

Abstract
Background and Purpose
The prevalence of smoking in schizophrenia patients is exceptionally high; it is not known why but many researchers suggest that smoking constitutes a form of self-medication. Among the symptoms of schizophrenia that may be improved by nicotine are cognitive deficits. Hence, we studied the effects of long-term nicotine administration on cognition in a genetic animal model of schizophrenia susceptibility, G72-transgenic (G72Tg) mice.
Experimental Approach
The effect of long-term nicotine or saline, administered by osmotic minipumps, on different cognitive domains was assessed in G72Tg mice and controls using a battery of behavioural tests. To investigate the mechanism underlying phenotypic differences, quantitative autoradiographic mapping of nACh receptor subtypes was performed in forebrain structures to explore effects of chronic nicotine exposure on nACh receptor density in wild-type (WT) and G72Tg mice.
Key Results
Genotype significantly affected the cognitive effects of chronic nicotine administration. Whereas chronic nicotine disrupted cognitive performance in WT mice, it was effective at restoring impaired prepulse inhibition, working memory and social recognition in G72Tg mice. However, long-term spatial learning was further impaired by nicotine in transgenic animals. In contrast, associative learning was protected by G72-expression against the adverse nicotine effects seen in WT animals. G72-expression did not decisively influence nicotine-induced up-regulation of the α4β2*subtype, whereas α7nACh receptor density was differentially altered by genotype or by a genotype·treatment interaction in specific brain areas, most notably hippocampal subregions.
Conclusions and Implications
Our data support the hypothesis that nicotine self-medication of schizophrenics improves cognitive symptoms, possibly by facilitating nicotine-induced α7nACh receptor activation in the hippocampus.
Keywords: short-term memory, nicotinic acetylcholine receptor, chronic nicotine, schizophrenia, cognition, G72 mouse model, self-medication, receptor autoradiography, susceptibility gene, G72/G30 gene locus
Introduction
Cognitive impairments represent a core symptom of schizophrenia and are a major cause of disability in schizophrenia patients. While hallucinations and delusions can be effectively treated with antipsychotic medication, no drug is currently available to improve the cognitive deficits. Schizophrenia-associated cognitive impairments affect several domains involving sensory information processing (Adler et al., 1998), attention, reasoning and certain other forms of non-affective learning and memory (Nuechterlein et al., 2008). The latter includes deficits in various dimensions of social cognition (Derntl and Habel, 2011), associative learning (Brambilla et al., 2011) and working memory (Gold et al., 2006; Nuechterlein et al., 2008). In particular, working memory dysfunctions are persistent and independent of psychotic symptoms (Timofeeva and Levin, 2011).
Schizophrenia is highly comorbid with tobacco smoking; heavy smoking and high nicotine dependence are more frequent in schizophrenic patients compared with the general population (De Leon and Diaz, 2005; Ziedonis et al., 2008). Alongside the established and well-known adverse effects of tobacco smoking, nicotine is described as improving cognitive symptoms in schizophrenia patients, including sensory motor gating (Adler et al., 1998), attention (Dépatie et al., 2002; Harris et al., 2004), spatial (Smith et al., 2001) and recognition memory (Myers et al., 2004). Hence, the hypothesis has been advanced that schizophrenic patients might smoke in order to self-medicate distressing cognitive symptoms (Kumari and Postma, 2005).
A large number of genome-wide association studies have identified SCZD7 on chromosome 13q32-q33 as a candidate region linked to the development of schizophrenia (see Detera-Wadleigh and McMahon, 2006, Drews et al., 2013 for review). SCZD7 comprises two genes, G72 and G30, which are transcribed from overlapping opposite DNA strands (Chumakov et al., 2002) and G72 transcripts encode a protein termed LG72 that appeared late during primate evolution and is only present in anthropoid primates. Moreover, the human protein of 153 amino acids is much larger than that of other primates. No protein product was detected for LG30 (Chumakov et al., 2002), and the transcripts are only weakly expressed in brain and testis. Schizophrenia has been associated with an elevated expression of G72, as increased G72 transcript levels have been shown in the forebrain structures of schizophrenic patients post-mortem (Korostishevsky et al., 2004). We, and others (Cheng et al., 2014), have recently generated a transgenic mouse model that carries a bacterial artificial chromosome of the human G72/G30 locus (G72Tg). These mice express all known splice variants of the G72 locus (Otte et al., 2009). In situ hybridization showed overall low levels of G72 expression throughout the brain but appreciable expression in granular cells of the cerebellum, cortex, olfactory bulb and dentate gyrus. Western blot analysis of two-dimensional gels from G72Tg mice also showed a LG72 protein in brain extracts. Strikingly, the G72Tg mice generated by us displayed several schizophrenia-related behavioural phenotypes, such as prepulse inhibition (PPI) deficits, cognitive impairments, increased sensitivity to the locomotor effects of phencyclidine, deficits in odour discrimination and impaired locomotor coordination (Otte et al., 2009; 2011,). The function of the LG72 protein is not entirely clear; as an evolutionarily novel protein no protein domains are recognizable by sequence analysis. It was first suggested that LG72 is an activator of d-amino-acid-oxidase, an enzyme involved in the degradation of d-serine (Chumakov et al., 2002). However, more recent studies have indicated that LG72 is a mitochondrial protein (Kvajo et al., 2008; Otte et al., 2011). Indeed, analysis of mitochondrial functions in G72Tg mice showed that the activity of complex I of the respriratorial chain and aconitase activity were reduced (Otte et al., 2011). We also found increased oxidative stress in cerebellum of these animals, with elevated levels of peroxidized lipids and proteins, increased expression of detoxifying glutathione transferases and reduced levels of glutathione (Otte et al., 2011). The increased oxidative stress probably contributed to some of the behavioural phenotypes as they were rescued by prolonged treatment with N-acetyl cysteine, which resulted in higher gluthathione levels.
In the present study, we investigated the effect of chronic nicotine on distinct cognitive domains in G72Tg mice. We observed mostly detrimental effects of chronic nicotine on cognition in wild-type (WT) mice, and a strong modulation of these effects in the G72Tg mice. To explore the mechanisms underlying these phenotypes, we analysed the most abundant nACh receptor subtypes (see Alexander et al., 2013b) in the fore and midbrain structures of experimental animals by quantitative receptor autoradiography. G72-expression did not markedly influence nicotine-induced up-regulation of the α4β2*subtype; however, α7nACh receptor density was differentially altered by genotype or genotype x treatment interaction in specific brain areas. In particular, we propose that the up-regulation of α7nACh receptor density in the dentate gyrus (DG) is a potential mediator of the beneficial effects of nicotine on cognitive deficits in G72Tg mice.
Methods
The drug/molecular target nomenclature used in this manuscript conforms to British Journal of Pharmacology's Concise Guide to PHARMACOLOGY (Alexander et al., 2013a).
Animals and reagents
Animals were group-housed under standard conditions with a 12:12 h reversed light/dark-cycle. G72Tg animals (Otte et al., 2009) were generated on a CD1 genetic background and maintained by crossing hemizygous G72Tg mice to CD1 WT mice. A total of 199 G72Tg mice and 208 WT littermates were balanced for sex in individual experiments. Two weeks before the implantation of an osmotic pump, animals were single-housed to analyse individual caloric demand. (-)-Nicotine hydrogen-tartrate salt ((-)-1-methyl-2(3-pyridyl)pyrrolidine; Sigma, St. Louis, MO, USA) was delivered in physiological saline (0.9%) for 14 days using Alzet® osmotic pumps, model 2002 (Charles River GmbH, Kißlegg, Germany). Doses dispensed from pumps were measured as free-base nicotine. We first performed experiments on working memory after administration of 16 and 24 mg kg−1 day−1, which produced nicotine plasma concentrations similar to those found in human smokers. As no significant behavioural effects were observed with the lower dose, all subsequent experiments were performed with 24 mg kg−1 day−1. Pumps were implanted s.c. under 2.0 v v−1 isoflurane/O2 anaesthesia using 0.2 mg kg−1 meloxicam i.m. for analgesia. Anaesthesia was evaluated by the absence of withdrawal reflexes. Behavioural experiments were performed during the active locomotor phase of the animals. For analysis of nicotine/cotinine plasma concentrations by GC/MS, blood was taken from the abdominal vein of individual animals at the end of chronic nicotine treatment (see supplemental methods). The work complies with current regulations covering animal experimentation in Germany and the EU (European Communities Council Directive 86/609/EEC) and was approved by local authorities (file-no.87-51.04.2010.A070). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Behavioural testing
Each mouse was exposed to a single behavioural test (single testing), with the exception of those tested for social memory on day 9 of nicotine treatment, who were also tested for sensory motor gating at days 6 and 14 of chronic nicotine treatment (see Supporting Information Figure S1).
Working memory test
Working memory performance was tested in a paired-trial alternation task on a Y-Maze, consisting of 44 × 12 × 10 cm spanning arms, illuminated with 100–150 lux at the start and 5–15 lux at the goal area. Food consumption was analysed for 10 days before minipump implantation (see Supporting Information Figure S1). Casein-pellets (Billaney Consultants Ltd, Kent, UK) were used for reward and placed in plastic dishes at the very end of the goal-arms. Minipumps containing saline or nicotine (24 mg kg day−1) were implanted for 14 days. Caloric intake of the animals was restricted to 80% 2 days following minipump implantation and remained restricted throughout the remaining 12 days of treatment. The day before training, mice were habituated for 2 min to the environmental setting. For the first training session (after 3 days of chronic nicotine/saline treatment), paired-trial alternation of entries to the two goal-arms of the Y-maze was forced by blocking of the sides for three subsequent days. Each day, animals had to pass three forced paired-trials with intertrial intervals of 10–20 s in a maximum time of 2 min or until the reward was gained. In order to avoid side preference, the entry-arm of the first run of discrete paired-trials was randomized. For the second training session (after 10 days of chronic nicotine/saline treatment), the entry of the second run of paired-trials was not forced and animals had to remember which arm has been visited before in order to gain the bait from the opposite arm. Subsequently, animals had to pass three to four paired-trials per day at a maximal time of 1 min until the mean error rate of the entire experimental group dropped below 30% for three consecutive days. Finally, the working memory test was performed without blocking of the sides of the Y-maze and mice had to pass three to four paired-trials per day (on days 10–13 from chronic nicotine/saline treatment). In this final test session, the percent of correct alternations was calculated for each individual animal from at least 10 discrete paired-trials and was performed independently for 10 and 20 s intertrial periods.
PPI test
PPI of the acoustic startle response was performed and analysed according to standard protocols (at days 6 and 14 of chronic nicotine/saline treatment; Otte et al., 2009; see supplemental methods and Supporting Information Figure S1).
Social recognition test
Social memory was analysed (at days 9 of chronic nicotine treatment) in a compartmentalized encounter test in an arena (44 × 44 × 26 cm), illuminated with 5–10 lux of white and 100–120 lux of red light. The ground of the arena was filled with bedding, collected from the home-cages of individual animals. Two cylindrical metal cages (8 × 10 cm) were placed in opposite corners of the arena, 5 cm from the walls. Two consecutive days previously, and on the day of testing, mice were habituated to the experimental setting for 10 min per day. A naïve, juvenile C57BL/6 mouse of the same sex was presented to the experimental animal in a metal cage. Any interaction with the object mouse was recorded for 5 min, using a camera, mounted 1 m above the centre of the maze. The social recognition was performed after intertrial-intervals of 2 or 4 h. In this test, a second, novel object mouse with the same characteristics as the first was introduced into the second cage. Interaction with both partners was recorded for 5 min. All video files were analysed (JWatcher V1.0, Macquarie University/UCLA, Australia) twice and data were re-evaluated by a second, independent observer for control.
Novel object preference test
To determine novel object preference in untreated mice, animals were first habituated to the arena for 5–10 min on three consecutive days before the test and then to two objects (LEGO building bricks) in three 6 min sessions spaced 10 min apart. After another 10 min interval, one of the familiar objects was replaced with a novel one of a different shape and colour. The mice were then left to explore the apparatus for 3 min. Nose point interactions with this object were assessed during a 3-min test session using the Ethovision software (Noldus Information Technology, Wageningen, Netherlands).
Operant conditioning
An operant conditioning paradigm was used to evaluate associative learning (on days 4 to 14 of chronic nicotine/saline treatment). Caloric intake of the animals was restricted to 80% 2 days before and for the 10 days of operant conditioning testing (see Supporting Information Figure S1). One to two days before operant conditioning, mice were habituated for 20 min to the operant chambers equipped with a single nose-poke sensor, signal-lights, a dispenser (TSE Systems GmbH, Bad Homburg, Germany) for casein-pellets, and bedding, collected from the home-cage of the test animal. The boxes were placed in individually ventilated, dark compartments. Conditioning sessions were conducted for 30 min per day, for 10 days under a fixed-ratio three schedule of positive reinforcement, using casein-pellets as a reward. Active phase of the nose-poke sensor was indicated by a yellow light persisting for a maximum of 30 s or until the reward was gained. Nose-poking in the active phase was confirmed by a green light and a casein-pellet was dispensed as soon as the sensor was activated three times. The active phase was followed by a 30 s time-out period, where nose-poking had no consequences. The maximum number of rewards was limited to 30 per session and animals were trained for 10 consecutive days. Phenomaster V4.0 software (TSE Systems GmbH) was used to analyse parameters, characteristic of associative learning.
Morris water maze test
Spatial learning of the animals was analysed (at days 4 to 14 of chronic nicotine treatment) according to standard protocols (Otte et al., 2011), with minor modifications (see supplemental methods and Supporting Information Figure S1).
Two-bottle choice preference test
One week before the test, animals were isolated and habituated to two 50 mL flasks, placed in the home-cage. To analyse rewarding properties of nicotine, animals had the choice of consuming either tap water offered in one flask, or nicotine solution offered in the second flask. Nicotine was added at concentrations of 5 μg mL−1 at days 1–3, 10 μg mL−1 at days 4–7, 25 μg mL−1 at days 8–10, 50 μg mL−1 at days 11–14 and 75 μg mL−1 at days 14–37 of the test (see Supporting Information Figure S1 for overview). The pH of the nicotine solution was adjusted to 7.0. In order to mask the slightly bitter taste of nicotine, 0.1% of saccharin was added to all solutions offered to the animals. Flask weight was measured before and after the solution had been offered to the animal for 3 days. The position of the nicotine-containing flasks was varied in order not to contaminate by side preference. Consumption was calculated by the loss of weight of individual flasks. The daily nicotine consumption was normalized by body weight. The ratio of nicotine to total fluid consumption was calculated.
Supplementary methods describe the analysis of basal activity measurements in the home-cage.
Quantitative receptor autoradiography
After chronic nicotine treatment for 14 days, mice were killed by cervical dislocation, their brains removed and snap-frozen at −20°C in isopentane on dry ice. Frozen female brains were sliced starting from bregma +3.2 in consecutive 20 μm coronal sections 300 μm apart, cutting sufficient sections for determining total and non-specific binding of three ligands, and thaw-mounted on gelatine-coated object slides. Quantitative autoradiography was performed in order to measure binding of α4β2* and α7nACh receptors using 100 pM [125I]-epibatidine (with and without cytisine) and 1 nM [125I]-α-bungarotoxin, respectively, according to established protocols (Orr-Urtreger et al., 1997; Besson et al., 2007; Metaxas et al., 2010; 2012,) with minor modifications (see supplemental methods).
Statistical analysis
Behavioural experiments were analysed using SPSS 20.0 software (IBM Corp., Armonk, New York, USA). Data were inspected for normality (Kolmogorov–Smirnov test) and homogeneity of variance (Levene's test). Parametric tests were used if the data met these criteria and replicates were equal between groups. Variables dependent on one or more factors were compared using univariate or multivariate analysis according to general linear models. Bonferroni corrections were made to adjust for multiple comparisons. All experiments were analysed for the factors treatment, sex and genotype. Repeated measures were inspected for sphericity (Mauchly's test). Non-spherical within-subject effects were subject to a Greenhouse-Geisser correction. Kruskal–Wallis was used for non-parametric tests of more than two independent groups, followed by Dunnet's-T3 multiple comparisons. Autoradiography data were analysed using Statistica 7 (StatSoft, Inc., Tulsa, OK, USA). Ligand binding was compared for multiple factors using two-or three-way anova, followed by Duncan's post hoc test where appropriate. Data are presented as mean ± SEM. Significance was accepted when *P < 0.05, **P < 0.01 and ***P < 0.001.
Results
Effect of chronic nicotine treatment on basal activity and consummatory behaviour of WT and G72Tg mice
Nicotine or saline was administered for 14 days using osmotic pumps implanted s.c. Treatment with 24 mg kg−1 day−1 produced mean plasma nicotine concentrations of 315.65 ± 83.7 μg L−1 and cotinine concentrations of 104.9 ± 6.9 μg·L−1 (Supporting Information Figure S2A–C). There was no difference between genotypes (see Supporting Information Table S1 for summary of statistical analysis). Concentrations were within the range of human smokers (nicotine: 6–498 μg L−1, cotinine: 4–96 μg L−1 in Shin et al., 2002; nicotine: 181–3702 μg L−1; cotinine: 21–4420 μg L−1 in Massadeh et al., 2009). As nicotine was described to act as an appetite suppressant in rodents (Chowdhury, 2014), we also analysed caloric intake in WT and G72Tg mice chronically treated with saline or nicotine, 24 mg kg−1 day−1, on days 7–14 following food restriction. Neither genotype, nor sex, nor treatment significantly affected food consumption (Supporting Information Table S1). A second group of animals, receiving 24 mg kg−1 day−1 nicotine, that had free access to food also had similar weights to the vehicle control group (Supporting Information Table S1). We also checked if the rewarding properties of nicotine might differ between genotypes in a long-term two-bottle choice test. Neither preference nor total amount of nicotine consumed (Supporting Information Table S1) was altered in G72Tg compared with WT mice of either sex. These data suggest that the rewarding properties of nicotine are not influenced by the expression of G72. As nicotine has sedative effects at high doses, we also tested home-cage activity of mice, chronically treated with nicotine or saline, administered by osmotic minipumps. However, we found neither treatment, nor sex, nor genotype effects on days 2–12 of treatment, indicating that the chronic treatment did not affect the animal's activity (Supporting Information Table S1). Thus, all of the behavioural studies were performed with a dose of 24 mg kg−1 day−1, except the working memory test, which was also done with a dose of 16 mg kg−1 day−1.
Effect of chronic nicotine treatment on working memory in WT and G72Tg mice
First we determined (i) whether working memory was defective in the G72Tg mouse model of schizophrenia and (ii) whether deficits were rescued by different doses of chronic nicotine. Working memory was tested in discrete paired-trial alternation tasks using a Y-maze. We initially trained food-restricted mice to retrieve food rewards placed at the end of two arms. For this purpose, mice had to enter both goal-arms alternately in two consecutive trials. Alternations were forced by blocking one of the two baited arms. During 7 days of forced alternation training (days 73–80), the latency to gain the reward significantly decreased over time (F(3,170) = 34.36, P < 0.001) and was neither affected by nicotine treatment nor genotype (Figure 1A). After the mean error rate dropped below 30%, the side blocks were removed and working memory was tested. In the memory test, trained animals had to choose one of two baited goal-arms upon a first run and to remember the choice in order to obtain the bait of the opposite arm during the second run. Analysis of correct choices in paired-trial alternation indicated that (i) working memory was defective in G72Tg animals and (ii) that increasing amounts of nicotine progressively, but antithetically affected both genotypes (Figure 1B). Multivariate statistical analysis revealed a significant (F(2,44) = 6.63, P = 0.003) interaction of treatment and genotype for correct alternations. Bonferroni post hoc test revealed that 24 mg kg−1 day−1 corrupted working memory in WT mice (P = 0.033), but ameliorated impairments in G72Tg animals (P = 0.021). These data suggest that chronic nicotine is able to restore working memory in defective G72Tg animals.
Figure 1.
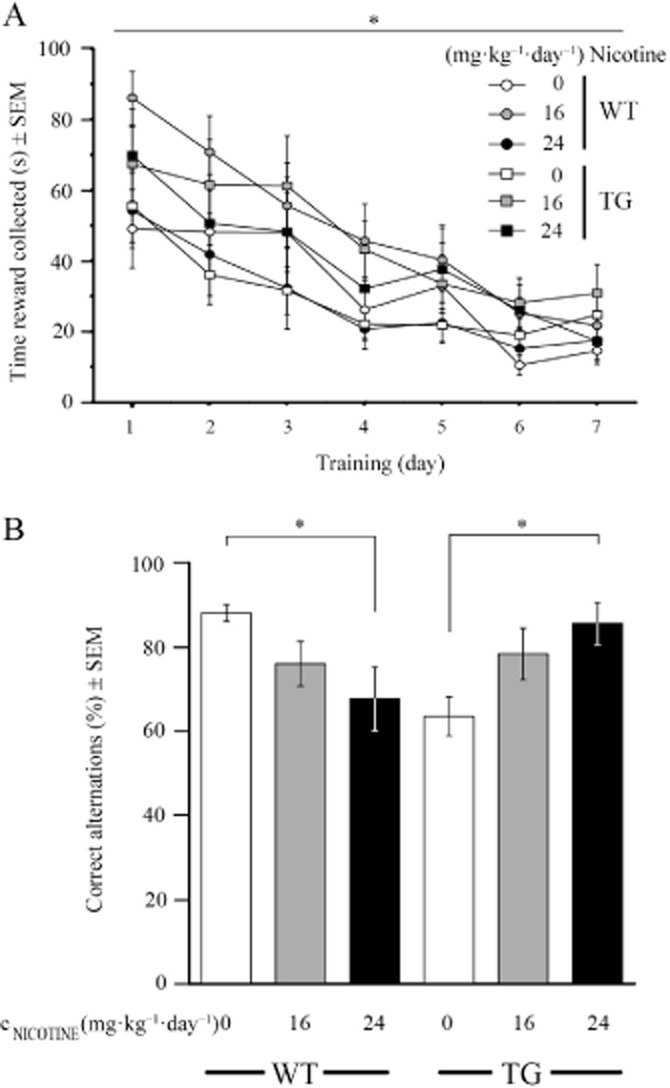
Chronic nicotine has beneficial effects on working memory in defective G72Tg, but impairs performance of WT mice. (A) Paired-trial alternation training on a Y-maze was initiated after 4 days of chronic treatment. Latency to gain the reward at the rear end of either goal arm was analysed in forced runs (postnatal day 74–77; NWTsaline = 10, NWTnicotine at 16 mg = 11, NWTnicotine at 24 mg = 10, NTGsaline = 8, NTGnicotine at 16 mg = 8, NTGnicotine at 24 mg = 9). (B) Working memory of trained mice (postnatal day 81–84) chronically exposed to saline, 16 or 24 mg kg−1 day−1 nicotine for 10–13 days was tested as correct alternation between both goal-arms in consecutive paired-trials with 10 s intertrials. *P < 0.05.
Effect of chronic nicotine treatment on PPI in WT and G72Tg mice
We investigated nicotine effects on sensory motor gating deficits of G72Tg mice using the PPI test at three different prepulse intensities of +5, +10 and +15 dB above background, after 6 and 14 days of chronic treatment with 24 mg kg−1 day−1 nicotine. Here, multivariate statistical analysis revealed a significant interaction of treatment, sex and genotype after 6 (F(7,71) = 6.41, P = 0.014 at +5 dB, F(7,71) = 3.07, P = 0.084 at +10 dB, F(7,71) = 6.47, P = 0.013 at +15 dB) and 14 days (F(7,71) = 6.51, P = 0.013 at +5 dB, F(7,71) = 11.01, P = 0.001 at +10 dB, F(7,71) = 5.133, P = 0.027 at +15 dB) of chronic nicotine treatment. Further comparison found no significant effects in males (Supporting Information Table S1), but a treatment × genotype interaction was observed in females (Supporting Information Table S1). Saline-infused G72Tg females were unable to suppress the startle response in the PPI test (Otte et al., 2009). However, 6 days of chronic nicotine produced a remarkable increase in PPI of G72Tg females, which was significant at different prepulse intensities (Figure 2A; treatment effect: F(1,17) = 5.54, P = 0.031 at +5 dB, F(1,17) = 4.32, P = 0.053 at +10 dB and F(1,17) = 9.63, P = 0.006 at +15 dB above background). Most strikingly, after 14 days of chronic nicotine treatment, sensory motor gating of G72Tg was indistinguishable from saline-treated WT females (Figure 2B; treatment effect: F(1,17) = 4.54, P = 0.048 at +5 dB, F(1,17) = 9.17, P = 0.008 at +10 dB and F(1,17) = 4.86, P = 0.041 at +15 dB). In contrast, in WT females nicotine significantly attenuated PPI at prepulse intensities of +10 dB (F(1,18) = 6.03, P = 0.024) after 14 days of treatment. Taken together, these data suggest that nicotine impairs sensory motor gating in WT, but improves deficits in G72Tg females.
Figure 2.
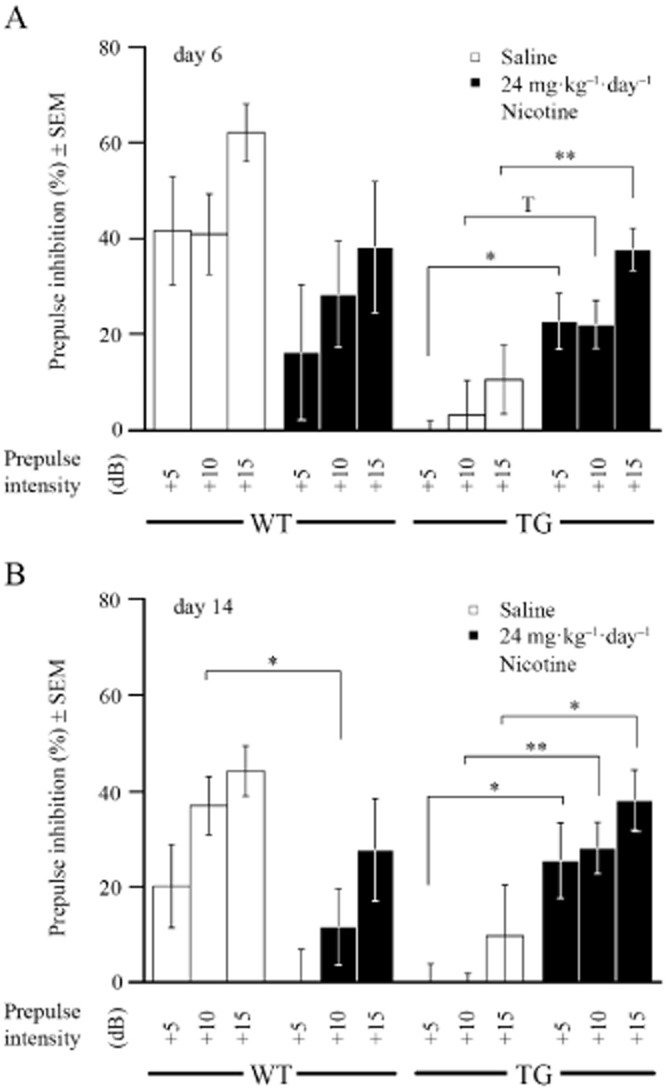
Chronic nicotine (24 mg kg−1 day−1) has beneficial effects on PPI in defective G72Tg, but impairs performance of WT female mice. (A) PPI of the acoustic startle response was analysed after 6 days of chronic nicotine (postnatal day 62; n = 10 each group) at prepulse intensities of +5, +10 and +15 dB above background (65 dB white noise). (B) Likewise, PPI was analysed after 14 days of treatment (postnatal day 70; n = 10 each group). *P ≤ 0.05; **P ≤ 0.01.
Effect of chronic nicotine treatment on social memory in WT and G72Tg mice
G72Tg animals and WT littermates of both sexes were repeatedly habituated to the experimental setup of the compartimentalized encounter test and sociability was analysed after 9 days of chronic treatment with 24 mg kg−1 day−1 nicotine. Social interaction was measured as time spent in voluntary investigation of an object mouse, presented in the cage. Interaction time was not significantly different between groups, indicating that neither G72-expression nor treatment affected sociability (Figure 3A).
Figure 3.
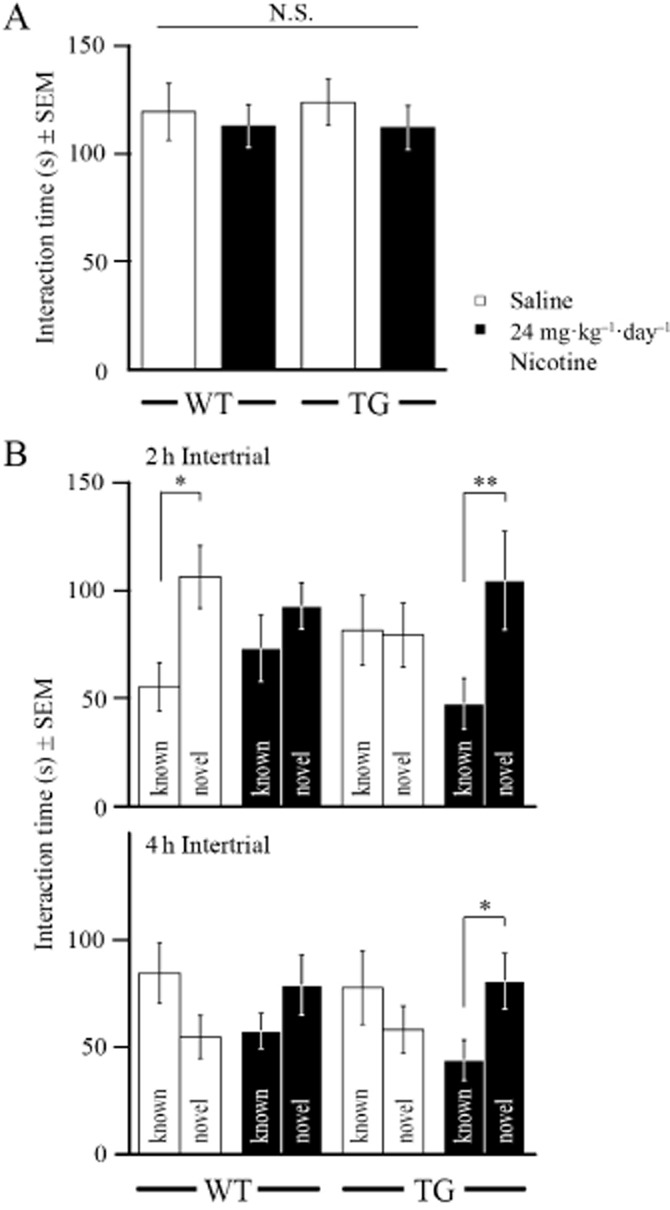
Chronic nicotine (24 mg·kg−1 day−1) for 9 days has beneficial effects on social memory of G72Tg, but not WT mice. (A) Social interaction with an unfamiliar mouse at 5 min-intervals of a compartimentalized encounter test was not significantly affected by sex, genotype or treatment (postnatal day 56, n = 20 each group). (B) Social recognition of the familiar mouse was measured as preference compared with a second, novel animal after 2 and 4 h intertrials (NTGsaline = 9, n = 10 for the other groups). *P ≤ 0.05; **P ≤ 0.01.
Two or 4 h after the initial conspecific exposure, social memory was measured as time in voluntary investigation of the familiar mouse, compared with a second, novel, unfamiliar mouse in another cage. Statistical analysis of social interaction time revealed a significant interaction of treatment, genotype and object mouse (F(15,60) = 4.54, P = 0.037 at 2 h, F(15,64) = 5.17, P = 0.026 at 4 h) and was followed by further inter-and inner-group comparison. Indeed, after 2 h, saline-treated WT animals spent twice as much time investigating the novel, rather than the familiar object mouse (F(1,18) = 7.00, P = 0.016; Figure 3B), indicating recognition of the familiar mouse. In contrast, saline-treated G72Tg mice did not display any preference at any intertrial period. However, nicotine increased interaction time with the novel object mouse (F(3,32) = 3.12, P = 0.087 for treatment × object interaction, F(1,16) = 4.83, P = 0.043 for object) in G72Tg mice to a level of saline-treated WT animals. Most notably, preference of nicotine-treated G72Tg animals for the novel object mouse was still significant after 4 h intertrials (F(3,36) = 4.28, P = 0.046 for treatment × object interaction, F(1,18) = 4.77, P = 0.042 for object), when WT mice failed to show social memory.
To determine if G72Tg mice simply responded more to novelty, we also performed a novel object preference test. After habituation to two objects in 3 × 6 min sessions, one of the objects was replaced with a different one and the interaction time with the novel object in a 3 min session was analysed. G72Tg and WT animals made a similar number of nose contacts with the novel object (WT: 5.1 ± 1.3, n = 9; G72Tg: 7.6 ± 1.9, n = 9; F(1,14) = 1.19, P = 0.293) and showed a similar nose contact time (WT: 1.8 ± 0.9 s; G72Tg: 2.1 ± 0.5 s; F(1,14) = 0.13 P = 0.720). These data suggest that G72Tg animals do not have an increased novelty preference. Rather, nicotine diminishes social memory in WT mice and particularly improves conspecific recognition in impaired G72Tg animals.
Effect of chronic nicotine treatment on associative learning in WT and G72Tg mice
An operant conditioning paradigm was used to study associative learning of food-restricted G72Tg and WT animals of both sexes and was initiated after 4 days of chronic treatment with 24 mg kg−1 day−1 nicotine or saline. Our control experiments indicated that neither nicotine treatment nor G72-expression influenced the appetite of mice undergoing restricted (Supporting Information Table S1) or free access to food protocols (Supporting Information Table S1), nor did it impair basal activity (Supporting Information Table S1), or affect motivation to perform the task (Supporting Information Table S1). Food was used to reward sensor activation when a light cue was presented. In 10 training days, both saline-treated WT and G72Tg animals progressively increased the amount of reward feeding per day (Figure 4A), indicating that both genotypes learned to associate the task with the reward. In contrast, in WT animals chronically treated with nicotine, the number of dispensed rewards remained constantly low over the entire training period. Finally, at training day 10 Kruskal–Wallis statistical analysis, followed by Dunnett's-T multiple comparisons revealed reward feeding to be significantly diminished by nicotine treatment in WT mice compared with saline WT controls (P < 0.001) and compared with saline-treated G72Tg mice: (P = 0.004; Figure 4B). These data indicate that nicotine abolishes establishment of associative memory in WT animals. In contrast, G72Tg mice chronically treated with nicotine still showed moderate learning of the task and achieved approximately 60% reward, which was not significantly different from control levels. These data suggest that G72-expression protects against adverse effects of chronic nicotine on associative learning. It is important to note that basal activity in sensor activation was not significantly different between groups, as analysed by the total number of nose-pokes on the first day of training (Supporting Information Table S1). However, as the total number of nose-pokes (Supporting Information Table S1) constantly doubles the amount required for reward feeding during 10 days of training, these data also suggest that animals associated the sensor activation task, but not the cued active phase period, with the reward.
Figure 4.
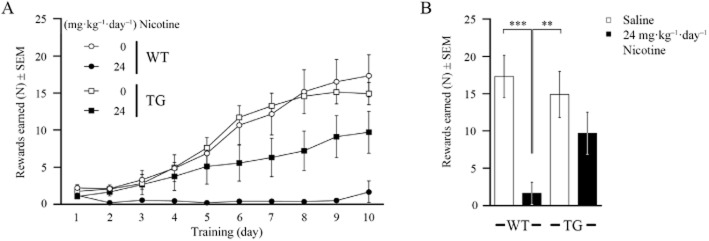
Chronic nicotine (24 mg kg−1 day−1) has adverse effects on associative learning of WT mice. (A) Associative learning was measured as number of rewards earned per day after 4 to 14 days of treatment in an operant conditioning fixed-ratio three schedule. (B) Associative memory was considered established at training day 10 (postnatal day 84; n = 20 each group). **P ≤ 0.01; ***P ≤ 0.001.
Effect of chronic nicotine treatment on spatial learning in WT and G72Tg mice
Memory acquisition training in the Morris water maze was started at day 7 of chronic treatment with 24 mg kg−1 day−1 nicotine or saline and performed for seven consecutive days. Saline-treated WT controls progressively decreased escape-distance to the hidden platform during the initial three to four training days (Figure 5A), indicating fast spatial learning. In saline-treated G72Tg animals, the reduction of path-length was delayed to 5–6 days, indicating deficits in spatial learning, as others have described (Otte et al., 2011). Surprisingly, irrespective of genotype, escape-distance to the hidden platform was most prolonged in nicotine-treated animals, but finally reached control levels at training day 7. Repeated measures analysis revealed a significant interaction of treatment × genotype × time (F(4,146) = 2.36, P = 0.048), and further comparison indicated a significant interaction of genotype and time in saline-treated (F(6,108) = 2.40, P = 0.033), but not nicotine-treated animals. These data suggest that chronic nicotine at doses of 24 mg kg−1 day−1 and G72-expression significantly impair, but does not abolish spatial learning. Animal velocity significantly decreased over time (F(4,145) = 41.21, P < 0.001), but was not affected by either nicotine treatment or genotype (Supporting Information Table S1).
Figure 5.
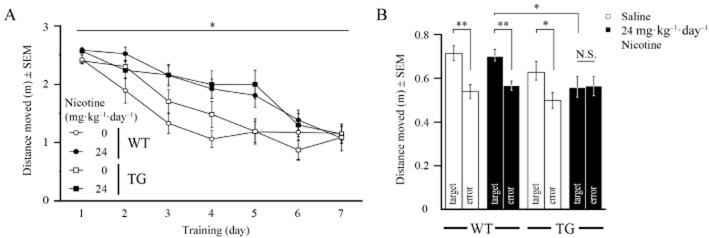
Chronic nicotine (24 mg kg−1 day−1) has adverse effects on long-term spatial memory. (A) Long-term spatial learning of mice (postnatal day 76–83; n = 10 each group) in the Morris water maze was initiated after 6 days of chronic nicotine. (B) At treatment day 14 (postnatal day 84), the hidden platform was removed and long-term memory was analysed comparing the average distance moved in the target quadrant. *P ≤ 0.05; **P ≤ 0.01.
After 14 days of treatment, the hidden platform was removed and long-term memory was analysed as preference for the former target quadrant (Figure 5B). Here, univariate statistical analysis, followed by further comparison revealed that saline-treated WT (F(1,18) = 12.88, P = 0.002), G72Tg (F(1,18) = 4.51, P = 0.048) and nicotine-treated WT mice (F(1,18) = 12.03, P = 0.003) significantly preferred the target quadrant. However, in nicotine-treated G72Tg animals, there was no difference in path-length in any quadrant. Intergroup comparison of nicotine-treated mice confirmed the significant effect of genotype on quadrant preference (F(1,18) = 5.66, P = 0.029). Thus, these data suggest that the combination of nicotine treatment and G72-expression impairs long-term memory formation.
Nicotine differentially alters nACh receptor density in distinct brain regions of WT and G72Tg mice
Lastly, we determined whether chronic treatment with 24 mg kg−1 day−1 of nicotine might also alter nACh receptor density in brain tissue of G72Tg compared with WT mice. We used female mice for these experiments, because they showed a stronger phenotype in the PPI test. Cytisine-sensitive [125I]-epibatidine binding was used to quantify α4β2*nACh receptor density (Whiteaker et al., 2000; Marks et al., 2002; Metaxas et al., 2010; 2012; 2013,,). Three-way anova found a significant effect of treatment (F(1,719) = 75.51, P < 0.001) and interaction of treatment × region (F(31,828) = 1.47, P = 0.030) for signal-density. Two-way anova in each brain region followed by Duncan's post hoc test revealed treatment within the same genotype to be significant in several areas. Irrespective of genotype, α4β2*nACh receptor binding was significantly up-regulated in many brain regions of nicotine-treated mice compared with saline controls (Figure 6A, Supporting Information Table S1). Increases were observed in the vertical diagonal band, the hypothalamus and many cortical regions, comprising the frontal association, prelimbic, primary and secondary motor, cingulate, somatosensory, piriform, retrosplenial, visual and auditory cortices. Irrespective of treatment, G72-expression significantly impaired α4β2*nACh receptor binding in the interpeduncular nucleus only (genotype effect: F(1,27) = 5.20, P = 0.031). While chronic nicotine treatment significantly increased α4β2*nACh receptor binding in the olfactory tubercles, medial septum, posterior and lateral dorsal thalamic nuclei of WT mice, up-regulation did not reach significance in transgenic animals suggesting a relatively minor effect of G72-introduction on α4β2*nACh receptor regulation by nicotine. In other brain regions expressing α4β2*nACh receptors, binding was not different between groups (Supporting Information Table S2). For control, cytisine-resistant and cytisine-sensitive binding was compared and shown to be equal in the medial habenula and interpeduncular nucleus, indicating that detection of other nACh receptor subtypes was restricted to those regions only (Supporting Information Figure S3B; Whiteaker et al., 2000; Marks et al., 2002). Non-specific binding was identical to film background (Supporting Information Figure S3A). These data suggest that chronic nicotine significantly increased α4β2*density in many brain regions irrespective of genotype and that G72-expression might modestly impair up-regulation in a few specific brain regions.
Figure 6.

G72-expression and nicotine treatment differentially alter nACh receptor density in various brain regions. (A) Data represent mean ± SEM cytisine-sensitive [125I]-epibatidine binding of α4β2*nACh receptors in WT and G72Tg mice (postnatal day 70–84) chronically treated with saline or 24 mg kg−1·day−1 nicotine (n = 4–6). (B) Computer-enhanced autoradiograms of total [125I]-α − bungarotoxin binding (fmol·mg−1 tissue equivalent) of α7nACh receptors in coronal brain sections of WT and G72Tg mice. The sections shown are from the level of the ventral dentate gyrus (brema −3.64 mm). Non-specific binding was indistinguishable from film background. (C) Data represent mean ± SEM [125I]-α − bungarotoxin binding to α7nACh receptors in WT and G72Tg mice (postnatal day 70–84) chronically treated with saline or nicotine (n = 4–6). AuCx, auditory cortex; BMA, basomedial amygdale; CA1, nucleus of the hippocampus; CgCx, cingulate cortex; DEn, dorsal endopiriform nucleus; DG, dentate gyrus; FrA, frontal association; Hyp, hypothalamus; IPn, interpeduncular nucleus; LD, laterodorsal thalamic nuclei; M1, primary motor cortex; M2, secondary motor cortex; MS, medial septum; Pir, piriform cortex; Po, posterior thalamic nuclei; PrL, prelimbic cortex; RS, retrosplenial cortex; SsCx, somatosensory cortex; Tu, olfactory tubercles; VDB, vertical diagonal band; ViCx, visual cortex; VLG, ventral medial geniculate; VMH, ventromedial hypothalamus. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
We used [125I]-α − bungarotoxin to label α7nACh receptors for receptor autoradiography (Figure 6B), as in brain tissue this ligand has been shown to specifically bind to this subtype (Orr-Urtreger et al., 1997). α7nACh receptor density was not significantly up-regulated by chronic nicotine treatment in individual genotypes. This was expected, as α7nACh receptors are known to have lower affinity for acetylcholine and therefore to be more resistant to up-regulation upon chronic nicotine exposure. However, three-way anova revealed a significant genotype effect (F(1,478) = 33.304, P < 0.001). Two-way anova for each brain region followed by Duncan's post hoc test found significant differences between WT and G72Tg mice. Irrespective of treatment, G72-expression significantly increased [125I]-α − bungarotoxin binding in the dorsal endopiriform (F(1,30) = 5.73, P = 0.023), ventral medial geniculate (F(1,24) = 4.46, P = 0.045) and the cornu ammonis region of the hippocampus (Figure 6C; F(1,25) = 4.30, P = 0.048). Moreover, we found α7nACh receptor density to be up-regulated in the DG (P = 0.049, Duncan's post hoc test) and ventromedial hypothalamus (VMH; P = 0.026, Duncan's post hoc test) of nicotine-treated G72Tg, compared with nicotine-treated WT mice. This significant up-regulation was not observed between saline-treated G72Tg and WT mice. In contrast, in the cingulate cortex (CgCx); and basomedial amygdala (BMA) nicotine abolished genotype-increased [125I]-α − bungarotoxin binding, seen in saline-treated G72Tg mice (genotype × treatment interaction: CgCx; F(1,28) = 4.42, P = 0.044 and BMA; F(1,22) = 6.87, P = 0.015 respectively). In other brain regions expressing α7nACh receptors, differences in autoradiographic ligand binding were negligible (Supporting Information Table S2). Non-specific binding was homogenous across each brain section (Supporting Information Figure S3C). Taken together, these data suggest that G72-expression might facilitate increased α7nACh receptor density in the cornu ammonis layer of the hippocampus, the dorsal endopiriform and the ventral medial geniculate irrespective of nicotine treatment and facilitates α7nACh receptor activation in the DG and VMH only following nicotine treatment.
Discussion
The present study demonstrates that chronic nicotine treatment enhanced several cognition-related functions that were impaired in G72Tg mice, a genetic animal model of schizophrenia. In contrast, chronic nicotine had detrimental effects on cognitive performance in WT controls. Interestingly, α7nACh receptors, which have been implicated in schizophrenia-related cognitive deficits (Levin, 2012), were differentially regulated in several brain regions of G72Tg mice either irrespective of nicotine treatment or only after nicotine treatment. Together, these data show that G72 modulates the behavioural and molecular effects of chronic nicotine exposure. Our findings support the hypothesis that the intense smoking behaviour observed in many schizophrenic patients might reduce specific cognitive impairments. It may therefore represent a form of self-medication, although the latter was not directly addressed in the present study by means of nicotine self-administration.
In order to investigate the impact of chronic heavy nicotine consumption on cognition, we first tested memory performance in mice receiving either 16 or 24 mg kg−1 day−1 of free-base nicotine for 14 days. We found that the higher dose completely restored working memory deficits in G72Tg mice, while impairing performance of WT controls. We then decided to continue the study with the higher dose only, because (i) the lower dose did not produce significant effects and (ii) the plasma nicotine and cotinine levels obtained with the higher dose was within the range observed in human smokers (Shin et al., 2002; Massadeh et al., 2009). Nicotine levels in our study were similar to those reported by others (AlSharari et al., 2013), but surprisingly, nicotine levels exceeded cotinine levels in our experiments, although the half-life of nicotine is typically shorter than that of cotinine (Florescu et al., 2009) and other rodent studies with lower doses of nicotine reported that cotinine levels were higher than nicotine after chronic treatment (Marks et al., 2004; Doura et al., 2008). This high nicotine level may be the result of low expression of cytochrome P450 2A6 (CYP2A6), the main nicotine degrading enzyme. It is known that chronic nicotine results in a down-regulation of CYP2A6 expression and, thus low nicotine metabolism, which in some cases can exceed cotinine levels in heavy smokers (see Massadeh et al., 2009). Accordingly, 24 mg kg−1 day−1 nicotine also attenuated PPI after 6 and 14 days of chronic treatment in defective G72Tg females at different prepulse intensities. This is in line with effects of chronic nicotine found in type III neuregulin1 heterozygous female mutant mice (Chen et al., 2008), and makes a strong case for nicotine to ameliorate deficits in sensory information processing caused by distinct schizophrenia susceptibility genes. Our focus on female G72Tg mice reflects the known sex differences that have been described for PPI in rodents (Ison and Allen, 2007). Furthermore, chronic nicotine treatment completely restored social recognition in impaired G72Tg animals after intertrial periods of 2 h and even 4 h. Taken together, beneficial effects of nicotine in female G72Tg mice were found on cognition-related functions, processing information in a rather short-term memory range.
These findings are in line with human studies reporting nicotine as enhancing related cognitive domains in abstinent schizophrenic smokers. Nicotine patches reversed spatial working memory impairment associated with haloperidol therapy in the delayed matching to sample test (Levin et al., 1996). Likewise, nicotine delivered via nasal spray improved delayed yes/no-recognition of visuospatial designs (Myers et al., 2004) and acute smoking re-instatement selectively enhanced PPI deficits in schizophrenic smokers (George et al., 2006). Most interestingly, as seen for strain-differences of rodents, PPI-enhancing nicotine effects were also highly genotype-dependent in humans, which demonstrates a strong genetic modulation, indicating the high predictive validity of animal PPI to model human sensorimotor gating phenomena. Thus, chronic nicotine in G72Tg mice resembled procognitive effects of acute nicotine administration on short-term memory processes in abstinent schizophrenic smokers.
In order to elucidate corresponding molecular mechanisms underlying the beneficial effects of nicotine in G72Tg mice, we focused on nACh receptor systems and whether they are differentially altered by nicotine in G72Tg compared with WT female mice. Although most behavioural tests showed no sex difference, we focused on females, as nicotine effects on PPI deficits were more pronounced in females than males. In the DG and VMH of G72Tg female mice, nicotine significantly increased α7nACh receptor binding, compared with nicotine-treated WT mice. In contrast, in the CgCx and BMA, nicotine treatment normalized the α7nACh receptor signal, which was elevated in G72Tg compared with WT mice under saline control conditions.
Indeed, α7nACh receptors are very likely to be involved in the ameliorative effects of nicotine in G72Tg mice. The α7nACh receptor gene is located in a region of chromosome 15, where linkage studies have found significant association with schizophrenia (Leonard and Freedman, 2006). Selective agonists of α7nACh receptors have already been demonstrated to be effective for pharmacotherapy of schizophrenia symptoms. For example, the α7nACh receptor agonist TC-5619 was shown to prevent impairments in PPI and sociability of the th(tk-)/th(tk-) mouse model for schizophrenia and object recognition in rats (Hauser et al., 2009). TC-5619 is currently in phase II, clinical trials for medication of schizophrenia symptoms (ClinicalTrials.gov identifier: NCT01488929). Furthermore, α7nACh receptor expression was significantly increased in the hippocampus of schizophrenic smokers (Mexal et al., 2010), indicating a possible target-region for the beneficial effects of nicotine. The hippocampus is well known for its pivotal role in spatial memory (Bird and Burgess, 2008; Sanderson and Bannerman, 2012) and lesions of the DG or perforant path subregions were shown to affect spatial learning, for example, in the modified Hebb-Williams maze (Lee and Kesner, 2004). Furthermore, α7nACh receptor signalling in this region is known to have a particular effect on cognitive performance (Levin, 2012). As α7 was shown to be the main nACh receptor subtype in the hippocampus, and receptor density was highest of all brain regions (Fabian-Fine et al., 2001), alteration of α7nACh receptor signalling (see Dajas-Bailador and Wonnacott, 2004 for review) might considerably affect information processing in this area. In accord with this, we found an increase in α7nACh receptor density in the DG of chronic nicotine-treated G72Tg compared with nicotine-exposed WT animals, which was accompanied by a restoration of short-term memory impairment in G72Tg mice. Further, the DG has been demonstrated to express the highest levels of G72 transcripts in the forebrain of transgenic animals (Otte et al., 2009). The fact that α7nACh receptor density is unaltered in the DG of saline-treated G72Tg compared with WT mice suggests that activation of α7nACh receptor signalling in the DG by endogenous ligands may not be involved in the impaired basal cognitive state of G72Tg animals. However, the increase of α7nACh receptor density in this area following chronic nicotine exposure in G72Tg mice could be an attractive explanation for the beneficial effect of chronic nicotine on short-term cognitive-related functions in schizophrenics, although direct causality remains to be demonstrated.
In contrast to the beneficial effects seen in G72Tg animals, chronic nicotine impaired working memory, PPI and social recognition of WT littermates. A focus on brain regions featuring genotypic differences in nACh receptor densities, which are also regulated by nicotine, has identified the CgCx to be involved in error detection, decision-making and learning tasks (Gehring and Taylor, 2004). It has also been shown to be involved in schizophrenia pathology, as in subjects with a high risk of psychotic incidence, the anterior part of the CgCx featured several structural and neurochemical abnormalities (Smieskova et al., 2010). We found that chronic nicotine produced an alteration of α7 nACh receptor binding in the CgCx, which was lower in saline-treated WT compared with G72Tg mice. As nACh receptors are mainly found on GABAergic interneurons in cortical regions (Timofeeva and Levin, 2011), enhanced α7 nACh receptor density might increase inhibition in the CgCx. The latter in turn might reduce activity of the CgCx and thereby contribute to cognitive deficits, although direct causality remains to be demonstrated. Additionally, chronic nicotine produced a genotype-independent increase of α4β2*nACh receptor density in the CgCx and many other cortical regions, also known to contribute to cognitive processing. Accordingly, increased α4β2*nACh receptor signalling in cortical interneurons (Timofeeva and Levin, 2011) might also increase inhibitory tone in those areas.
In our experiments, nicotine-induced impairments of long-term spatial learning in the Morris water maze, were independent of genotype. This is in line with findings in isolated NMRI mice (a strain closely related to CD1), which also showed impaired learning upon daily s.c. administrations of nicotine before water maze acquisition training (Moragrega et al., 2003). These data suggest that irrespective of disease state, chronic nicotine consumption might, in general, have adverse long-term effects on cognition. The latter is supported by studies examining associations between smoking and cognitive decline in a decade-range, for example, the Whitehall cohort study (Sabia et al., 2012). In this study, smokers in transition from midlife to old age were shown to experience faster decline in many cognitive functions, compared with non-smokers.
In conclusion, we found chronic nicotine ameliorated the impairment of short-term cognition-related functions caused by the G72 susceptibility gene of schizophrenia in mice. We found G72-expression to significantly affect the density of α7nACh receptors in specific brain regions, which collectively might contribute to cognitive performance in a complex interplay. Most interestingly, α7nACh receptor density was significantly increased in the DG of chronically nicotine-treated G72Tg versus nicotine-treated WT animals. This effect was not observed in saline-treated mice. In contrast, a general adverse effect of nicotine was found on long-term spatial learning of both genotypes and cognitive performance of WT animals. These results suggest that the beneficial cognitive effect of nicotine in G72-related schizophrenics might be due to increased α7nACh receptor signalling upon nicotine exposure in the DG subregion of the hippocampus. Our findings might therefore also recommend screening of schizophrenic patients for G72 and other susceptibility genes, (i) in order to regulate smoking behaviour, and (ii) for the treatment of cognitive deficits with α7nACh receptor-targeted pharmacotherapy of responders carrying a particular genetic predisposition.
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft (DFG), Priority Programme 1226 and a UK funded MRC/ESRC studentship grant (IK, HK, AB). We thank Dreisow ML, Markert A, Schrage H and Erxlebe E for active support, Prof. Kühn-Velten, Bremen for nicotine analytics.
Glossary
- BMA
basomedial amygdale
- CgCx
cingulate cortex
- CYP2A6
cytochrome P450 2A6
- DG
dentate gyrus
- G72Tg
G72-transgenic (mice)
- PPI
prepulse inhibition
- VMH
ventromedial hypothalamus
- WT
wild-type
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12578
Time schedule of behavioural experiments.
{kind=link}
Nicotine and cotinine concentrations were analysed in individual and pooled samples of blood plasma of G72Tg (n = 8) and WT (n = 10) animals, treated with 24 mg kg-1 day-1 of free-base nicotine for 14 days. Multivariate statistical analysis according to general linear models revealed no significant differences. Also shown are the nicotine/cotinine ratios from individual animals.
{kind=link}
(A) α4β2*nACh receptor binding is up-regulated by chronic nicotine treatment irrespective of genotype. Computer-enhanced colour autoradiograms of total and cytisine-resistant binding, following saline or nicotine treatment in WT and G72Tg mice. Coronal brain sections were cut at the level of the frontal cortex (bregma 1.98), caudate (bregma 1.10), dorsal hippocampus (bregma .1.46), substantia nigra (bregma .3.40) and the interpeduncular nucleus (bregma .3.64). Specific α4β2*nACh receptor binding (fmol·mg-1 tissue equivalent) was calculated following subtraction of (B) cytisine-resistant from (A) total [125I]-epibatidine images. Adjacent sections were incubated with 300 μM (-)-nicotine hydrogen tartrate to calculate nonspecific binding (NSB), which was indistinguishable from background. (C) Computer-enhanced autoradiograms of α7nACh receptor binding, following saline or nicotine treatment in WT and G72Tg mice. Coronal brain sections were cut at the level of the caudate (bregma 1.10), dorsal (bregma.1.46), and ventral hippocampus (bregma .3.64). Total α7nACh receptor binding (fmol·mg-1 tissue equivalent) was determined by incubation with [125I]-α. bungarotoxin. Adjacent sections were incubated with 1 mM nicotine (non-specific binding; NSB).
Table for the description of statistics, in order of their appearance.
G72-expression and nicotine treatment did not significantly alter nACh receptor density. Brain sections were bound with [125I]-epibatidine for quantitative α4β2* and [125I]-α . bungarotoxin for α7nACh receptor autoradiography. Ligand binding (fmol·mg-1 tissue equivalent) was calculated from five to nine replicates per group as value ± SEM. AcbC, nucleus accumbens core; AcbSh, nucleus accumbens shell; AV, anteroventricular thalamic nuclei; BLA, basolateral amygdale; CA2/3, cornu ammonis region 2 and 3; CPu, caudate putamen; DLG, dorsal geniculate nucleus; DM, dorsomedial hypothalamus; fr, fasiculus retroflexus; Hip, hippocampus; InG, intermediategrey layer of the superior colliculus; LH, lateral hypothalamus; InG, intermediategrey layer of the superior colliculus; M1, primary motor cortex; MHb, medial habenual; S, subiculum; SN, substantia nigra; SuG, superficial grey layer of the superior colliculus; VLG, ventral medial geniculate; VTA, ventral tegmental area; ZI, zona incerta.
References
- Adler LE, Olincyn A, Waldo M, Harris JQ, Qriffith J, Stevens K, et al. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr Bull. 1998;24:189–202. doi: 10.1093/oxfordjournals.schbul.a033320. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013a;170:1449–1867. doi: 10.1111/bph.12447. CGTP Collaborators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ. The Concise Guide to PHARMACOLOGY 2013/14: Ligand-Gated Ion Channels. Br J Pharmacol. 2013b;170:1582–1606. doi: 10.1111/bph.12446. CGTP Collaborators. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlSharari SD, Akbarali HI, Abdullah RA, Shahab O, Auttachoat W, Ferreira GA, et al. Novel insights on the effect of nicotine in a murine colitis model. J Pharmacol Exp Ther. 2013;344:207–217. doi: 10.1124/jpet.112.198796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson M, Granon S, Mameli-Engvall M, Cloez-Tayarani I, Maubourguet N, Cormier A, et al. Long-term effects of chronic nicotine exposure on brain nicotinic receptors. Proc Natl Acad Sci U S A. 2007;104:8155–8160. doi: 10.1073/pnas.0702698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird CM, Burgess N. The hippocampus and memory: insights from spatial processing. Nat Rev Neurosci. 2008;9:182–194. doi: 10.1038/nrn2335. [DOI] [PubMed] [Google Scholar]
- Brambilla P, Cerruti S, Bellani M, Perlini C, Ferro A, Marinelli V, et al. Shared impairment in associative learning in schizophrenia and bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1093–1099. doi: 10.1016/j.pnpbp.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Johnson MA, Lieberman MD, Goodchild RE, Schobel S, Lewandowski N, et al. Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J Neurosci. 2008;28:6872–6883. doi: 10.1523/JNEUROSCI.1815-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Hattori E, Nakajima A, Woehrle NS, Opal MD, Zhang C, et al. Expression of the G72/G30 gene in transgenic mice induces behavioral changes. Mol Psychiatry. 19:175–183. doi: 10.1038/mp.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury P. Endocrine and metabolic regulation of body mass by nicotine: role of growth hormone. Ann Clin Lab Sci. 2014;20:415–419. 1990. [PubMed] [Google Scholar]
- Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, et al. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci U S A. 2002;99:13675–13680. doi: 10.1073/pnas.182412499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dépatie L, O'Driscoll GA, Holahan AL, Atkinson V, Thavundayil JX, Kin NN, et al. Nicotine and behavioral markers of risk for schizophrenia: a double-blind, placebo-controlled, cross-over study. Neuropsychopharmacology. 2002;27:1056–1070. doi: 10.1016/S0893-133X(02)00372-X. [DOI] [PubMed] [Google Scholar]
- De Leon J, Diaz FJ. A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophr Res. 2005;76:135–157. doi: 10.1016/j.schres.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Derntl B, Habel U. Deficits in social cognition: a marker for psychiatric disorders? Eur Arch Psychiatry Clin Neurosci. 2011;261(Suppl. 2):S145–S149. doi: 10.1007/s00406-011-0244-0. [DOI] [PubMed] [Google Scholar]
- Detera-Wadleigh SD, McMahon FJ. G72/G30 in schizophrenia and bipolar disorder: review and meta-analysis. Biol Psychiatry. 2006;60:106–114. doi: 10.1016/j.biopsych.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Doura MB, Gold AB, Keller AB, Perry DC. Adult and periadolescent rats differ in expression of nicotinic cholinergic receptor subtypes and in the response of these subtypes to chronic nicotine exposure. Brain Res. 2008;1215:40–52. doi: 10.1016/j.brainres.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews E, Otte DM, Zimmer A. Involvement of the primate specific gene G72 in schizophrenia: from genetic studies to pathomechanisms. Neurosci Biobehav Rev. 2013;37:2410–2417. doi: 10.1016/j.neubiorev.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, et al. Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florescu A, Ferrence R, Einarson T, Selby P, Soldin O, Koren G. Methods for quantification of exposure to cigarette smoking and environmental tobacco smoke: focus on developmental toxicology. Ther Drug Monit. 2009;31:14–30. doi: 10.1097/FTD.0b013e3181957a3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring WJ, Taylor SF. When the going gets tough, the cingulate gets going. Nat Neurosci. 2004;7:1285–1287. doi: 10.1038/nn1204-1285. [DOI] [PubMed] [Google Scholar]
- George TP, Termine A, Sacco KA, Allen TM, Reutenauer E, Vessicchio JC, et al. A preliminary study of the effects of cigarette smoking on prepulse inhibition in schizophrenia: involvement of nicotinic receptor mechanisms. Schizophr Res. 2006;87:307–315. doi: 10.1016/j.schres.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Gold JM, Fuller RL, Robinson BM, McMahon RP, Braun EL, Luck SJ. Intact attentional control of working memory encoding in schizophrenia. J Abnorm Psychol. 2006;115:658–673. doi: 10.1037/0021-843X.115.4.658. [DOI] [PubMed] [Google Scholar]
- Harris JG, Kongs S, Allensworth D, Martin L, Tregellas J, Sullivan B, et al. Effects of nicotine on cognitive deficits in schizophrenia. Neuropsychopharmacology. 2004;29:1378–1385. doi: 10.1038/sj.npp.1300450. [DOI] [PubMed] [Google Scholar]
- Hauser TA, Kucinski A, Jordan KG, Gatto GJ, Wersinger SR, Hesse RA, et al. TC-5619: an alpha7 neuronal nicotinic receptor-selective agonist that demonstrates efficacy in animal models of the positive and negative symptoms and cognitive dysfunction of schizophrenia. Biochem Pharmacol. 2009;78:803–812. doi: 10.1016/j.bcp.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison JR, Allen PD. Pre-but not post-menopausal female CBA/CaJ mice show less prepulse inhibition than male mice of the same age. Behav Brain Res. 2007;185:76–81. doi: 10.1016/j.bbr.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korostishevsky M, Kaganovich M, Cholostoy A, Ashkenazi M, Ratner Y, Dahary D, et al. Is the G72/G30 locus associated with schizophrenia? single nucleotide polymorphisms, haplotypes, and gene expression analysis. Biol Psychiatry. 2004;56:169–176. doi: 10.1016/j.biopsych.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Kumari V, Postma P. Nicotine use in schizophrenia: the self medication hypotheses. Neurosci Biobehav Rev. 2005;29:1021–1034. doi: 10.1016/j.neubiorev.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Kvajo M, Dhilla A, Swor DE, Karayiorgou M, Gogos JA. Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol Psychiatry. 2008;13:685–696. doi: 10.1038/sj.mp.4002052. [DOI] [PubMed] [Google Scholar]
- Lee I, Kesner RP. Encoding versus retrieval of spatial memory: double dissociation between the dentate gyrus and the perforant path inputs into CA3 in the dorsal hippocampus. Hippocampus. 2004;14:66–76. doi: 10.1002/hipo.10167. [DOI] [PubMed] [Google Scholar]
- Leonard S, Freedman R. Genetics of chromosome 15q13-q14 in schizophrenia. Biol Psychiatry. 2006;60:115–122. doi: 10.1016/j.biopsych.2006.03.054. [DOI] [PubMed] [Google Scholar]
- Levin ED. α7-Nicotinic receptors and cognition. Curr Drug Targets. 2012;13:602–606. doi: 10.2174/138945012800398937. [DOI] [PubMed] [Google Scholar]
- Levin ED, Wilson W, Rose JE, McEvoy J. Nicotine-haloperidol interactions and cognitive performance in schizophrenics. Neuropsychopharmacology. 1996;15:429–436. doi: 10.1016/S0893-133X(96)00018-8. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Whiteaker P, Grady SR, Picciotto MR, McIntosh JM, Collins AC. Characterization of [(125) I]epibatidine binding and nicotinic agonist-mediated (86) Rb(+) efflux in interpeduncular nucleus and inferior colliculus of beta2 null mutant mice. J Neurochem. 2002;81:1102–1115. doi: 10.1046/j.1471-4159.2002.00910.x. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Rowell PP, Cao J-Z, Grady SR, McCallum SE, Collins AC. Subsets of acetylcholine-stimulated 86Rb+ efflux and [125I]-epibatidine binding sites in C57BL/6 mouse brain are differentially affected by chronic nicotine treatment. Neuropharmacology. 2004;46:1141–1157. doi: 10.1016/j.neuropharm.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Massadeh AD, Gharaibeh AA, Omari KW. A single-step extraction method for the determination of nicotine and cotinine in Jordanian smokers' blood and urine samples by RP-HPLC and GC–MS. J Chromatogr Sci. 2009;47:170–177. doi: 10.1093/chromsci/47.2.170. [DOI] [PubMed] [Google Scholar]
- Metaxas A, Bailey A, Barbano MF, Galeote L, Maldonado R, Kitchen I. Differential region-specific regulation of alpha4beta2* nAChRs by self-administered and non-contingent nicotine in c57bl/6j mice. Addict Biol. 2010;15:464–479. doi: 10.1111/j.1369-1600.2010.00246.x. [DOI] [PubMed] [Google Scholar]
- Metaxas A, Keyworth H, Yoo J, Chen Y, Kitchen I, Bailey A. The stereotypy-inducing and OCD-like effects of chronic ‘binge’ cocaine are modulated by distinct subtypes of nicotinic acetylcholine receptors. Br J Pharmacol. 2012;167:450–464. doi: 10.1111/j.1476-5381.2012.02023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metaxas A, Al-Hasani R, Farshim P, Tubby K, Berwick A, Ledent C, et al. Genetic deletion of the adenosine A receptor prevents nicotine-induced upregulation of alpha7, but not alpha4beta2* nicotinic acetylcholine receptor binding in the brain. Neuropharmacology. 2013;71:228–236. doi: 10.1016/j.neuropharm.2013.03.023. [DOI] [PubMed] [Google Scholar]
- Mexal S, Berger R, Logel J, Ross RG, Freedman R, Leonard S. Differential regulation of α7 nicotinic receptor gene (CHRNA7) expression in schizophrenic smokers. J Mol Neurosci. 2010;40:185–195. doi: 10.1007/s12031-009-9233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moragrega I, Carrasco MC, Vicens P, Redolat R. Spatial learning in male mice with different levels of aggressiveness: effects of housing conditions and nicotine administration. Behav Brain Res. 2003;147:1–8. doi: 10.1016/s0166-4328(03)00112-8. [DOI] [PubMed] [Google Scholar]
- Myers CS, Robles O, Kakoyannis AN, Sherr JD, Avila MT, Blaxton TA, et al. Nicotine improves delayed recognition in schizophrenic patients. Psychopharmacology (Berl) 2004;174:334–340. doi: 10.1007/s00213-003-1764-8. [DOI] [PubMed] [Google Scholar]
- Nuechterlein KH, Green MF, Kern RS, Baade LE, Barch DM, Cohen JD, et al. The MATRICS consensus cognitive battery, part 1: test selection, reliability, and validity. Am J Psychiatry. 2008;165:203–213. doi: 10.1176/appi.ajp.2007.07010042. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Goldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, et al. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otte DM, Bilkei-Gorzó A, Filiou MD, Turck CW, Yilmaz O, Holst MI, et al. Behavioral changes in G72/G30 transgenic mice. Eur Neuropsychopharmacol. 2009;19:339–348. doi: 10.1016/j.euroneuro.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Otte DM, Sommersberg B, Kudin A, Guerrero C, Albayram O, Filiou MD, et al. N-acetyl cysteine treatment rescues cognitive deficits induced by mitochondrial dysfunction in G72/G30 transgenic mice. Neuropsychopharmacology. 2011;36:2233–2243. doi: 10.1038/npp.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabia S, Elbaz A, Dugravot A, Head J, Shipley M, Hagger-Johnson G, et al. Impact of smoking on cognitive decline in early old age: the Whitehall II cohort study. Arch Gen Psychiatry. 2012;69:627–635. doi: 10.1001/archgenpsychiatry.2011.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson DJ, Bannerman DM. The role of habituation in hippocampus-dependent spatial working memory tasks: evidence from GluA1 AMPA receptor subunit knockout mice. Hippocampus. 2012;22:981–994. doi: 10.1002/hipo.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HS, Kim JG, Shin YJ, Jee SH. Sensitive and simple method for the determination of nicotine and cotinine in human urine, plasma and saliva by gas chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;769:177–183. doi: 10.1016/s1570-0232(02)00007-7. [DOI] [PubMed] [Google Scholar]
- Smieskova R, Fusar-Poli P, Allen P, Bendfeldt K, Stieglitz RD, Drewe J, et al. Neuroimaging predictors of transition to psychosis-a systematic review and meta-analysis. Neurosci Biobehav Rev. 2010;34:1207–1222. doi: 10.1016/j.neubiorev.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Smith RC, Infante M, Ali A, Nigam S, Kotsaftis A. Effects of cigarette smoking on psychopathology scores in patients with schizophrenia: an experimental study. Subst Abus. 2001;22:175–186. doi: 10.1080/08897070109511457. [DOI] [PubMed] [Google Scholar]
- Timofeeva OA, Levin ED. Glutamate and nicotinic receptor interactions in working memory: importance for the cognitive impairment of schizophrenia. Neuroscience. 2011;195:21–36. doi: 10.1016/j.neuroscience.2011.08.038. [DOI] [PubMed] [Google Scholar]
- Whiteaker P, Jimenez M, McIntosh JM, Collins AC, Marks MJ. Identification of a novel nicotinic binding site in mouse brain using [(125)I]-epibatidine. Br J Pharmacol. 2000;131:729–739. doi: 10.1038/sj.bjp.0703616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziedonis D, Hitsman B, Beckham JC, Zvolensky M, Adler LE, Audrain-McGovern J, et al. Tobacco use and cessation in psychiatric disorders: National Institute of Mental Health report. Nicotine Tob Res. 2008;10:1691–1715. doi: 10.1080/14622200802443569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Time schedule of behavioural experiments.
Nicotine and cotinine concentrations were analysed in individual and pooled samples of blood plasma of G72Tg (n = 8) and WT (n = 10) animals, treated with 24 mg kg-1 day-1 of free-base nicotine for 14 days. Multivariate statistical analysis according to general linear models revealed no significant differences. Also shown are the nicotine/cotinine ratios from individual animals.
(A) α4β2*nACh receptor binding is up-regulated by chronic nicotine treatment irrespective of genotype. Computer-enhanced colour autoradiograms of total and cytisine-resistant binding, following saline or nicotine treatment in WT and G72Tg mice. Coronal brain sections were cut at the level of the frontal cortex (bregma 1.98), caudate (bregma 1.10), dorsal hippocampus (bregma .1.46), substantia nigra (bregma .3.40) and the interpeduncular nucleus (bregma .3.64). Specific α4β2*nACh receptor binding (fmol·mg-1 tissue equivalent) was calculated following subtraction of (B) cytisine-resistant from (A) total [125I]-epibatidine images. Adjacent sections were incubated with 300 μM (-)-nicotine hydrogen tartrate to calculate nonspecific binding (NSB), which was indistinguishable from background. (C) Computer-enhanced autoradiograms of α7nACh receptor binding, following saline or nicotine treatment in WT and G72Tg mice. Coronal brain sections were cut at the level of the caudate (bregma 1.10), dorsal (bregma.1.46), and ventral hippocampus (bregma .3.64). Total α7nACh receptor binding (fmol·mg-1 tissue equivalent) was determined by incubation with [125I]-α. bungarotoxin. Adjacent sections were incubated with 1 mM nicotine (non-specific binding; NSB).
Table for the description of statistics, in order of their appearance.
G72-expression and nicotine treatment did not significantly alter nACh receptor density. Brain sections were bound with [125I]-epibatidine for quantitative α4β2* and [125I]-α . bungarotoxin for α7nACh receptor autoradiography. Ligand binding (fmol·mg-1 tissue equivalent) was calculated from five to nine replicates per group as value ± SEM. AcbC, nucleus accumbens core; AcbSh, nucleus accumbens shell; AV, anteroventricular thalamic nuclei; BLA, basolateral amygdale; CA2/3, cornu ammonis region 2 and 3; CPu, caudate putamen; DLG, dorsal geniculate nucleus; DM, dorsomedial hypothalamus; fr, fasiculus retroflexus; Hip, hippocampus; InG, intermediategrey layer of the superior colliculus; LH, lateral hypothalamus; InG, intermediategrey layer of the superior colliculus; M1, primary motor cortex; MHb, medial habenual; S, subiculum; SN, substantia nigra; SuG, superficial grey layer of the superior colliculus; VLG, ventral medial geniculate; VTA, ventral tegmental area; ZI, zona incerta.