

Abstract
Background
Biological WWTPs must be functionally stable to continuously and steadily remove contaminants which rely upon the activity of complex microbial communities. However, knowledge is still lacking in regard to microbial community functional structures and their linkages to environmental variables.
Aims
To investigate microbial community functional structures of activated sludge in wastewater treatment plants (WWTPs) and to understand the effects of environmental factors on their structure.
Methods
12 activated sludge samples were collected from four WWTPs in Beijing. A comprehensive functional gene array named GeoChip 4.2 was used to determine the microbial functional genes involved in a variety of biogeochemical processes such as carbon, nitrogen, phosphorous and sulfur cycles, metal resistance, antibiotic resistance and organic contaminant degradation.
Results
High similarities of the microbial community functional structures were found among activated sludge samples from the four WWTPs, as shown by both diversity indices and the overlapped genes. For individual gene category, such as egl, amyA, lip, nirS, nirK, nosZ, ureC, ppx, ppk, aprA, dsrA, sox and benAB, there were a number of microorganisms shared by all 12 samples. Canonical correspondence analysis (CCA) showed that the microbial functional patterns were highly correlated with water temperature, dissolved oxygen (DO), ammonia concentrations and loading rate of chemical oxygen demand (COD). Based on the variance partitioning analyses (VPA), a total of 53% of microbial community variation from GeoChip data can be explained by wastewater characteristics (25%) and operational parameters (23%), respectively.
Conclusions
This study provided an overall picture of microbial community functional structures of activated sludge in WWTPs and discerned the linkages between microbial communities and environmental variables in WWTPs.
Introduction
Biological activated sludge process is the most widely used biological process for treating municipal and industrial wastewater. By enriching selected functional microorganisms in the activated sludge of wastewater treatment plant (WWTP), microbial activities in the sludge community are accelerated, enabling removal of oxygen-depleting organics, toxics, and nutrients. Biological WWTPs must be functionally stable to continuously and steadily remove contaminants which rely upon the activity of complex microbial communities [1], [2]. Therefore, a better understanding of the microbial community structure and functional genes of activated sludge in WWTPs can help elucidate the mechanisms of biological pollutant removal and improve the treatment performance and operational stability [3], [4].
Although microbial communities of activated sludge in WWTPs have been intensively studied [5]–[11], most of such efforts have been focused only on microbial phylogenetic composition. Knowledge is still lacking in regard to microbial community functional structures and their linkages to environmental variables. To date, there have been only a limited number of studies that characterized the overall functional profiles and metabolic pathways in the activated sludge of WWTPs using high-throughput sequencing [12], [13].
Functional gene arrays, which contain genes encoding key enzymes involved in a variety of biogeochemical cycling processes [14], can be used to study the overall microbial functional potentials of activated sludge in WWTPs. GeoChip 4.2, a powerful functional gene array, contains 83,992 oligonucleotide (50-mer) probes targeting 152,414 genes in 410 gene categories involved in nitrogen, carbon, sulfur and phosphorus cycling, metal resistance, antibiotic resistance as well as organic contaminant degradation [15], [16]. GeoChip has been widely used to examine microbial community functional structures in various environmental samples, such as soil and sediments [17]–[19], groundwater [20], acid mine drainage[21], deep-sea water [15], ocean crust [15], and hydrothermal vent [22].
In this study, 12 activated sludge samples from four WWTPs in Beijing were analyzed by GeoChip 4.2 to address the following two questions: (i) What were the microbial community functional structures of activated sludge in WWTPs? (ii) How do environmental factors and operational parameters affect microbial community functional structures? Our results revealed high similarities of microbial functional communities among activated sludge samples from the four WWTPs, and microbial functional potentials were highly correlated with water temperature, dissolved oxygen (DO), ammonia concentrations and loading rate of chemical oxygen demand (COD).
Materials and Methods
WWTPs and Sampling
Anaerobic/anoxic/aerobic (A2O) is the most commonly used process for wastewater treatment in China, thus we examined four A2O WWTPs in Beijing. Activated sludge samples were taken from the aeration tank of each WWTP once a day for three consecutive days in the summer of 2011. Therefore, three replicate samples were available from each WWTP, resulting in a total of 12 activated sludge samples. No specific permits were required for the described field studies. We confirm that: i) the locations were not privately-owned or protected in any way; and ii) the field studies did not involve endangered or protected species.
For microbial community analysis, each sample was dispensed into a sterile Eppendorf tube and centrifuged at 14,000 g for 10 min. After decanting the supernatant, the pellet was stored at −80°C prior to further analysis. Meanwhile, various chemical parameters such as COD, total nitrogen (TN), ammonia, total phosphorus (TP), pH and conductivity were analyzed according to standard methods [23]. Concentrations of chromium, cobalt, nickel, copper, zinc, and cadmium were measured by inductively coupled plasma mass spectrometry (ICP-MS)(Thermo Fisher Scientific, Waltham, MA, USA).
DNA Purification and GeoChip Hybridization
Microbial genomic DNA was extracted from the activated sludge samples by combining freeze-thawing and sodium dodecyl sulfate (SDS) for cell lysis as previously described [24]. Crude DNA was purified using the Wizard SV Genomic DNA Purification Kit (Promega, Madison WI, USA). The quality of purified DNA was assessed based on the ratios of 260/280 nm and 260/230 nm absorption measured by an ND-1000 spectrophotometer (Nanodrop Inc., Wilmington, DE, USA) and agarose gel electrophoresis. The quantity of community DNA was determined with Quant-It PicoGreen kit (Invitrogen, Carlsbad, CA, USA).
Purified DNA (1 μg) was labeled with Cy5, purified and resuspended in 10 μl hybridization solution as previously described (Lu et al. 2012). Then it was hybridized with GeoChip 4.2 on a MAUI hybridization station (BioMicro, Salt Lake City, UT, USA) at 42°C with 40% formamide for 16 hours. After hybridization, microarrays were scanned (NimbleGen MS200, Madison, WI, USA) at a laser power of 100%. The ImaGene version 6.0 (Biodiscovery, El Segundo, CA) was then used to determine the intensity of each spot, and identify poor-quality spots. The GeoChip raw data has been uploaded to the NCBI GEO with the accession number GSE54055.
Data Analysis
Raw data from ImaGene were submitted to Microarray Data Manager (http://ieg.ou.edu/microarray/) and analyzed using the data analysis pipeline with the following major steps: (i) The spots flagged as 1 or 3 by Imagene and with a signal to noise ratio (SNR) less than 2.0 were removed as low quality spots; (ii) After removing the bad spots, normalized intensity of each spot was calculated by dividing the signal intensity of each spot by the mean intensity of the microarray; (iii) If any of replicates had more than two times the standard deviation, this replicate was moved as an outlier [18].
Hierarchical cluster analysis was performed using the unweighted pairwise average-linkage clustering algorithm in the CLUSTER software and visualized in TREEVIEW software [25]. Canonical correspondence analysis (CCA) was used to examine the relationships of microbial communities and environmental variables. The significant environmental variables were identified by a forward selection procedure using Monte Carlo permutations ((999 permutations with a p-value<0.05). In addition, Variance inflation factors (VIFs; a measure for cross-correlation of explanatory variables) were checked and eliminated if VIFs were more than 20 [26].Based on partial CCA, variance partitioning analysis (VPA) was performed to attribute the variation observed in the microbial communities to the environmental variables. CCA and VPA were performed by the vegan package in R 2.14.0 (R Development Core Team, 2011).
Results
Performance of WWTPs
Details of the influent and effluent characteristics and operational parameters of the four WWTPs are listed in Table S1 and S2 in File S1. Chemical oxygen demand (COD) in the influents ranged from 257 to 452 mg/L, while total nitrogen (TN) concentrations were between 46 and 64 mg/L. The COD removal efficiencies were more than 90% in all systems, and TN removal efficiencies varied from 68% to 75%. The removal efficiencies of COD and TN indicated that the four WWTPs were functionally stable.
Overall Microbial Community Functional Structure
Microbial community functional structures of 12 activated sludge samples from four WWTPs were analyzed with GeoChip 4.2 A total of 29,124 functional genes were detected. For individual samples, the numbers of detected genes were between 18,847 and 24,268 (Table 1). Hierarchical cluster analysis showed that the three replicate samples from each WWTP were clustered together, and the distances among the three replicate samples was always <2 (Figure 1). The samples from different WWTPs were well separated, and the distances among the four WWTPs were generally>2.
Table 1. Percentage of overlapping genes and diversity indices of all functional genes in activated sludge samples.
| sample | GBD_1 | GBD_2 | GBD_3 | QH_1 | QH_2 | QH_3 | XHM_1 | XHM_2 | XHM_3 | XJH_1 | XJH_2 | XJH_3 |
| GBD_1 | ||||||||||||
| GBD_2 | 84.67% | |||||||||||
| GBD_3 | 89.16% | 86.65% | ||||||||||
| QH_1 | 69.41% | 71.45% | 71.96% | |||||||||
| QH_2 | 67.39% | 69.49% | 69.22% | 85.78% | ||||||||
| QH_3 | 65.12% | 68.03% | 67.38% | 81.68% | 80.53% | |||||||
| XHM_1 | 79.10% | 80.08% | 81.10% | 72.82% | 69.85% | 67.72% | ||||||
| XHM_2 | 75.56% | 78.38% | 77.87% | 71.42% | 68.90% | 68.33% | 85.82% | |||||
| XHM_3 | 77.18% | 78.64% | 79.68% | 74.95% | 72.89% | 71.38% | 88.14% | 82.43% | ||||
| XJH_1 | 73.92% | 77.03% | 74.81% | 71.96% | 71.38% | 71.08% | 75.72% | 76.30% | 76.37% | |||
| XJH_2 | 74.11% | 76.22% | 74.91% | 72.62% | 72.29% | 72.01% | 74.94% | 75.19% | 76.71% | 89.15% | ||
| XJH_3 | 67.12% | 69.41% | 67.89% | 70.21% | 71.17% | 70.94% | 67.74% | 69.71% | 69.96% | 79.99% | 80.27% | |
| Richnessa | 23064 | 22434 | 23554 | 20862 | 20610 | 19688 | 24268 | 22831 | 23413 | 20774 | 20838 | 18847 |
| Hb | 10.1 | 10.0 | 10.1 | 10.0 | 9.9 | 9.9 | 10.1 | 10.0 | 10.1 | 10.0 | 10.0 | 9.9 |
| 1/Dc | 23301.2 | 22682.7 | 23805.7 | 21049.3 | 20777.9 | 19855.9 | 24452.9 | 23022.1 | 23624.7 | 20959.6 | 21033.5 | 18984.4 |
| Evennessd | 0.996 | 0.996 | 0.996 | 0.996 | 0.995 | 0.995 | 0.994 | 0.995 | 0.995 | 0.995 | 0.995 | 0.995 |
Detected gene number.
Shannon–Weiner index, higher number represents higher diversity.
Reciprocal of Simpson's index, higher number represents higher diversity.
Evenness index.
Figure 1. Hierarchical cluster analysis of functional genes in 12 activated sludge samples from four WWTPs named GBD, QH, XHM and XJH.
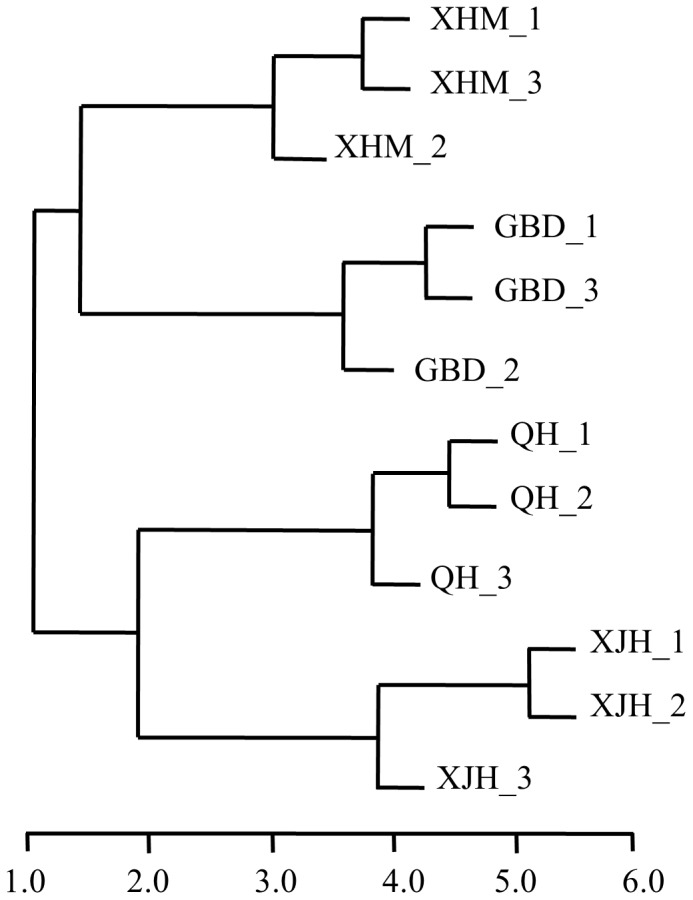
Each sample is named after WWTP plant with “_1”, “_2”, or “_3” that indicates one of three replicate samples from each plant.
To assess α-diversity of microbial communities, Shannon-Weaver index (H), Simpson's (1/D) and evenness were calculated (Table 1). The values of H were very similar across the 12 samples, ranging from 9.9 to 10.1. Consistently, the Simpson's (1/D) and evenness did not show significant differences among these four plants.
The overlapping genes were also determined. A large percentage (65–89%) of detected genes were shared among the samples (Table 1), indicative of high similarities of microbial community functional structures among activated sludge of four WWTPs. In consistency to the results of hierarchical clustering analysis, the percentages of the overlapping genes in samples from the same WWTP were higher than those from different WWTPs. For example, sample GBD_1 had 85% and 89% of the genes in common with sample GBD_2 and GBD_3 respectively, but 65% in common with QH_3.
At the phylogenetic level, 87.6–88.7% genes were derived from bacteria, 1.9–2.0% from archaea, and 8.7–9.7% from fungi. Proteobacteria was the predominant phylum of bacteria, constituting 50.8–52.2% of all detected microorganisms. Actinobacteria, Firmicutes, Cyanobacteria and Bacteroidetes were the subdominant bacterial groups, each containing 14.8–15.5%, 4.6–5.1%, 1.8–2.1% and 1.3–1.6% of detected microorganisms, respectively. Ascomycota was the most abundant phylum of fungi, constituting 6.3–7.4% of all detected microorganisms, followed by Basidiomycota (1.5–1.7%) as the second most abundant fungal group. In addition, Euryarchaeota, containing about 1.5% of detected microorganisms, was the most predominant phylum within archaea. (Table S3 in File S1). Taken together, these results indicated overall functional structures as well as phylogenetic diversities of microbial communities within WWTPs appeared to be quite high.
Carbon Cycling Genes
Carbon cycling, especially carbon degradation, is a crucial biochemical process in WWTPs. The rate of carbon degradation depends on a number of factors, including the availability and types of carbon substrates as well as the microbial community [27]. Among detected carbon cycling genes, 3,375 genes were involved in the degradation of complex carbon substrates such as cellulose, starch, hemicelluloses, chitin and lignin. Generally, relative abundances and numbers of all detected carbon degradation genes were similar among activated sludge of four WWTPs (Figure 2 and Table S4 in File S1).
Figure 2. The normalized signal intensity of detected key genes involved in carbon cycling.

The signal intensity for each functional gene was the average of the signal intensity from all the replicates. All data are presented as mean ± SE.
Endoglucanase are responsible for the initial steps of cellulose degradation. A total of 41 egl genes encoding endoglucanase were detected, and about half of them (20 genes) were commonly shared by all samples (Figure S1 in File S1). Nearly all of the egl genes were derived from isolated microorganisms, including the probes from the sequenced 149121623 (protein id number) of Methylobacterium sp. 4–46, 197934254 of Streptomyces sviceus ATCC 29083, and 171059025 of Leptothrix cholodnii SP-6.
α-Amylase functions to hydrolyze starch to yield glucose and maltose, which plays an important role in starch degradation. A total of 366 amyA genes encoding α-amylase were detected, among which 156 genes were commonly shared across all samples. Almost all the detected amyA genes were derived from isolated organisms such as 113897923 of Herpetosiphon aurantiacus ATCC 23779, 50842594 of Propionibacterium acnes KPA171202, 240169491 of Mycobacterium kansasii ATCC 12478.
Xylanase is an enzyme that breaks down hemicellulose, which is a major component of plant cell walls. 33 xylanase genes were detected, and 13 of them were shared by all samples (Figure S2 in File S1). Most of xylanase genes were from isolates such as 113935746 of Caulobacter sp. K31, 116098389 of Lactobacillus brevis ATCC 367, 68230042 of Frankia sp. EAN1pec, etc. Also a total of 422 genes encoding chitin degradative enzymes, including endochitinase, acetylglucosaminidase and exochitinase, were found in all 12 samples. For genes encoding endochitinase, 99 out of 239 genes were shared by all samples.
A total of 268 genes related to lignin degradation were detected, including genes encoding glyoxal oxidase (glx), ligninase (lip), manganese peroxidase (mnp) and phenol oxidase (lcc). Many of these genes were detected across all samples. For example, 13 out of 28 lip genes were shared by all samples and all of them were derived from isolates (Figure S3 in File S1). Similarly, almost all genes of glx, mnp and lcc were from isolated microorganisms rather than uncultured microorganisms.
Nitrogen Cycling Genes
As nitrogen removal is one of the most important functions in WWTPs, we examined the nitrogen cycling genes and their relationship to nitrogen concentrations. A total of 2,055 genes involved in ammonification, nitrification, denitrification and assimilatory nitrate/nitrite reduction were detected by GeoChip 4.2. Similar to carbon degradation genes, the relative abundances of detected nitrogen cycling genes were similar among activated sludge of four WWTPs (Figure 3).
Figure 3. The normalized signal intensity of detected key genes involved in nitrogen cycling.

The signal intensity for each functional gene was the average of the signal intensity from all the replicates. All data are presented as mean ± SE.
Of 197 detected nirS genes encoding nitrite reductases, 65 were present in all samples. 7 genes were derived from isolates, while the majority of genes were derived from environmental clones in various environments. The results of Mantel test showed that the abundance of nirS genes was positively correlated to the influent concentrations of TN (r = 0.489, P<0.01) and ammonia (r = 0.498, P<0.01) (Table S5 in File S1).
For nirK genes encoding nitrite reductases, 184 genes were detected. Among them, 62 genes were present in all samples. Most of them were from laboratory clones, while only 10 genes were derived from isolates. The results of Mantel test indicated that nirK genes were positively correlated to the concentrations of influent TN (r = 0.421, P<0.01) and ammonia (r = 0.358, P<0.05) (Table S5 in File S1).
For nosZ genes encoding nitrous oxide reductases, 110 genes were detected. Among them, 43 were shared by all samples. 94 nosZ genes were derived from uncultured microorganisms and 16 were from cultured organisms, including 39935130 of Rhodopseudomonas palustris CGA009, 83816541 of Salinibacter ruber DSM 13855, 82947026 of Magnetospirillum magneticum AMB-1, etc (Figure 4). The results of Mantel test analysis showed abundances of nosZ genes were positively correlated with the concentrations of influent TN (r = 0.348, P<0.05) and ammonia (r = 0.338, P<0.01) (Table S5 in File S1).
Figure 4. The hierarchical cluster analysis of nosZ genes encoding nitrous oxide reductases.

The protein id number and its derived organism for each gene are indicated. The color intensity of each panel shows the normalized signal intensity of individual genes, referring to color key at the top right.
295 ureC genes encoding ureases were detected, and 128 genes were shared by all samples. Unlike other nitrogen genes, most of the ureC genes were form cultured microorganisms and only 9 from uncultured bacteria. The results of Mantel test showed a positive correlation between ureC genes and the concentrations of influent TN (r = 0.463, P<0.01) and ammonia (r = 0.393, P<0.01) (Table S5 in File S1).
Only four hzo genes encoding hydrazine oxidoreductases were detected, including two genes (208605292 and 224712632) from Candidatus Brocadia sp. and two genes (224712622 and 208605290) from uncultured Planctomycete. No significant correlation between hzo genes and the concentrations of influent TN (P>0.05) and ammonia (P>0.05) (Table S5 in File S1) was observed.
Phosphorus Cycling Genes
Phosphorus removal from WWTPs is a key factor in preventing eutrophication of surface waters. Genes encoding exopolyphosphatase (ppx) for inorganic polyphosphate degradation, polyphosphate kinase (ppk) for catalyzing the formation of polyphosphate from ATP and phytase for phytate degradation were detected among all samples (Figure 5).
Figure 5. The normalized signal intensity of detected key genes involved in phosphorus cycling.

The signal intensity for each functional gene was the average of the signal intensity from all the replicates. All data are presented as mean ± SE.
A total of 90 of 246 detected ppx genes were shared by all samples, including genes derived from Leptothrix cholodnii SP-6 (171060286), Streptomyces coelicolor A3(2) (21221778), Erythrobacter sp. NAP1 (85709358). Nevertheless, there was no significant correlation between ppx genes and the concentrations of influent TP (Table S5 in File S1). For ppk, 57 of 165 detected genes were shared by all samples. Significantly positive correlation was observed between ppk gene abundances and the concentrations of influent TP (r = 0.242, P<0.05) (Table S5 in File S1). In addition, a total of 27 phytase genes were detected. Among them, 7 were shared by all samples (Figure S4 in File S1). The results of Mantel tests suggested that there was no significant correlation between phytase genes and influent TP (Table S5 in File S1).
Sulfur Cycling Genes
A total of 1,241 genes involved in sulfite reductase, adenylylsulfate reductase, sulphur oxidation, sulfide oxidation were detected. The relative abundances of sulfur cycling genes were presented in Figure 6 . Among them, there were 46 aprA genes encoding dissimilatory adenosine-5′-phosposulfate reductase, a key enzyme involved in microbial sulfate reduction and sulfur oxidation. 15 of 22 aprA genes present in all 12 samples were derived from isolates (Figure S5 in File S1). In addition, 327 dsrA genes encoding dissimilatory sulfite reductase were detected. Of the 100 genes shared by all samples, most genes were derived from uncultured microorganisms except for 18 genes from isolates such as 107784967 of Desulfovibrio desulfuricans, 13992710 of Bilophila wadsworthia and 83573380 of Moorella thermoacetica ATCC 39073. For sox genes encoding sulfite oxidase, 77 of 205 detected genes were shared by all samples, of which most were derived from isolated microorganisms such as 56677633 of Silicibacter pomeroyi DSS-3, 255258527 of Sideroxydans lithotrophicus ES-1, 214028622 of Ruegeria sp. R11.
Figure 6. The normalized signal intensity of detected key genes involved in sulfur cycling.

The signal intensity for each functional gene was the average of the signal intensity from all the replicates. All data are presented as mean ± SE.
Degradation of Organic Contaminants
Organic contamination is a concern and a lot of research has been undertaken to examine the role of microorganisms in the degradation and remediation of organic contaminants [14]. In total, 7,012 genes from 165 gene categories involved in the degradation of organic contaminants related to aromatics, chlorinated solvents, herbicides and pesticides were detected. The relative abundances of keg genes involved in organic contaminant degradation were present in Figure 7 . Among them, 24 benAB genes encoding benzoate 1, 2-dioxygenases were detected across all samples and 19 were present in all samples. The most abundant genes were derived from Burkholderia xenovorans LB400 (91779181), Mycobacterium sp. MCS (108768762), Rhodococcus sp. RHA1 (110818905).
Figure 7. The normalized signal intensity of detected key genes involved in organic contaminant degradation.

The signal intensity for each functional gene was the average of the signal intensity from all the replicates. All data are presented as mean ± SE.
Metal Resistance Genes
Various heavy metals were often detected in WWTPs as a result of societal industrialization and modernization. Resistance genes of Ag, As, Cd, Co, Cr, Cu, Hg, Pb, Ni, Te and Zn were detected in this study. A substantial number (3,381) of genes involved in metal resistance were detected in all the samples, of which the vast majority was derived from isolated microorganisms. Notably, relative abundances of detected metal resistance genes were similar among activated sludges of four WWTPs (Figure S6 in File S1). In addition, the results of Mantel test showed that most of metal resistance genes, such as chrA, nreB, copA, cueO, zntA, cadA and cadBD, were positively correlated to the concentrations of Cr, Ni, Cu, Cu, Zn, Cd and Cd in WWTPs, respectively. In contrast, other metal resistance genes (corC, cusA, zitB) did not show significant correlations to the respective metal concentrations (Table S5 in File S1).
Antibiotic Resistance
Antibiotic resistance is a growing concern worldwide as more and more pathogens develop resistance to commonly used antibiotics. 9 gene categories families related to antibiotic resistance were detected in this study, including five transporters (ATP-binding cassette, multidrug toxic compound extrusion, major facilitator superfamily, Mex, and small multidrug resistance efflux pumps), three β-lactamase genes and several genes involved in tetracycline resistance (Figure S7 in File S1). A total of 1,072 antibiotic resistance genes were detected. Of 92 tetracycline resistance genes, 39 were shared by all samples and most of them were derived from isolated microorganisms, such as 214028433 of Ruegeria sp. R11, 259487609 of Aspergillus nidulans FGSC A4, 254407454 of Streptomyces sviceus ATCC 29083, etc. Since antibiotic resistance is often associated with metal resistance, Mantel test was performed to examine the correlation between heavy metal resistance genes and antibiotic resistance genes. The results indicated that the whole metal resistance genes were positively correlated to the whole antibiotic resistance genes (r = 0.325, P<0.01).
Relationship of Environmental Factors to the Microbial Communities.
CCA was performed to correlate environmental variables with microbial community functional structure and determine major environmental variables in shaping the microbial community structure. On the basis of variance inflation factors, four variables were selected: DO, temperature, ammonia and COD (Figure 8). The results of CCA showed that the model was significant (P<0.05), suggesting that four variables were major environmental factor influencing microbial community functional structure. DO showed a strong positive correlation with the first axis and a negative correlation with the second axis, while ammonia and COD loading rate showed a strong positive correlation with the second axis and a negative correlation with the first axis. Temperature showed a strong negative correlation with both the first and second axes. Microbial community functional structures in WWTPs GBD and XHM were primarily linked to DO, while QH was linked to temperature. Ammonia and COD loading rate seemed to be major factors linking to microbial community functional structures in XJH.
Figure 8. Canonical correspondence analysis (CCA) of GeoChip data and environmental variables.

Environmental variables were chosen based on significance calculated from individual CCA data and variance inflation factors (VIFs).
Variance partitioning analyses (VPA) was further performed to assess contributions of wastewater characteristics and operational parameters to microbial community variances. Wastewater characteristics included influent concentrations of COD, TN, ammonia, TP, pH and conductivity. Operational parameters consisted of DO, temperature, hydraulic retention time (HRT) and mixed liquor suspended solids (MLSS). The results showed that 53% of total variances could be explained by these variables (Figure S8 in File S1). Wastewater characteristics and operational parameters can independently explain 25% and 23% of the variation of microbial communities, respectively. Interactions among the two major components (5%) appeared to be small.
Discussion
In this study, microbial community functional structures in activated sludges of four WWTPs were analyzed by GeoChip 4.2. A total of 29,124 functional genes involved in carbon, nitrogen, phosphorous and sulfur cycles, metal resistance, antibiotic resistance and organic contaminant degradation were detected. It is reasonable to expect that the high diversity of functional genes can increase the functional redundancy, which is ensured by the presence of a pool of species able to perform the same ecological function. That is to say, many species share similar traits in activated sludge, therefore the loss of a few species has little impact on the system performance. Functional redundancy of microbial community in activated sludge is important to ensure the system stability when individual species are lost due to environmental changes, such as significant changes in the influent wastewater composition [4].
For individual gene category, such as egl, amyA, lip, nirS, nirK, nosZ, ureC, ppx, ppk, aprA, dsrA, sox and benAB, high diversity were detected. For example, 110 nosZ genes were detected in this study. nosZ gene encodes nitrous oxide reductase that is the only enzyme to mediate the conversion of N2O to N2 during denitrification process, which makes the nosZ gene an important molecular marker to trace complete denitrification [28]. This high diversity of nosZ genes ensures the presence of different species with similar function mediating the conversion of N2O to N2, which could lead to a more stable denitrification in WWTPs. Similarly, the high diversity of egl, amyA, etc. can increase the functional redundancy, and ensure the stable operation of plants.
GeoChip data revealed high similarities among microbial community functional structures within activated sludge of four WWTPs as measured by diversity indices and overlapped genes. This can be partly explained by the fact that all the WWTPs studied were located in the same city (Beijing), used to treat similar domestic wastewater, and operated with the same anaerobic/anoxic/aerobic (A2O) process of similar operating parameters (COD and TN loading rate and MLSS concentration). For each gene category such as egl, amyA, lip, nirS, nirK, nosZ, ureC, ppx, ppk, aprA, dsrA, sox, benAB, there were a number of microorganisms commonly shared by all 12 samples, suggesting that there could be a core microbial community in activated sludge of four WWTPs. This agrees with the findings of zhang et al.[29], who suggested, based on pyrosequencing surveys of 14 WWTPs, that some core populations were shared by multiple samples. Similar results were also reported by other studies [30], [31].
Wastewater contains a broad spectrum of organic contaminants resulting from the mixing of wastewaters from different sources. Starch, cellulose, chitin and lignin could be the major carbon and energy sources for various microbial populations. Thus, we hypothesized that microbial communities would be abundant in functional genes involved in starch, cellulose, chitin and lignin. Our GeoChip data validated the hypothesis by evidence of detecting a wide variety of these genes across all samples such as egl, amyA, lip, xylanase genes, etc. Also, in WWTPs, biological nitrification coupled with denitrification is widely used to remove ammonia from wastewater. Nitrification is the main supply pathway of NO3 − for denitrification, and thus it is reasonable to expect the genes involved in nitrogen cycle would be tightly linked with NH4 + and NO3 − concentration. The results of Mantel test supported this expectation (Table S5 in File S1).
Understanding the factors that shape microbial community structure in WWTPs could potentially enhance treatment performance and control [11]. Of the 9 operational and environmental variables tested in this study, CCA ordination analysis indicated that DO was an important variable influencing microbial community structures. This agreed with the findings of Wells et al. [32], which suggested that DO was one of the most influential variables on microbial community structures of ammonia-oxidizing bacteria (AOB). Similar results have also been obtained in several lab-scale bioreactors by Park et al. [33].
Our results indicated that water temperature was also significantly linked to the microbial community structures in WWTPs, which was consistent with previous observations that water temperature was an important factor in shaping the microbial community structures. Ebrahimi et al. [34] found that, in two sequencing batch reactors (SBR) operated at 20°C, 30°C and 35°C, bacterial richness and diversity were clearly altered. In a membrane bioreactor treating domestic sewage, Ebie et al. [35] revealed shifts in AOB lineages in response to temperature changes by using denaturing gradient gel electrophoresis. Several other studies also demonstrated that temperature imposed a strong selective pressure on microbial communities of activated sludge [36]–[38].
In addition to DO and water temperature, ammonia concentrations within WWTPs were strongly and significantly linked to microbial community functional structures. Some previous studies have demonstrated the importance of ammonia concentration within bioreactor. In three gravel biofilters treating saline wastewater, Gregory et al. [39] observed that increasing ammonia concentration significantly influenced community structures of total bacteria and AOB. Miura et al. [40] found that ammonia concentration was an important structuring factor based on the DGGE analysis of AOB communities in four pilot-scale wastewater treatment systems with different ammonia concentrations.
The importance of COD loading rate for microbial community structures has been shown by previous studies. Pholchan et al. [41] reported that in laboratory-scale activated sludge reactors, high COD loading rate resulted in an increase and decrease of community diversity of total bacteria and AOB, respectively. In addition, Zhou et al. [42] showed that organic carbon (equivalent to COD loading rates) significantly affected microbial diversity in soil.
VPA was performed to attribute variations observed in microbial community functional structures to wastewater characteristics and operational parameters. VPA results showed that the deterministic wastewater characteristics and operational factors explained 53% of variances of microbial community functional structures, thus 47% of the variance was attributed to unknown factors. Some of them may result from unknown environmental variables or additional factors such as stochastic dispersal and immigration processes [43], [44], protozoan grazing [45], phage predation [46] and chaotic behavior [47], [48], which may play an influential role in shaping microbial communities in WWTPs.
Like other microarray technologies, GeoChip only detects genes that are represented on the array. Therefore, GeoChip 4.2 used in this study may not cover all of functional genes of microbial communities, though it contains as many as 83,992 probes targeting 152,414 genes involved in major microbial biogeochemical processes. Also, in our study, the DNA rather than cDNA from RNA was used for GeoChip hybridization. It is argued that DNA-based community analysis detects microbes irrespective of their viability or metabolic activity. Furthermore, DNA could persist extracellularly in environments after cell death, and it could result in biased population profiles [49].
In conclusion, GeoChip 4.2 was used to evaluate the microbial functional genes of activated sludge in four WWTPs. Our results revealed high similarities of microbial community functional structures among activated sludges of four WWTPs as measured by diversity indices and overlapped genes. CCA analysis indicated that microbial community functional structures were highly correlated to the water temperature, DO, ammonia concentrations and COD loading rate. Finally, a total of 53% of microbial community variations from GeoChip data were explainable by wastewater characteristics and operational parameters.
Supporting Information
This includes Tables S1–S5 and Figures S1–S8. Table S1 Summary of overall operating parameters of four WWTPs. Table S2 Metal concentrations within the four WWTPs. Table S3 Phylogenetic classification based on GeoChip data. Table S4 Numbers of detected genes involved in carbon cycling and total gene numbers present on GeoChip. Table S5 The relationship of different gene categories to related environmental variables revealed by Mantel test. Figure S1 Hierarchical cluster analysis of egl genes. Figure S2 The hierarchical cluster analysis of Xylanase genes. Figure S3 The hierarchical cluster analysis of lip genes. Figure S4 Hierarchical cluster analysis of phytase genes. Figure S5 The hierarchical cluster analysis of aprA genes. Figure S6 The normalized signal intensity of detected key genes involved in metal resistance. Figure S7 The normalized signal intensity of detected key genes involved in Antibiotic resistance. Figure S8 Variation partitioning analysis of microbial diversity explained by wastewater characteristics (W) and operational parameters (O).
(DOC)
Funding Statement
This study was supported by National Natural Science Foundation of China funds (51078207 and 51178239) and the special fund of State Key Joint Laboratory of Environment Simulation and Pollution Control (12L03ESPC). This study made use of the GeoChip and associated computational pipelines funded by ENIGMA-Ecosystems and Networks Integrated with Genes and Molecular Assemblies through the Office of Science, Office of Biological and Environmental Research, the U. S. Department of Energy under Contract No. DE-AC02-05CH11231. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Gentile ME, Jessup CM, Nyman JL, Criddle CS (2007) Correlation of functional instability and community dynamics in denitrifying dispersed-growth reactors. Appl Environ Microb 73: 680–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang XH, Wen XH, Yan HJ, Ding K, Hu M (2011) Community dynamics of ammonia oxidizing bacteria in a full-scale wastewater treatment system with nitrification stability. Front Environ Sci Engin China 5: 92–98. [Google Scholar]
- 3. Rittmann BE, Hausner M, Loffler F, Love NG, Muyzer G, et al. (2006) A vista for microbial ecology and environmental biotechnology. Environ Sci Technol 40: 1096–1103. [DOI] [PubMed] [Google Scholar]
- 4. Briones A, Raskin L (2003) Diversity and dynamics of microbial communities in engineered environments and their implications for process stability. Curr Opin Biotech 14: 270–276. [DOI] [PubMed] [Google Scholar]
- 5. Akarsubasi AT, Eyice O, Miskin I, Head IM, Curtis TP (2009) Effect of sludge age on the bacterial diversity of bench scale sequencing batch reactors. Environ Sci Technol 43: 2950–2956. [DOI] [PubMed] [Google Scholar]
- 6. Dytczak MA, Londry KL, Oleszkiewicz JA (2008) Activated sludge operational regime has significant impact on the type of nitrifying community and its nitrification rates. Water Res 42: 2320–2328. [DOI] [PubMed] [Google Scholar]
- 7. Huang ZH, Gedalanga PB, Asvapathanagul P, Olson BH (2010) Influence of physicochemical and operational parameters on Nitrobacter and Nitrospira communities in an aerobic activated sludge bioreactor. Water Res 44: 4351–4358. [DOI] [PubMed] [Google Scholar]
- 8. López-Vázquez CM, Hooijmans CM, Brdjanovic D, Gijzen HJ, van Loosdrecht MCM (2008) Factors affecting the microbial populations at full-scale enhanced biological phosphorus removal (EBPR) wastewater treatment plants in The Netherlands. Water Res 42: 2349–2360. [DOI] [PubMed] [Google Scholar]
- 9. Miura Y, Hiraiwa MN, Ito T, Itonaga T, Watanabe Y, et al. (2007) Bacterial community structures in MBRs treating municipal wastewater: relationship between community stability and reactor performance. Water Res 41: 627–637. [DOI] [PubMed] [Google Scholar]
- 10. Wang XH, Wen XH, Yan HJ, Ding K, Zhao F, et al. (2011) Bacterial community dynamics in a functionally stable pilot-scale wastewater treatment plant. Bioresource Technol 102: 2352–2357. [DOI] [PubMed] [Google Scholar]
- 11. Wang XH, Wen XH, Criddle C, Wells G, Zhang J, et al. (2010) Community analysis of ammonia-oxidizing bacteria in activated sludge of eight wastewater treatment systems. J Environ Sci-China 22: 627–634. [DOI] [PubMed] [Google Scholar]
- 12. Sanapareddy N, Hamp TJ, Gonzalez LC, Hilger HA, Fodor AA, et al. (2009) Molecular Diversity of a North Carolina Wastewater Treatment Plant as Revealed by Pyrosequencing. Appl Environ Microb 75: 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ye L, Zhang T, Wang T, Fang Z (2012) Microbial structures, functions, and metabolic pathways in wastewater treatment bioreactors revealed using high-throughput sequencing. Environ Sci Technol 46: 13244–13252. [DOI] [PubMed] [Google Scholar]
- 14. He Z, Deng Y, Van Nostrand JD, Tu Q, Xu M, et al. (2010) GeoChip 3.0 as a high-throughput tool for analyzing microbial community composition, structure and functional activity. ISME J 4: 1167–1179. [DOI] [PubMed] [Google Scholar]
- 15. Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, et al. (2010) Deep-Sea Oil Plume Enriches Indigenous Oil-Degrading Bacteria. Science 330: 204–208. [DOI] [PubMed] [Google Scholar]
- 16. Lu Z, Deng Y, Van Nostrand JD, He Z, Voordeckers J, et al. (2012) Microbial gene functions enriched in the Deepwater Horizon deep-sea oil plume. ISME J 6: 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou JZ, Kang S, Schadt CW, Garten CT (2008) Spatial scaling of functional gene diversity across various microbial taxa. Proc Natl Acad Sci USA 105: 7768–7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He ZL, Xu MY, Deng Y, Kang SH, Kellogg L, et al. (2010) Metagenomic analysis reveals a marked divergence in the structure of belowground microbial communities at elevated CO(2). Ecol Lett 13: 564–575. [DOI] [PubMed] [Google Scholar]
- 19. Van Nostrand JD, Wu LY, Wu WM, Huang ZJ, Gentry TJ, et al. (2011) Dynamics of Microbial Community Composition and Function during In Situ Bioremediation of a Uranium-Contaminated Aquifer. Appl Environ Microb 77: 3860–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hemme CL, Deng Y, Gentry TJ, Fields MW, Wu LY, et al. (2010) Metagenomic insights into evolution of a heavy metal-contaminated groundwater microbial community. ISME J 4: 660–672. [DOI] [PubMed] [Google Scholar]
- 21. Xie JP, He ZL, Liu XX, Liu XD, Van Nostrand JD, et al. (2011) GeoChip-Based Analysis of the Functional Gene Diversity and Metabolic Potential of Microbial Communities in Acid Mine Drainage. Appl Environ Microb 77: 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang F, Zhou H, Meng J, Peng X, Jiang L, et al. (2009) GeoChip-based analysis of metabolic diversity of microbial communities at the Juan de Fuca Ridge hydrothermal vent. Proc Natl Acad Sci USA 106: 4840–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Association) AAPH (2012) Standard Methods for the Examination of Water and Waste Water. 22th ed. APHA, Washington DC, USA.
- 24. Zhou JZ, Bruns MA, Tiedje JM (1996) DNA recovery from soils of diverse composition. Appl Environ Microb 62: 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95: 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.ter B, F CJ, Smilauer P (2002) CANOCO Reference Manual and CanoDraw for Windows User's Guide: Software for canonical community ordination (version 4.5). Microcomputer Power, Ithaca, New York, USA.
- 27. Wu L, Kellogg L, Devol AH, Tiedje JM, Zhou J (2008) Microarray-based characterization of microbial community functional structure and heterogeneity in marine sediments from the gulf of Mexico. Appl Environ Microb 74: 4516–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bergaust L, Bakken LR, Frostegard A (2011) Denitrification regulatory phenotype, a new term for the characterization of denitrifying bacteria. Biochem Soc Trans 39: 207–212. [DOI] [PubMed] [Google Scholar]
- 29. Zhang T, Shao MF, Ye L (2012) 454 Pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J 6: 1137–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang X, Hu M, Xia Y, Wen X, Ding K (2012) Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in china. Appl Environ Microb 78: 7042–7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xia SQ, Duan LA, Song YH, Li JX, Piceno YM, et al. (2010) Bacterial Community Structure in Geographically Distributed Biological Wastewater Treatment Reactors. Environ Sci Technol 44: 7391–7396. [DOI] [PubMed] [Google Scholar]
- 32. Wells GF, Park HD, Yeung CH, Eggleston B, Francis CA, et al. (2009) Ammonia-oxidizing communities in a highly aerated full-scale activated sludge bioreactor: betaproteobacterial dynamics and low relative abundance of Crenarchaea. Environ Microbiol 11: 2310–2328. [DOI] [PubMed] [Google Scholar]
- 33. Park HD, Noguera DR (2004) Evaluating the effect of dissolved oxygen on ammonia-oxidizing bacterial communities in activated sludge. Water Res 38: 3275–3286. [DOI] [PubMed] [Google Scholar]
- 34. Ebrahimi S, Gabus S, Rohrbach-Brandt E, Hosseini M, Rossi P, et al. (2010) Performance and microbial community composition dynamics of aerobic granular sludge from sequencing batch bubble column reactors operated at 20 A degrees C, 30 A degrees C, and 35 A degrees C. Appl Microbiol Biot 87: 1555–1568. [DOI] [PubMed] [Google Scholar]
- 35. Ebie Y, Matsumura M, Noda N, Tsuneda S, Hirata A, et al. (2002) Community analysis of nitrifying bacteria in. an advanced and compact Gappel-Johkasou by FISH and PCR-DGGE. Water Sci Technol 46: 105–111. [PubMed] [Google Scholar]
- 36. Li T, Bo L, Yang F, Zhang S, Wu Y, et al. (2012) Comparison of the removal of COD by a hybrid bioreactor at low and room temperature and the associated microbial characteristics. Bioresource Technol 108: 28–34. [DOI] [PubMed] [Google Scholar]
- 37. Szukics U, Abell GCJ, Hodl V, Mitter B, Sessitsch A, et al. (2010) Nitrifiers and denitrifiers respond rapidly to changed moisture and increasing temperature in a pristine forest soil. Fems Microbiol Ecol 72: 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Urakawa H, Tajima Y, Numata Y, Tsuneda S (2008) Low temperature decreases the phylogenetic diversity of ammonia-oxidizing archaea and bacteria in aquarium biofiltration systems. Appl Environ Microb 74: 894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gregory SP, Shields RJ, Fletcher DJ, Gatland P, Dyson PJ (2010) Bacterial community responses to increasing ammonia concentrations in model recirculating vertical flow saline biofilters. Ecol Eng 36: 1485–1491. [Google Scholar]
- 40. Lydmark P, Almstrand R, Samuelsson K, Mattsson A, Sorensson F, et al. (2007) Effects of environmental conditions on the nitrifying population dynamics in a pilot wastewater treatment plant. Environ Microbiol 9: 2220–2233. [DOI] [PubMed] [Google Scholar]
- 41. Pholchan MK, Baptista JD, Davenport RJ, Curtis TP (2010) Systematic study of the effect of operating variables on reactor performance and microbial diversity in laboratory-scale activated sludge reactors. Water Res 44: 1341–1352. [DOI] [PubMed] [Google Scholar]
- 42. Zhou JZ, Xia BC, Treves DS, Wu LY, Marsh TL, et al. (2002) Spatial and resource factors influencing high microbial diversity in soil. Appl Environ Microb 68: 326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Curtis TP, Sloan WT (2006) Towards the design of diversity: stochastic models for community assembly in wastewater treatment plants. Water Sci Technol 54: 227–236. [DOI] [PubMed] [Google Scholar]
- 44. Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, et al. (2006) Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ Microbiol 8: 732–740. [DOI] [PubMed] [Google Scholar]
- 45. Petropoulos P, Gilbride KA (2005) Nitrification in activated sludge batch reactors is linked to protozoan grazing of the bacterial population. Can J Microbiol 51: 791–799. [DOI] [PubMed] [Google Scholar]
- 46. Kunin V, He S, Warnecke F, Peterson SB, Martin HG, et al. (2008) A bacterial metapopulation adapts locally to phage predation despite global dispersal. Genome Res 18: 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Graham DW, Knapp CW, Van Vleck ES, Bloor K, Lane TB, et al. (2007) Experimental demonstration of chaotic instability in biological nitrification. ISME J 1: 385–393. [DOI] [PubMed] [Google Scholar]
- 48. Ofiteru ID, Lunn M, Curtis TP, Wells GF, Criddle CS, et al. (2010) Combined niche and neutral effects in a microbial wastewater treatment community. Proc Natl Acad Sci USA 107: 15345–15350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nogales B, Moore ER, Llobet-Brossa E, Rossello-Mora R, Amann R, et al. (2001) Combined use of 16 S ribosomal DNA and 16 S rRNA to study the bacterial community of polychlorinated biphenyl-polluted soil. Appl Environ Microbiol 67: 1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This includes Tables S1–S5 and Figures S1–S8. Table S1 Summary of overall operating parameters of four WWTPs. Table S2 Metal concentrations within the four WWTPs. Table S3 Phylogenetic classification based on GeoChip data. Table S4 Numbers of detected genes involved in carbon cycling and total gene numbers present on GeoChip. Table S5 The relationship of different gene categories to related environmental variables revealed by Mantel test. Figure S1 Hierarchical cluster analysis of egl genes. Figure S2 The hierarchical cluster analysis of Xylanase genes. Figure S3 The hierarchical cluster analysis of lip genes. Figure S4 Hierarchical cluster analysis of phytase genes. Figure S5 The hierarchical cluster analysis of aprA genes. Figure S6 The normalized signal intensity of detected key genes involved in metal resistance. Figure S7 The normalized signal intensity of detected key genes involved in Antibiotic resistance. Figure S8 Variation partitioning analysis of microbial diversity explained by wastewater characteristics (W) and operational parameters (O).
(DOC)