

Abstract
Germline mutations in the succinate dehydrogenase genes (SDHA, SDHB, SDHC, and SDHD) are established as causes of pheochromocytoma/paraganglioma, renal carcinoma, and gastrointestinal stromal tumor. It has recently been suggested that pituitary adenomas may also be a component of this syndrome. We sought to determine the incidence of SDH mutation in pituitary adenomas. We performed screening immunohistochemistry for SDHB and SDHA on all available pituitary adenomas resected at our institution from 1998 to 2012. In those patients with an abnormal pattern of staining, we then performed SDH mutation analysis on DNA extracted from paraffin-embedded tissue, fresh frozen tissue, and peripheral blood. One of 309 adenomas (0.3%) demonstrated an abnormal pattern of staining, a 30 mm prolactin-producing tumor from a 62-year-old man showing loss of staining for both SDHA and SDHB. Examination of paraffin-embedded and frozen tissues confirmed double-hit inactivating somatic SDHA mutations (c.725_736del and c.989_990insTA). Neither of these mutations was present in the germline. We conclude that, although pathogenic SDH mutation may occur in pituitary adenomas and can be identified by immunohistochemistry, it appears to be a very rare event and can occur in the absence of germline mutation. SDH-deficient pituitary adenomas may be larger and more likely to produce prolactin than other pituitary adenomas. Unless suggested by family history and physical examination, it is difficult to justify screening for SDH mutations in pituitary adenomas. Surveillance programs for patients with SDH mutation may be tailored to include the possibility of pituitary neoplasia; however, this is likely to be a low-yield strategy.
Key Words: succinate dehydrogenase, SDHB, SDHA, pituitary adenoma
The reported prevalence of pituitary adenomas is up to 1 in 1000.1 At least 5% are hereditary, and there are clear associations with multiple endocrine neoplasia type 1 (MEN1, associated with MEN1 mutation), familial isolated pituitary adenoma (often associated with AIP mutation), Carney complex (often associated with PRKAR1A mutation), and MEN4 (associated with CDKN1B mutation).2,3 However, there remain cases of hereditary pituitary adenoma for which no clear syndromic or genetic cause has been identified.
The succinate dehydrogenase (SDH) genes SDHA, SDHB, SDHC, and SDHD encode the protein subunits of the mitochondrial complex II, a key respiratory enzyme that links the Krebs cycle and the electron transport chain.4 These genes also function as tumor-suppressor genes, and germline SDH mutations are associated with a tumor syndrome characterized by pheochromocytoma/paraganglioma,5 a unique subtype of gastrointestinal stromal tumor (GIST) known as SDH-deficient GIST6 and a distinctive type of renal carcinoma.7 It is noteworthy that loss of immunohistochemical (IHC) staining for SDHB has been consistently identified in pheochromocytomas/paragangliomas,8,9 GISTs,6,10–14 and renal carcinomas7,15 associated with SDH mutation regardless of which SDH subunit is mutated. In addition to loss of SDHB staining, negative staining for SDHA also occurs in pheochromocytomas/paragangliomas16 and GISTs17,18 associated with SDHA mutation. To date, SDHA mutation has not been reported in association in with renal carcinoma. Tumors that show negative staining for SDHB are known as succinate dehydrogenase deficient, and IHC for SDHB and SDHA is used routinely to screen patients presenting with compatible tumors for germline SDH mutation.4
There is now emerging evidence that pituitary adenomas may also be associated with SDH mutation. Briefly, 35 cases of coexistent pheochromocytoma/paraganglioma and pituitary adenoma in individuals or kindreds have been reported,3,19–26 and second-hit inactivation has been demonstrated by either loss of heterozygosity19 or acquired mutation23 in 2 pituitary adenomas arising in the setting of germline SDH mutation. However, to date, the evidence linking SDH mutation and pituitary neoplasia has been based on case reports, and the incidence and clinical significance of SDH mutation in pituitary adenomas is unknown.
In this study, we sought to estimate the incidence and clinicopathologic associations of SDH mutation in pituitary adenomas.
METHODS
Patient and Tumor Samples
The computerized database of the Department of Anatomical Pathology Royal North Shore Hospital was searched for all pituitary adenomas resected during the calendar years 1998 to 2012 with material available in archived formalin-fixed paraffin-embedded (FFPE) blocks.
The original slides were reviewed to confirm the diagnosis and select areas of definite tumor for tissue microarray (TMA) construction. The TMA was constructed with duplicate 1 mm cores of neoplastic tissue from all available cases.
Immunohistochemistry
IHC for SDHB and SDHA was performed on the TMA sections using commercially available mouse monoclonal antibodies against both SDHB (ABCAM ab14714, clone 21A11, dilution of 1 in 100) and SDHA (Mitosciences Abcam MS204, Clone 2E, dilution of 1 in 1000) as previously described.6,7,9,17,23 IHC was interpreted independently by 2 observers with extensive experience in interpreting these stains (A.J.G. and C.W.T.) who were blinded to all clinical and pathologic features. Cases with definite granular cytoplasmic staining were classified as positive. Cases with absent cytoplasmic staining in the presence of an internal positive control of non-neoplastic cells were classified as negative. If there was any uncertainty in interpreting the staining on TMA sections (for example due to weak or absent internal positive controls) or if the staining pattern was anything other than definite, strong, diffuse, granular, and cytoplasmic then IHC was repeated on whole sections.
Mutation Analysis
Mutation analysis was performed in all tumors exhibiting loss of SDHB or SDHA staining by IHC. DNA was extracted from FFPE tissue blocks of macrodissected neoplastic tissue and, where available, from fresh frozen neoplastic tissue and whole blood both prospectively banked at the time of surgery (QIAamp DNA FFPE tissue kit and QIAamp DNA blood minikit; Qiagen, Melbourne, Vic., Australia). Mutation analysis of the entire coding sequence, including exon-intron boundaries, was performed for the 15 exons of SDHA (NCBI Ref Seq: NM_004168.2). Primer sequences were specifically designed to avoid amplification of the 3 pseudogenes (SDHAP1, SDHAP2, SDHAP3) as described previously,17 and human control genomic DNA (Promega) was used to confirm that none of the pseudogenes were amplified. In some instances, 2 rounds of polymerase chain reaction were required to amplify the DNA from paraffin-embedded tissues. Mutations were confirmed by sequencing of 2 independent polymerase chain reactions. Mutation analysis of SDHB, SDHC, and SDHD was not performed as there were no tumors found to exhibit loss of SDHB alone by IHC.
This study was approved by the Northern Sydney Local Health District Human Research Ethics Committee.
RESULTS
A total of 336 adenomas from 314 patients were identified from the time period 1998 to 2012. Twenty-seven (8%) of the adenomas were excluded due to insufficient tissue, leaving a study cohort of 309 adenomas from 287 patients. The mean age at surgery was 57 (range, 17 to 88) years; 164 (54%) patients were male.
A total of 299 adenomas demonstrated diffuse strong granular cytoplasmic staining for both SDHA and SDHB on the TMA sections (Fig. 1). One of 309 adenomas (0.3%) showed negative staining for both SDHB and SDHA. This negative staining was identified by both observers and was present when staining was repeated on whole sections. This was a clinically nonfunctioning pituitary adenoma in a 62-year-old man with no relevant personal or family history to suggest syndromic disease. On preoperative MRI, the tumor demonstrated extensive cystic change. It was 30 mm in diameter and extended from the pituitary fossa to the suprasellar cistern to compress the optic pathways. Despite being clinically nonfunctioning, by IHC the adenoma demonstrated diffuse strong prolactin expression with intense paranuclear (Golgi) accentuation (Fig. 2B)—a staining pattern indicative of the sparsely granulated variant of prolactinoma. It is noteworthy that all the neoplastic tissue highlighted by prolactin expression demonstrated loss of SDHB and SDHA expression implying that SDH deficiency was an early clonal event. There was strong positive staining for SDHB in the non-neoplastic pituitary and tumor-associated endothelial cells, which served as an internal positive control (Figs. 2C, D).
FIGURE 1.
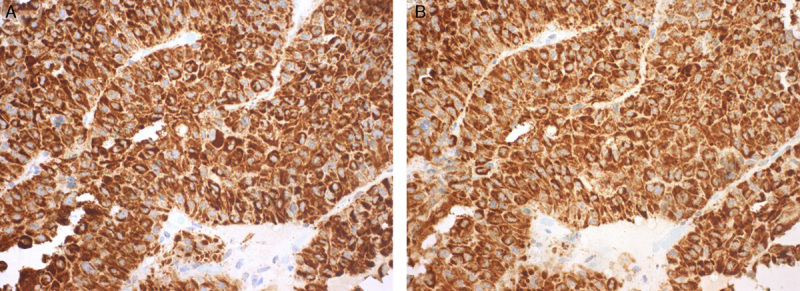
Most of the pituitary adenomas demonstrated readily recognizable diffuse and strong granular cytoplasmic staining for both SDHA (A) and SDHB (B) (A and B, IHC).
FIGURE 2.
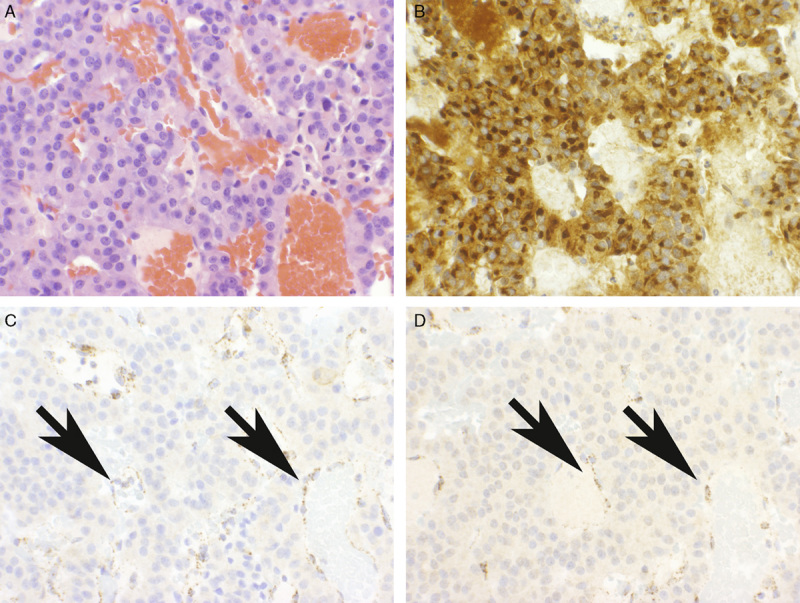
Pituitary adenoma demonstrating loss of staining for SDHB and SDHA. The neoplastic area (A), which demonstrates uniform strong prolactin expression (B), demonstrates completely absent staining for both SDHA (C) and SDHB (D). Note: The non-neoplastic endothelial cells (arrows) demonstrate strong positive staining for both SDHA and SDHB and serve as a positive internal control (A, H&E; B, Prolactin; C and D, IHC).
Nine adenomas demonstrated less intense but still recognizably positive staining on the TMA sections. Staining of these adenomas was confirmed to be unequivocally diffusely positive when repeated on whole mount sections (Fig. 3).
FIGURE 3.
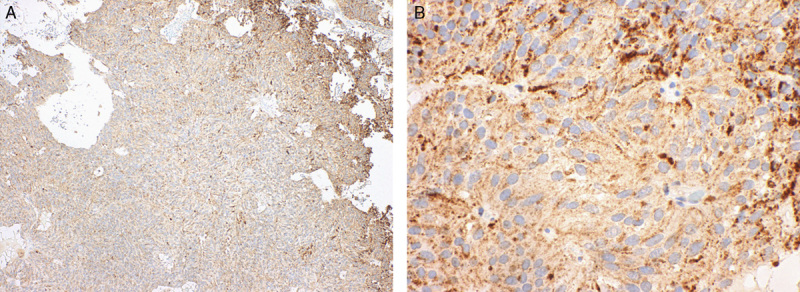
Nine pituitary adenomas demonstrated less intense staining but were still recognizably positive. For example, in this case, although positive staining was not readily apparent at low power (A), at high power (B) distinct granular staining was apparent (A and B SDHB IHC)
Both macrodissected FFPE and fresh frozen tumor tissue from the SDHA/B-negative pituitary adenoma underwent targeted sequencing for SDHA. In both the FFPE and frozen tissue the same 2 inactivating mutations were identified—a deletion in exon 6 (c.725_736del) and an insertion in exon 8 (c.989_990insTA). The entire coding region of germline SDHA obtained from the blood sample was sequenced and did not reveal any mutations (Fig. 4).
FIGURE 4.
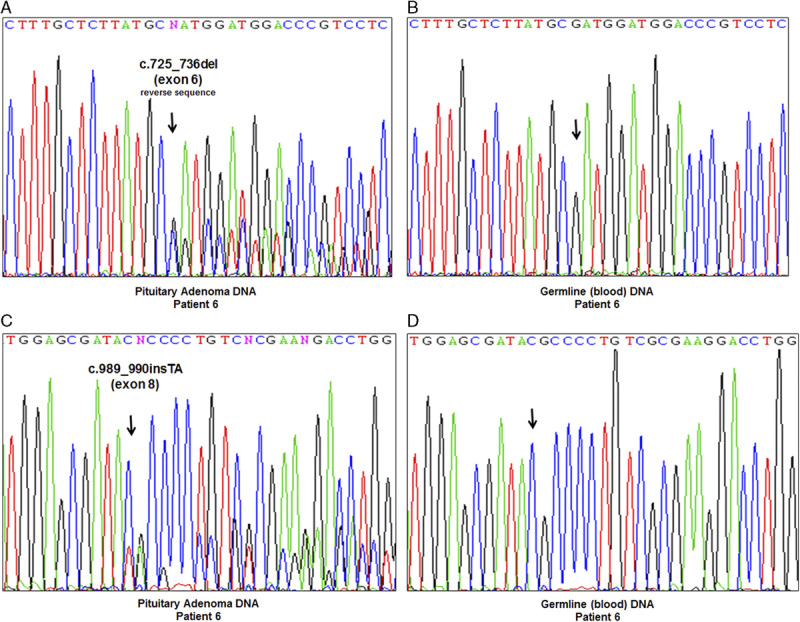
Sequencing chromatograms of the SDHA-mutated pituitary adenoma and the patient's corresponding blood. A, Region of exon 6 harboring the c.725_736del mutation (A and C, pituitary adenoma; B and D, blood). B, Region of exon 8 harboring the c.989_990insTA mutation (A and C, pituitary adenoma; C and D, blood). Germline (blood) analysis of the entire coding region of SDHA did not reveal any mutations.
The patient is currently well with no evidence of recurrence 45 months after surgery.
DISCUSSION
There have been 8 previously reported cases of pituitary adenoma occurring in association with confirmed germline SDH mutation,20,21,23–26 comprising 1 adenoma associated with mutation of SDHA,23 3 with SDHB mutation,20,21 2 with SDHC mutation,21,26 and 2 with SDHD mutation24,25 (summarized in Table 1). The causal link between SDH mutation and pituitary neoplasia is strengthened by: the presence of confirmed double-hit inactivation in SDH in this and 2 other pituitary adenomas23,25 and the fact that all the neoplastic cells in this case demonstrated loss of expression of SDHA and SDHB by IHC (implying that SDH inactivation is an early clonal event); the fact that SDH is an established tumor-suppressor gene in pheochromocytoma/paraganglioma, GIST, and renal carcinoma; and the coexistence of pituitary adenoma and pheochromocytoma/paraganglioma in 27 other reported cases, albeit without confirmation of SDH mutation.19
TABLE 1.
Reported Cases of Pituitary Adenoma Arising in the Setting of Confirmed SDH Mutation
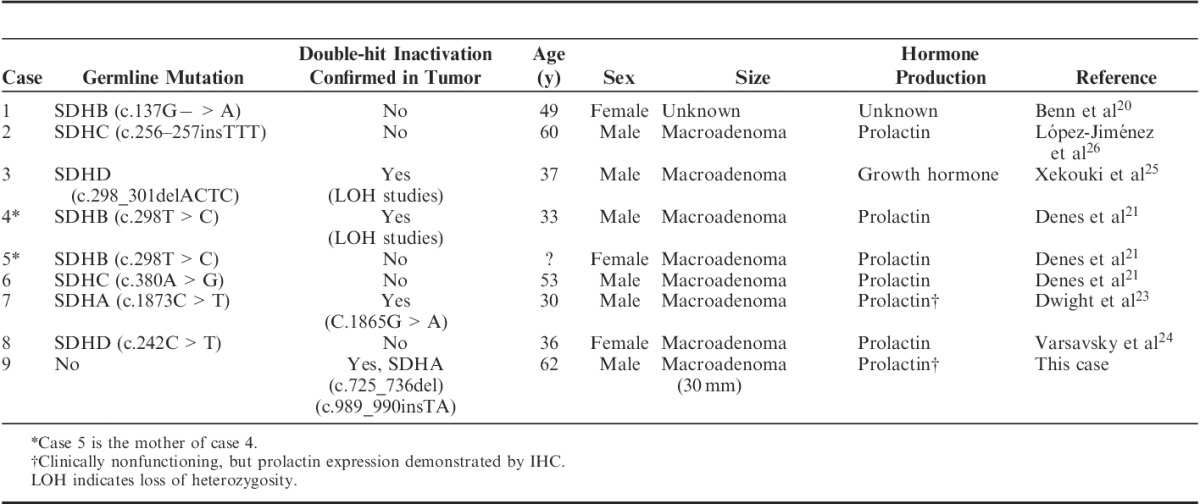
A clinical phenotype of SDH-deficient pituitary neoplasia is now emerging. Including this case, of the 9 pituitary adenomas reported in association with confirmed SDH mutation, the mean age has been 45 years (range, 30 to 62 y), and 6 have occurred in men. Seven have had hormone production documented clinically or by IHC, of which 6 have been prolactin-producing macroadenomas21,24,26 and 1 a growth hormone–secreting macroadenoma.25 In addition, the clinically nonfunctioning pituitary macroadenoma arising in the setting of germline SDHA mutation, which we recently described,23 also demonstrated positive IHC staining for prolactin (previously unreported data).
Taken together the findings suggest that SDH-deficient pituitary adenomas may occur at a young age, may commonly be macroadenomas, may more commonly produce prolactin (on the basis of clinical or IHC findings), and may show a slight male predisposition. However, prolactin-producing adenomas in male patients tend to be larger at presentation than in female patients, and prolactinomas in general do not usually come to surgery unless they are large or not responsive to medical therapy. Furthermore, relatively few SDH-deficient pituitary tumors have been reported to date. Therefore, more cases will need to be identified before a strong phenotype-genotype correlation for SDH-deficient pituitary tumors can emerge to further investigate this suggestion that prolactin-producing macroadenomas in young male patients are more likely to be SDH deficient.
Presuming that IHC for SDHB is as sensitive for SDH deficiency in pituitary adenomas as it is in other tumors, our study indicates that SDH-deficient pituitary adenomas are rare, accounting for only 0.3% of unselected pituitary adenomas. Furthermore, germline SDH mutation presenting with pituitary neoplasia is extremely rare (none in 309 consecutive adenomas). This is in contrast to pheochromocytoma/paraganglioma, in which SDH deficiency occurs in up to 15% of cases arising in an unselected population (3% in adrenal pheochromocytomas and up to 40% in extra-adrenal parangangliomas),9 and gastric GIST, in which SDH deficiency occurs in 5% to 7.5% of all cases,6,13 and more in keeping with the low incidence of succinate dehydrogenase deficiency in renal cancer, which is estimated to be 0.6%.27 On this basis it is hard to justify IHC screening for SDHB in all pituitary tumors as has been suggested for all pheochromocytomas/paragangliomas as well as GISTs and renal carcinomas with compatible morphology.4,6–8,15 Instead, we would recommend that IHC screening be considered only in pituitary adenomas with suggestive clinical features such as a young age at onset (particularly if a large prolactin-producing adenoma) or a personal or family history suggestive of syndromic disease.
The very low incidence of SDH deficiency in pituitary adenomas has important implications for the development of surveillance guidelines for individuals known to carry SDH mutations. Currently, we recommend annual physical examination with blood pressure documentation and fasting fractionated plasma metanephrine and normetanephrine as well as a third yearly MRI from the base of the skull to the coccyx, including kidneys and adrenal glands.23 Because this field of imaging does not encompass the pituitary, we have considered extending the field to include the sellar region in patients already scheduled for an MRI scan. However, given the high incidence of clinically insignificant pituitary incidentalomas,28 the benefits of this approach will have to be carefully weighed against the risk of overdiagnosing and overtreating clinically insignificant incidental pituitary adenomas. In the interim, we recommend that the possibility of pituitary disease, particularly associated with hyperprolactinemia, be specifically considered in the annual history and physical examination for patients with known SDH mutation.
The occurrence of 2 somatic SDHA mutations in the absence of a germline mutation is very unusual. In other tumors associated with SDH deficiency, somatic inactivation of SDH in the absence of a germline mutation is a very rare event. In fact, we are only aware of 2 reports of somatic mutation in SDH occurring in pheochromocytoma/paraganglioma (1 SDHB and 1 SDHD) in the absence of germline mutation and none in GIST or renal cancer.29,30 Although the identification of SDH mutation in a tumor is usually considered prima facie evidence of germline SDH mutation,4,6,8,9,12,13 we caution that this is not always the case and that inactivation of the SDH genes can occur as a truly somatic event. This reinforces that IHC for SDHB, which is also negative in the syndromic but nonhereditary association of gastric GIST, paraganglioma, and pulmonary chondroma known as Carney Triad,6,10,12,13 should only be considered a screening test to triage formal genetic testing for the SDH genes rather than proof of germline mutation.
In conclusion, it appears that pituitary adenomas are a legitimate albeit very rare component of the hereditary syndromes associated with SDH mutation, and therefore the possibility of pituitary neoplasia should be considered clinically in patients with SDH mutation who are under surveillance. SDH-deficient pituitary adenomas may be more frequently large and prolactin producing, may show a slight male sex preponderance, and may more commonly occur at a younger age. However, the issue of whether these associations represent a strong phenotype-genotype correlation awaits confirmation in further cases. Although somatic inactivation of the SDH genes in the absence of a germline mutation does occur in pituitary adenomas, it appears to be a rare event, and IHC for SDHA and SDHB can be used to triage formal genetic testing in pituitary adenomas when clinical suspicion of hereditary disease arises.
Footnotes
Conflicts of Interest and Source of Funding: Supported by the Hillcrest Foundation and the Pheo-Para Alliance. The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
REFERENCES
- 1.Daly AF, Rixhon M, Adam C, et al. High prevalence of pituitary adenomas: a cross-sectional study in the province of Lieve, Belgium.J Clin Endocrinol Metab. 2006;91:4769–4775 [DOI] [PubMed] [Google Scholar]
- 2.Beckers A, Aaltonen LA, Daly AF, et al. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene.Endocr Rev. 2013;34:239–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beckers A.Means, motive, and opportunity: SDH mutations are suspects in pituitary tumors.J Clin Endocrinol Metab. 2013;98:2274–2276 [DOI] [PubMed] [Google Scholar]
- 4.Gill AJ.Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia.Pathology. 2012;44:285–292 [DOI] [PubMed] [Google Scholar]
- 5.Fishbein L, Nathanson KL.Pheochromocytoma and paraganglioma: understanding the complexities of the genetic background.Cancer Genet. 2012;205:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gill AJ, Chou A, Vilain R, et al. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types.Am J Surg Pathol. 2010;34:636–644 [DOI] [PubMed] [Google Scholar]
- 7.Gill AJ, Pachter NS, Chou A, et al. Renal tumors associated with germline SDHB mutation show distinctive morphology.Am J Surg Pathol. 2011;35:1578–1585 [DOI] [PubMed] [Google Scholar]
- 8.van Nederveen FH, Gaal J, Favier J, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis.Lancet Oncol. 2009;10:764–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gill AJ, Benn DE, Chou A, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC and SDHD in paraganglioma-phaeochromocytoma syndromes.Hum Pathol. 2010;41:805–814 [DOI] [PubMed] [Google Scholar]
- 10.Janeway KA, Kim SY, Lodish M, et al. NIH Pediatric and Wild-Type GIST Clinic Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations.Proc Natl Acad Sci USA. 2011;108:314–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gill AJ, Chou A, Vilain RE, et al. “Pediatric type” Gastrointestinal stromal tumors are SDHB negative (“type 2”) GISTs.Am J Surg Pathol. 2011;35:1245–1247 [DOI] [PubMed] [Google Scholar]
- 12.Gaal J, Stratakis CA, Carney JA, et al. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors.Mod Pathol. 2011;24:147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miettinen M, Wang ZF, Sarlomo-Rikala M, et al. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age.Am J Surg Pathol. 2011;35:1712–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chou A, Chen J, Clarkson A, et al. Succinate dehydrogenase-deficient GISTs are characterized by IGF1R overexpression.Mod Pathol. 2012;25:1307–1313 [DOI] [PubMed] [Google Scholar]
- 15.Gill AJ, Pachter NS, Clarkson A, et al. Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome.N Engl J Med. 2011;364:885–886 [DOI] [PubMed] [Google Scholar]
- 16.Korpershoek E, Favier J, Gaal J, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas.J Clin Endocrinol Metab. 2011;96:E1472–E1476 [DOI] [PubMed] [Google Scholar]
- 17.Dwight T, Benn DE, Clarkson A, et al. Loss of SDHA expression identifies SDHA mutations in succinate dehydrogenase deficient gastrointestinal stromal tumors.Am J Surg Pathol. 2013;37:226–233 [DOI] [PubMed] [Google Scholar]
- 18.Miettinen M, Killian JK, Wang ZF, et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation.Am J Surg Pathol. 2013;37:234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xekouki P, Stratakis CA.Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/or Krebs cycle defects?Endocr Relat Cancer. 2012;19:33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes.J Clin Endocrinol Metab. 2006;91:827–836 [DOI] [PubMed] [Google Scholar]
- 21.Denes J, Swords F, Xekouki P, et al. Familial pituitary adenoma and paraganglioma syndrome—a novel type of multiple endocrine neoplasia [meeting abstracts].Endocr Rev. 2012;33:OR41–OR42 [Google Scholar]
- 22.Boguszewski CL, Fighera TM, Bornschein A, et al. Genetic studies in a coexistence of acromegaly, pheochromocytoma, gastrointestinal stromal tumor (GIST) and thyroid follicular adenoma.Arq Bras Endocrinol Metabol. 2012;56:507–512 [DOI] [PubMed] [Google Scholar]
- 23.Dwight T, Mann K, Benn D, et al. Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma.J Clin Endocrinol Metab. 2013;98:E1103–E1108 [DOI] [PubMed] [Google Scholar]
- 24.Varsavsky M, Sebastián-Ochoa A, Torres Vela E.Coexistence of a pituitary macroadenoma and multicentric paraganglioma: a strange coincidence.Endocrinol Nutr. 2013;60:154–156 [DOI] [PubMed] [Google Scholar]
- 25.Xekouki P, Pacak K, Almeida M, et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: a new association for SDH?J Clin Endocrinol Metab. 2012;97:E357–E366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.López-Jiménez E, de Campos JM, Kusak EM, et al. SDHC mutation in an elderly patient without familial antecedents.Clin Endocrinol (Oxf). 2008;69:906–910 [DOI] [PubMed] [Google Scholar]
- 27.Miettinen M, Sarlomo-Rikala M, Cue PM, et al. Mapping of succinate dehydrogenase losses in 2258 epithelial neoplasms.Appl Immunohistochem Mol Morphol. 2013;25[Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freda PU, Beckers AM, Katznelson L, et al. Pituitary incidentaloma: an Endocrine Society Clinical Practice Guideline.J Clin Endocrinol Metab. 2011;96:894–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gimm O, Armanios M, Dziema H, et al. Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial phaeochromocytoma.Cancer Res. 2000;60:6822–6825 [PubMed] [Google Scholar]
- 30.van Nederveen FH, Korpershoek E, Lenders JW, et al. Somatic SDHB mutation in extraadrenal pheochromocytoma.N Engl J Med. 2007;357:306–308 [DOI] [PubMed] [Google Scholar]