of drugs, which apparently have binding sites on different sides of the hERG channel. In contrast, the CI value calculated for the erythromycin-clotrimazole combination (0.8) suggests an additive or slightly synergistic effect.
Conclusion
The results of the present study suggest distinct, but allosterically linked, binding sites for erythromycin and the pore channel blockers, thioridazine and terfenadine, on opposite sides of the hERG channel. Occupancy of the external erythromycin binding site by submaximal blocking concentrations dramatically reduced the affinity for binding to the pore site. Interestingly, the reduction in affinity induced by occupancy of the external binding site was greater for terfenadine than for thioridazine. This may imply that the reduction in affinity for the pore binding site is at least partially dependent upon the configuration of the pore-blocking molecule. One could envision conformational changes in the pore binding site induced by binding of erythromycin at an external site, which result in different pore binding affinities dependent upon the three-dimensional configuration of the blocking molecule. This hypothesis requires further study to understand the structure-activity relationship of this effect.
As block of the hERG channel is the primary mechanism for QT prolongation by thioridazine and terfenadine, these results suggest that co-administration of erythromycin with pore-blocking drugs such as thioridazine or terfenadine may actually impart some reduction in their cardiac liability. Further in vivo experiments are needed to test this hypothesis.
Acknowledgments
The author would like to thank Sean Ekins, Ph.D. for his helpful comments.
Glossary
- hERG
human ether-a-go-go-related gene product
Conflict of interest
None.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Crumb W, Cavero I. Patch-clamping human cardiac ion channels for evaluating cardiac electrophysiological effects of drugs. Curr Protoc Pharmacol. 2002;10.8:1–17. doi: 10.1002/0471141755.ph1008s20. [DOI] [PubMed] [Google Scholar]
- Crumb W, Ekins S, Sarazan R, Wikel J, Wrighton S, Carlson C, et al. Effects of antipsychotic drugs on I(to), I(Na), I(sus), I(K1), and hERG. QT prolongation, structure activity relationship, and network analysis. Pharm Res. 2006;23:1133–1143. doi: 10.1007/s11095-006-0070-7. [DOI] [PubMed] [Google Scholar]
- Curtis L, Ostbye T, Sendersky V, Hutchison S, LaPointe N, Khatib S, et al. Prescription of QT prolonging drugs in a cohort of about 5 million outpatients. Am J Med. 2003;114:135–141. doi: 10.1016/s0002-9343(02)01455-9. [DOI] [PubMed] [Google Scholar]
- Duncan R, Ridley J, Dempsey C, Leishman D, Leaney J, Hancox J, et al. Erythromycin block of the HERG K + channel: accessibility to F656 and Y652. Biochem Biophys Res Commun. 2006;341:500–506. doi: 10.1016/j.bbrc.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Ekins S, Crumb W, Sarazan R, Wikel J, Wrigton S. Three dimensional quantitative structure activity relationship for the inhibition of the hERG (human ether-a-go-go related gene) potassium channel. J Pharmacol Exp Ther. 2002;301:427–434. doi: 10.1124/jpet.301.2.427. [DOI] [PubMed] [Google Scholar]
- Friemel A, Zunkler B. Interactions at human ether-a-go-go-related gene channels. Toxicol Sci. 2010;114:346–355. doi: 10.1093/toxsci/kfq011. [DOI] [PubMed] [Google Scholar]
- Hreiche R, Plante I, Drolet B, Morissette P, Turgeon J. Lengthening of cardiac repolarization in isolated guinea pigs hearts by sequential or concomitant administration of two IKr blockers. J Pharm Sci. 2011;100:2469–2481. doi: 10.1002/jps.22437. [DOI] [PubMed] [Google Scholar]
- Kamiya K, Niwa R, Morishima M, Honjo H, Sanguinetti MC. Molecular determinants of hERG channel block by terfenadine and cisapride. J Pharmacol Sci. 2008;108:301–307. doi: 10.1254/jphs.08102fp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe N. Frequency of high-risk use of QT-prolonging medications. Pharmacoepidemiol Drug Saf. 2006;15:361–368. doi: 10.1002/pds.1155. [DOI] [PubMed] [Google Scholar]
- Milnes J, Witchel H, Leaney J, Leishman D, Hancox J. hERG K+ channel blockade by the antipsychotic drug thioridazine: an obligatory role for the S6 helix residue F656. Biochem Biophys Res Commun. 2006;351:273–280. doi: 10.1016/j.bbrc.2006.10.039. [DOI] [PubMed] [Google Scholar]
- Mitcheson J. Drug binding to HERG channels: evidence for a ‘non-aromatic’ binding site for fluvoxamine. Br J Pharmacol. 2003;139:883–884. doi: 10.1038/sj.bjp.0705336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanat S, Carlton C, Crumb W, Agrawal K, Clarkson C. Characterization of the inhibitory effects of erythromycin and clarithromycin on the HERG potassium channel. Mol Cell Biochem. 2003;254:1–7. doi: 10.1023/a:1027309703313. [DOI] [PubMed] [Google Scholar]
- Valentin J, Pollard C, Lainée P, Hammond T. Value of non-clinical cardiac repolarization assays in supporting the discovery and development of safer medicines. Br J Pharmacol. 2010;159:25–33. doi: 10.1111/j.1476-5381.2009.00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Cao Z, Yin S, Dai C, Wu Y, Li W. Interaction simulation of hERG K+ channel with its specific BeKm-1 peptide: insights into the selectivity of molecular recognition. J Proteome Res. 2007;6:611–620. doi: 10.1021/pr060368g. [DOI] [PubMed] [Google Scholar]
- Zhang M, Korolkova Y, Liu J, Jiang M, Grishin E, Tseng G. BeKm-1 is a HERG-specific toxin that shares the structure with ChTx but the mechanism of action with ErgTx1. Biophys J. 2003;84:3022–3036. doi: 10.1016/S0006-3495(03)70028-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhou Z, Gong Q, Makielski J, January C. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res. 1999;84:989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]