

Abstract
The molecular regulation of horn growth in ruminants is still poorly understood. To investigate this process, we collected 1019 hornless (polled) animals from different cattle breeds. High-density SNP genotyping confirmed the presence of two different polled associated haplotypes in Simmental and Holstein cattle co-localized on BTA 1. We refined the critical region of the Simmental polled mutation to 212 kb and identified an overlapping region of 932 kb containing the Holstein polled mutation. Subsequently, whole genome sequencing of polled Simmental and Holstein cows was used to determine polled associated genomic variants. By genotyping larger cohorts of animals with known horn status we found a single perfectly associated insertion/deletion variant in Simmental and other beef cattle confirming the recently published possible Celtic polled mutation. We identified a total of 182 sequence variants as candidate mutations for polledness in Holstein cattle, including an 80 kb genomic duplication and three SNPs reported before. For the first time we showed that hornless cattle with scurs are obligate heterozygous for one of the polled mutations. This is in contrast to published complex inheritance models for the bovine scurs phenotype. Studying differential expression of the annotated genes and loci within the mapped region on BTA 1 revealed a locus (LOC100848215), known in cow and buffalo only, which is higher expressed in fetal tissue of wildtype horn buds compared to tissue of polled fetuses. This implicates that the presence of this long noncoding RNA is a prerequisite for horn bud formation. In addition, both transcripts associated with polledness in goat and sheep (FOXL2 and RXFP2), show an overexpression in horn buds confirming their importance during horn development in cattle.
Introduction
Permanent horns (Figure 1A) are a typical feature of domesticated ruminants like cattle, sheep and goats. Consisting of an outer keratin-layer and a bony pneumatized core [1] they are important for the animal's self-defense in wildlife. Nevertheless there is evidence for the existence of hornless cattle until back to ancient times, as for example shown in several Old Egyptian tomb sceneries [2]. Besides the absence of horn growth (polledness, Figure 1C) these animals may show atypical eyelashes and defects of the genital tract in males [3].
Figure 1. Horn phenotypes in Simmental cattle.
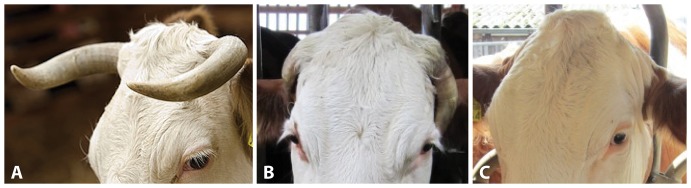
Normally horned cow (A), cow with loosely attached small horns termed scurs (B), smooth polled cow showing the typical peaked shape of the proximal frontal bone (C).
Since there is no need for self-defense in modern intensive cattle production, and new housing systems as free stalls with headlock barriers have been established, horns have become an undesired trait in beef and dairy cattle. To avoid the risk of injury to humans and animals, most of the cattle are dehorned at a young age. Due to animal welfare [4] and economic reasons, efforts are taken to breed hornless (polled) cattle. There are several cattle breeds such as Angus and Galloway, which are fixed for the autosomal dominant polled allele [5]–[7]. In other breeds natural polledness has until now just been accomplished in a small part of the populations by cross breeding or selection of polled individuals.
The polled locus (P) has previously been mapped on the proximal end of BTA 1 [8]–[13] corresponding to a region on HSA 21 [14]. The apparent causative mutation for polledness in various beef or dual-purpose cattle breeds with a Celtic origin has been described as a structural sequence variant, a complex insertion-deletion affecting an intergenic region of BTA 1 [15]. Studying the polled mutation in cattle with a Friesian origin like Holstein and Jersey showed allelic heterogeneity. A second polled associated haplotype has been detected and four intergenic candidate variants, three SNPs and an 80 kb duplication, are discussed as possibly causative for the Friesian polledness [15]. Recently, a SNP within intron 3 of IFGR2 has been described to be in perfect association with polledness in Holstein cattle by another group [16]. A further dominantly inherited polled phenotype accompanied by congenital malformations has been shown to be caused by a ZEB2 mutation in French Charolais cattle [17]. This report confirms the assumption that other mutations beside the characterized polled alleles on BTA 1 may affect horn growth in cattle. In goats, the polled intersex syndrome (PIS) is caused by the deletion of an 11.7 kb DNA element located at goat chromosome 1q43, which affects the transcription of at least two flanking genes, FOXL2 and a noncoding RNA [18], [19]. A polymorphism at RXFP2 explains horn variability in sheep [20], [21].
A second locus affecting horn growth in cattle is called Scurs (Sc) [5], [6]. This interacting second mutation causes scurs in cattle which carry the polled mutation. Scurs are corneous growths of different sizes from crusts up to big horn-like formations, which develop in the same area as horns but are not firmly attached to the skull (Figure 1B). In Angus and Galloway scurs have been described to develop depending on the sex and polled genotype [6]. Whereas homozygous polled animals develop scurs only if they also carry the Sc mutation in a homozygous state, heterozygous polled females develop scurs only if they carry the Sc mutation in a homozygous state. Heterozygous polled males develop scurs in the presence of one or two Sc alleles [6]. The mode of inheritance of the scurs mutation is still under debate [6], [22]. One report stated that the scurs locus maps to BTA 19 in Canadian beef cattle based on linkage analysis [23]. This was not confirmed in French Charolais cattle showing polledness and scurs [22]. A recent study in French Charolais cattle showing a scurs-like phenotype revealed a dominantly inherited causative TWIST1 mutation in the absence of the polled mutation [24].
Comparative studies were performed to uncover differential gene expression associated with the development of horns and scurs in cattle [3], [25]. Microarray based differential expression studies of epidermal and dermal tissues from the skull region of newborn calves revealed no differentially expressed gene in the region of the polled locus on BTA 1 [25]. Recently, differential gene expression analysis has been performed in horn buds and frontal skin of horned and polled fetuses 90 days post-coitum using quantitative RT-PCR [3]. This study was focussed on the expression of known genes involved in ruminant horn growth and annotated genes of the polled locus on BTA 1. An increased expression of a long intergenic noncoding RNA (LincRNA#1) located at BTA 1 and a reduced expression of RXFP2 was shown in polled fetuses indicating their role during horn bud agenesis. In addition, OLIG2 located at BTA 1 and FOXL2 showed different expression levels between the horn buds and frontal skin in polled as well as in wildtype fetuses.
In the present study we describe our efforts to identify the causative variants for polledness in the Simmental and Holstein breed, which were carried out independently from the recently published studies [3], [15]. Furthermore, we analysed the gene expression to evaluate differentially expressed transcripts in wildtype and polled fetuses during development.
Results
Two different polled associated haplotypes are co-localized on BTA 1
We performed genome-wide homozygosity mapping based on genotypes of 19 progeny tested homozygous polled (PP) Simmental bulls genotyped with illumina's bovine HD BeadChip comprising 777,962 SNP markers. Thereby, a 441 kb interval of shared homozygosity (BTA 1: 1,582,828–2,023,687; UMD 3.1 assembly) was identified within the previously mapped polled interval. We searched for possible recombinations by manual comparison of the SNP data of 108 genotyped obligate heterozygous polled Simmental cattle with the previously defined polled associated 441 kb haplotype present in the homozygous polled animals. We identified one copy of the entire polled haplotype in each animal and no evidence for further crossing over events. Subsequently, SNP genotypes of nine progeny tested PP bulls from other beef and dual-purpose breeds were included in the analysis. In seven out of nine bulls homozygosity for the identical 441 kb polled associated haplotype seen in Simmental was found. However, a polled Blonde d'Aquitaine bull carried a recombinant haplotype adjusting the proximal border of the critical interval to BTA 1: 1,684,495. A Braunvieh bull showed a recombination at BTA 1: 1,896,112 refining the distal border of the polled interval. Thus – assuming that it resides on the common haplotype block – the causative polled mutation in these beef breeds is located within a 212 kb interval on BTA 1 (Figure 2). The same approach was applied for three progeny tested homozygous polled (PP) and 29 obligate heterozygous polled (Pp) bulls of the Holstein breed. Within the previously mapped region on BTA 1 the three PP Holstein bulls shared a homozygous haplotype block of 1,606 kb (BTA 1: 903,971–2,509,966). Twenty-eight out of the 29 Pp bulls carried one copy of this haplotype. One of the heterozygous Holstein bulls had a recombinant copy of this polled associated haplotype and in comparison to the PP bulls it was homozygous for the opposite SNP alleles at several positions proximal of BTA 1: 1,578,430 (Figure 2). Thus, we mapped the polled mutation within the Holstein breed to a 932 kb region on BTA 1. Taken together, the analyses revealed two different polled associated haplotypes overlapping within the same chromosomal region on BTA 1.
Figure 2. Homozygosity mapping on BTA 1.

SNP genotypes of BTA 1 markers are presented as vertical bars. The dark grey segments represent homozygous blocks with shared alleles. A total of 28 progeny tested homozygous polled bulls belonging to beef and dual-purpose breeds of Celtic origin are shown on the left (SI: Simmental, LI: Limousin, CH: Charolais; HF: Hereford, PG: Pinzgauer; BA: Blonde d'Aquitaine, BV: Braunvieh). Some recombinations were observed in more than one animal, the number of animals is displayed above the chromosome bar. The haplotype analysis suggests the position of the Celtic polled mutation within a 212 kb interval shown in red. The annotated genes and loci on the BTA 1 segment (UMD3.1 assembly) are shown in the center. Three progeny tested homozygous polled bulls and a single heterozygous polled bull belonging to the Holstein (HO) breed of Friesian origin are shown on the right. The 932 kb critical region of the Friesian polled mutation is indicated in blue.
Whole genome re-sequencing reveals candidate causal mutations for polledness
We performed whole genome re-sequencing of one homozygous polled (PP) Simmental (39.3x mean coverage), one heterozygous (Pp) Simmental (21.8x mean coverage) and 1 horned Simmental cattle (23.7x mean coverage). SNPs, short insertions and deletions, and structural variants were called with respect to the UMD3.1 cow reference genome sequence of a horned Hereford cow. The PP cow carried 121 homozygous sequence variants within the mapped 212 kb interval. Subsequent filtering for variants that were heterozygous in the Pp cow and absent in the horned Simmental cow revealed four associated intergenic variants. The variants were further compared with 33 cow genomes of various horned breeds and 2 genomes of hornless Galloways, one PP and one Pp, that had been sequenced in our laboratory in the course of other ongoing studies. Assuming that the mutant allele of the causative variant should be completely absent in horned cattle and present in the polled Galloways, a single polled associated variant at BTA 1: 1,706,044 was identified. Sanger sequencing of this variant revealed a duplication of 208 bp (BTA 1: 1,705,837–1,706,044), inserted after 10 bp of the wildtype sequence at BTA 1: 1,706,054 in combination with a 6 bp deletion (BTA 1: 1,706,055–1,706,060) designated as complex insertion-deletion (indel, Figure S1A). Subsequently, this indel variant was genotyped in a larger cohort of polled and horned cattle from different breeds using fragment length analysis (Figure S1B). Genotyping 2,329 cattle from 30 breeds this complex indel was found to be in perfect association with the polled locus in various breeds like Angus, Galloway, Blonde d'Aquitaine, Braunvieh, Hereford, Norwegian Red and Pinzgauer (Table S1). Among 403 polled Simmental cattle we identified one single hornless cow with a crust at one side, that did not carry the variant indel allele. In addition, we observed two polled Limpurger animals with scurs and one polled Yak, which also did not carry the variant indel allele. A total of 239 out of 252 polled Limousin and Limousin crosses and 15 out of 16 polled Charolais cattle carried one or two copies of the variant indel allele. The remaining 14 polled Limousin and Charolais animals carried a copy of the second polled associated haplotype found in Holstein cattle (see below). In contrast, among polled Holstein cattle only six out of 167 genotyped animals were found to carry the variant indel allele.
To detect possible causal variants for the second polled associated haplotype the whole genome of a single homozygous PP Holstein cow was sequenced at 10.1x mean coverage. Within the critical 932 kb interval there were 1,769 variants. Subtracting variants observed in the 33 horned control cattle from different breeds, there were 191 intergenic variants left (Table S2). For subsequent genotyping of more than 400 horned and more than 80 polled Holstein cattle (Table S2) we focused on the 43 variants located within the critical region of 441 kb initially mapped in Simmental cattle, as we assumed that the second polled mutation in Holstein should affect the same chromosome segment as the Simmental mutation and as it is the segment within which the (Holstein) polled mutation had also been mapped in a previous publication [3]. This approach enabled us to exclude 9 variants as they occurred also in horned animals. Finally there were 34 variants left that showed perfect association with polledness in Holstein cattle: 31 SNPs, a 1 bp deletion, a short 5 bp deletion/12 bp insertion, and an 80 kb genomic duplication (Table S2). As the large duplication was identified by other groups before [15], we re-analyzed it by Sanger sequencing to determine its precise structure. It represents a tandem duplication of 80,129 bp (BTA 1: 1,909,352–1,989,480) combined with two 2 bp deletions (BTA 1: 1,909,354–1,909,355delTG and BTA 1: 1,909,396–1,909,397delTG) in the duplicated copy (Figure S2). In addition to these 34 variants there were still 148 variants located within the distal region of the mapped 932 kb haplotype. Beside in hornless Holstein cattle the polled Holstein haplotype was found in 20 Pp Limousin and Charolais beef cattle, the aforementioned 14 animals not carrying the Simmental indel plus six Limousin cattle carrying both polled mutations, the indel and the Holstein haplotype, thus being compound polled PP homozygotes. The three hornless cattle of the Simmental and Limpurger breed and the polled Yak mentioned above were tested negative for the presence of the sequence variants associated with polledness in Holstein. Finally, we genotyped a recently published IFNGR2 SNP (BTA 1: 1,390,292, Table S3) supposed to be perfectly associated with the Holstein polled mutation [16] in 160 polled Holstein cattle and showed that 4 polled animals (one hornless cow with two polled offspring and one progeny-tested polled bull) did not carry the variant SNP allele. These animals were genotyped as heterozygous carriers of several polled associated Holstein variants and did not carry the previously identified indel of polled Simmental cattle. In addition, the variant IFNGR2 allele was also present in heterozygous state in one of the sequenced control genomes of a horned Brown Swiss cow.
Male and female cattle with scurs are always heterozygous Pp
In regard to a possible influence of the individual polled genotype on the expression of scurs we determined the polled genotype in 207 scurred animals. The animals were tested for the indel mutation found in beef and dual-purpose breeds and for the C>A SNP at BTA 1: 1′768′587 associated to the polled mutation in Holstein cattle. Animals were classified as scurred if they showed crusts or horn-like formations that were not firmly attached to the skull. This clearly revealed that regardless of the kind of polled mutation and regardless of the breed and sex, scurred animals were heterozygous Pp whereas all the homozygous polled animals were smoothly polled without any visible signs of scurs (Table 1).
Table 1. Relation of the polled genotype in regard to the expression of scurs in animals of different breeds carrying the indel variant or the variants associated with the hornless mutation derived in the Holstein breed.
| breed | phenotype | male | male | female | female | total | total |
| Pp | PP | Pp | PP | Pp | PP | ||
| with indel variant | scurred | ||||||
| Angus | 1 | 1 | 2 | ||||
| Braunvieh | 1 | 1 | |||||
| Blonde d'Aquitaine | 1 | ||||||
| Charolais | 4 | 4 | |||||
| Holstein | 5 | 5 | |||||
| Limousin | 13 | 16 | 29 | ||||
| Simmental | 38 | 92 | 130 | ||||
| total | 63 | 109 | 172 | ||||
| with indel variant | polled | ||||||
| Angus | 4 | 4 | |||||
| Braunvieh | 3 | 1 | 3 | 1 | |||
| Charolais | 2 | 1 | 2 | 1 | |||
| Galloway | 1 | 8 | 1 | 2 | 8 | ||
| Holstein | 1 | 1 | |||||
| Limousin | 42 | 11 | 110 | 29 | 152 | 40 | |
| Pinzgauer | 6 | 6 | |||||
| Simmental | 23 | 51 | 119 | 68 | 142 | 119 | |
| total | 78 | 72 | 234 | 97 | 312 | 169 | |
| with Holstein polled variants | scurred | ||||||
| Holstein | 30 | 3 | 33 | ||||
| Limousin | 1 | 1 | 2 | ||||
| total | 31 | 4 | 35 | ||||
| with Holstein polled variants | polled | ||||||
| Charolais | 1 | 1 | |||||
| Pinzgauer | 12 | 3 | 75 | 18 | 87 | 21 | |
| Limousin | 2 | 8 | 10 | ||||
| total | 14 | 4 | 83 | 18 | 97 | 22 |
Gene expression studies reveal known and new horn development specific candidates
Initially, we performed total mRNA sequencing (RNA-Seq) of skin biopsies derived from one polled and one horned fetus to study possible effects of the polled mutation on gene expression. The horned fetus with ∼32 cm crown-rump length, corresponding to an age of approximately 150 days post fertilization [26], and macroscopically visible horn buds was confirmed to be wildtype for both, the Simmental indel and the Holstein associated variants. The polled fetus, ∼35 cm from crown to rump and therefore approximately 158 days post fertilization, with a smooth skin without any visible horn buds, was homozygous PP for the Simmental indel. Both fetuses were tested wildtype for the C>A SNP at BTA 1: 1′768′587 and the 80 kb duplication associated with polledness in Holstein. Biopsies from the horn bud of the wildtype fetus and from that same skin area in the PP fetus were used. Mapping the obtained sequence reads to the UMD3.1 cow assembly showed a large number of differentially expressed genes (Table S5). Among the differentially expressed (P value <0.05) transcripts were three genes located within the critical region on BTA 1 (OLIG1, OLIG2, C1H21orf62) and two genes known to be involved in horn development of sheep and goat (RXFP2 and FOXL2). Visual inspection of the mapped reads within the critical BTA 1 region harboring the polled mutations revealed evidence for a spliced transcript at position BTA 1: 1,898,100 to 1,899,400 (spliced between position BTA 1: 1,898,193 and 1,899,327; Figure S3). These reads were present in the horned fetus only and correspond to an uncharacterized locus annotated as LOC100848215. The splicing pattern of the obtained sequence reads are similar to those of three ESTs (EH130782, EH138227, EV693397) used for the computer predicted annotation at UCSC genome browser [27]. Cross-species dataset comparison revealed known transcripts of this locus in cow and buffalo only (Figure S4).
In a second experiment, the gene expression analysis was extended to samples of different developmental stages. Quantitative RT-PCR (qRT-PCR) was performed for six transcripts that were found to be differentially expressed in the aforementioned experiment. Biopsies were taken from horn bud tissue and frontal skin of 21 fetuses measuring 6.8 up to 44 cm from crown to rump (Figure 3). According to the developmental stages, the fetuses were divided into eight case-control groups of matching age. The youngest group comprised two wildtype fetuses and five fetuses carrying at least one copy of the Simmental indel, the older groups consisted of one wildtype and polled fetus each. All the polled fetuses were carrying a copy of the indel variants, none of the fetuses carried the variants associated with polledness in Holstein. Expression was detectable for five out of the six examined transcripts expression, while it was not possible to amplify the OLIG1 transcripts in the younger fetuses using different sets of primers. C1H21orf62 was found to be less expressed in horn buds compared to frontal skin regardless of the polled genotype (Figure S5). OLIG2 was found to be higher expressed in the early developmental stages compared to older fetuses (Figure S6), whereas no visible difference between the genotypes could be observed across the different stages. FOXL2 was found to be strongly over-expressed in all the wildtype horn buds compared to the respective wildtype frontal skin area (Figure 3A). The expression of FOXL2 decreased with increasing age in wildtype fetuses. Within the polled fetuses FOXL2 showed the same tendency to be higher expressed in the “horn bud” tissue biopsies compared to frontal skin, but the findings in the polled fetuses were not as strong and consistent as in the wildtype fetuses. In general, the expression levels of FOXL2 were lower in the polled fetuses. As seen in the RNA-Seq experiment before, RXFP2 was found to be highly expressed in wildtype horn buds. Expression was also detectable at lower levels in polled fetuses, where it was higher expressed in the area of the horn bud compared to frontal skin (Figure 3B). The LOC100848215 was generally found to be less expressed in the polled fetuses than in the wildtype fetuses regardless of the tissue. In two individual fetuses, a 70 days old PP fetus and a 140 days old Pp fetus, the expression pattern was comparable with those of the age-matching wildtype fetuses. Expression levels in three out of five homozygous polled fetuses were below the detection threshold. In the horned fetuses we observed a trend of decreasing LOC100848215 expression level with increasing age of the fetuses and a higher expression in the horn bud compared to the frontal skin (Figure 3C).
Figure 3. Gene expression study based on RT-PCR.

Relative expression of FOXL2 (A), RXFP2 (B), and LOC100848215 (C) transcripts in fetal skin and horn bud biopsies of different developmental stages and different polled genotypes. Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are normalized to the average of three control genes and shown as relative expression in relation to the frontal skin sample of the youngest horned fetus.
Discussion
Mapping and mutation analysis
Using high-density SNP genotyping to fine map the bovine polled mutation showed the presence of two independent polled associated haplotypes in different cattle breeds. Haplotype analyses revealed a 212 kb polled associated haplotype present in many beef and dual-purpose cattle breeds like Simmental, Angus, Galloway. Thus we were able to improve previous mappings based on illumina's bovineSNP50 BeadChip, where the polled locus had been mapped on a 381 kb interval in a mixed breed approach including Holstein and beef breeds [13], [15] and to a 400 kb interval including four different polled haplotypes from Holstein and Charolais cattle respectively [3]. The small size of the mapped haplotypes suggests that the polled mutation occurred a long time ago as stated by Allais-Bonnet et al. [3]. In contrast to previous studies [3], [15] we mapped the polled mutation in Holstein cattle separately and ended up with a 932 kb polled associated interval. This critical interval includes the mapped interval of 212 kb in polled beef cattle and contains 14 annotated genes and loci.
Whole genome re-sequencing of selected polled and horned individuals from the Simmental and Holstein breed was used for mutation analysis. Filtering of variants present in controls of different breeds was performed to identify sequence variants private to polled animals. In Simmental, we were left with only one single private variant, a complex indel within the critical interval. This confirms other recently published results [3], [15]. Using Sanger sequencing, for the first time the precise architecture of this sequence variant was determined (Figure S1). Genotyping more than 2,000 animals the indel was found to be in perfect association with polledness in Angus, Galloway, Blonde d'Aquitaine, Braunvieh, Hereford, Norwegian Red and Pinzgauer cattle. As the indel variant is present in polled Scottish beef breeds like Angus and Galloway, it is very likely that the polled allele in beef and related dual-purpose breeds originates in these Scottish breeds and was introduced through crossbreeding to other breeds long time ago. Therefore, we agree with Medugorac et al. who designated this polled mutation as Celtic [15]. In Limousin and Charolais cattle the majority of polled animals carry the indel variant, nevertheless there are some polled and scurred animals, which do not carry the indel but do carry the polled associated haplotype of Holstein cattle, indicating that polledness in these breeds obviously is due to different introgression events from different breeds. In Limousin, we found six polled animals being compound homozygotes carrying the Simmental indel as well as the Holstein polled haplotype. This indicates that both polled alleles segregate within these two breeds. In Simmental cattle, there was one hornless cow out of 403 polled animals which did not carry a copy of the indel variant. Based on pedigree data this cow was found to have horned parents and the owner confirmed that this animal never has been dehorned. Therefore, we speculate that its polledness represents a phenocopy e.g. due to mosaicism without an effect on the germ cell line as no transmission of the polled trait has been observed in a total of 22 offspring of this cow. In addition, two hornless Limpurger cattle, a bull with horned parents and its male offspring, both scurred neither carry the indel variant and nor the polled associated Holstein haplotype. Transmission of the polled phenotype from father to son suggests the presence of an inherited de novo mutation leading to polledness. Recently, also in the French Charolais cattle population independent polled phenotypes have been reported. Capitan et al. states to have additional cases of newly occurring horn anomalies, which cannot be explained by the known polled associated variants, indicating that de novo mutations affecting horn growth sporadically occur [17], [24]. The genotyped individual polled yak from Switzerland doesn't carry any of the polled variants reported in this paper. This indicates the presence of an independent polled mutation. According to a recent study the polled mutation in domestic yaks was mapped to the same genomic region on BTA 1 [28].
In the critical Holstein polled interval of 931 kb there are a total of 182 private associated candidate variants. Due to the publication of another group [3], the identical phenotype, the initial mapping and the findings in other breeds, we considered the overlapping BTA 1 segment more likely to contain the causal mutation assuming that this independent mutation affects the same gene or regulatory element. Therefore we focused on genotyping the variants within a 441 kb region originally mapped in Simmental and still ended up with a total of 34 variants associated with polledness in Holstein. The majority of these variants (Table S2) were detected and reported to be ruled out by Allais-Bonnet [3] by genotyping eight of them in recombinant animals. Nevertheless in our cohort we genotyped all of them and they were perfectly associated with polledness in more than 80 polled Holstein and not present in over 400 horned (mainly Holstein) cattle. At the moment it is impossible to conclusively prove, which variant is responsible for polledness in Holstein. However, we speculate that, due to its large structure and possible regulatory effect, an 80 kb duplication is most likely the causative variant. Nevertheless the causative variant in Holstein could theoretically also be one of the 181 further sequence variants. A recently published IFNGR2 associated SNP [16] could be clearly ruled out as it was not perfectly associated with the polled phenotypes in our Holstein cohort (Table S3). In addition it maps outside of the critical region on BTA 1.
Polled and scurs
Two types of horns are known in cattle: regular horns attached to the skull and so called scurs or “wiggle horns” in German, which refers to small loosely attached horns. For the first time we were able to compare the accurate polled genotype with recorded horn phenotype details. The fact that all 207 scurred animals are heterozygous for one of the polled mutations indicates epistasis of the polled and scurs loci. Polled is epistatic over scurs and in homozygous P/P animals scurs cannot be expressed. A total of 191 homozygous polled cattle showed no signs of scurs. Therefore, our data did not confirm previous models where homozygous polled cattle being homozygous for the scurs mutation were described to develop scurs [6]. Comparing the expression of scurs in heterozygous polled animals between male and female, it is conspicuous that especially in the Holstein breed more than two third of the male sampled Pp animals show scurs whereas in female almost all of the Pp animals are smooth polled. This tendency is apparent in the Simmental breed as well, but not as striking as in the Holstein breed (Table 1). This observation has to be taken with caution as the sampling was not representative for the whole population and there might be a bias, as we were specifically looking for scurred individuals. Although previous studies indicate an autosomal locus on BTA19 [23], an X-chromosomal recessive mode of inheritance would explain these observations. In different published inheritance models [5]–[7], female individuals are thought to develop scurs only if they are homozygous, but males also in the heterozygous state. It might be assumed that these males are in fact hemizygous. Nevertheless, there are still several inconsistencies with this inheritance model reported in the literature [6]. Many of those arise from the absence of scurs that would have been expected based on the pedigree. This might yet be explained by misclassified phenotypes or incomplete penetrance, especially in males. However, there is no clear evidence for X-linked inheritance. Alternatively, genetic heterogeneity, as reported or proposed for other cattle populations [22], [24], might also explain the observed mode of inheritance.
Expression differences during development between polled and wildtype fetuses
The Simmental polled variant observed in almost all hornless beef and dual-purpose cattle is located in an intergenic region and therefore the question of the functional consequences remains open. In polled goats a genomic deletion affects the transcription of neighbouring genes including a previously unknown long noncoding RNA [19]. First histological changes of bovine horn buds take place at a neck-rump length of 5.3 cm, with approximately 70 days of gestation, as a thickening of the epidermis [29], indicating that key genes involved in horn development already have to be expressed in the first trimester of gestation. To study possible regulatory effects we collected samples of developing cattle fetuses of eight different developmental stages from 70 to 175 days with and without visible horn buds. Genotyping confirmed the presence of one or two copies of the indel mutation in the polled fetuses. In an initial RNA-Seq experiment we identified a large number of differentially expressed genes by comparing the expression patterns in horn bud tissue of a single polled and a single horned fetus, both at approximately 5 months of gestation. Subsequently we performed qRT-PCR based gene expression studies to evaluate differential expressed transcripts in wildtype and polled fetuses during development. We focused on transcripts of two genes (FOXL2 and RXFP2) affected by polled causing mutations in small ruminants and the genes of the mapped polled region, that were found to be differentially expressed in the RNA-Seq experiment (OLIG1, OLIG2, C1H21orf62) plus a single uncharacterized locus (LOC100848215) of the polled associated region on BTA 1.
The genes FOXL2 and RXFP2 were clearly down-regulated in the polled horn bud compared to the wildtype horn bud in the RNA-Seq experiment. Quantitative RT-PCR confirmed these findings, as these two genes were higher expressed in wildtype horn buds compared to all the other tissues in different stages of the fetal development. Interestingly there was also a differential expression within the polled samples, where both of the genes were higher expressed in the polled horn bud than in the polled frontal skin. Allais-Bonnet et al. [3] showed the same tendency in 90 days old fetuses, where RXFP2 was described to be up-regulated in wildtype horn buds compared to Pp horn buds and FOXL2 was described to be up-regulated in horn buds of both genotypes compared to frontal skin. The fact that these genes are known to be involved in horn development in goat and sheep [19], [21] together with the expression patterns we found in wildtype and polled fetuses, suggests their involvement during horn bud formation in cattle.
The BTA 1 genes OLIG1 and OLIG2 encode transcription factors supposedly involved in oligodendrocyte differentiation and maturation [30]–[32]. We found them to be up-regulated in the wildtype horn bud in the RNA-Seq experiment. In further qRT-PCR expression studies in younger fetuses it was not possible to detect expression of OLIG1. For OLIG2 we couldn't confirm the findings of the RNA-Seq experiment in younger fetuses. OLIG2 was found to be higher expressed in younger stages generally, but there was no constant expression difference between genotypes or tissues across different stages. This stands in contrast to a recent report, where OLIG2 was described to be higher expressed in frontal skin than in horn buds from 90 days old fetuses [3]. The C1H21orf62, a protein coding gene of unknown function, was the only positional candidate shown to be up-regulated in the polled fetal tissue compared to the wildtype horn bud tissue in the RNA-Seq experiment. Quantitative RT-PCR didn't confirm this finding as no constant difference in expression pattern between polled and wildtype fetuses was observed across the studied developmental stages. This is in agreement with Allais-Bonnet et al. who also did not observe differential expression of C1H21orf62 [3]. Nevertheless qRT-PCR indicates that C1H21orf62 is generally higher expressed in frontal skin than in horn buds regardless of the polled genotype.
In a previous study Allais-Bonnet et al. [3] reported two long noncoding RNA (LincRNA#1 and LincRNA#2) both located within the mapped polled region on BTA 1. The annotated locus LOC100848368 corresponds perfectly to LincRNA#1 whereas only 4.7 kb in the 3′ region of the 74 kb spanning LincRNA#2 overlaps to the four annotated exons of LOC100848215. The LincRNA#1 was described to be significantly overexpressed in horn bud of polled fetuses compared to frontal skin of the polled fetuses and compared to the horn bud and frontal skin of wildtype fetuses whereas the LincRNA#2 was not reported to be differentially expressed using qRT-PCR [3]. In the fetal samples of our study LincRNA#1 (or LOC100848368) was not detectable, neither by RNA-Seq nor by qRT-PCR using the published primers [3]. Interestingly, our RNA-Seq experiment revealed some spliced reads covering two exons of LOC100848215 in the wildtype horn bud sample only. Quantitative RT-PCR confirmed the presence of LOC100848215 expression in our samples, especially in both horn bud and frontal skin tissues of wildtype fetuses. In the samples of polled fetuses LOC100848215 expression was usually lower and even not detectable at all in biopsies from three out of five homozygous polled (PP) fetuses. Allais-Bonnet et al. [3] designated the position of LincRNA#2 based on several spliced ESTs, which span more than 74 kb. If primers of that study (whose sequences were not published) were designed within the 5′ region, the actual transcripts could possibly have been missed, as our RNA-Seq data shows reads in the distal segment of the last two annotated exons only (BTA 1: 1,898,100–1,899,400). Long noncoding RNAs have the potential to play important regulatory roles, as their sequence structure allows them to regulate transcription in an allele- and locus-specific manner [33]. A recent deep transcriptome sequencing study revealed a large number of long noncoding RNAs in bovine skin samples, revealing an unexplored reservoir of novel possibly functional RNAs [34]. So far there are no known functions of the LOC100848215 and interestingly ESTs are found for cow and buffalo only (Figure S4). Therefore this RNA seems to be specific for horned ruminants. The physical proximity of LOC100848215 to the sites of the different polled mutations (Figure 2) and its decreased or even absent expression in the presence of one or two copies of the polled allele together with its apparent ruminant specificity makes it a very interesting candidate. Therefore we speculate that this long noncoding RNA might play a significant role in horn growth. As the presence of the dominant polled allele inhibits horn bud formation probably by influencing the expression of genes required for horn development (FOXL2, RXFP2, LOC100848215) the functional effect could be due to haploinsufficiency of the involved genes. The performed RNA-Seq experiment indicates that numerous other transcripts are differently expressed between wildtype horned and polled fetuses. Therefore we plan to perform future more comprehensive RNA-Seq experiments comparing the expression pattern across different developmental stages and between different genotypes to specify these findings.
Conclusion
We independently identified a complicated indel confirming the recently published Celtic polled mutation as the causative variant for polledness in Simmental and other beef and dual-purpose cattle. In addition, we confirmed the presence of an independent polled associated haplotype in Holstein cattle corresponding to the assumed Friesian polled allele. In addition to the four reported possible causative variants we provide a list of 182 sequence variants perfectly associated with the polled mutation in Holstein cattle. We found evidence for sporadic occurrence of de novo mutations leading to polledness. Comparing the genotypes of about 400 polled cattle with the recorded phenotype details we found that homozygous polled animals are always smooth polled. In contrast all animals showing scurs are heterozygous for one of the polled alleles. RNA-Seq of fetal horn bud tissue of a horned and a polled fetus revealed a large number of differentially expressed genes, including some of the previously known positional and functional candidates. Quantitative RT-PCR of skin and horn bud biopsies from different fetal stages implicates an important role of RXFP2 and FOXL2 in ruminant horn development and suggests a key role of the ruminant specific transcript LOC100848215 for horn bud formation in cattle.
Materials and Methods
Ethics Statement
All animal work was conducted according to the national and international guidelines for animal welfare. The collection of fetal tissue was done at a local slaughterhouse, as a low number of pregnant cows are routinely slaughtered. Blood sampling was done with owner consent. The whole study was approved by the “Cantonal Committee for Animal Experiments” (Canton of Bern; permits BE78/12).
Material
We collected blood, hair root, tissue or semen samples from 1,019 polled cattle belonging to 14 different breeds. For phenotyping we inspected and palpated the area on the cattle's forehead, where horns usually grow and recorded any scabs, corneous growths or horn-like formations. We observed the development of different forms of scurs in a total of 207 animals born as polled. The DNA of 1,501 horned cattle from 28 different breeds was taken from the archive of the Institute of Genetics.
DNA was either isolated from EDTA-blood using the Nucleon Bacc2 kit (GE Healthcare) or from sperm straw, hair roots or ear punch biopsies using QIAGEN's DNeasy kit according to the manufacturers' instruction.
A total of 23 fetuses (Table S4) were collected at a governmentally authorized slaughterhouse, inspecting the uteri of the slaughtered cows. Fetal horn buds were biopsied using a 4 mm or 6 mm biopsy punch and immediately stored in RNA-later (Ambion). After storage at 4°C for 48 hours they were kept at −20°C. Total RNA was isolated using Qiagen RNeasy Kit including a DNase treatment and subsequently stored at −80°C.
SNP genotyping
High-density SNP genotyping was performed using the illumina BovineHD BeadChip with 777,962 SNP markers at GeneSeek [35].
Whole genome re-sequencing
We prepared fragment libraries with 250 bp insert size and collected one to three lanes of illumina HiSeq2000 paired-end reads (2×100 bp) obtaining about 200 million tags per lane. We mapped the reads to the cow reference genome Bos_taurus_UMD_3.1 with the Burrows-Wheeler Aligner (BWA) version 0.5.9-r16 [36] with default settings. After sorting the mapped reads by the coordinates of the sequence with Picard tools, we labeled the PCR duplicates also with Picard tools [37]. We used the Genome Analysis Tool Kit (GATK version 0591, [38]) to perform local realignment and to produce a cleaned BAM file. Variants calls were then made with the unified genotyper module of GATK. Variant data for each sample were obtained in variant call format (version 4.0) as raw calls for all samples and sites flagged using the variant filtration module of GATK. Variant calls that failed to pass the following filters were labeled accordingly in the call set: (i) Hard to Validate MQ0≥4 & ((MQ0/(1.0 * DP)) >0.1); (ii) strand bias (low Quality scores) QUAL <30.0 || (Quality by depth) QD <5.0 || (homopolymer runs) HRun >5 || (strand bias) SB >0.00; (iii) SNP cluster window size 10. The snpEFF software [39] together with the recent ensembl cow genome annotation was used to predict the functional effects of detected variants. IGV-viewer software [40] was used for manual inspection of sequence variants.
Genotyping of candidate variants
For genotyping of SNPs and small InDels we applied Sanger sequencing. Primers (Table S6) were designed with Primer3 software [41] after masking of repetitive sequences with RepeatMasker [42]. PCR products were amplified using AmpliTaqGold360Mastermix (LifeTechnologies) and directly sequenced on an ABI3730 capillary sequencer (LifeTechnologies) after treatment with exonuclease I and shrimp alkaline phosphatase. The sequence data were analyzed with Sequencher 5.1 software. For genotyping of the 80 kb duplication we set the forward primer at the end of the duplicated sequence and the reverse primer at the beginning of the duplicated sequence (Figure S2), therefore only in the mutant allele a PCR-product was amplified, which was detected on a 1% agarose gel. The polled associated indel detected in Simmental cattle was genotyped using fragment analysis, setting the forward primer at the end of the duplicated sequence and the fluorescently labeled reverse primer in the region afterwards (Figure S1). PCR products were amplified using QIAGEN Multiplex PCR Kit (Qiagen) and the fragment length of the PCR products was directly analyzed with an ABI 3730 capillary sequencer (LifeTechnologies) and the Genemapper-software (LifeTechnologies).
RNA-Seq
We prepared two fragment libraries with 350 bp insert size following illumina's TruSeq Stranded mRNA Sample Preparation Guide and collected a quarter of a lane of illumina HiSeq2000 paired-end reads (2×100 bp) obtaining 70248709 (polled) and 85385646 (horned) tags per library. We mapped the reads to the cattle reference genome (Bostaurus_UMD3.1) using the spliced alignment program TopHat2 (version 2.0.4) with default parameters [43]. Read counting was carried out using HTSeq-count (version 0.5.3p9) and differential gene expression analysis was performed using DEseq software [44].
Quantitative PCR of fetal tissue
cDNA was synthesized using the First Strand cDNA synthesis kit (GE Healthcare) and 1 μg total RNA. Primers were taken from the literature [3] or designed as described above spanning exon junctions if possible (Table S7). Quantitative RT-PCR was performed in triplicates using 10 μl Power Sybr Green reaction mix (LifeTechnologies), 6.4 μl H2O, 0.8 μl primer (10 pmol/μl) and 2 μl cDNA (∼16 ng). The reaction was performed on an ABI 7300 Real-Time PCR System (LifeTechnologies). Cycle threshold (Ct) values were normalized to three endogenous control genes (GART, HPRT1 and RPLP0, Figure S7, Figure S8, Figure S9). Relative quantification was calculated using the 2−ΔΔCt method [45].
Supporting Information
Characterization of the polled associated insertion-deletion ( indel ). Schematic representation of the duplication and deletion on BTA 1 (A). Sequence details and primers used for genotyping are shown. Fragment length analysis showing three different genotypes (B).
(TIF)
Characterization of the polled associated 80 kb duplication. Region of the 80 kb duplication (taken from the UCSC genome browser), segments conserved in other species are shown as colored bars (A). Schematic illustration of the 80 kb duplication, both 2 bp deletions are indicated in red, primers used to determine the presence of the duplication are shown as black arrows (B).
(TIF)
RNA-Seq data of horn bud tissue from a wildtype and polled fetus at LOC100848215 . Screenshot of the mapped reads displayed in the igv viewer BTA 1 UMD3.1: 1897536–1899936. Presence of spliced reads in the wildtype sample (above) in contrast absence of reads in the polled fetus (below).
(TIF)
Cross-species comparison of LOC100848215 associated EST's, showing expression of this sequence in ruminants only (buffalo).
(TIF)
Gene expression study of C1H21orf62 based on RT-PCR. Expression of C1H21orf62 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Gene expression study of OLIG2 based on RT-PCR. Expression of OLIG2 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Gene expression study of FOXL2 based on RT-PCR. Expression of FOXL2 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Gene expression study of RXFP2 based on RT-PCR. Expression of RXFP2 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Gene expression study of LOC100848215 based on RT-PCR. Expression of LOC100848215 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Complex indel associated with polledness in Simmental cattle, genotyped in 2,329 animals.
(DOCX)
Sequence variants of polled associated 932 kb haplotype in Holstein. Animals carrying the polled associated indel found in Simmental are not included. The remaining associated variants are highlighted in grey and the excluded variants shown in red.
(PDF)
Genotypes of the IFNGR2 SNP at BTA 1 UMD3.1: 1,390,292 (reference allele: G , variant allele: A ). Genotypes of 161 polled Holstein and 55 horned control cattle.
(DOCX)
Fetuses used for RNA-Seq and RT-PCR. Age was estimated based on the relation between crown-rump length and time of gestation described by Schnorr and Kressin [26]. They were genotyped for the indel variant associated with polledness in beef and dual-purpose breeds and for the C>A SNP at BTA 1: 1′768′587 associated with polledness in the Holstein breed.
(PDF)
Differential gene expression in horn bud biopsies of a wildtype and a polled fetus.
(PDF)
Primer used for genotyping of candidate variants.
(PDF)
Primer used for qRT-PCR.
(PDF)
Acknowledgments
The authors thank the cattle breeders and breeding associations (Simmental Suisse, Rinderzuchtverband Salzburg, Züchtervereinigung Limpurger Rind) and the insemination stations Göpelgenetik, Swissgenetics, and Selectstar, Triple-Genetics-Service, Besamungsverein Nordschwaben, Besamungsstation Greifenberg, Rinderproduktion Berlin-Brandenburg, Masterrind for providing samples. The Next Generation Sequencing Platform of the University of Bern is acknowledged for performing the sequencing experiments. The authors thank Michèle Ackermann, Doreen Becker, Brigitta Colomb, Carolina Duart and Muriel Fragnière for expert technical assistance. The authors are grateful to Tosso Leeb and all the staff of the Institute of Genetics for helpful discussions.
Funding Statement
This study was funded by grant no. 31003A_141228 from the Swiss National Science Foundation. The authors further acknowledge the Karl Eibl-Stiftung, Neustadt, Germany, for financial support, as well as the German Federal Ministry of Education and Research (project FUGATO-plus GENOTRACK, grant no. 0315134A), and the KMSH (Kompetenzzentrum Milch–Schleswig-Holstein, Kiel, Germany) for partly funding the genotyping. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Dyce KM, Sack WC, Wensing CJG (2002) Textbook of veterinary anatomy. Elsevier.359 p. [Google Scholar]
- 2.Strouhal E (1997) Life of the Ancient Egyptians. Liverpool University Press. 128 p. [Google Scholar]
- 3. Allais-Bonnet A, Grohs C, Medugorac I, Krebs S, Djari A, et al. (2013) Novel insights into the bovine polled phenotype and horn ontogenesis in Bovidae . PLOS ONE 8: e63512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Graf B, Senn M (1999) Behavioural and physiological responses of calves to dehorning by heat cauterization with or without local anaesthesia. Appl Anim Behav Sci 62: 153–171. [Google Scholar]
- 5. White WT, Ibsen HL (1936) Horn inheritance in Galloway-Holstein cattle crosses. J Genet 32: 33–49. [Google Scholar]
- 6. Long CR, Gregory KE (1978) Inheritance of the horned, scurred, and polled condition in cattle. J Hered 69: 395–400. [Google Scholar]
- 7. Brem G, Karnbaum B, Rosenberg E (1982) Zur Vererbung der Hornlosigkeit beim Fleckvieh. Bayer Landwirtsch Jahrb 59: 688–695. [Google Scholar]
- 8. Georges M, Drinkwater R, King T, Mishra A, Moore SS, et al. (1993) Microsatellite mapping of a gene affecting horn development in Bos taurus . Nat Genet 4: 206–210. [DOI] [PubMed] [Google Scholar]
- 9. Schmutz SM, Marquess FL, Berryere TG, Moker JS (1995) DNA marker-assisted selection of the polled condition in Charolais cattle. Mamm Genome 6: 710–713. [DOI] [PubMed] [Google Scholar]
- 10. Brenneman RA, Davis SK, Sanders JO, Burns BM, Wheeler TC, et al. (1996) The polled locus maps to BTA1 in a Bos indicus x Bos taurus cross. J Hered 87: 156–161. [DOI] [PubMed] [Google Scholar]
- 11. Harlizius B, Tammen I, Eichler K, Eggen A, Hetzel DJ (1997) New markers on bovine chromosome 1 are closely linked to the polled gene in Simmental and Pinzgauer cattle. Mamm Genome 8: 255–257. [DOI] [PubMed] [Google Scholar]
- 12. Drögemüller C, Wöhlke A, Momke S, Distl O (2005) Fine mapping of the polled locus to a 1-MB region on bovine chromosome 1q12. Mamm Genome 16: 613–620. [DOI] [PubMed] [Google Scholar]
- 13. Seichter D, Russ I, Rothammer S, Eder J, Förster M, et al. (2012) SNP-based association mapping of the polled gene in divergent cattle breeds. Anim Genet 43: 595–598. [DOI] [PubMed] [Google Scholar]
- 14. Drögemüller C, Wöhlke A, Leeb T, Distl O (2005) A 4 Mb high resolution BAC contig on bovine chromosome 1q12 and comparative analysis with human chromosome 21q22. Comp Funct Genom 6: 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Medugorac I, Seichter D, Graf A, Russ I, Blum H, et al. (2012) Bovine polledness - an autosomal dominant trait with allelic heterogeneity. PLOS ONE 7: e39477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glatzer S, Merten N, Dierks C, Wöhlke A, Philipp U, et al. (2013) A single nucleotide polymorphism within the interferon gamma receptor 2 gene perfectly coincides with polledness in Holstein cattle. PLOS ONE 8: e67992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Capitan A, Allais-Bonnet A, Pinton A, Marquant-Le Guienne B, Le Bourhis D, et al. (2012) A 3.7 Mb deletion encompassing ZEB2 causes a novel polled and multisystemic syndrome in the progeny of a somatic mosaic bull. PLOS ONE 7: e49084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaiman D, Schibler L, Oustry-Vaiman A, Pailhoux E, Goldammer T, et al. (1999) High-resolution human/goat comparative map of the goat polled/intersex syndrome (PIS): the human homologue is contained in a human YAC from HSA3q23. Genomics 56: 31–39. [DOI] [PubMed] [Google Scholar]
- 19. Pailhoux E, Vigier B, Chaffaux S, Servel N, Taorit S, et al. (2001) A 11.7-kb deletion triggers intersexuality and polledness in goats. Nat Genet 29: 453–458. [DOI] [PubMed] [Google Scholar]
- 20. Johnston S, Beraldi D, McRae A, Pemberton J, Slate J (2010) Horn type and horn length genes map to the same chromosomal region in Soay sheep. Heredity 104: 196–205. [DOI] [PubMed] [Google Scholar]
- 21. Johnston S, McEwan J, Pickering N, Kijas J, Beraldi D, et al. (2011) Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol Ecol 20: 2555–2566. [DOI] [PubMed] [Google Scholar]
- 22. Capitan A, Grohs C, Gautier M, Eggen A (2009) The scurs inheritance: new insights from the French Charolais breed. BMC Genet 10: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Asai M, Berryere TG, Schmutz SM (2004) The scurs locus in cattle maps to bovine chromosome 19. Anim Genet 35: 34–39. [DOI] [PubMed] [Google Scholar]
- 24. Capitan A, Grohs C, Weiss B, Rossignol MN, Reverse P, et al. (2011) A newly described bovine type 2 scurs syndrome segregates with a frame-shift mutation in TWIST1. PLOS ONE 6: e22242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mariasegaram M, Reverter A, Barris W, Lehnert S, Dalrymple B, et al. (2010) Transcription profiling provides insights into gene pathways involved in horn and scurs development in cattle. BMC Genomics 11: 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schnorr B, Kressin M (2011) Embryologie der Haustiere. Enke. 288 p. [Google Scholar]
- 27.Homepage UCSC genome browser (2013) Available: http://genome.ucsc.edu. Accessed 2013 Nov 20.
- 28. Liu WB, Liu J, Liang CN, Guo X, Bao PJ, et al. (2014) Associations of single nucleotide polymorphisms in candidate genes with the polled trait in Datong domestic yaks. Anim Genet 45: 138–141. [DOI] [PubMed] [Google Scholar]
- 29.Rüsse I, Sinowatz F (1991) Lehrbuch der Embryologie der Haustiere. Parey 473 p. [Google Scholar]
- 30. Ligon KL, Fancy SP, Franklin RJ, Rowitch DH (2006) Olig gene function in CNS development and disease. Glia 54: 1–10. [DOI] [PubMed] [Google Scholar]
- 31. Niu J, Mei F, Wang L, Liu S, Tian Y, et al. (2013) Phosphorylated olig1 localizes to the cytosol of oligodendrocytes and promotes membrane expansion and maturation. Glia 60: 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mei F, Wang H, Liu S, Niu J, Wang L, et al. (2013) Stage-specific deletion of Olig2 conveys opposing functions on differentiation and maturation of oligodendrocytes. J Neurosci. 33: 8454–8462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee JT (2012) Epigenetic regulation by long noncoding RNAs. Science 338: 1435–1439. [DOI] [PubMed] [Google Scholar]
- 34. Weikard R, Hadlich F, Kuehn C (2013) Identification of novel transcripts and noncoding RNAs in bovine skin by deep next generation sequencing. BMC Genomics 14: 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Homepage GeneSeek (2013) Available: http://www.neogen.com/Agrigenomics. Accessed 2013 Nov 20.
- 36. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Homepage picard tools (2013) Available: http://sourceforge.net/projects/picard/. Accessed 2013 Nov 20.
- 38. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cingolani P, Platts A, Coon M, Nguyen T, Wang L, et al. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6: 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Homepage IGV-viewer 2.1.25 software (2013) Available: http://www.broadinstitute.org/igv/. Accessed 2013 Nov 20.
- 41.Homepage Primer3 (2013) Available: http://bioinfo.ut.ee/primer3-0.4.0. Accessed 2013 Nov 20.
- 42.Homepage Repeat Masker Server (2013) Available: http://www.repeatmasker.org. Accessed 2013 Nov 20.
- 43. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Homepage DESeq software (2013) Available: http://bioconductor.org/packages/release/bioc/html/DESeq.html. Accessed 2013 Nov 20.
- 45. Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Characterization of the polled associated insertion-deletion ( indel ). Schematic representation of the duplication and deletion on BTA 1 (A). Sequence details and primers used for genotyping are shown. Fragment length analysis showing three different genotypes (B).
(TIF)
Characterization of the polled associated 80 kb duplication. Region of the 80 kb duplication (taken from the UCSC genome browser), segments conserved in other species are shown as colored bars (A). Schematic illustration of the 80 kb duplication, both 2 bp deletions are indicated in red, primers used to determine the presence of the duplication are shown as black arrows (B).
(TIF)
RNA-Seq data of horn bud tissue from a wildtype and polled fetus at LOC100848215 . Screenshot of the mapped reads displayed in the igv viewer BTA 1 UMD3.1: 1897536–1899936. Presence of spliced reads in the wildtype sample (above) in contrast absence of reads in the polled fetus (below).
(TIF)
Cross-species comparison of LOC100848215 associated EST's, showing expression of this sequence in ruminants only (buffalo).
(TIF)
Gene expression study of C1H21orf62 based on RT-PCR. Expression of C1H21orf62 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Gene expression study of OLIG2 based on RT-PCR. Expression of OLIG2 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Gene expression study of FOXL2 based on RT-PCR. Expression of FOXL2 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Gene expression study of RXFP2 based on RT-PCR. Expression of RXFP2 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Gene expression study of LOC100848215 based on RT-PCR. Expression of LOC100848215 after normalization to GART (A), HPRT1 (B) and RPLP0 (C). Different fetal stages are divides in eight groups of estimated age (d). Wildtype fetuses are marked with the shape of a horned cow head, fetuses carrying the polled mutation are marked with the shape of a polled cow head, whereas each icon designates one fetus. For each individual a biopsy of the horn bud area (H) and a biopsy of the frontal skin (S) were studied. Expression levels in wildtype fetuses are shown in blue, those of heterozygous Pp polled fetuses in orange and those of homozygous PP polled fetuses in dark red. Expression levels are shown as relative expression in relation to the wildtype frontal skin of the youngest fetus.
(TIF)
Complex indel associated with polledness in Simmental cattle, genotyped in 2,329 animals.
(DOCX)
Sequence variants of polled associated 932 kb haplotype in Holstein. Animals carrying the polled associated indel found in Simmental are not included. The remaining associated variants are highlighted in grey and the excluded variants shown in red.
(PDF)
Genotypes of the IFNGR2 SNP at BTA 1 UMD3.1: 1,390,292 (reference allele: G , variant allele: A ). Genotypes of 161 polled Holstein and 55 horned control cattle.
(DOCX)
Fetuses used for RNA-Seq and RT-PCR. Age was estimated based on the relation between crown-rump length and time of gestation described by Schnorr and Kressin [26]. They were genotyped for the indel variant associated with polledness in beef and dual-purpose breeds and for the C>A SNP at BTA 1: 1′768′587 associated with polledness in the Holstein breed.
(PDF)
Differential gene expression in horn bud biopsies of a wildtype and a polled fetus.
(PDF)
Primer used for genotyping of candidate variants.
(PDF)
Primer used for qRT-PCR.
(PDF)