

Abstract
Background and Purpose
In contrast to T-cell priming in the periphery, therapeutic strategies targeting the initiation step of T-cell trafficking into the CNS have not been extensively investigated. In this study, we examined the effect of NSC-87877, a potent Src homology 2-containing protein tyrosine phosphatase 2 (SHP-2) inhibitor, on experimental autoimmune encephalomyelitis (EAE) and elucidated its unique mechanism of action.
Experimental Approach
C57BL/6 mice were immunized with myelin oligodendrocyte glycoprotein35–55 and monitored for clinical severity of disease and histopathological features in the CNS. Levels of cytokines in serum were measured by elisa. Effects of NSC-87877 on expressions of chemokines and cytokines in the CNS were determined by quantitative PCR.
Key Results
NSC-87877-treated mice developed conventional TH1 and TH17 responses, but were highly resistant to the induction of EAE. NSC-87877 decreased the accumulation of lymphocytes in the CNS and increased the functional expression of chemokine receptor CXCR7 on CD8+ T-cells. Adoptive transfer of T-cells from 2D2-transgenic mice restored EAE susceptibility in NSC-87877-treated mice, indicating that NSC-87877 only targets the initial migration of pioneer T-cells. Furthermore, T-cell-conditioned SHP-2-deficient mice treated with NSC-87877 were no longer resistant to EAE, suggesting that inhibition of SHP-2 contributes to the amelioration of EAE by NSC-87877.
Conclusions and Implications
NSC-87877 almost completely abolished the development of EAE by blocking the initial infiltration of pioneer CD8+ T-cells into the uninflamed CNS. These results reveal a critical role for SHP-2 in regulating EAE pathogenesis and indicate that NSC-87877 is a potential candidate for the treatment of relapsing-remitting multiple sclerosis.
Keywords: SHP-2, pioneer CD8+ T-cells, CXCR7, EAE, multiple sclerosis
Introduction
Multiple sclerosis (MS) is an inflammatory disease of the CNS that affects approximately 400 000 young and middle-aged adults in the USA and 2.5 million individuals worldwide (Noseworthy et al., 2000; Pelletier and Hafler, 2012). Although its prevalence is only approximately 1 case per 1000 individuals in Europe and the USA, MS places a heavy burden on both societies and individuals, as it severely impairs a persons physical and social functions in the most productive years of their life (Kobelt et al., 2000; Petermann and Korn, 2011; Pelletier and Hafler, 2012). MS is classified into four subtypes: relapsing-remitting MS, secondary progressive MS, primary progressive MS and progressive-relapsing MS. In more than 80% of patients the disease is initially manifest as a relapsing-remitting condition with unpredictable clinical exacerbations of neurological symptoms (Miller and Rhoades, 2012; Miller, 2012). The goal of MS therapy is to decrease the annual relapse rate and to prevent the progression of disease and accumulation of physical disabilities (Fox, 2010; Fox and Rhoades, 2012). Current disease-modifying treatments for relapsing-remitting MS, such as IFN-β, glatiramer acetate and natalizumab, have shown marginal efficacy or have significant safety concerns (De Jager and Hafler, 2007; Singer et al., 2011; Chen et al., 2012; Fox and Rhoades, 2012; Saidha et al., 2012). Thus, novel therapeutic strategies or new drugs are urgently required for the treatment of relapsing-remitting MS.
In MS and its corresponding animal model, experimental autoimmune encephalomyelitis (EAE) (Hemmer et al., 2006), aberrant autoreactive T-cells combined with a dysfunctional immune system play an important role (Hafler, 2004; Qin et al., 2010). Multiple lines of evidence indicate that CD4+ T helper cells (particularly TH1 and TH17 cells) are involved in the pathogenesis of EAE and MS at different phases/stages and sites of disease (Bettelli et al., 2004; Tzartos et al., 2008). Although traditional studies have suggested the almost exclusive role of CD4+ T-cells in MS, several lines of evidence indicate that CD8+ T-cells are also potentially important in this disease (Steinman, 2001; Neumann et al., 2002; Lassmann and Ransohoff, 2004; Johnson et al., 2010; Mars et al., 2011; Saxena et al., 2011). CD8+ T-cells are activated in the periphery and then migrate into the CNS, where they reactivate, undergo clonal expansion and cause targeted cell death (Kivisakk et al., 2004; Lehner and Cresswell, 2004; Zang et al., 2004; Friese and Fugger, 2005). Other studies have suggested that as compared with CD4+ T-cells, CD8+ T-cells from patients with relapsing-remitting MS in the acute disease phase display increased adhesiveness and ability to migrate across the blood–brain barrier (BBB) to be recruited into the inflamed brain (Battistini et al., 2003; Johnson et al., 2010). The development of the disease not only requires the initial priming of naive autoreactive T-cells in the periphery, but also critically depends on the ability of antigen-specific T-cells to access the CNS, particularly the uninflamed CNS, to initiate tissue inflammation (Ransohoff, 2009; Reboldi et al., 2009; Jain et al., 2010). A ‘two-wave hypothesis’ has been proposed to define the effector stage of EAE (Zepp et al., 2011). In the first wave, antigen-specific ‘pioneer’ T-cells cross the BBB and the ensuing restimulation and activation allows other leukocytes to infiltrate and accumulate. Many molecular determinants have been found to control the migration of lymphocytes to the inflamed CNS. For example, VLA-4 (very late antigen-4) has a key role in controlling the migration of lymphocytes across the BBB by interacting with the adhesion molecule VCAM (vascular cell adhesion molecule) (Yednock et al., 1992; Reboldi et al., 2009). However, the key factors that control the first step of migration have not been identified (Carrithers et al., 2000).
It is also well established that chemokine receptors (see Alexander et al., 2013), which are expressed on different subsets of T-cells, provide specificity for the entry of lymphocytes into the CNS (Bromley et al., 2008; Reboldi et al., 2009). Previous research indicated that the CXC chemokine receptor 7 (CXCR7), which is an alternative receptor for CXC chemokine ligand 12 (CXCL12), is critical for mediating CXCL12 internalization at endothelial barriers of the CNS (Cruz-Orengo et al., 2011b). Additionally, other studies suggest that ligand binding to the decoy receptor CXCR7 does not initiate typical activation of G protein signalling pathways, but does activate MAPK signalling through the recruitment of β-arrestin (Zabel et al., 2009; Rajagopal et al., 2010). Previous findings also indicate that CXCR7 functions primarily to sequester CXCL12 and thus regulate CXCR4 signalling (Boldajipour et al., 2008). All these findings suggest an important role for chemokines especially CXCL12 and its decoy receptor CXCR7 in the initation of cell migration into the uninflamed CNS.
The two Src homology 2 domain-containing protein tyrosine phosphatases (SHP-1 and SHP-2) are important for the regulation of many different signal transduction pathways (Irandoust et al., 2009). Generally speaking, SHP-1 is involved in the negative regulation of cell development and activation (Deng et al., 2002), while SHP-2 has dual functions in various types of disease (Chan and Feng, 2007; Bard-Chapeau et al., 2011). It is reported that loss of SHP-1 enhances T-cell responses and exacerbates EAE (Deng et al., 2002). Moreover, mice expressing a mutant form of SHPS-1, a docking protein to recruit and activate SHP-1, are resistant to EAE (Tomizawa et al., 2007). However, the potential role of SHP-2 in EAE has not been investigated before now.
Recently, the molecular basis of the first steps in CNS autoimmunity has been extensively studied with the hope of identifying a new therapeutic approach for MS treatment. In the present study, we evaluated the role of SHP-2 in the initial steps of EAE using a potent SHP-2 inhibitor NSC-87877. The unprecedented therapeutic efficacy of NSC-87877 in EAE made it possible to identify the key cell types and critical factors responsible for the initial infiltration of effector cells into the uninflamed CNS. This study provides compelling evidence that CXCR7 expressed on pioneer CD8+ T-cells is the key factor in the treatment of autoimmune disease and has important implications for the development of a therapy for relapsing-remitting MS.
Methods
Mice
Specific pathogen-free (SPF), 6–8-week-old female C57BL/6 and myelin oligodendrocyte glycoprotein (MOG)-specific 2D2 transgenic mice (C57BL/6 background) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). Shp2-floxed mouse was a kind gift from Dr Gensheng Feng at Department of Pathology, School of Medicine, University of California, San Diego, and Dr Yuehai Ke at School of Medicine, Zhejiang University. All the mice were kept under SPF conditions throughout the experiments. The animals were housed, five per cage with food and water ad libitum, on a 12 h light/dark cycle with lights on at 06:00 h and controlled (22-23°C) temperature. Animal welfare and experimental procedures were carried out strictly in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, USA) and the related ethical regulations of our university. All efforts were made to minimize animals’ suffering and to reduce the number of animals used. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Generation of T-cell conditional SHP-2 knockout (cSHP-2 KO) mice
cSHP-2 KO mice (C57BL/6 background) were generated by crossing shp-2flox/flox mice with CD4-Cre transgenic mice (Wu et al., 2012).
Reagents
The SHP-2 inhibitor NSC-87877 and PHPS1 (phenyl hydrazono pyrazolone sulfonate 1) were purchased from Calbiochem (La Jolla, CA, USA). Mouse CD4 and CD8 MicroBeads were purchased from Miltenyi (Bergisch Gladbach, Germany). Anti-CD4-APC was purchased from BD Pharmingen (San Diego, CA, USA). Anti-CD8-FITC and anti-B220-FITC were purchased from eBioscience (San Diego, CA, USA). CXCL12 (SDF-1α) was purchased from Millipore (Billerica, MA, USA). Incomplete Freund's adjuvant, Pertussis toxin and sodium fluorescein were purchased from Sigma-Aldrich (St Louis, MO, USA). MOG residues 35–55 (MOG35–55) was purchased from Chinapeptide (Shanghai, China), both displayed a purity >95%. Heat-killed H37Ra strain of Mycobacterium tuberculosis was purchased from Difco Laboratories (Detroit, MI, USA). All other chemicals were purchased from Sigma-Aldrich.
EAE induction and drug administration
For standard EAE, female C57BL/6 mice were immunized s.c. with 200 μg MOG35–55 emulsified in incomplete Freund's adjuvant containing 6 mg mL−1 Mycobacterium tuberculosis. Pertussis toxin (400 ng per mouse) in PBS was administered i.v. on the day of immunization and 48 h later. Mice were examined daily and scored for disease severity using the following scale: 0 = no symptoms; 1 = tail weakness; 2 = paraparesis (incomplete paralysis of one or two hind limbs); 3 = paraplegia (complete paralysis of two hind limbs); 4 = paraplegia with forelimb weakness or paralysis; and 5 = moribund or dead. For treatment, NSC-87877 or vehicle (PBS) was administered i.p. at 2.5 mg kg−1 daily according to different therapy protocols. For adoptive transfer EAE, CD8+ T-cells from EAE mice on day 10 post immunization were cultured in the presence of MOG35–55 for 3 days. The resulting cell blasts (5 × 107 cells per mouse) were adoptively transferred into irradiated (400 rad) C57BL per six recipients. The recipient mice also received Pertussis toxin (400 ng per mouse) i.v. on the day of immunization and 48 h later. In some experiments, naive T-cells (1 × 106) from 2D2-transgenic mice were transferred i.v. into mice 24 h before immunization.
Histopathology
Mice were killed by cervical dislocation. Spinal cords for histological analysis were removed from mice and immediately fixed in 4% paraformaldehyde. Paraffin-embedded 5 μm sections of spinal cord were stained with H&E or Luxol fast blue and then examined by light microscopy.
Mononuclear cell (MNC) purification
For purification of infiltrating MNCs from the spinal cord, mice were anaesthetized with 50 mg kg−1 pentobarbital sodium i.p. and perfused through the left cardiac ventricle with 30 mL PBS. Spinal cords were separately pressed through a 40 μm cell strainer to obtain a single-cell suspension. MNCs were purified by discontinuous gradient centrifugation in 75%/40% Percoll (GE Healthcare, Piscataway, NJ, USA).
T-cell proliferation and cytokine measurement
Lymph node-derived T-cells (3 × 105 per well) isolated from EAE mice were restimulated with MOG35–55 for up to 72 h. Cell proliferation was assayed by incorporation of [methyl-3H]-thymidine (ICN Pharmaceuticals, Costa Mesa, CA, USA) at 0.5 μCi per well during the last 8 h of incubation, and the uptake was measured as counts min−1. Culture supernatants of MOG-reactive T-cells were collected 48 h later and evaluated for IFN-γ, IL-17, IL-6, TNF-α, IL-4, IL-2 or IL-10 production using the Cytometric Bead Array (CBA) cytokine assay kit (BD Biosciences) according to the manufacturer's instructions.
Flow cytometry
For surface-marker staining, cells were incubated with fluorochrome conjugated Abs to CD4, CD8, B220 at recommended dilution or isotype control Abs for 30 min on ice. Flow cytometric analysis was performed on BD FACSCalibur (BD Biosciences) and results were analysed using FlowJo software (Tree Star, Ashland, OR, USA).
Determination of BBB permeability
BBB permeability was assessed by measuring tissue content of sodium fluorescein, as described previously (Esaki et al., 2010). In brief, mice were injected i.v. with 200 μL 5% sodium fluorescein in PBS. Thirty minutes later mice were anaesthetized and perfused with a minimum of 50 mL of heparin (1000 U L−1) in PBS. The spinal cord was then removed and homogenized, and the homogenate was mixed with an equal volume of 80% trichloroacetic acid before centrifugation at 10 000 × g for 10 min. The supernatant was diluted with 0.8 volume of 5 M NaOH and then measured for fluorescence at excitation and emission wavelengths of 485 and 538 nm respectively. Sodium fluorescein standard solutions (1–1000 ng mL−1) were used to calculate the tissue content, which was normalized to the total amount of protein in the homogenate.
Quantitative real-time PCR
Total RNA was extracted from cells using Trizol Reagent (Invitrogen, Carlsbad, CA, USA). One microgram of RNA was reversely transcribed to cDNA. The mRNA expression was determined by real-time PCR using iQ SYBR Green Supermix (Bio-Rad, Richmond, CA, USA). Mouse β-actin gene was used as endogenous control for sample normalization. Results are presented as fold increases relative to the expression of mouse β-actin. Sequences of PCR primer pairs are shown in Supporting Information Table S1.
Mouse genome microarray analysis
CD8+ T-cells were obtained from lymph nodes of EAE mice treated with PBS or NSC-87877 on day 10 post immunization. All the subsequent technical procedures, including RNA isolation, quality control, concentration measurement of RNA, cDNA synthesis, labelling of cRNA, hybridization and scanning of the arrays, were done at Gene Company Limited (Shanghai, China). Affymetrix Mouse Genome 430 2.0 Array (a total six chips were used in this study; three chips for each group) was used to probe the global gene expression profile in mice following NSC-87877 treatment. Briefly, double-stranded cDNA was synthesized from total RNA, with a T7 RNA polymerase promoter site added to its 3′ end. Biotinylated cRNAs were generated from cDNAs in vitro and amplified using the BioArray T7 RNA polymerase labelling kit (Affymetrix, Santa Clara, CA, USA). After purification of cRNAs, 20 μg of cRNA was fragmented at 94°C for 35 min. Approximately 12.5 μg of fragmented cRNA was used in a 250 μL hybridization mixture containing herring-sperm DNA, plus bacterial and phage cRNA controls to serve as internal controls for hybridization efficiency. Aliquots (200 μL) of the mixture were hybridized to arrays for 18 h at 45°C in a GeneChip Hybridization Oven 640 (Affymetrix). Each array was washed and stained with streptavidin-phycoerythrin and amplified with biotinylated anti-streptavidin antibody on the GeneChip Fluidics Station 450 (Affymetrix). Arrays were scanned with the GeneArray G7 scanner to obtain image and signal intensities. Upon comparison of the two groups, lists of genes that were either induced or suppressed > twofold between the NSC-87877-and vehicle-treated group are presented.
Migration assay
Three μm Transwells® (Corning, Acton, MA, USA) were used to determine chemokine-specific migration patterns of T-cells. CD8+ T-cells were harvested as previously described. Recombinant chemokine ligand CXCL12 (100 ng mL−1, 1% FBS DMEM) was placed in the bottom well. CD8+ T-cells (3 × 105) were put into the upper chambers precoated with fibronectin. After incubation at 37°C for 4 h, cells that migrated from the top well through the filter were counted using standard microscopy.
Western blot
Proteins were extracted in lysis buffer (30 mM Tris, pH 7.5, 150 mM NaCl, 1 mM PMSF, 1 mM sodium orthovanadate, 1% Nonidet P-40, 10% glycerol, and phosphatase and protease inhibitors). The proteins were then separated by SDS-PAGE and electrophoretically transferred onto PVDF membranes. The membranes were probed with antibodies overnight at 4°C, and then incubated with a HRP-coupled secondary antibody. Detection was performed using a LumiGLO chemiluminescent substrate system (Cell Signaling Technology).
Statistics
Data are expressed as means ± SEM. Statistical analyses were performed using one-way anova followed by Student's two-tailed t-test. P < 0.05 was considered significant.
Results
SHP-2 inhibitor almost completely abolishes the incidence of EAE in mice
To determine the role of SHP-2 in controlling inflammation during the pathogenesis of EAE, a SHP-2 competitive inhibitor, NSC-87877, was utilized in this study. NSC-87877 (2.5 mg kg−1) was i.p. administered to mice daily from the day of immunization onward. After being immunized with a s.c. injection of MOG residues 35–55 (MOG35–55), the mice developed a monophasic disease characterized by ascending paralysis from day 10 after immunization. The pretreatment with NSC-87877 resulted in a substantial delay in the onset of the disease and a significant reduction in EAE severity, accompanied by markedly reduced inflammation and demyelination in the affected spinal cord as compared with the vehicle-treated mice (Figure 1A,B). Notably, most of the NSC-87877-treated mice were completely resistant to the development of EAE, and only a few (one of eight) developed minimal disease symptoms. However, administration of NSC-87877 starting from day 13 did not show such efficacy (Figure 1C). This huge difference between these two regimens indicates that a targeted blockade of SHP-2 mainly affects the trafficking rather than the effectiveness of leukocytes involved in the EAE. In addition, we performed experiments in which NSC-87877 was administered to mice from day 8. As a result, we found that administration of NSC-87877 starting from day 8 also showed some efficacy (Supporting Information Figure S1). These results suggest that the major effect of NSC-87877 is on the migratory properties of leukocytes, but not on their activation.
Figure 1.

SHP-2 inhibition significantly ameliorates the clinical signs and disease progression of EAE. Active EAE was induced in C57BL/6 mice by flank s.c. immunization with 200 μg of peptide MOG35–55. (A) SHP-2 inhibitor NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from day 0 post immunization. Values are shown as the mean ± SEM of eight mice per group. (B) Spinal cord sections harvested from vehicle-or NSC-87877-treated EAE mice on day 25 post immunization were analysed for degree of inflammation by H&E and for demyelination by Ponceau 2R-fast green (original magnification 200×). (C) NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from day 13 post immunization. Values are shown as the mean ± SEM of six mice per group. (D) Active EAE was induced in C57BL/6 mice. SHP-2 inhibitor PHPS1 (1 mg kg−1) or vehicle was administered i.p. daily from the day of immunization. Values are shown as the mean ± SEM of five mice per group.
To determine whether the preventative effect of NSC-87877 against EAE was associated with SHP-2, another specific SHP-2 inhibitor, PHPS1, was administered to MOG-induced EAE mice. As a result, PHPS1-treated mice (1 mg kg−1 day−1) displayed the same reduction in disease severity as NSC-87877-treated mice did (Figure 1D).
SHP-2 inhibition leads to decreased inflammation in the CNS without altering peripheral T-cell responses during EAE
To investigate the mechanism by which NSC-87877 controls EAE development and progression, we examined the antigen-specific responses of peripheral T-cells isolated from EAE mice. As shown in Supporting Information Figure S2A and B, vehicle-and NSC-87877-treated mice (starting from day 0) exhibited comparable levels of cytokines in both serum and supernatant, including TH1 cytokines IFN-γ and TNF-α, TH2 cytokines IL-4 and IL-6, regulatory cytokine IL-10, and TH17 cytokine IL-17. Similar results were obtained with draining lymph node T-cells in response to MOG35–55 peptide in vitro (Supporting Information Figure S2C). These results suggest that NSC-87877 does not affect the production of TH1/TH17/TH2/Treg cytokines in vivo and in vitro. In addition, no significant difference was observed in MOG35–55-specific T-cell proliferation in vitro and in vivo between the vehicle-and NSC-87877-treated groups (Supporting Information Figure S3A,B). These data strongly suggest that the effect of in vivo NSC-87877 administration on EAE does not involve the modulation of peripheral antigen-specific immune responses. Next, we assessed the ability of NSC-87877-treated T-cells to polarize into effector T-cells in vitro. We stimulated naive cells under the TH1-, TH17-or Treg-polarizing conditions. As expected, NSC-87877 did not inhibit the production of either IFN-γ in TH1-polarized T-cells, IL-17 in TH17-polarized T-cells, or IL-10 in Treg-polarized T-cells (Supporting Information Figure S3C).
Next, we wondered whether NSC-87877-induced decrease in CNS inflammation was related to lymphocyte infiltration and chemokine expression during EAE. To investigate this, spleen and spinal cords were collected from EAE mice at a point when clinical symptoms peaked in the vehicle control group. We found that, on day 16, cell numbers in the spleen were greatly decreased in EAE mice but were up-regulated following NSC-87877 treatment (Figure 2A). In addition, the proportions of CD4+ and CD8+ T lymphocytes in infiltrated cells from the brains and spinal cords of NSC-87877-treated mice were much lower than those of vehicle-treated mice (Figure 2B) on day 16 after MOG immunization. Also, real-time RT-PCR analysis of mRNA isolated from the spinal cord showed that the expressions of CCL2, CCL3, CCL5, CXCL10, IFN-γ, IL-17, IL-1β, GM-CSF and IL-6 in the CNS were almost completely eliminated in the NSC-87877-treated group compared to the control group (Figure 2C).
Figure 2.

SHP-2 inhibitor NSC-87877 modulates the expression of associated factors in the spinal cord. (A and B) EAE was induced in C57BL/6 mice and NSC-87877 was administered to mice as in Figure 1A. (A) Numbers of spleen cells from vehicle-and NSC-87877-treated mice after immunization are shown. (B) Relative proportions of spinal cord infiltrating CD4+ and CD8+ T lymphocytes from vehicle-and NSC-87877-treated mice on day 16 post immunization are displayed. (C) The expressions of inflammatory factors in the spinal cord from vehicle-and NSC-87877-treated mice on day 16 post immunization were measured via quantitative real-time RT-PCR. (D) RT-PCR analysis of ICAM-1, VCAM-1, LFA-1 and VLA-4 mRNA expression in the spinal cord from vehicle-and NSC-87877-treated mice on day 16 post immunization. (E) NSC-87877 was administered to mice as in Figure 1A, sodium fluorescein was injected i.v. on day 16 post immunization, and the spinal cord was dissected 30 min later and evaluated for fluorescein incorporation. Data are expressed as mg sodium fluorescein mg−1 of spinal cord protein. (F) The mRNA levels of occludin, ZO-1, claudin 3, and claudin 5 in the spinal cord from vehicle-and NSC-87877-treated mice on day 16 post immunization were measured by RT-PCR. The housekeeping gene β-actin mRNA was used to normalize the relative amounts of mRNA. Values are given as the mean ± SEM. *P < 0.05; **P < 0.01 versus EAE.
Furthermore, the expressions of tight-junction proteins and cell adhesion molecules were evaluated in the lumbar spinal cord from vehicle-and NSC-87877-treated EAE mice. The expressions of the adhesion molecules ICAM-1 (intercellular adhesion molecule 1) and VCAM-1 were up-regulated following EAE immunization. Interestingly, NSC-87877 treatment selectively down-regulated ICAM-1 mRNA level in the CNS and LFA-1 (lymphocyte function-associated antigen 1) mRNA level in the lymph node (Figure 2D).
A pivotal step for triggering CNS inflammation is the disruption of the BBB. Therefore, we examined the permeability of BBB in NSC-87877-treated EAE mice on day 16 after immunization. Mice were injected i.v. with sodium fluorescein followed by quantitative analysis of fluorescein incorporation in the spinal cord 30 min later. The increased uptake of fluorescein into the CNS as a result of EAE was blocked almost completely by daily administration of NSC-87877 (Figure 2E). The expressions of tight junction-associated factors including occludin, ZO-1 (tight junction protein 1) and claudin 3 were also recovered from the down-regulation in the EAE (Figure 2F), suggesting that NSC-87877 may contribute to the preservation of BBB integrity, and thereby impede the entry of inflammatory factors and cells into the CNS.
Blocking the initial migration of pioneer T-cells into the CNS is responsible for the preventative effect of NSC-87877 in EAE
To examine whether NSC-87877 affects the initial migration of pioneer T-cells into the CNS, we adoptively transferred naive 2D2 T-cells into recipient C57BL/6 mice followed by immunization with MOG35–55 24 h later. T-cells from 2D2-transgenic mice were MOG-specific T-cells. After immunizing mice with MOG35–55 in complete Freund's adjuvant, these MOG-reactive T-cells differentiated into effector cells and migrated into the uninflamed CNS quickly. As shown in Figure 3, adoptive transfer of T-cells from 2D2-transgenic mice almost completely abolished the preventative effect of NSC-87877 and restored EAE susceptibility in NSC-87877-treated mice. Furthermore, to confirm the possibility that NSC-87877 affects the migratory activity of lymphocyte, we treated the 2D2 donors with NSC-87877 for 10 days before the transfer of their cells. We found that the adoptive transfer of T-cells from NSC-87877-pretreated 2D2 mice significantly restored the inhibitory effect of NSC-87877 in EAE mice (Supporting Information Figure S4).
Figure 3.
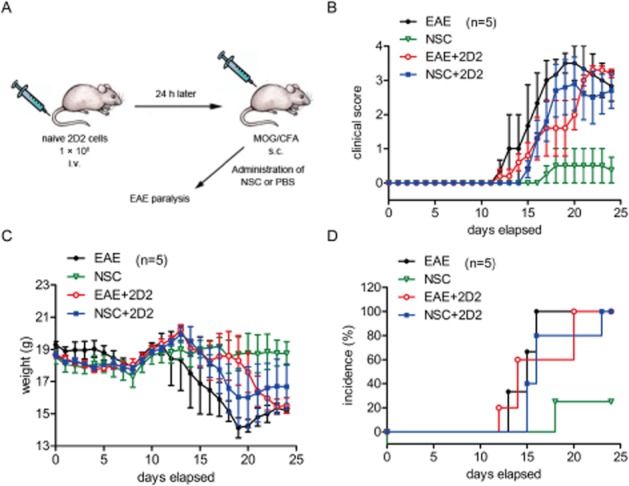
Transfer of 2D2 T-cells reconstitutes EAE susceptibility in NSC-87877-treated mice. (A) C57BL/6 mice were adoptively transferred with naive 2D2 T-cells and then immunized with MOG33–55 24 h later for EAE induction. NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from the day of immunization. Mice were observed daily for EAE severity (B), weight change (C) and incidence (D). Values are given as the mean ± SEM of five mice per group.
Primed CD8+ T-cells are critical for the main inhibitory effect of NSC-87877 on the induction of EAE
The next experiments were performed to clarify the phenotypic characteristics of these lymph node cells in the circulation, which were blocked out by the BBB. Our results showed that lymph node cells from both vehicle-treated and NSC-87877-treated mice contained CD4+ T-cells, CD8+ T-cells and B-cells. The number of CD4+ and CD8+ T-cells but not B-cells greatly decreased on day 10 (before onset of disease), suggesting that T-cells may exit the lymph nodes to play a role as pro-inflammatory cells. Treatment with NSC-87877 significantly inhibited this decrease in CD8+, but not CD4+ T-cells, on day 16 (peak of disease) (Figure 4A), indicating that primed CD8+ T-cells are blocked in the periphery by NSC-87877. Then we examined the functional status of CD8+ T-cells from mice treated with NSC-87877. On day 10 after immunization, CD8+ T-cells were isolated from vehicle-and NSC-87877-treated mice and we found that there was no differences in the ability of these two groups of CD8+ T-cells to proliferate (Supporting Information Figure S5A), cytokine production (Supporting Information Figure S5B) of respond to MOG35–55 stimulation. In addition, comparable expression levels of cytotoxicity-associated genes including perforin1, FasL and granzyme B were observed in CD8+ T-cells isolated from the vehicle-and NSC-87877-treated groups (Supporting Information Figure S5C). These results suggest that the effects of NSC-87877 on EAE are executed through inhibition of CD8+ T-cell migration into the CNS.
Figure 4.

Blocking the infiltration of CD8+ T-cells into the CNS is responsible for the amelioration of EAE by NSC-87877. (A) Cells harvested from draining lymph nodes of vehicle-or NSC-87877-treated mice on day 10 (before onset of disease) or day 16 (peak of disease) post immunization were stained for CD4, CD8 and B220 followed by flow cytometric analysis. Cell numbers were calculated and displayed as percentages. **P < 0.01. (B, C) Ag-specific donor CD8+ T-cells were obtained from draining lymph nodes of vehicle-or NSC-87877-treated mice on day 10 post immunization. Then the cells were cultured in the presence of MOG35–55 for 72 h. Cultured cells were then adoptively transferred into normal C57BL/6 recipients (5 × 107 per mouse), and the EAE severity (D), weight change (E) and incidence (F) were observed daily. Values are given as the mean ± SEM of six mice per group.
The relevance of the effects of NSC-87877 on CD8+ T-cells for EAE development in mice was further confirmed by adoptive transfer of MOG-reactive CD8+ T-cells from EAE mice into normal recipient C57BL/6 mice. Recipients of CD8+ T-cells from NSC-87877-treated mice showed almost no incidence of EAE (Figure 4D–F).
NSC-87877 up-regulates chemokine receptor CXCR7 expression by priming CD8+ T-cells
Next we examined what happened in CD8+ T-cells after the treatment of NSC-87877 in EAE mice. We compared the mRNA expression of CD8+ T-cells isolated from vehicle-and NSC-87877-treated mice on day 10 after immunization using the Mouse Genome 430 2.0 Array (Figure 5A). Both the quantitative real-time PCR results of the microarray (Figure 5B) and the immunofluorescence detection of CXCR7 and CD8+ T-cells in the spinal cords of mice with EAE (Figure 5C) showed that NSC-87877 significantly increased the expression of chemokine receptor CXCR7, but not the various other factors, as compared with vehicle control. Previous studies showed that CXCR7 (an alternative receptor for CXCL12) functions primarily to sequestrate CXCL12 and thus negatively regulates CXCR4 signalling (Boldajipour et al., 2008; Zabel et al., 2009; Rajagopal et al., 2010). As shown in Figure 5D, NSC-87877 significantly inhibited the ability of T-cells to migrate towards CXCL12, demonstrating an increase in the functional surface expression of CXCR7 in CD8+ T-cells from NSC-87877-treated mice. Moreover, CXCL12 increased JNK phosphorylation, but not NF-κB p65 phosphorylation in CD8+ T-cells from immunized mice and NSC-87877 markedly reduced this phosphorylation of JNK induced by CXCL12 (Figure 5E).
Figure 5.

Increased expression of CXCR7 accounts for the block of the infiltration of CD8+ T-cells into the CNS by SHP-2 inhibitor NSC-87877 in EAE. (A) CD8+ T-cells were isolated from vehicle-and NSC-87877-treated mice on day 10 post immunization. The mRNA expressions were detected by Mouse Genome 430 2.0 Array. (B) The results of the microarray assay were verified by quantitative real-time RT-PCR analysis. The samples used for validation were isolated from CD8+ T-cells of vehicle-and NSC-87877-treated mice on day 10 after immunization. (C) Confocal imaging detection of CXCR7 (red) and CD8 (green) within the spinal cords of EAE mice treated with vehicle (top) versus NSC-87877 (bottom). Nuclei were counterstained with DAPI (blue). (D) CD8+ T-cells derived from EAE mice were isolated by magnetic bead cell sorting and analysed for cell surface chemokine receptor expression using an in vitro migration assay. (E) p-JNK and p-p65 were assessed by immunoblotting from total cell lysates. The results represent one of similar results from three independent experiments. Densitometric analysis was performed on JNK phosphorylation in three blots and presented as the ratio of phospho over total. Values are given as the mean ± SEM of three different experiments. **P < 0.01, NS, no significance.
SHP-2 is required for the amelioration of EAE by NSC-87877
The effect of NSC-87877 on the development of EAE as mentioned earlier can be rationally linked to the inhibition of SHP-2. To further validate this link, we constructed T-cell cSHP-2 KO mice for further investigation. However, C57BL/6 mice lacking SHP-2 in CD4+ and CD8+ T-cells (Supporting Information Figure S6A) developed the same severity of clinical signs as the wild-type mice did (Figure 6A). Namely, the incidence and maximal disease scores of EAE during the acute phase (between days 10 and 20 post immunization) did not differ between cSHP-2 KO mice and wild-type mice. It should be noted that when we followed the progression of EAE for over 50 days, we observed a relapsing-remitting course starting from day 30 in wild-type mice, while mice with SHP-2-deficient T-cells developed only an acute phase without relapses (Supporting Information Figure S6B,C), The discrepancy between cSHP-2 KO mice and NSC-87877-treated mice may be attributed to multiple targets of NSC-87877 on various cell types other than T-cells, as SHP-2 is a ubiquitous phosphatase involved in a variety of cells/signal processes. However, we also found that NSC-87877-treated cSHP-2 KO mice were no longer completely, but only partially resistant to the development of EAE (Figure 6A–C). The NSC-87877-induced decrease in T-cell migration, increase in CXCR7 expression on CD8+ T-cells as well as down-regulation of JNK phosphorylation in response to CXCL12 were also completely reversed in cSHP-2 KO mice, respectively (Figure 6D–F). Collectively, these observations suggest that SHP-2-regulated entry of CD8+ T-cells into the CNS is required for the initiation of EAE.
Figure 6.

SHP-2 is required for the amelioration of EAE by NSC-87877. cSHP-2 KO mice were subjected to MOG-induced EAE. NSC-87877 (2.5 mg kg−1) or vehicle was administered i.p. daily from the day of immunization. The EAE severity (A), weight change (B) and incidence (C) were observed daily. Values are given as the mean ± SEM of five mice per group. (D) CD8+ T-cells derived from EAE groups of knockout mice were isolated by magnetic bead cell sorting and analysed for cell surface chemokine receptor expression using an in vitro migration assay. (E) Confocal imaging detection of CXCR7 (red) and CD8 (green) within the spinal cords. Nuclei were counterstained with DAPI (blue). (F) p-JNK was assessed by immunoblotting from total cell lysates. The results represent one of similar results from three independent experiments. Densitometric analysis was performed on JNK phosphorylation in three blots and presented as the ratio of phospho over total. Values are given as the mean ± SEM of three different experiments. **P < 0.01, NS, no significance.
Taken together, our results suggest a novel mode for the pathogenesis of EAE in that SHP-2 regulates the initial triggering step of EAE. In brief, the inhibition of SHP-2 triggers an increased expression of CXCR7 on CD8+ T-cells. CXCR7 sequestrates CXCL12 and thus negatively regulates CXCR4 signalling, which contributes to the amelioration of EAE (Figure 7).
Figure 7.
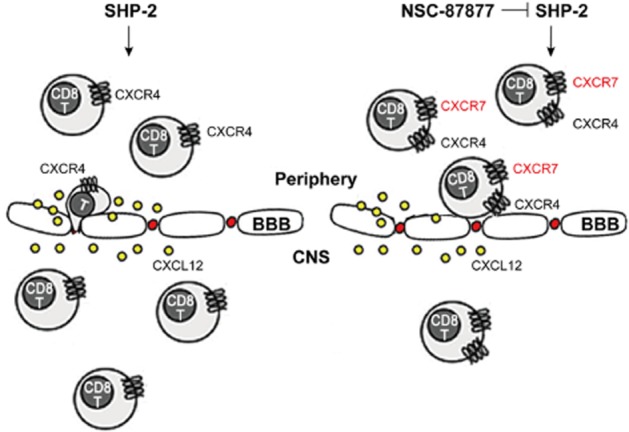
A novel mode for SHP-2-mediated regulation of the initial triggering step of EAE pathogenesis. NSC-87877, a potent SHP-2 inhibitor, increased the expression of CXCR7 on CD8+ T-cells. CXCR7 sequestrated CXCL12 and thus negatively regulated CXCR4 signalling, which contributes to the decreased CNS accumulation of lymphocytes and amelioration of EAE.
Discussion and conclusions
In contrast to an effect on T-cell priming in the periphery, therapeutic strategies targeting the initiation step of T-cell trafficking across the BBB have not been extensively investigated (Ransohoff, 2009; Reboldi et al., 2009; Jain et al., 2010). In the present study, we addressed how NSC-87877, a potent SHP-2 inhibitor, regulates the inflammatory process leading to EAE pathogenesis and demonstrated, for the first time, that it is very effective at reducing the occurrence of EAE (Figure 1). Importantly, we found that NSC-87877 almost completely prevented EAE development by blocking the accumulation of inflammatory cells in the CNS. Collectively our findings suggest that CXCR7 expressed on CD8+ T-cells is essentially upregulated in the presence of SHP-2 inhibition and negatively regulates the function of these cells, which are critical for the initiation of EAE.
To explain the potent efficacy of NSC-87877 against EAE, we first examined the effect of NSC-87877 on the production of IFN-γ and/or IL-17, which are involved in the pathogenesis of EAE (Bettelli et al., 2004; Hemmer et al., 2006; Komiyama et al., 2006; Steinman, 2007). However, when mice were immunized with MOG35–55 peptide plus Pertussis toxin, NSC-87877-treated mice failed to develop EAE symptoms, but still developed conventional TH1 and TH17 responses. In particular, the response to MOG35–55 was similarly elevated in the periphery T-cells from NSC-87877-and vehicle-treated mice. Noteably all measured parameters on the TH1 and TH17 responses in the periphery were comparable between NSC-87877-and vehicle-treated mice as well as between NSC-87877-and vehicle-treated T-cells isolated from EAE mice either on day 10 (before onset of disease, data not shown) or day 16 (peak of disease, Supporting Information Figures S1 and S2). It should be noted that the efficacy of NSC-87877 against EAE varied enormously because of the different administration regimens. The treatment of NSC-87877 starting from day 0 post immunization almost completely blocked the incidence of EAE while that starting from approximately day 13 did not (Figure 1C). These findings suggest that NSC-87877 treatment does not alter the encephalitogenic response of periphery T-cells but affects their ability to migrate into the CNS and cause inflammation. In other words, NSC-87877 may attenuate EAE development primarily by retarding the initial infiltration of inflammatory cells into the uninflamed CNS. In fact, the relative proportions of infiltrating CD4+ and CD8+ T lymphocytes in the lumbar spinal cord on day 16 post immunization were greatly decreased in the NSC-87877-treated mice (Figure 2A).
However, chemokines including CCL2, CCL3 and CXCL10, and integrins such as LFA-1 and VLA-4 (Yednock et al., 1992; Baron et al., 1993; Reboldi et al., 2009) have been reported to be important for T-cell entry and subsequent disease pathogenesis and we found that NSC-87877 almost completely decrease the expressions of various inflammatory factors including CCL2, CCL3, CCL5, CXCL10, TNF-α, IFN-γ, IL-17, IL-1β and IL-6 in the spinal cord (Figure 2C). In addition, NSC-87877 treated mice had markedly decreased mRNA levels of ICAM-1 in CNS and LFA-1 in lymph nodes at the peak of disease (Figure 2D). These results suggest that an inhibitory effect on the above chemokines, cytokines and adhesion molecules may contribute to the dramatic effect of NSC-87877 on EAE. Hence, NSC-87877 could abolish EAE progression by inhibiting the migration of effector T-cells into the CNS, rather than affecting their priming in lymph nodes or their entry into the circulation.
Previously published descriptions of the two-wave hypothesis and pioneer lymphocytes have emphasized the importance of migration in the initial phases of EAE (Reboldi et al., 2009). Consistent with the two-wave model, we found here that the transfer of naive 2D2 T-cells into NSC-87877-treated mice was sufficient to reconstitute the EAE susceptibility (Figure 3). It is reported that the transcription factor RUNX1 is a positive regulator of T-lymphocyte differentiation and the phosphorylation of RUNX1 can negatively regulate RUNX1 activity. At the same time, the tyrosine phosphatase SHP-2 binds directly to RUNX1 and contributes to RUNX1 tyrosine dephosphorylation (Huang et al., 2012). Hence, treatment with NSC-87877, an inhibitor of SHP-2, could increase the phosphorylation of RUNX1, which could impair T-cell differentiation. Moreover, SHP-2 has been implicated in the regulation of CD4 and CD8 expression in thymocytes (Nguyen et al., 2006). However, the results shown in Figure 4A indicate that the expressions of CD4 and CD8 were not changed by administration of NSC-87877 for 10 days. This result, together with our findings that no significant difference was observed in MOG-specific T-cell proliferation and cytokine production between vehicle-and NSC-87877-treated group (Supporting Information Figures S2 and S3), suggesta that NSC-87877 mainly targets the initial migration of pioneer T-cells into the CNS during the initial phases of EAE.
The next question we investigated was how NSC-87877 affects the migration step of T-cells into the CNS? We further examined the phenotypic characteristics of the pioneer lymphocytes. Draining lymph nodes collected from NSC-87877-treated EAE mice on day 16 post immunization showed a significantly decreased percentage of CD8+ T-cells compared with vehicle-treated mice (Figure 4A). Published research has described ‘pioneer’ lymphocytes as specific activated T-cells that migrate into non-inflamed CNS in the initial phases of EAE. These pioneer cells could cross through choroid plexus in a P-selectin-dependent way (TH1) or in a CCR6-dependent way (TH17) (Carrithers et al., 2000; Reboldi et al., 2009). However, these interactions seem to play a partial role in the initial penetration of pioneer T-cells. Also there has been no identification of a specific cluster of T-cells (TH1 vs. TH17 cells, or CD4+ vs. CD8+ T-cells) that has an advantage in entering the CNS (Hickey, 2000). In the present study, our results suggest that CD8+ T-cells are likely to be involved in the pioneer population and NSC-87877 blocks these CD8+ T-cells from infiltrating into the uninflamed CNS.
Additionally, the results of the microarray assay performed with CD8+ T-cells on day 10 post immunization indicated that NSC-87877 treatment resulted in an evident increase in the expression of the chemokine receptor CXCR7, which led to a decrease in T-cell migration in response to chemokine CXCL12 (also known as SDF-1α). It has been shown that CXCL12 prevents the migration of infiltrating cells from the perivascular space into parenchymal (McCandless et al., 2006), thereby limiting inflammation. Furthermore, CXCL12 was reported to redirect the polarization of effector Th1 cells into IL-10-producing T-cells, thereby suppressing ongoing EAE (Meiron et al., 2008). However, a truncated form of CXCL12 that antagonized CXCR4 reduced CD4+ T-cells accumulation in the CNS and inhibited EAE in mice (Kohler et al., 2008). These different published results suggest that CXCL12 is not always protective in EAE and its localization is of importance for regulating neuroinflammation. In our experiments, NSC-87877 treatment resulted in an increase in the expression of CXCR7, which led to a decrease in T-cell migration in response to CXCL12. The findings obtained here also suggest that in contrast to CXCR7 expression in the CNS tissue (Cruz-Orengo et al., 2011a,b2011b), CXCR7 expressed on CD8+ T-cells primarily functioned as a decoy receptor that sequestrated CXCL12 to down-regulate CXCR4 signalling. Adoptive transfer of those CD8+ T-cells from NSC-87877-treated mice yielded significantly less severe EAE (Figure 4D–F), which further confirmed our hypothesis.
Although the critical factor targeted by NSC-87877, which is responsible for its effect on CXCR7, still remains unknown, we provide compelling evidence that supports the key role of SHP-2 in mediating the efficacy of NSC-87877 in EAE. SHP-1 has been shown to be a negative regulator of EAE pathogenesis (Deng et al., 2002). Reduced SHP-1 activity leads to a stronger T-cell response and exacerbates the clinical signs of EAE (Tamir et al., 2000; Deng et al., 2002; Wasserman et al., 2008). Moreover, direct administration of PHPS1, a specific inhibitor of SHP-2, markedly ameliorated EAE (Figure 1D). These findings shed light on the critical role of SHP-2 in an autoimmune disease setting, which is different from that of SHP-1. Our results clearly showed that NSC-87877 induced a SHP-2 loss-of-function effect in EAE as cSHP-2 KO mice treated with NSC-87877 developed severe EAE as compared with wild-type mice. Thus, our findings indicate that NSC-87877 may act through SHP-2 to activate CXCR7, thereby modulating the ability of pioneer CD8+ T lymphocytes to accumulate in the uninflamed CNS.
To our knowledge, the data presented herein are the first to demonstrate that SHP-2 regulates chemokine receptor CXCR7 expression on CD8+ T-cells. The finding that NSC-87877 blocked the entry of T-cells into the CNS during the early phase of EAE indicates the potential involvement of the molecular determinant SHP-2 in the initial triggering step of EAE pathogenesis. Given that relapsing-remitting MS may be consistent with distinct waves of migration, the discoveries described herein support the use of NSC-87877 as a potential therapeutic agent for human relapsing-remitting MS. Although therapeutic approaches utilizing small molecules like NSC-87877 are rare among current MS therapies, the present study provides a promising strategy/direction for the development of more effective disease-modifying treatments for human relapsing-remitting MS.
Acknowledgments
We greatly thank Dr Gensheng Feng at University of California, San Diego, and Dr Yuehai Ke at Zhejiang University who kindly presented us the Shp2-floxed mice as a gift for generating Shp2-floxed/CD4-Cre mice. We also thank Dr Wenjie Guo for his helpful advice; Cheng Qian for his technical assistance; and Feifei Tao for her language proofreading.
Glossary
- BBB
blood–brain barrier
- cSHP-2 KO
conditional SHP-2 knockout mice
- CXCL
CXC chemokine ligand
- CXCR
CXC chemokine receptor
- EAE
experimental autoimmune encephalomyelitis
- MOG
myelin oligodendrocyte glycoprotein; MS, multiple sclerosis
- NSC-87877
8-hydroxy-7-[(6-sulfo-2-naphthyl)azo]-5-quinolinesulfonic acid
- SHP-2
Src homology 2-containing protein tyrosine phosphatase 2
- TH
T helper
Conflicts of interest
The authors have no financial conflicts of interest.
Funding: This work was supported by the Science Fund for Creative Research Groups of NSFC (No. 81121062), the National Natural Science Foundation of China (Nos. 90913023, 81173070, 91229109, 81273528, 81330079) and the National Science & Technology Major Project (No. 2012ZX09304-001).
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12565
Figure S1 SHP-2 inhibitor NSC-87877 ameliorates the clinical signs and disease progression of EAE. Active EAE was induced in C57BL/6 mice by flank s.c. immunization with 200 μg of peptide MOG35–55. NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from day 8 post immunization. Values are shown as the mean ± SEM of five mice per group.
Figure S2 SHP-2 inhibitor NSC-87877 does not affect production of TH1/TH17/TH2/Treg cytokines in vivo and in vitro. Active EAE was induced in C57BL/6 mice by flank s.c. immunization with 200 μg of peptide MOG35–55. SHP-2 inhibitor NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from day 0 post immunization. (A) Comparable cytokines release in serum from vehicle- and NSC-87877- treated EAE mice. Cytokines in serum from EAE mice were measured by CBA cytokine assay on day 16 after immunization. (B) Th1 and Th17 cells in lymphocytes derived from vehicle- and NSC-87877-treated EAE mice at day 10. (C) Lymphocytes derived from vehicle- and NSC-87877-treated EAE mice at day 10 after immunization were restimulated with 10 μg mL−1 MOG35–55 peptide for 48 h. Cytokine production was determined by CBA cytokine assay. (D) Lymphocytes were isolated from untreated EAE mice and examined for MOG3–55-reactive cytokine production in the presence or absence of 10 μM NSC-87877. Supernatants were analysed for the indicated cytokines. Data are means ± SEM of three independent experiments.
Figure S3 SHP-2 inhibitor NSC-87877 treatment does not affect MOG35–55-specific T-cell proliferation as well as TH1, TH17 and Treg cell differentiation. (A) Lymphocytes derived from vehicle- and NSC-87877-treated EAE mice at day 10 after immunization were restimulated with MOG35–55 peptide (10 μg mL−1) for 72 h. Proliferation was measured by [3H]- thymidine incorporation. (B) Lymphocytes were isolated from untreated EAE mice and examined for proliferation stimulated with MOG35–55 in the presence or absence of 10 μM NSC-87877. (C) Naive CD4+ T-cells were purified by MACS (magnetic-activated cell sorting) and then were differentiated into TH1 (10 ng mL−1 IL-12, 1 μg mL−1 anti-IL-4), TH17 (20 ng mL−1 IL-6, 1 ng mL−1 TGF-β, 10 μg mL−1 anti-IFN-γ, 1 μg mL−1 anti-IL-4) or Treg (1 ng mL−1 TGF-β, 50 U mL−1 IL-2) cells in the presence of 10 μM NSC-87877 in vitro. The levels of cytokines were analysed by elisa. Data are means ± SEM of three independent experiments.
Figure S4 Transfer of 2D2 T-cells reconstitutes EAE susceptibility in NSC-87877-treated mice. 2D2 mice were treated with NSC-87877 for 10 days. C57BL/6 mice were adoptively transferred with these pretreated 2D2 T-cells and then immunized with MOG33–55 24 h later for EAE induction. NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from the day of immunization. Mice were observed daily for EAE severity. Values are given as the mean ± SEM.
Figure S5 Functional status of CD8+ T-cells from mice treated with NSC-87877. On day 10 after immunization, CD8+ T-cells were isolated from vehicle- and NSC-87877- treated mice. (A) CD8+ T-cells were restimulated with MOG35–55 peptide (10 μg mL−1) for 72 h. Proliferation was measured by [3H]-thymidine incorporation. (B) CD8+ T-cells were restimulated with 10 μg mL-1 MOG35–55 peptide for 48 h. Cytokine production was determined by elisa. (C) The expressions of genes in CD8+ T-cells were measured via quantitative real-time RT-PCR.
Figure S6 Attenuated relapses of EAE in in cSHP-2 KO mice. (A) CD4+ and CD8+ T-cells were isolated from the conditional SHP-2-deficient mice. Cells were lysed for Western blot analysis of SHP-2. (B,C) Active EAE was induced in wild-type and cSHP-2 KO mice by flank s.c. immunization with 200 μg of peptide MOG35–55. Mice were observed daily for EAE severity (B) and weight change (C). Data are means ± SEM of 10 mice in each group.
Table S1 Sequences of PCR primer pairs used in the present study.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard-Chapeau EA, Li S, Ding J, Zhang SS, Zhu HH, Princen F, et al. Ptpn11/Shp2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell. 2011;19:629–639. doi: 10.1016/j.ccr.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron JL, Madri JA, Ruddle NH, Hashim G, Janeway CA., Jr Surface expression of alpha 4 integrin by CD4 T cells is required for their entry into brain parenchyma. J Exp Med. 1993;177:57–68. doi: 10.1084/jem.177.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistini L, Piccio L, Rossi B, Bach S, Galgani S, Gasperini C, et al. CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: a critical role for P-selectin glycoprotein ligand-1. Blood. 2003;101:4775–4782. doi: 10.1182/blood-2002-10-3309. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200:79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldajipour B, Mahabaleshwar H, Kardash E, Reichman-Fried M, Blaser H, Minina S, et al. Control of chemokine-guided cell migration by ligand sequestration. Cell. 2008;132:463–473. doi: 10.1016/j.cell.2007.12.034. [DOI] [PubMed] [Google Scholar]
- Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol. 2008;9:970–980. doi: 10.1038/ni.f.213. [DOI] [PubMed] [Google Scholar]
- Carrithers MD, Visintin I, Kang SJ, Janeway CA., Jr Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain. 2000;123(Pt 6):1092–1101. doi: 10.1093/brain/123.6.1092. [DOI] [PubMed] [Google Scholar]
- Chan RJ, Feng GS. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood. 2007;109:862–867. doi: 10.1182/blood-2006-07-028829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SJ, Wang YL, Fan HC, Lo WT, Wang CC, Sytwu HK. Current status of the immunomodulation and immunomediated therapeutic strategies for multiple sclerosis. Clin Dev Immunol. 2012;2012:970789. doi: 10.1155/2012/970789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Orengo L, Chen YJ, Kim JH, Dorsey D, Song SK, Klein RS. CXCR7 antagonism prevents axonal injury during experimental autoimmune encephalomyelitis as revealed by in vivo axial diffusivity. J Neuroinflammation. 2011a;8:170. doi: 10.1186/1742-2094-8-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Orengo L, Holman DW, Dorsey D, Zhou L, Zhang P, Wright M, et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med. 2011b;208:327–339. doi: 10.1084/jem.20102010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Hafler DA. New therapeutic approaches for multiple sclerosis. Annu Rev Med. 2007;58:417–432. doi: 10.1146/annurev.med.58.071105.111552. [DOI] [PubMed] [Google Scholar]
- Deng C, Minguela A, Hussain RZ, Lovett-Racke AE, Radu C, Ward ES, et al. Expression of the tyrosine phosphatase SRC homology 2 domain-containing protein tyrosine phosphatase 1 determines T cell activation threshold and severity of experimental autoimmune encephalomyelitis. J Immunol. 2002;168:4511–4518. doi: 10.4049/jimmunol.168.9.4511. [DOI] [PubMed] [Google Scholar]
- Esaki Y, Li Y, Sakata D, Yao C, Segi-Nishida E, Matsuoka T, et al. Dual roles of PGE2-EP4 signaling in mouse experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:12233–12238. doi: 10.1073/pnas.0915112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox EJ. Alemtuzumab in the treatment of relapsing-remitting multiple sclerosis. Expert Rev Neurother. 2010;10:1789–1797. doi: 10.1586/ern.10.135. [DOI] [PubMed] [Google Scholar]
- Fox EJ, Rhoades RW. New treatments and treatment goals for patients with relapsing-remitting multiple sclerosis. Curr Opin Neurol. 2012;25(Suppl):S11–S19. doi: 10.1097/01.wco.0000413320.94715.e9. [DOI] [PubMed] [Google Scholar]
- Friese MA, Fugger L. Autoreactive CD8+ T cells in multiple sclerosis: a new target for therapy? Brain. 2005;128:1747–1763. doi: 10.1093/brain/awh578. [DOI] [PubMed] [Google Scholar]
- Hafler DA. Multiple sclerosis. J Clin Invest. 2004;113:788–794. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmer B, Nessler S, Zhou D, Kieseier B, Hartung HP. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat Clin Pract Neurol. 2006;2:201–211. doi: 10.1038/ncpneuro0154. [DOI] [PubMed] [Google Scholar]
- Hickey WF. P selectin, pioneer cells and the path to inflammation. Brain. 2000;123(Pt 6):1073–1074. doi: 10.1093/brain/123.6.1073. [DOI] [PubMed] [Google Scholar]
- Huang H, Woo AJ, Waldon Z, Schindler Y, Moran TB, Zhu HH, et al. A Src family kinase-Shp2 axis controls RUNX1 activity in megakaryocyte and T-lymphocyte differentiation. Genes Dev. 2012;26:1587–1601. doi: 10.1101/gad.192054.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irandoust M, van den Berg TK, Kaspers GJ, Cloos J. Role of tyrosine phosphatase inhibitors in cancer treatment with emphasis on SH2 domain-containing tyrosine phosphatases (SHPs) Anticancer Agents Med Chem. 2009;9:212–220. doi: 10.2174/187152009787313864. [DOI] [PubMed] [Google Scholar]
- Jain P, Coisne C, Enzmann G, Rottapel R, Engelhardt B. Alpha4beta1 integrin mediates the recruitment of immature dendritic cells across the blood–brain barrier during experimental autoimmune encephalomyelitis. J Immunol. 2010;184:7196–7206. doi: 10.4049/jimmunol.0901404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TA, Jirik FR, Fournier S. Exploring the roles of CD8(+) T lymphocytes in the pathogenesis of autoimmune demyelination. Semin Immunopathol. 2010;32:197–209. doi: 10.1007/s00281-010-0199-7. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivisakk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B, et al. Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol. 2004;55:627–638. doi: 10.1002/ana.20049. [DOI] [PubMed] [Google Scholar]
- Kobelt G, Jonsson L, Henriksson F, Fredrikson S, Jonsson B. Cost–utility analysis of interferon beta-1b in secondary progressive multiple sclerosis. Int J Technol Assess Health Care. 2000;16:768–780. doi: 10.1017/s0266462300102041. [DOI] [PubMed] [Google Scholar]
- Kohler RE, Comerford I, Townley S, Haylock-Jacobs S, Clark-Lewis I, McColl SR. Antagonism of the chemokine receptors CXCR3 and CXCR4 reduces the pathology of experimental autoimmune encephalomyelitis. Brain Pathol. 2008;18:504–516. doi: 10.1111/j.1750-3639.2008.00154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Ransohoff RM. The CD4-Th1 model for multiple sclerosis: a critical [correction of crucial] re-appraisal. Trends Immunol. 2004;25:132–137. doi: 10.1016/j.it.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Lehner PJ, Cresswell P. Recent developments in MHC-class-I-mediated antigen presentation. Curr Opin Immunol. 2004;16:82–89. doi: 10.1016/j.coi.2003.11.012. [DOI] [PubMed] [Google Scholar]
- McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS. limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol. 2006;177:8053–8064. doi: 10.4049/jimmunol.177.11.8053. CXCL12. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mars LT, Saikali P, Liblau RS, Arbour N. Contribution of CD8 T lymphocytes to the immuno-pathogenesis of multiple sclerosis and its animal models. Biochim Biophys Acta. 2011;1812:151–161. doi: 10.1016/j.bbadis.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N. CXCL12 (SDF-1α) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J Exp Med. 2008;205:2643–2655. doi: 10.1084/jem.20080730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AE, Rhoades RW. Treatment of relapsing-remitting multiple sclerosis: current approaches and unmet needs. Curr Opin Neurol. 2012;25(Suppl):S4–S10. doi: 10.1097/01.wco.0000413319.87092.19. [DOI] [PubMed] [Google Scholar]
- Miller E. Multiple sclerosis. Adv Exp Med Biol. 2012;724:222–238. doi: 10.1007/978-1-4614-0653-2_17. [DOI] [PubMed] [Google Scholar]
- Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25:313–319. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- Nguyen TV, Ke Y, Zhang EE, Feng GS. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol. 2006;177:5990–5996. doi: 10.4049/jimmunol.177.9.5990. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Medical progress: multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. N Engl J Med. 2012;366:339–347. doi: 10.1056/NEJMct1101691. [DOI] [PubMed] [Google Scholar]
- Petermann F, Korn T. Cytokines and effector T cell subsets causing autoimmune CNS disease. FEBS Lett. 2011;585:3747–3757. doi: 10.1016/j.febslet.2011.03.064. [DOI] [PubMed] [Google Scholar]
- Qin X, Guo BT, Wan B, Fang L, Lu L, Wu L, et al. Regulation of Th1 and Th17 cell differentiation and amelioration of experimental autoimmune encephalomyelitis by natural product compound berberine. J Immunol. 2010;185:1855–1863. doi: 10.4049/jimmunol.0903853. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Kim J, Ahn S, Craig S, Lam CM, Gerard NP, et al. Beta-arrestin-but not G protein-mediated signaling by the ‘decoy’ receptor CXCR7. Proc Natl Acad Sci U S A. 2010;107:628–632. doi: 10.1073/pnas.0912852107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM. Immunology: in the beginning. Nature. 2009;462:41–42. doi: 10.1038/462041a. [DOI] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- Saidha S, Eckstein C, Calabresi PA. New and emerging disease modifying therapies for multiple sclerosis. Ann N Y Acad Sci. 2012;1247:117–137. doi: 10.1111/j.1749-6632.2011.06272.x. [DOI] [PubMed] [Google Scholar]
- Saxena A, Martin-Blondel G, Mars LT, Liblau RS. Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett. 2011;585:3758–3763. doi: 10.1016/j.febslet.2011.08.047. [DOI] [PubMed] [Google Scholar]
- Singer B, Ross AP, Tobias K. Oral fingolimod for the treatment of patients with relapsing forms of multiple sclerosis. Int J Clin Pract. 2011;65:887–895. doi: 10.1111/j.1742-1241.2011.02721.x. [DOI] [PubMed] [Google Scholar]
- Steinman L. Myelin-specific CD8 T cells in the pathogenesis of experimental allergic encephalitis and multiple sclerosis. J Exp Med. 2001;194:F27–F30. doi: 10.1084/jem.194.5.f27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- Tamir I, Dal Porto JM, Cambier JC. Cytoplasmic protein tyrosine phosphatases SHP-1 and SHP-2: regulators of B cell signal transduction. Curr Opin Immunol. 2000;12:307–315. doi: 10.1016/s0952-7915(00)00092-3. [DOI] [PubMed] [Google Scholar]
- Tomizawa T, Kaneko Y, Saito Y, Ohnishi H, Okajo J, Okuzawa C, et al. Resistance to experimental autoimmune encephalomyelitis and impaired T cell priming by dendritic cells in Src homology 2 domain-containing protein tyrosine phosphatase substrate-1 mutant mice. J Immunol. 2007;179:869–877. doi: 10.4049/jimmunol.179.2.869. [DOI] [PubMed] [Google Scholar]
- Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. 2008;172:146–155. doi: 10.2353/ajpath.2008.070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman HA, Beal CD, Zhang Y, Jiang N, Zhu C, Evavold BD. MHC variant peptide-mediated anergy of encephalitogenic T cells requires SHP-1. J Immunol. 2008;181:6843–6849. doi: 10.4049/jimmunol.181.10.6843. [DOI] [PubMed] [Google Scholar]
- Wu X, Guo W, Wu L, Gu Y, Gu L, Xu S, et al. Selective sequestration of STAT1 in the cytoplasm via phosphorylated SHP-2 ameliorates murine experimental colitis. J Immunol. 2012;189:3497–3507. doi: 10.4049/jimmunol.1201006. [DOI] [PubMed] [Google Scholar]
- Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- Zabel BA, Wang Y, Lewen S, Berahovich RD, Penfold ME, Zhang P, et al. Elucidation of CXCR7-mediated signaling events and inhibition of CXCR4-mediated tumor cell transendothelial migration by CXCR7 ligands. J Immunol. 2009;183:3204–3211. doi: 10.4049/jimmunol.0900269. [DOI] [PubMed] [Google Scholar]
- Zang YC, Li S, Rivera VM, Hong J, Robinson RR, Breitbach WT, et al. Increased CD8+ cytotoxic T cell responses to myelin basic protein in multiple sclerosis. J Immunol. 2004;172:5120–5127. doi: 10.4049/jimmunol.172.8.5120. [DOI] [PubMed] [Google Scholar]
- Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232–239. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 SHP-2 inhibitor NSC-87877 ameliorates the clinical signs and disease progression of EAE. Active EAE was induced in C57BL/6 mice by flank s.c. immunization with 200 μg of peptide MOG35–55. NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from day 8 post immunization. Values are shown as the mean ± SEM of five mice per group.
Figure S2 SHP-2 inhibitor NSC-87877 does not affect production of TH1/TH17/TH2/Treg cytokines in vivo and in vitro. Active EAE was induced in C57BL/6 mice by flank s.c. immunization with 200 μg of peptide MOG35–55. SHP-2 inhibitor NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from day 0 post immunization. (A) Comparable cytokines release in serum from vehicle- and NSC-87877- treated EAE mice. Cytokines in serum from EAE mice were measured by CBA cytokine assay on day 16 after immunization. (B) Th1 and Th17 cells in lymphocytes derived from vehicle- and NSC-87877-treated EAE mice at day 10. (C) Lymphocytes derived from vehicle- and NSC-87877-treated EAE mice at day 10 after immunization were restimulated with 10 μg mL−1 MOG35–55 peptide for 48 h. Cytokine production was determined by CBA cytokine assay. (D) Lymphocytes were isolated from untreated EAE mice and examined for MOG3–55-reactive cytokine production in the presence or absence of 10 μM NSC-87877. Supernatants were analysed for the indicated cytokines. Data are means ± SEM of three independent experiments.
Figure S3 SHP-2 inhibitor NSC-87877 treatment does not affect MOG35–55-specific T-cell proliferation as well as TH1, TH17 and Treg cell differentiation. (A) Lymphocytes derived from vehicle- and NSC-87877-treated EAE mice at day 10 after immunization were restimulated with MOG35–55 peptide (10 μg mL−1) for 72 h. Proliferation was measured by [3H]- thymidine incorporation. (B) Lymphocytes were isolated from untreated EAE mice and examined for proliferation stimulated with MOG35–55 in the presence or absence of 10 μM NSC-87877. (C) Naive CD4+ T-cells were purified by MACS (magnetic-activated cell sorting) and then were differentiated into TH1 (10 ng mL−1 IL-12, 1 μg mL−1 anti-IL-4), TH17 (20 ng mL−1 IL-6, 1 ng mL−1 TGF-β, 10 μg mL−1 anti-IFN-γ, 1 μg mL−1 anti-IL-4) or Treg (1 ng mL−1 TGF-β, 50 U mL−1 IL-2) cells in the presence of 10 μM NSC-87877 in vitro. The levels of cytokines were analysed by elisa. Data are means ± SEM of three independent experiments.
Figure S4 Transfer of 2D2 T-cells reconstitutes EAE susceptibility in NSC-87877-treated mice. 2D2 mice were treated with NSC-87877 for 10 days. C57BL/6 mice were adoptively transferred with these pretreated 2D2 T-cells and then immunized with MOG33–55 24 h later for EAE induction. NSC-87877 (2.5 mg kg−1) or vehicle control was administered i.p. daily from the day of immunization. Mice were observed daily for EAE severity. Values are given as the mean ± SEM.
Figure S5 Functional status of CD8+ T-cells from mice treated with NSC-87877. On day 10 after immunization, CD8+ T-cells were isolated from vehicle- and NSC-87877- treated mice. (A) CD8+ T-cells were restimulated with MOG35–55 peptide (10 μg mL−1) for 72 h. Proliferation was measured by [3H]-thymidine incorporation. (B) CD8+ T-cells were restimulated with 10 μg mL-1 MOG35–55 peptide for 48 h. Cytokine production was determined by elisa. (C) The expressions of genes in CD8+ T-cells were measured via quantitative real-time RT-PCR.
Figure S6 Attenuated relapses of EAE in in cSHP-2 KO mice. (A) CD4+ and CD8+ T-cells were isolated from the conditional SHP-2-deficient mice. Cells were lysed for Western blot analysis of SHP-2. (B,C) Active EAE was induced in wild-type and cSHP-2 KO mice by flank s.c. immunization with 200 μg of peptide MOG35–55. Mice were observed daily for EAE severity (B) and weight change (C). Data are means ± SEM of 10 mice in each group.
Table S1 Sequences of PCR primer pairs used in the present study.