

Abstract
Sorafenib—a broad kinase inhibitor—is a standard therapy for advanced hepatocellular carcinoma (HCC), and has been shown to exert anti-fibrotic effects in liver cirrhosis, a precursor of HCC. However, the effects of sorafenib on tumor desmoplasia—and its consequences on treatment resistance — remain unknown. We demonstrate that sorafenib has differential effects on tumor fibrosis versus liver fibrosis in orthotopic models of HCC in mice. Sorafenib intensifies tumor hypoxia, which increases stromal-derived factor 1α (SDF1α) expression in cancer and stromal cells, and subsequently Gr-1+ myeloid cell infiltration. The SDF1α/CXCR4 pathway directly promotes hepatic stellate cell (HSC) differentiation and activation via MAP kinase pathway. This is consistent with the association between SDF1α expression with fibrotic septa in cirrhotic liver tissues as well as with desmoplastic regions of human HCC samples. We demonstrate that after treatment with sorafenib, SDF1α increased the survival of HSCs and their α-SMA and Collagen I expression, thus increasing tumor fibrosis. Finally, we show that Gr-1+ myeloid cells mediate HSC differentiation/activation in a paracrine manner. CXCR4 inhibition using AMD3100 in combination with sorafenib treatment prevents the increase in tumor fibrosis—despite persistently elevated hypoxia—in part by reducing Gr-1+ myeloid cell infiltration, and inhibits HCC growth. Similarly, antibody blockade of Gr-1 reduces tumor fibrosis and inhibited HCC growth when combined with sorafenib treatment.
Conclusion
Blocking SDF1α/CXCR4 or Gr-1+ myeloid cell infiltration may reduce hypoxia-mediated HCC desmoplasia and increase the efficacy of sorafenib treatment.
Keywords: hepatocellular carcinoma, hypoxia, collagen I, hepatic stellate cell, α-smooth muscle actin
INTRODUCTORY STATEMENT
Hepatocellular carcinoma (HCC) almost exclusively arises in cirrhotic livers, and the preexisting chronic inflammation and fibrosis fuel hepatocarcinogenesis and HCC growth (1–3). Fibrosis is the consequence of hepatic stellate cell (HSC) activation and proliferation, and myofibroblast differentiation leading to increased collagen deposition (4). This dual pathology of the liver contributes to an aggressive and systemic treatment-refractory characteristic in HCCs (1). Recently, the tyrosine kinase inhibitor (TKI) sorafenib has emerged as the first systemic therapy for HCC. Sorafenib is an antiangiogenic drug that has a broad tyrosine kinase inhibition spectrum (5). However, despite this progress, the mortality rate from HCC remains high, making this disease the third leading cause of cancer-related death worldwide (6–10).
Sorafenib is widely considered as an anti-angiogenic/anti-vascular drug through inhibition of VEGF receptors (VEGFRs) and platelet-derived growth factor receptors (PDGFRs). However, more potent and selective anti-VEGF agents or more broad antiangiogenic agents (e.g., VEGFR/FGFR and anti-VEGFR/PDGFR inhibitors) have failed so far to match the efficacy of sorafenib in phase III trials in HCC (10–13). Moreover, anti-angiogenic therapy has not led to tumor regression in patients or in experimental models in mice: The benefit seen with sorafenib in HCC patients is likely due to a transient delay in HCC growth, after which most tumors resume their growth (10). Whereas the mechanisms of acquired resistance to sorafenib and other anti-VEGF inhibitors in HCC remain unknown, it is likely that tumor stroma-mediated survival pathways might play a key role (1, 10). Of these, increased hypoxia has been proposed as a mechanism of resistance to multitargeted TKI therapy (14–17). The challenge is to identify the key molecular pathways regulating stroma-mediated resistance to sorafenib treatment in HCC.
Hypoxia and other cellular stresses can promote the expression of the chemokine stromal-derived factor 1 alpha (SDF1α or CXCL12) and of its receptor CXCR4 (18–22). In clinical studies, we showed that SDF1α level increased in plasma circulation in HCC patients after treatment with sunitinib or cediranib (both anti-VEGFR and anti-PDGFR TKIs) (23, 24). Moreover, we showed that elevated circulating levels of SDF1α correlated with poor treatment outcome in HCC patients after sunitinib treatment (23). Systemic activation of SDF1α/CXCR4 axis is known to mediate intra-tumoral infiltration of inflammatory cells, including Gr-1+ myeloid (CD11b+) cells (25–28). Gr-1+ myeloid cells can drive tumor recurrence after anti-VEGF therapy in various tumor models (29). Finally, clinical correlative data also strongly suggest that the effects on multi-targeted TKI treatment on tumor vasculature and on myeloid cells may mediate the response and resistance therapy in HCC patients (23, 30). However, a causal role of Gr-1+ myeloid cells in HCC resistance to anti-angiogenic treatment has not been characterized. Furthermore, a mechanistic understanding of the interplay between treatment-induced hypoxia, SDF1α/CXCR4 pathway activation, and Gr-1+ myeloid cell infiltration and tumor fibrosis in HCC is currently lacking. Here, we examined in orthotopic HCC models whether the SDF1α/CXCR4 pathway is activated and causally related to Gr-1+ myeloid cell infiltration and tumor-associated fibrosis and, ultimately, to sorafenib resistance.
EXPERIMENTAL PROCEDURES
Cells and Materials
We used the C3H mouse-derived HCC cell line HCA-1 (31). Human hepatic stellate cells (HSCs; Catalog #5300) were purchased from ScienCell Research Laboratories (San Diego, CA). These primary HSCs have been previously characterized (32). We purchased AMD3100 and FR180204 from Sigma (St. Louis, MO), AZD6244 and sorafenib from Selleck Chemicals (Houston, TX), recombinant PDGF-B and SDF1α from R&D system (Minneapolis, MN), and anti-Gr-1 antibody (LeafTH purified anti-mouse Gr-1 antibody) from Biolegend (San Diego, CA). For co-culture experiments, we isolated Gr-1+ myeloid cells from enzymatically digested HCA-1 tumors.
Animals
To induce liver fibrosis, we treated 5-week-old male C3H or Mst1−/−Mst2F/− mice with carbon tetrachloride (CCl4, 16 %v/v in olive oil, 100μL gavage, 3 times per week) prior to tumor implantation/induction. HCA-1 cells were orthotopically implanted in mice 2 weeks after the last CCl4 treatment, as previously described (33). Mst1−/−Mst2F/− mice develop spontaneous HCCs after i.v. injection of Ad-Cre (34), and were a kind gift from Dr. Nabeel Bardeesy (MGH). All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86–23 revised 1985).
Treatment studies
Mice were treated daily by gavage with sorafenib (40mg/kg) or vehicle (PBS) alone. AMD3100 (10mg/kg/day) was delivered continuously using Alzet micro-osmotic pumps (DURECT Corporation, Cupertino, CA) over 2 weeks.
Cell viability assays
We assessed cell viability using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay (Sigma).
Western blot analysis
Western blotting was performed using antibodies against collagen I and α-SMA (Abcam, Cambridge, MA), and phosphorylated (p)-ERK, ERK, p-AKT and AKT (Cell Signaling, Danvers, MA).
siRNA knockdown
ON-TARGETplus CXCR4 siRNA and non-targeting control siRNA were purchased from Dharmacon (Lafayette, CO) and used for transfection of HCA-1 cells.
Immunohistochemistry
For murine samples, we used antibodies against α-SMA, CAIX (Abcam), collagen I (LF-67, kindly provided by Dr. L. Fisher, National Institute of Dental Research), and SDF1α (BioVision). For apoptosis detection, frozen tumor sections were stained using TACS™ TdT Kit (R&D Systems, Minneapolis, MN). To assess expression level, we measured the fluorescently stained area for each marker and normalized it to DAPI area (used as measure of cellularity in viable tumor regions), as previously described (28). In addition, we performed immunostaining for SDF1α in tumor samples from patients who underwent HCC resection. Hepatic fibrosis in the non-HCC liver area was evaluated according to the Laennec system as previously described (35). Co-localization between SDF1α expression and desmoplasia was assessed in 10 randomly selected high-power fields at 4x magnification.
Flow cytometry
We used fluorescently labeled rat monoclonal antibodies anti-mouse CD45-PE-Cy7, Gr-1-APC and CD11b-APC-Cy7 (BD Biosciences) to perform flow cytometric analysis in digested tumor tissue, as previously described (36).
Quantitative RT-PCR
We determined the relative gene expression of SDF1α, CXCR4, MCP-1, 5-LO, TGF-α, TGF-β, PDGF-α, PDGF-β, MMP9, MMP13, and β-actin in tumor-infiltrating Gr-1+ cells using specific primers (Table S1), Real-Time SYBR Green PCR master mix (Applied Biosystems, Branchburg, NJ) and the Stratagene Mx3000P QPCR System, as previously described (36).
Statistical analysis
Comparisons between treatment groups were performed using the Mann-Whitney U-test. A p-value of less than 0.05 was considered to denote statistical significance. (See more details in Supplemental Experimental Methods.)
RESULTS
Sorafenib treatment increases hypoxia and SDF1α expression in orthotopic HCC models
To model the clinical features of HCC, we first induced liver fibrosis in mice by CCl4 treatment for 11 weeks (Fig. S1). Then, we generated orthotopic tumors by intrahepatic HCA-1 cell implantation in C3H mice or by inducing spontaneous HCC using Cre-adenovirus (Ad-Cre) i.v. injection in Mst1−/− Mst2F/− mice (Fig. S2). When tumors became established, we treated the mice with sorafenib for 14 days and then measured the changes in tissue oxygenation. Sorafenib treatment significantly increased the hypoxic tissue fraction—measured by carbonic anhydrase IX (CAIX) immunostaining—when compared to control-treated tumors (Fig. 1A, B). In contrast, hypoxic tissue surface area did not increase in the surrounding fibrotic liver tissue after treatment (Fig. 1E).
Figure 1. Tumor hypoxia and SDF1α expression are increased after sorafenib treatment in orthotopic HCC.

A, Representative immunofluorescence staining of CAIX and SDF1α in HCA-1 tumor implanted in mice with liver fibrosis. Images are 636μm across. B,C, The hypoxic tumor tissue fraction (B) and SDF1α expression (C) increased significantly after sorafenib treatment in orthotopic HCA-1 tumors in mice (n=5–9). D, Immunofluorescence imaging analysis showed that SDF1α expression is increased after 14 days of sorafenib treatment in spontaneous HCCs in Mst1−/− Mst2F/− transgenic mice. The increased hypoxia and SDF1α expression persisted when sorafenib treatment was combined with AMD3100 (n=5–9). E,F, Expression of CAIX (E) and SDF1α (F) in the liver was not significantly changed after sorafenib, AMD3100 or combination treatment (n=4–7). The number of random regions of interest used for quantification is shown in parentheses. Data are presented as mean ± SEM; *P<0.05, **P<0.01.
To examine the consequence of treatment-induced increase in tumor hypoxia, we next evaluated whether the increase in SDF1α expression—suggested by studies in HCC patients (23, 24)—is recapitulated in these HCC models. Indeed, we found a 2-fold elevation in SDF1α expression after sorafenib treatment in HCA-1 tumor grafts and in spontaneous HCCs in Mst1−/− Mst2F/− mice (Fig. 1A–D, Fig. S3). In contrast, SDF1α expression did not significantly increase in the fibrotic liver after sorafenib treatment (Fig. 1F).
To confirm that hypoxia is the driver of SDF1α expression in cancer and stromal cells, we cultured HCA-1 cells as well as hepatic stellate cells (HSCs) for 48hr in hypoxic (1% O2) or normoxic (21% O2) conditions and measured SDF1α expression by qPCR. Exposure to hypoxic conditions increased SDF1α expression in both HCC cells and HSCs (Fig. S4).
To evaluate the effect of SDF1α/CXCR4 pathway inhibition on tumor tissue oxygenation and SDF1α expression after treatment, we combined sorafenib treatment with the CXCR4 inhibitor AMD3100 (Sigma, 10mg/kg/day). Interestingly, the combination therapy further increased hypoxic tissue fraction and SDF1α expression in orthotopic HCA-1 tumor grafts and spontaneous HCCs in Mst1−/− Mst2F/− mice when compared to sorafenib treatment alone (Fig. 1, Fig. S3).
Inhibition of CXCR4 prevents the increase in HCC fibrosis after sorafenib in vivo
To establish if fibrosis is associated with elevated SDF1α expression in HCC patients, we examined surgical specimens for HCC patients with cirrhosis. We observed that SDF1α expression co-localized with extracellular matrix components/fibrotic septa in both HCC and cirrhotic liver tissues (Fig. 2). Thus, we next evaluated the effect of sorafenib treatment with or without the inhibition of SDF1α/CXCR4 pathway on fibrosis, a hallmark of HCC progression (4), in the mouse models. To this end, we measured the expression of collagen I as well as the number of α SMA+ myofibroblasts after sorafenib treatment both in the tumor region and in the surrounding liver tissue. Of note, the α SMA+ myofibroblasts were also positive for GFAP (Fig. S5). IHC analyses revealed that the HCCs growing in the face of sorafenib treatment showed significantly increased tumor desmoplasia. Treatment resulted in increased intra- and peri-tumoral collagen I and α-SMA+ myofibroblasts infiltration in orthotopic HCA-1 HCCs in mice with CCl4-induced liver fibrosis and in spontaneous HCCs in Mst1−/− Mst2F/− mice (Fig. 3A–E). Of note, sorafenib treatment reduced collagen I expression levels in the surrounding liver tissue in mice with CCl4-induced liver fibrosis (Fig. 3F).
Figure 2. Immunostaining for SDF1α in liver tissue samples from HCC patients.

Paraffin embedded tissue sections were stained for SDF1α by immunohistochemistry. A,B SDF1α expression in non-malignant liver tissue (A) was consistently lower than in malignant HCC tumor area (B). Both in cirrhotic liver tissue (C) and in HCC nodules (D), SDF1α expression was colocalized with ECM components/fibrous septa supporting the potential pro-fibrotic role of SDF1α in human hepatic fibrogenesis and HCC desmoplasia. E, Specimens of tumors that re-occurred after transcatheter arterial chemoembolization and radiofrequency ablation – both of which are known to cause severe hypoxia – consistently showed high expression of SDF1α, which was also associated with more pronounced desmoplasia in HCC area. Error bars represent score ± SEM (n=10 regions of interest per sample).
Figure 3. Sorafenib treatment selectively increases tumor-associated fibrosis in HCC in an SDF1α/CXCR4 pathway-dependent manner.

A,B, Sorafenib significantly increased the collagen I content (A) and the number of α-SMA+ myofibroblasts (B) in orthotopic HCA-1 tumors implanted in mice with fibrotic livers. The analysis was performed using immunofluorescence in tumor tissue, and maker-positive area was normalized to the area of DAPI (nuclear stain) (N=5–7). C–D, Sorafenib treatment significantly increased the collagen I content (C and D) and the expression of α-SMA (D) (detected by Western blotting) in spontaneously arising HCCs (N=7–8). E, Representative images of immunofluorescence of Collagen I staining in spontaneous HCCs. Images are 636μm across. Addition of AMD3100 to sorafenib treatment prevented the increase in collagen I content and the number of α-SMA+ myofibroblasts in HCCs (A–E). F, In tumor-bearing mice, sorafenib significantly reduced the collagen I content in the surrounding liver tissue in mice with underlying liver fibrosis to levels comparable to those seen in livers from non-CCl4-treated mice (N=4–8). The number of random regions of interest used for quantification is shown in parentheses. *p<0.05; **p<0.01; Data are shown as mean ± SEM.
To assess the effect of SDF1α/CXCR4 pathway inhibition on tumor-associated fibrosis after sorafenib treatment, we tested the combined administration of sorafenib with the CXCR4 inhibitor AMD3100. This combination therapy prevented the increase in collagen I expression and α SMA+ myofibroblasts after sorafenib treatment in spontaneous and implanted HCCs, without significantly changing collagen I expression in the fibrotic liver in CCl4-treated mice (Fig. 3). Of note, this effect was seen despite a persistent increase in hypoxia and SDF1α expression in the HCCs treated with sorafenib and AMD3100 (Fig. 1A–D). Thus, CXCR4 inhibition can prevent the pro-fibrotic effects of sorafenib treatment in HCC in vivo, despite persistent tumor hypoxia.
SDF-1α/CXCR4 axis directly mediates hepatic stellate cell (HSC) differentiation to myofibroblasts in HCC despite PDGFR blockade by sorafenib
We next examined the role of SDF1α/CXCR4 axis in selective promotion of tumor fibrosis after sorafenib treatment in vitro. To determine if increased SDF1α expression in the tumor could mediate the increase in tumor fibrosis after sorafenib treatment, we first exposed primary HSCs to recombinant (r)SDF1α in the presence or absence of sorafenib or rPDGF-B. Consistent with previous reports (37), we found that rPDGF-B stimulated proliferation and α-SMA expression in HSCs (Fig. 4A, B). Sorafenib treatment prevented the effects of rPDGF-B and led to significant decrease in HSC viability (as evidenced by an increase in cleaved caspase 3 expression and in the number of apoptotic HSCs) and inhibited α-SMA and collagen I expression (Fig. 4A–C). These results indicate that sorafenib treatment may directly reduce liver fibrosis by blocking PDGFR pathway in HSCs. Exposure to rSDF1α induced HSC differentiation into myofibroblast, as evidenced by dose-dependent increases in α-SMA and collagen I expression as well as in ERK and Akt activation (Fig. 4D). We then evaluated whether SDF1α can drive HSC differentiation and activation of HSCs in the face of PDGFR blockade by sorafenib treatment. We found that SDF1α increased cell proliferation and viability, and promoted the differentiation of HSCs despite sorafenib treatment (Fig. 4A–C).
Figure 4. SDF1α/CXCR4 axis promotes HSC to myofibroblast differentiation in the face of PDGFR blockade by sorafenib.

A, Exposure to recombinant PDGF-B increased HSC proliferation, while treatment with sorafenib reduced the viability of HSCs. Exposure to recombinant SDF1α increased the viability of HSCs despite PDGFR inhibition using sorafenib treatment, in a dose-dependent manner. HSC viability was measured by MTT assay (N=6 experimental repeats). B, Exposure to recombinant SDF1α increased the expression of α-SMA and collagen I and reduced cleaved caspase-3 expression (evaluated by Western blotting) despite sorafenib treatment, in a dose-dependent manner. C, Exposure to recombinant SDF1α increased viability of HSCs despite sorafenib treatment (N=3–5 experimental repeats). D, Exposure to recombinant SDF1α upregulated collagen I and α-SMA expression levels as well as ERK and AKT activation in HSCs, consistent with their myofibroblast differentiation. Inhibition of CXCR4 with AMD3100 (D) or using siRNA (E) prevented the effects of SDF1α. F, ERK inhibition with FR-180204 (2μM) decreased α-SMA expression in HSCs treated with recombinant SDF1α. Data are presented as mean ± SEM.
Finally, to determine if SDF1α/CXCR4 axis mediates these effects in HSCs, we used pharmacologic and genetic approaches to inhibit CXCR4. To this end, we cultured HSCs in the presence of rSDF1α with or without the CXCR4 inhibitor AMD3100 or with or without CXCR4 expression knockdown using siRNA. In both settings, inhibition of CXCR4 prevented the effects of SDF1α on HSC differentiation and activation (i.e., prevented the increase in α-SMA and collagen I expression induced by SDF1α in HSCs) (Fig. 4D, E, Fig. S6). The inhibitory effects were mediated in part by preventing the activation of the MAPK pathway activation by SDF1α, as shown by ERK inhibition with FR180204 (2μM) or MEK inhibition with AZD6244 in HSCs treated with rSDF1α (Fig. 4D–F, Fig. S7).
Gr1+ myeloid cell infiltration increases in HCC after sorafenib in an SDF1α/CXCR4 dependent manner
We next examined the effects of treatment with sorafenib—with or without inhibition of CXCR4—on inflammatory cell infiltration in HCC. To this end, we evaluated enzymatically digested HCC tissue by flow cytometric analysis. We found that the number of Gr-1+ myeloid cells increased by over 2-fold in HCA-1 transplanted HCC models and in spontaneous HCCs in Mst1−/− Mst2F/− mice after sorafenib treatment (Fig. 5A, B). In contrast, we found that sorafenib treatment reduced the accumulation of Gr-1+ myeloid cells in the surrounding fibrotic liver tissue (Fig. 5C). Finally, inhibition of CXCR4 using AMD3100 in combination with sorafenib decreased the number of tumor-infiltrating Gr-1+ myeloid cells to levels comparable to control-treated spontaneous and transplanted HCCs (Fig. 5A–B).
Figure 5. Intratumoral infiltration of Gr1+ myeloid cells is increased after sorafenib (SOR) treatment, and is prevented by CXCR4 inhibition in HCC.
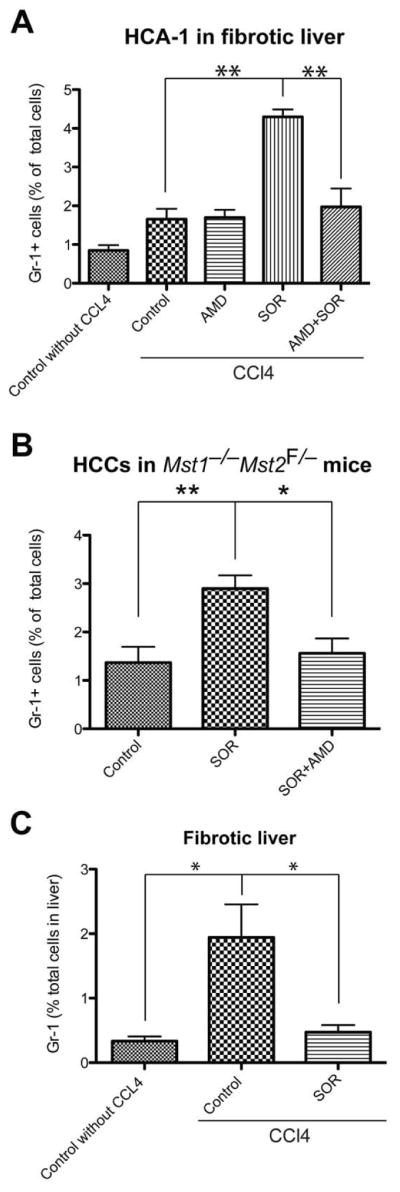
A–B, After SOR treatment, the number of 7AAD–CD11b+Gr1+ monocytes (measured by flow cytometry) significantly increased in HCA-1 tumors growing C3H mice with liver fibrosis (A) of as well as in spontaneous HCCs in Mst1−/− Mst2F/− transgenic mice (B). Treatment with the CXCR4 inhibitor AMD3100 prevented this effect (A–B). C, The number of 7AAD–CD11b+Gr1+ myeloid cells was significantly reduced in the fibrotic liver tissues from sorafenib-treated mice. Data are shown as percentages of the total number of cells evaluated in enzymatically digested tissue. Data are presented as mean ± SEM (N=5–14 mice per group). **P<0.01.
Gr1+ myeloid cell infiltration increases fibrosis in HCC after sorafenib in a SDF1α/CXCR4 dependent manner
We next tested whether direct Gr-1+ myeloid cell blockade could prevent the increase in fibrosis after sorafenib treatment in HCC. To achieve this, we used an anti-Gr-1-blocking antibody with or without sorafenib treatment in mice with CCl4-induced liver fibrosis with orthotopically implanted HCA-1 tumors. Gr-1 blockade prevented the increase in Gr-1+ myeloid cell infiltration in HCC after sorafenib treatment to levels comparable to tumors from control-treated mice (Fig. 6A). Moreover, inhibition of Gr-1+ myeloid cell infiltration also prevented the increase in HCC-associated fibrosis seen after sorafenib treatment (i.e., decreased the levels of collagen I and α–SMA expression) (Fig. 6B, C). To examine the mechanisms by which Gr-1+ myeloid cells promote fibrosis, we isolated HCC-infiltrating Gr-1+ (Ly-6G/Ly-6C) myeloid cells from HCA-1 digested tumor tissue by way of magnetic separation, and co-cultured them with HSCs – either with or without direct contact. In both co-culture systems, Gr-1+ cells stimulated collagen I and α–SMA expression (measured in HSCs) in a dose-dependent manner (Fig. 6D, Fig. S8A). This indicates that soluble factors released by Gr-1+ myeloid cells promote fibrosis. To determine whether the effects of Gr-1+ myeloid cells on HSCs can overcome the PDGFR blockade by sorafenib, we repeated the co-culture experiments in the presence or absence of sorafenib and rPDGF-B. We found that Gr-1+ myeloid cells can drive, in a dose-dependent manner, HSC differentiation and activation despite effective blockade of PDGFR by sorafenib treatment (Fig. 6E, Fig. S8B). To examine the pro-fibrotic factors involved in this paracrine interaction, we sorted Gr1+ myeloid cells from digested HCC tissue from mice treated with sorafenib or vehicle control and then extracted mRNA. We next performed real-time PCR analysis to measure the expression of pro-fibrotic cytokines (4). Gr-1+ myeloid cells from sorafenib-treated tumors showed higher expression of several pro-fibrotic factors (5-lipoxygenase (5-LO), PDGF-B and MCP-1 as well as of SDF1α and CXCR4) compared to Gr-1+ myeloid cells from control-treated tumors (Table S2). To evaluate if SDF1α/CXCR4 axis mediates not only Gr-1+ myeloid cell infiltration in HCC but also their paracrine interaction with HSCs leading to fibrosis, we inhibited CXCR4 with AMD3100 in the co-culture systems described above. AMD3100 treatment prevented the upregulation of collagen I and α-SMA expression in HSCs co-cultured with tumor-derived Gr-1+ myeloid cells, in a dose-dependent manner (Fig. 6F). Taken together, these data show that SDF1α/CXCR4 pathway mediates both the increased infiltration of Gr-1+ myeloid cells in HCC after by sorafenib treatment as well as their pro-fibrotic effects on HSCs.
Figure 6. Gr-1+ myeloid cell infiltration increases fibrosis in HCC after sorafenib in an SDF1α/CXCR4 pathway-dependent manner.

A–C, Depletion of Gr-1+ myeloid cells using systemic therapy with anti-Gr-1 blocking antibodies prevents the shift towards a pro-fibrotic environment after sorafenib treatment. Sorafenib significantly increased the number of 7AAD–CD11b+Gr1+ monocytes (A) (N=3–7 mice per group), the collagen I content (B) and the number of α-SMA+ myofibroblasts (C) in orthotopic HCA-1 tumors growing in mice with liver fibrosis, evaluated by immunohistochemistry. Combining anti-Gr-1 antibodies with sorafenib treatment prevented these effects. Quantification was performed in 5–12 confocal microscopy images per mouse. D–F, Tumor-infiltrating Gr-1+ myeloid cells directly mediate HSC differentiation to myofibroblasts via SDF1α/CXCR4 axis. Co-culture with tumor-tissue isolated Gr1+ myeloid cells (obtained using magnetic beads) increased collagen I and α-SMA expression levels as well as ERK activation in HSCs (D). Co-culture with Gr1+ myeloid cells increased collagen I and α-SMA expression in HSCs despite sorafenib treatment, in a dose-dependent manner (E). Recombinant PDGF-B and sorafenib alone were used as positive and negative control, respectively, for HSC differentiation (E). Inhibition of CXCR4 with AMD3100 prevented the increase in collagen I, α-SMA, and p-ERK expression in HSCs co-cultured with Gr-1+ myeloid cells (F). Data are presented as mean ± SEM. * P<0.05.
Inhibition in CXCR4 or Gr-1 in combination with sorafenib inhibits HCC growth compared to sorafenib alone
Finally, given the SDF1α mediation of pro-fibrotic and pro-inflammatory effects in HCC after sorafenib treatment, we next evaluated the specific impact of fibrosis on HCC growth after treatment with sorafenib or the CXCR4 inhibitor AMD3100. First, we found that the growth of spontaneous and grafted HCC was accelerated in mice with fibrotic liver compared to mice with normal liver (Fig. 7A, Fig. S2). AMD3100 treatment alone showed no significant inhibition of HCC growth. However, when combined with sorafenib, AMD3100 induced a significant additional tumor growth inhibition of orthotopic HCA-1 tumors in immunocompetent C3H mice with underlying liver fibrosis (Fig. 7A). Moreover, inhibition of CXCR4 with AMD3100 in combination of sorafenib induced a significant increase in cell apoptosis compared to sorafenib or AMD3100 alone in both grafted and spontaneous orthotopic HCC models (up to 20% in HCA-1 growing in fibrotic liver) (Fig. 7B–E). Of note, the liver function parameters (ALT, AST and ALP) remained unchanged after combination treatments (not shown). Finally, combination of anti-Gr-1 antibody with sorafenib also induced a significant delay in HCC growth compared to sorafenib alone (Fig. 7F).
Figure 7. Sorafenib induces a modest growth delay in HCC, and CXCR4 inhibition or blockade of Gr-1 synergize with sorafenib treatment by increasing cell apoptosis.

A, Sorafenib (SOR) treatment induces a minor but significant delay in orthotopic HCC models after 14 days of treatment. HCA-1 tumor growth was delayed by sorafenib in syngeneic C3H mice with underlying liver fibrosis (N=6–14 mice per group). Whereas treatment with the CXCR4 inhibitor AMD3100 (AMD) had no effect as monotherapy, AMD3100 with sorafenib induces a more significant tumor growth delay. B, Representative images of immunofluorescence TUNEL in HCA-1 tumors. Images are 636μm across. C, Cell apoptosis, measured by TUNEL, increased after sorafenib treatment in HCA-1 tumors (N=6–11). D, Representative images of immunofluorescence of TUNEL in spontaneous HCCs. Images are 636μm across. E, Cell apoptosis, measured by TUNEL, increased after sorafenib treatment in spontaneously arising HCCs in Mst1−/− Mst2F/− transgenic mice (N=6–11). Cell apoptosis was significantly increased when sorafenib treatment was combined with AMD3100 in both tumor models. F, Combining anti-Gr-1 antibodies with sorafenib treatment induced a significant tumor growth delay of orthotopic HCA-1 tumors growing in mice with liver fibrosis (N=7–13 mice per group). Data are presented as mean ± SEM; *P<0.05, **P<0.01.
DISCUSSION
Despite its inhibitory effect on hepatic fibrogenesis, the efficacy of sorafenib treatment in HCC may be thwarted ensuing increase in hypoxia that leads to increased tumor desmoplasia and inflammation. We show that although sorafenib can reduce chemically induced liver fibrosis in mice, its anti-vascular effects in tumors lead to increased hypoxia, inflammation and fibrosis in the tumor tissue. Our results confirm the anti-fibrotic effects seen in cirrhotic livers with sorafenib and other TKIs (vatalanib) (38, 39), but also reveal that tumor-associated fibrosis/desmoplasia is increased after sorafenib treatment. This indicates a potential role of hypoxia-induced tumor fibrosis during development of resistance to sorafenib treatment in HCC.
PDGF-B is a pro-fibrotic growth factor whose signaling is blocked by sorafenib (5, 40, 41). However, the conversion of HSCs to myofibroblasts during hepatic fibrogenesis or the activation of fibroblasts during development of desmoplasia in malignant tumors (such as breast cancer) may be directly mediated by other pro-fibrotic and pro-inflammatory factors such as SDF1α (42–47). We demonstrate here in mouse models of HCC that increased tumor-associated fibrosis is due to increased myofibroblast infiltration and differentiation mediated by the SDF1α CXCR4 pathway (Fig. 8). Our data show that SDF1α can directly induce HSC differentiation and proliferation via MAPK activation after sorafenib treatment. We also show that SDF1α can counteract the anti-fibrotic effects of sorafenib via PDGFR-inhibition – thus leading to increased tumor-associated fibrosis. This led us to test whether CXCR4 blockade could prevent the increase in fibrosis in HCC after sorafenib treatment. Indeed, addition of CXCR4 to sorafenib treatment prevented the increase in tumor-associated fibrosis in the face of persistent hypoxia. Moreover, this combination therapy significantly inhibited HCC growth compared to sorafenib alone.
Figure 8. Differential impact of sorafenib on liver versus tumor-associated fibrosis mediated by SDF1α/CXCR4 axis and Gr-1+ cells in HCC.

The differential effects of sorafenib are the result of increased intratumoral hypoxia, leading to elevated SDF1α expression and Gr-1+ myeloid cell infiltration. Blocking CXCR4 prevents Gr-1+ myeloid cell infiltration and hepatic stellate cells differentiation and activation, and synergizes with the anti-tumor effects of sorafenib.
Increased SDF1α expression can also lead to accumulation of tumor-promoting (pro-angiogenic and immune-suppressive) inflammatory cells (18, 25, 48, 49). We previously demonstrated that CXCR4 is critical for myeloid cell infiltration in tumors and can compensate for VEGFR1 inhibition in bone marrow-derived cells by inhibiting CXCR4 with pharmacologic agents and in genetic models (28). Indeed, we detected an increased infiltration in Gr-1+ myeloid cells in HCC after sorafenib treatment. Paracrine interactions between stellate cells and inflammatory cells leading to liver fibrosis are also critical in viral hepatitis and in pancreatic malignancies (43–45, 50). Here, we demonstrate that Gr-1+ myeloid cells from sorafenib-treated tumors showed higher expression of multiple pro-fibrotic factors, including SDF1α and CXCR4, compared to Gr-1+ myeloid cells from control-treated tumors. Furthermore, we show Gr-1+ cells directly stimulated the differentiation of HSCs and the CXCR4 blockade prevented the upregulation of collagen I and α-SMA expression in HSCs co-cultured with tumor-derived Gr-1+ myeloid cells. It indicates that SDF1α/CXCR4 axis plays an important role mediating not only Gr-1+ myeloid cell infiltration in HCC but also their paracrine interaction with HSCs leading to fibrosis. Finally, antibody blockade of Gr-1 reduced Gr-1+ myeloid cell infiltration, tumor desmoplasia, and HCC growth.
In conclusion, targeting the SDF1α/CXCR4 pathway or Gr-1+ myeloid cell infiltration may be an effective approach to block hypoxia-induced HCC desmoplasia and overcome resistance to sorafenib therapy in HCC.
Supplementary Material
Acknowledgments
Financial Support: This study was supported by NIH grant P01-CA080124. DGD’s work has been supported through NIH grants R01-CA159258, R21-CA139168 and Proton Beam/Federal Share Program, and the American Cancer Society grant 120733-RSG-11-073-01-TBG. RKJ’s work has been supported through NIH grants R01-CA126642 and Proton Beam/Federal Share Program, and the Department of Defense Breast Cancer Innovator Award W81XWH-10-1-0016. TR’s work is supported by a Max Kade Fellowship.
We thank D Nguyen and C Smith for outstanding technical support, Drs. JA Engelman and Dr. CH Benes (MGH) for useful discussions and Dr. Bardeesy for providing the Mst1−/− Mst2F/− transgenic mice.
List of Abbreviations
- HCC
hepatocellular carcinoma
- SDF1α
stromal-derived factor 1 alpha
- CXCR4
C-X-C receptor type 4
- HSC
hepatic stellate cell
- MAPK
mitogen-activated protein kinase
- Gr-1
myeloid differentiation antigen
- TKI
tyrosine kinase inhibitor
- VEGFR
vascular endothelial growth factor receptor
- PDGFR
platelet-derived growth factor receptor
- FGFR
basic fibroblast growth factor receptor
- CXCL12
C-X-C ligand 12
- PBS
phosphate buffered solution
- Ad-Cre
Cre-expressing adenovirus
- CCl4
carbon tetrachloride
- α–SMA
alpha smooth muscle actin
- MCP-1
monocyte chemoattractant protein-1
- 5-LO
5-lipoxygenase
- TGF
transforming growth factor
- MMP
matrix metalloproteinase
- CAIX
carbonic anhydrase 9
Contributor Information
Yunching Chen, Email: yunching@mx.nthu.edu.tw.
Yuhui Huang, Email: yhuang@steele.mgh.harvard.edu.
Thomas Reiberger, Email: reiberger@steele.mgh.harvard.edu.
Annique M. Duyverman, Email: anniquep@gmail.com.
Peigen Huang, Email: peigen@steele.mgh.harvard.edu.
Rekha Samuel, Email: rekhasamuel@mcvellore.ac.in.
Lotte Hiddingh, Email: lhiddingh@hotmail.com.
Sylvie Roberge, Email: sylvie@steele.mgh.harvard.edu.
Christina Koppel, Email: christina@steele.mgh.harvard.edu.
Gregory Y. Lauwers, Email: glauwers@partners.org.
Andrew X. Zhu, Email: azhu@partners.org.
Rakesh K. Jain, Email: jain@steele.mgh.harvard.edu.
Dan G. Duda, Email: duda@steele.mgh.harvard.edu.
References
- 1.Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology. 2013;144:512–527. doi: 10.1053/j.gastro.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hui CK, Leung N, Shek TW, Yao H, Lee WK, Lai JY, Lai ST, et al. Sustained disease remission after spontaneous HBeAg seroconversion is associated with reduction in fibrosis progression in chronic hepatitis B Chinese patients. Hepatology. 2007;46:690–698. doi: 10.1002/hep.21758. [DOI] [PubMed] [Google Scholar]
- 3.Luedde T, Schwabe RF. NF-kappaB in the liver--linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 5.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 6.Almhanna K, Philip PA. Safety and efficacy of sorafenib in the treatment of hepatocellular carcinoma. Onco Targets Ther. 2009;2:261–267. doi: 10.2147/ott.s5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5:215–219. doi: 10.1016/s1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 8.Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 9.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 10.Zhu AX, Duda DG, Sahani DV, Jain RK. HCC and angiogenesis: possible targets and future directions. Nat Rev Clin Oncol. 2011;8:292–301. doi: 10.1038/nrclinonc.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng A, Kang Y, Lin D, Park J, Kudo M, Qin S, Omata S, et al. Phase III trial of sunitinib versus sorafenib in advanced hepatocellular carcinoma. J Clin Oncol. 2011;(suppl):abstr 4000. doi: 10.1200/JCO.2012.45.8372. [DOI] [PubMed] [Google Scholar]
- 12.Eckel F, von Delius S, Mayr M, Dobritz M, Fend F, Hosius C, Schleyer E, et al. Pharmacokinetic and clinical phase II trial of imatinib in patients with impaired liver function and advanced hepatocellular carcinoma. Oncology. 2005;69:363–371. doi: 10.1159/000089990. [DOI] [PubMed] [Google Scholar]
- 13.Kaseb AO, Hanbali A, Cotant M, Hassan MM, Wollner I, Philip PA. Vascular endothelial growth factor in the management of hepatocellular carcinoma: a review of literature. Cancer. 2009;115:4895–4906. doi: 10.1002/cncr.24537. [DOI] [PubMed] [Google Scholar]
- 14.Loges S, Mazzone M, Hohensinner P, Carmeliet P. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell. 2009;15:167–170. doi: 10.1016/j.ccr.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Sennino B, McDonald DM. Controlling escape from angiogenesis inhibitors. Nat Rev Cancer. 2012;12:699–709. doi: 10.1038/nrc3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–2218. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duda DG, Kozin SV, Kirkpatrick ND, Xu L, Fukumura D, Jain RK. CXCL12 (SDF1alpha)-CXCR4/CXCR7 pathway inhibition: an emerging sensitizer for anticancer therapies? Clin Cancer Res. 2011;17:2074–2080. doi: 10.1158/1078-0432.CCR-10-2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 20.Friand V, Haddad O, Papy-Garcia D, Hlawaty H, Vassy R, Hamma-Kourbali Y, Perret GY, et al. Glycosaminoglycan mimetics inhibit SDF-1/CXCL12-mediated migration and invasion of human hepatoma cells. Glycobiology. 2009;19:1511–1524. doi: 10.1093/glycob/cwp130. [DOI] [PubMed] [Google Scholar]
- 21.Schimanski CC, Bahre R, Gockel I, Muller A, Frerichs K, Horner V, Teufel A, et al. Dissemination of hepatocellular carcinoma is mediated via chemokine receptor CXCR4. Br J Cancer. 2006;95:210–217. doi: 10.1038/sj.bjc.6603251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang ZL, Zeng ZC, Tang ZY, Fan J, Zhuang PY, Liang Y, Tan YS, et al. Chemokine receptor CXCR4 expression in hepatocellular carcinoma patients increases the risk of bone metastases and poor survival. BMC Cancer. 2009;9:176. doi: 10.1186/1471-2407-9-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu AX, Sahani DV, Duda DG, di Tomaso E, Ancukiewicz M, Catalano OA, Sindhwani V, et al. Efficacy, safety, and potential biomarkers of sunitinib monotherapy in advanced hepatocellular carcinoma: a phase II study. J Clin Oncol. 2009;27:3027–3035. doi: 10.1200/JCO.2008.20.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu AX, Ancukiewicz M, Supko JG, Sahani DV, Blaszkowsky LS, Meyerhardt JA, Abrams TA, et al. Efficacy, Safety, Pharmacokinetics, and Biomarkers of Cediranib Monotherapy in Advanced Hepatocellular Carcinoma: A Phase II Study. Clin Cancer Res. 2013;19:1557–1566. doi: 10.1158/1078-0432.CCR-12-3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, Song H, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Littlepage LE, Egeblad M, Werb Z. Coevolution of cancer and stromal cellular responses. Cancer Cell. 2005;7:499–500. doi: 10.1016/j.ccr.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 27.Sutton A, Friand V, Brule-Donneger S, Chaigneau T, Ziol M, Sainte-Catherine O, Poire A, et al. Stromal cell-derived factor-1/chemokine (C-X-C motif) ligand 12 stimulates human hepatoma cell growth, migration, and invasion. Mol Cancer Res. 2007;5:21–33. doi: 10.1158/1541-7786.MCR-06-0103. [DOI] [PubMed] [Google Scholar]
- 28.Hiratsuka S, Duda DG, Huang Y, Goel S, Sugiyama T, Nagasawa T, Fukumura D, et al. C-X-C receptor type 4 promotes metastasis by activating p38 mitogen-activated protein kinase in myeloid differentiation antigen (Gr-1)-positive cells. Proc Natl Acad Sci U S A. 2011;108:302–307. doi: 10.1073/pnas.1016917108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 30.Zhu AX, Duda DG, Ancukiewicz M, di Tomaso E, Clark JW, Miksad R, Fuchs CS, et al. Exploratory analysis of early toxicity of sunitinib in advanced hepatocellular carcinoma patients: kinetics and potential biomarker value. Clin Cancer Res. 2011;17:918–927. doi: 10.1158/1078-0432.CCR-10-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tofilon PJ, Basic I, Milas L. Prediction of in vivo tumor response to chemotherapeutic agents by the in vitro sister chromatid exchange assay. Cancer Res. 1985;45:2025–2030. [PubMed] [Google Scholar]
- 32.Das A, Shergill U, Thakur L, Sinha S, Urrutia R, Mukhopadhyay D, Shah VH. Ephrin B2/EphB4 pathway in hepatic stellate cells stimulates Erk-dependent VEGF production and sinusoidal endothelial cell recruitment. Am J Physiol Gastrointest Liver Physiol. 2010;298:G908–915. doi: 10.1152/ajpgi.00510.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim W, Seong J, Oh HJ, Koom WS, Choi KJ, Yun CO. A novel combination treatment of armed oncolytic adenovirus expressing IL-12 and GM-CSF with radiotherapy in murine hepatocarcinoma. J Radiat Res. 2011;52:646–654. doi: 10.1269/jrr.10185. [DOI] [PubMed] [Google Scholar]
- 34.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim SU, Oh HJ, Wanless IR, Lee S, Han KH, Park YN. The Laennec staging system for histological sub-classification of cirrhosis is useful for stratification of prognosis in patients with liver cirrhosis. J Hepatol. 2012;57:556–563. doi: 10.1016/j.jhep.2012.04.029. [DOI] [PubMed] [Google Scholar]
- 36.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109:17561–17566. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bai Q, An J, Wu X, You H, Ma H, Liu T, Gao N, et al. HBV promotes the proliferation of hepatic stellate cells via the PDGF-B/PDGFR-beta signaling pathway in vitro. Int J Mol Med. 2012;30:1443–1450. doi: 10.3892/ijmm.2012.1148. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Lui EL, Friedman SL, Li L, Ye T, Chen Y, Poon RT, et al. PTK787/ZK22258 attenuates stellate cell activation and hepatic fibrosis in vivo by inhibiting VEGF signaling. Lab Invest. 2009;89:209–221. doi: 10.1038/labinvest.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Gao J, Zhang D, Zhang J, Ma J, Jiang H. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol. 2010;53:132–144. doi: 10.1016/j.jhep.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 40.Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Gentilini A, Caligiuri A, et al. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol. 1996;148:785–800. [PMC free article] [PubMed] [Google Scholar]
- 41.Lederle W, Stark HJ, Skobe M, Fusenig NE, Mueller MM. Platelet-derived growth factor-BB controls epithelial tumor phenotype by differential growth factor regulation in stromal cells. Am J Pathol. 2006;169:1767–1783. doi: 10.2353/ajpath.2006.060120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107:20009–20014. doi: 10.1073/pnas.1013805107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitra P, Shibuta K, Mathai J, Shimoda K, Banner BF, Mori M, Barnard GF. CXCR4 mRNA expression in colon, esophageal and gastric cancers and hepatitis C infected liver. Int J Oncol. 1999;14:917–925. doi: 10.3892/ijo.14.5.917. [DOI] [PubMed] [Google Scholar]
- 44.Terada R, Yamamoto K, Hakoda T, Shimada N, Okano N, Baba N, Ninomiya Y, et al. Stromal cell-derived factor-1 from biliary epithelial cells recruits CXCR4-positive cells: implications for inflammatory liver diseases. Lab Invest. 2003;83:665–672. doi: 10.1097/01.lab.0000067498.89585.06. [DOI] [PubMed] [Google Scholar]
- 45.Wald O, Pappo O, Safadi R, Dagan-Berger M, Beider K, Wald H, Franitza S, et al. Involvement of the CXCL12/CXCR4 pathway in the advanced liver disease that is associated with hepatitis C virus or hepatitis B virus. Eur J Immunol. 2004;34:1164–1174. doi: 10.1002/eji.200324441. [DOI] [PubMed] [Google Scholar]
- 46.Hong F, Tuyama A, Lee TF, Loke J, Agarwal R, Cheng X, Garg A, et al. Hepatic stellate cells express functional CXCR4: role in stromal cell-derived factor-1alpha-mediated stellate cell activation. Hepatology. 2009;49:2055–2067. doi: 10.1002/hep.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sawitza I, Kordes C, Reister S, Haussinger D. The niche of stellate cells within rat liver. Hepatology. 2009;50:1617–1624. doi: 10.1002/hep.23184. [DOI] [PubMed] [Google Scholar]
- 48.Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475:226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 49.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 50.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.