

Abstract
Objective
We previously demonstrated that NADPH oxidase 4 (Nox4) mediates increased monocyte priming and chemotaxis under conditions of diabetic metabolic stress (DMS), and emerging data indicate that Group VIA Phospholipase A2 (iPLA2β) also participates in regulating monocyte chemotaxis. Here we examined relationships between iPLA2β expression and Nox4 action in mouse peritoneal macrophages subjected to DMS.
Approach and Results
Increased iPLA2β expression and activity were observed in macrophages from low density lipoprotein receptor knockout (LDLR−/−) mice that were fed a high fat diet (HFD), and this was associated with time-dependent increases in atherosclerotic lesion size and macrophage content. Incubating macrophages with 30 mM D-glucose (HG), 100 μg/ml LDL, or both (HG+LDL) induced a robust increase in iPLA2β expression and activity and in cell migration in response to monocyte chemoattractant protein-1 (MCP-1). The increases in iPLA2β activity and cell migration were prevented by a bromoenol lactone (BEL) iPLA2β suicide inhibitor or an iPLA2β antisense oligonucleotide. Incubating macrophages under conditions that mimic DMS ex vivo resulted in increased Nox4 expression and activity and H2O2 generation compared to controls. BEL prevented those effects without affecting Nox2 expression. Nox4 inhibition eliminated DMS-induced acceleration of macrophage migration. Lysophosphatidic acid (LPA) restored Nox4 expression, H2O2 generation, and migration to BEL-treated cells, and an LPA receptor antagonist abrogated iPLA2β-mediated increases in Nox4 expression.
Conclusions
Taken together, these observations identify iPLA2β and LPA derived from its action as critical in regulating macrophage Nox4 activity and migration in the diabetic state in vivo and under similar conditions ex vivo.
Keywords: Phospholipase A2, NADPH oxidase, Macrophages, Migration, Diabetes
INTRODUCTION
Diabetes mellitus is associated with an increased risk of cardiovascular disease,1 and individuals with diabetes have an absolute risk of major coronary events similar to that of those without diabetes who have established coronary artery disease.2 In the evolution of type 2 diabetes, insulin resistance eventuates in beta cell failure that results in conditions that we have designated “diabetic metabolic stress” (DMS) characterized by hyperglycemia, hyperlipidemia, and hyperinsulinemia. Although molecular mechanisms that link diabetes to atherosclerosis and coronary artery disease are incompletely understood, enhanced recruitment of macrophages into inflamed vascular sites, such as coronary arteries, occurs in DM, and increased cellular oxidative stress contributes to this phenomenon.3–6
Reactive oxygen species (ROS) can be generated by mitochondrial metabolism or as byproducts of oxygenases such as cytochrome P450 monooxygenases, arachidonate dioxygenases, and the uncoupling of nitric oxide synthases, inter alia. In addition, NADPH oxidases (Nox) are now recognized to be enzymes that produce ROS as a primary product rather than as a byproduct,7 and seven human and animal Nox variants are now recognized. These include Nox1 - 5, Duox1, and Duox2, and they share the ability to transport electrons across the plasma membrane and to generate O2•−, which is then dismutated to yield hydrogen peroxide (H2O2). The Nox2 isoform was originally designated phagocytic Nox gp91phox and was believed to be the predominant ROS producer in phagocytes, and its role in the respiratory bust of neutrophils and macrophages has been studied extensively,8, 9
We recently demonstrated that Nox4 is also a major inducible source of ROS in human and mouse macrophages but that Nox isoforms other than Nox2 and Nox4 are barely detectable in these cells.10 Our most recent data demonstrated that Nox4, but not Nox2, contributes to increases in monocyte chemotaxis and macrophage recruitment under conditions of DMS in vivo and similar conditions ex vivo,11 but little is known about upstream regulatory machinery that governs Nox4 expression. The Group VIA Phospholipase A2 (iPLA2β) is a member of a superfamily of PLA2 enzymes that hydrolyze the sn-2 ester bond of phospholipids to release a fatty acid (e.g., arachidonic acid) and a 2-lysophospholipid (e.g., 2-lysophosphatidylcholine, LPC, or 2-lysophosphatidic acid, LPA).12 Metabolites of arachidonic acid (eicosanoids, e.g. prostaglandins and leukotrienes) and of lysophospholipids (e.g., platelet-activating factor and endocannabinoids) serve as important inflammatory mediators 13, and both arachidonic acid and lysophospholipids, including LPA, also have intrinsic mediator functions.14, 15
iPLA2β has been reported to be an important participant in macrophage chemotactic responses to chemoattractants, including Monocyte Chemattractant Protein-1 (MCP-1).16 We demonstrated previously that transgenic mice that overexpress iPLA2β specifically in vascular smooth muscle cells develop exaggerated neointima formation in a carotid artery ligation model of vascular injury and atherogenesis and that the resultant lesions exhibit increased content of macrophages and inflammatory cytokines compared to those of control mice.17 In addition, emerging data reveal increased iPLA2β expression in the tissues of humans and animals with diabetes,18–20 but the mechanism underlying this phenomenon or whether it involves Nox4 has not previously been examined to our knowledge. To address this issue, we fed a Western high fat diet (HFD) to mice rendered deficient in the low density lipoprotein receptor (LDLR−/−) by homologous recombination and compared them to mice fed a standard chow maintenance diet. We also studied isolated mouse peritoneal macrophages ex vivo that were incubated in medium containing a high glucose (HG) concentration, added low density lipoprotein (LDL), or both in experiments that examined: 1) iPLA2β expression and activity under conditions that mimic DMS; 2) any relationship between expression and activity of iPLA2β and Nox4; and 3) effects of the potential iPLA2β products arachidonic acid and lysophosphatidic acid on Nox4 expression and activity.
MATERIALS AND METHODS
The Materials and Methods sections are available in the online-only Data Supplement.
RESULTS
Increased iPLA2β expression and activity in macrophages from mice subjected to metabolic stress
We fed LDLR−/− mice a Western high fat diet (HFD) for 8, 16, or 24 weeks to induce metabolic stress characterized by hyperglycemia and hyperlipidemia. Age-matched mice fed a standard chow maintenance diet served as controls. As summarized in Table, consumption of the HFD resulted in a time-dependent increase in body weight, white fat pad mass, and concentrations of blood glucose, plasma total cholesterol, plasma triglycerides, and plasma insulin. Atherosclerotic lesion area and macrophage content also increased in a time-dependent manner in the HFD-fed mice (Table). Figure 1 illustrates that peritoneal macrophages from HFD-fed mice exhibited a time-dependent increase in iPLA2β immunoreactive protein (Panel A) and iPLA2β enzymatic activity (Panel B). In addition, the HFD-fed mice exhibited a time-dependent increase in aortic sinus atherosclerotic lesion area and macrophage content, and both parameters exhibited positive linear correlations with peritoneal macrophage iPLA2β specific enzymatic activity (Panels C and D). Similar correlations were observed between iPLA2β specific activity and atherosclerotic lesion in aortic arch (data not shown). These findings suggest that increased iPLA2β expression in macrophages of mice with diabetes may promote their migration into the sub-endothelial space of inflamed arteries to accelerate atherogenesis.
Table.
Characteristics of LDLR−/− mice fed a high fat diet (HFD)
| Weeks of HFD | 0 | 8 | 16 | 24 |
|---|---|---|---|---|
| Body weight (g) | 25.3 ± 1.2 | 36.6 ± 1.7* # | 43.8 ± 2.0* | 48.2 ± 2.5* |
| White fat pad mass (g) | 1.7 ± 0.2 | 2.6 ± 0.2* # | 3 ± 0.3* | 3.6 ± 0.2* |
| Blood glucose | 103.6 ± 6.1 | 156.1 ± 8.9* # | 201.5 ± 11.3* | 233.3 ± 13.6* |
| Plasma TC (mg/ml) | 210 ± 6.2 | 520 ± 9.0* # | 625 ± 7.5* | 630 ± 10.6* |
| Plasma TG (mg/ml) | 129 ± 6.1 | 196 ± 5.5* # | 256 ± 8.5* | 276 ± 11.8* |
| Plasma insulin (ng/ml) | 0.6 ± 0.2 | 7.5 ± 3.0* | 10.9 ± 2.1* | 11.6 ± 2.6* |
| Macrophages in lesion (mm2) | 0.04 ± 0.01 | 0.23 ± 0.06*# | 0.57 ± 0.1* | 1.26 ± 0.5* |
| Atherosclerotic lesion (mm2) | 0.09 ± 0.03 | 0.61 ± 0.15*# | 1.5 ± 0.4* | 3.6 ± 0.6* |
Note: LDLR−/− mice fed a HFD for the indicated intervals develop obesity and a metabolic syndrome characterized by hyperglycemia and hyperlipidemia. Body weight, epididymal fat pad mass, and concentrations of blood glucose and plasma total cholesterol (TC), triglycerides (TG), and insulin were measured as described in Materials and Methods. Macrophages recruited into aortic sinus atherosclerotic lesions are quantified as CD-68 positive staining area (mm2). Aortic sinus atherosclerotic lesion area pertains to the area inside the elastic lamina (mm2). Data are presented as mean ± SEM. An asterisk (*) denote a p value < 0.05 for the difference between the indicated condition and the HFD 0 week condition, which pertains to mice fed a maintenance diet. A hashtag symbol (#) denotes a p value < 0.01 for the difference between the indicated condition and HFD-fed mice at 16 or 24 weeks (n = 9).
Figure 1. Expression and activity of iPLA2β increase in peritoneal macrophages from High Fat Diet (HFD)-fed mice compared to controls fed a standard chow maintenance diet (MD) and correlate positively with atherosclerotic lesion area and macrophage content.

iPLA2β immunoreactive protein expression and specific enzymatic activity were determined for peritoneal macrophages isolated from LDLR−/− mice fed a HFD for the indicated intervals. A, Western blotting quantification of iPLA2β immunoreactive protein. An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition compared to MD-fed control mice (AIN-93G, F3156, BioServ) that are represented by the group designated HFD 0 wk. A hashtag symbol (#) denotes a p value < 0.05 for the difference between the indicated condition compared to the remaining groups (n = 9). B, iPLA2β specific enzymatic activity. An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition compared to control mice, as in Panel A, and a hashtag symbol (#) denotes a p value < 0.05 for the difference between the indicated condition compared to mice fed HFD for 16 or 24 weeks (n = 9). C, Regression plot of the relationship between peritoneal macrophage iPLA2β specific enzymatic activity and aortic sinus atherosclerotic lesion macrophage content reflected by CD-68 positive staining area (R2 = 0.938, P < 0.001). Grey, white and dark triangles indicate mice fed a HFD for 0, 8, or 16 weeks, respectively. D, Regression plot of the relationship between peritoneal macrophage iPLA2β specific enzymatic activity and total aortic sinus atherosclerotic lesion area (R2 = 0.903, p < 0.001). Grey, white, and dark circles indicate mice fed a HFD for 0, 8, or 16 weeks, respectively.
Metabolic stress enhances expression and activity of iPLA2β in macrophages ex vivo
To determine whether the changes in macrophage iPLA2β expression that occurred in vivo in mice with diet-induced diabetes could be mimicked by exposure of macrophages to high concentrations of glucose and lipids ex vivo, peritoneal macrophages were isolated from C57BL/6J mice and cultured (24 hr) in medium containing normal level glucose (5.5 mM), HG (30 mM), LDL (100 μg/ml), or both (HG+LDL). The non-metabolized L-glucose (LG) enantiomer was used as an osmotic control. As illustrated in Figure 2, incubation of macrophages with HG induced a ten-fold rise in content of immunoreactive iPLA2β protein (Panel A) and an eight-fold rise in iPLA2β specific enzymatic activity that was sensitive to inhibition by BEL and suppressed by an iPLA2β antisense oligonucleotide (Panel B). The increased expression of iPLA2β occurred as early as 6 hr after initiating exposure to HG and persisted at the same level for at least 48 hr (Panel A). Addition of LDL to the incubation medium also induced a rise in macrophage iPLA2β specific enzymatic activity that was similar in magnitude to that produced by HG, and the combination of HG and LDL in the incubation medium induced an additive rise in iPLA2β. Thus, exposure ex vivo to incubation medium containing high concentrations of glucose and lipoprotein induces a rise in macrophage iPLA2β expression and activity that is similar to that which occurs in vivo in the setting of hyperglycemia and hyperlipidemia that characterize diabetes. Notably, macrophage cell death was not observed upon incubation in medium with HG and bromoenol lactone (BEL) (25 μM) under the conditions of these experiments (Figure 2D).
Figure 2. Expression and activity of iPLA2β increase in peritoneal macrophages from cultured in the presence of high concentrations of D-glucose or lipoproteins.
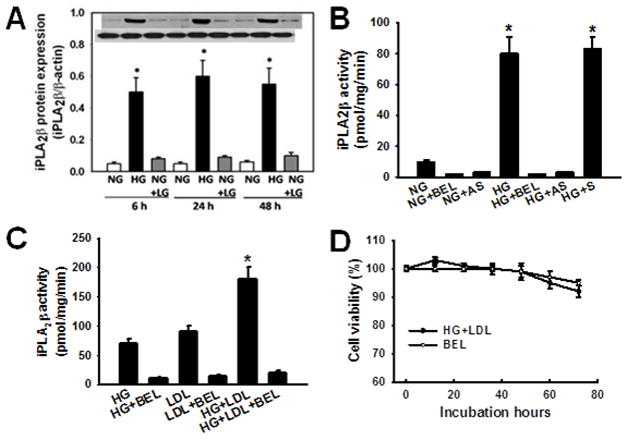
iPLA2β immunoreactive protein expression and specific enzymatic activity were determined as in Figure 1 for mouse peritoneal macrophages cultured in medium supplemented with 5 mM D-glucose (“NG”) or 30 mM D-glucose (“HG”) without or with 100 μg/mL LDL (“LDL”) for the indicated intervals. The enantionmer L-glucose (“LG”) is not metabolized by mammalian cells and was used as an osmotic control at final concentration of 30 mM. A, Quantitative immunoblotting of expression of iPLA2β protein by macrophages cultured under conditions described above. An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition compared to NG (n = 3). B, iPLA2β specific enzymatic activity in macrophages cultured with NG or HG and assayed without or with the iPLA2β inhibitor BEL (25 μM) or an iPLA2β antisense (AS) or sense (S) oligonucleotide. An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition compared to NG or HG (n = 3). C, iPLA2β specific enzymatic activity in macrophages cultured with HG and without or with LDL and assayed without or with BEL. An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition vs. HG or LDL (n = 3). D, A time course of cell viability of peritoneal macrophages, as assessed by trypan blue exclusion, upon incubation in medium supplemented with 30 mM D-glucose (HG) plus LDL (100 μg/mL) without or with BEL (25 μM). No cell death was detectable within 48 hr of incubation under these conditions, and viability exceeded 90% at 72 hr without a significant difference between groups. (n = 3).
Metabolic stress-induced enhancement of macrophage migration in response to Monocyte Chemoattractant Protein-1 (MCP-1) requires participation of iPLA2β
To determine whether iPLA2β participates in the acceleration of the macrophage migratory response that occurs under conditions of diabetic metabolic stress, the migration of isolated mouse peritoneal macrophages was measured in a Boyden chamber chemotaxis assay after 24 hr of incubation under various test conditions. Macrophages exposed to medium containing HG exhibited about a 3-fold increase in migration in response to MCP-1 compared to control macrophages incubated in medium with normal glucose concentration (Figure 3). Macrophages incubated in normal glucose medium supplemented with LDL exhibited migratory responses to MCP-1 similar to those of macrophages incubated with HG alone (Figure 3A). Incubation in HG medium supplemented with LDL resulted in macrophage migratory responses to MCP-1 that were additive (six-fold over control) compared to those that resulted from incubation with either HG or LDL alone (Figure 3A). Either pharmacologic inhibition of iPLA2β activity with BEL or suppression of iPLA2β expression with antisense oligonucleotides prevented the effects of supplementing the incubation medium with HG, LDL, or both to stimulate macrophage migratory responses to MCP-1, although a sense oligonucleotide had no effect (Figure 3A). Restoration of iPLA2β expression to BEL-treated macrophages with adenoviral vectors reversed the inhibition of MCP-1-induced migration of macrophages that had been incubated in medium supplemented with both HG and LDL (Figure 3B). Moreover, peritoneal macrophages isolated from iPLA2β-knockout (KO) mice prepared by homologous recombination exhibited a much diminished enhancement of MCP-1-induced migration after incubation in medium supplemented with both HG and LDL compared to wild type macrophages (Figure 3C). These observations demonstrate that the enhanced MCP-1-induced migration that occurs with macrophages incubated with high concentrations of glucose and LDL requires the participation of iPLA2β.
Figure 3. Culture of peritoneal macrophages ex vivo in medium supplemented with high concentrations of glucose or lipoproteins results in an enhanced migratory response to MCP-1 in which iPLA2β participates.
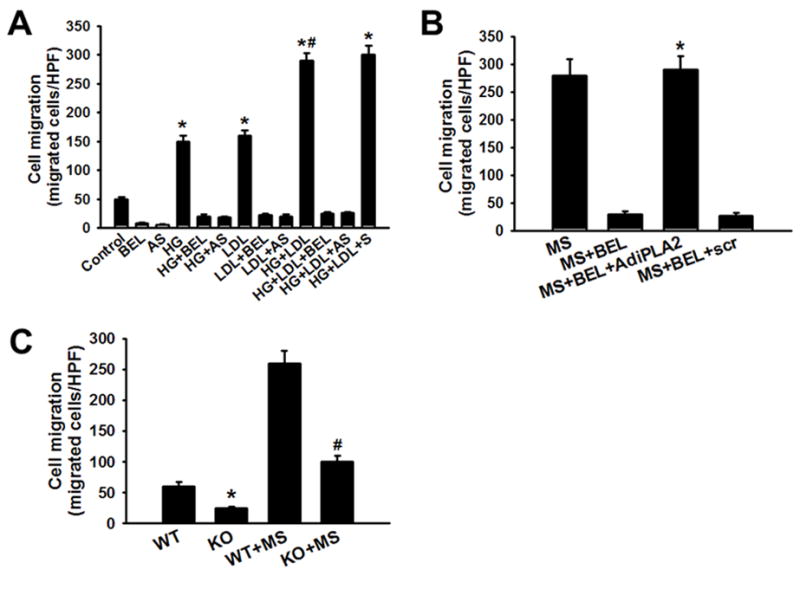
Mouse peritoneal macrophages were incubated (24 hr) in medium supplemented with 5 mM D-glucose (Control) or 30 mM D- glucose (HG) without or with 100 μg/mL LDL (LDL). The HG + LDL condition is designated “MS” for “metabolic stress.” Macrophage migratory responses to MCP-1 were then assayed in a Boyden chamber in the absence or presence of BEL and/or iPLA2β antisense (AS) or sense (S) oligonucleotides. A, Culture with HG and/or LDL increases macrophage migration, and this is suppressed by BEL or AS. An asterisk (*) denotes a p value < 0.01 vs. the Control condition. A hashtag symbol (#) denotes no statistically significant difference between the indicated condition and the HG+LDL+S condition. B, Adnenoviral vector driven overexpression of iPLA2β (AdiPLA2) restores an enhanced migratory response to MCP-1 to BEL-treated macrophages. An asterisk (*) denotes either a p value < 0.01 for the indicated condition compared to the other BEL-treated groups or a lack of a statistically significant difference compared to the MS condition. C, MCP-1 induced migration of peritoneal macrophages isolated from wild type (WT) or iPLA2β-knockout (KO) mice after incubation in NG or MS medium. An asterisk (*) denotes a p value < 0.01 compared to WT, and a hashtag symbol (#) denotes a p value < 0.01 vs. WT+MS (n = 3).
Nox4 expression and activity increase in macrophages incubated under conditions of metabolic stress, and this requires the participation of iPLA2β
Accumulating evidence suggests that increased cellular ROS generation occurs in diabetes, and we have previously demonstrated that increased Nox4 activity and the resultant ROS generation contribute to the enhancement of macrophage migration and accumulation at inflammatory sites that occur under conditions of metabolic stress.11 To determine whether iPLA2β participates in these processes, ROS production in DCFH-DA-loaded macrophages was measured by monitoring DCF fluorescence by FACS and was found to rise markedly in macrophages incubated in medium supplemented with HG and LDL compared to control macrophages incubated in normal glucose medium (Figure 4A). Moreover, this response was suppressed by the iPLA2β BEL, which also inhibited basal ROS production. We found that intracellular H2O2 levels were six-fold higher in macrophages incubated in medium supplemented with HG and LDL compared to macrophages incubated in normal glucose medium, and that both basal and (HG + LDL)-stimulated H2O2 levels were greatly reduced in BEL-treated macrophages (Figure 4B). Similar results were obtained when H2O2 was measured by an Amplex Red assay (Online Figure V). Consistent with reports that Nox4 is the predominant source of macrophage H2O2 generation,21–23 we found that incubation in medium supplemented with HG and LDL resulted in a rise in immunoreactive Nox4 and iPLA2β protein levels, and we also found that BEL blocked the rise in Nox4 protein but did not affect the rise in iPLA2β protein (Figure 4C). The RT-PCR analyses revealed that Nox4 mRNA levels rose in parallel with Nox4 immunoreactive protein in macrophages incubated in medium supplemented with HG and LDL, as illustrated in Online Figure III, and Nox4 enzymatic activity also rose under these conditions (Figure 4D). The latter effect was blocked by BEL but restored to BEL-pretreated macrophages by infection with an adenoviral vector that drives expression of iPLA2β (Figure 4D). Because cytosolic SOD could affect measurements of Nox activity in whole cell homogenates, Nox activity was also measured in isolated total membrane fractions by the lucigenin method, and the results of these experiments were concordant with those from whole cell homogenates, as illustrated in Online Figure VI. These observations suggest that increased Nox4 expression and the resultant ROS production in macrophages incubated in medium supplemented with HG and LDL requires signals derived from iPLA2β. Consistent with this interpretation, we found that adenoviral vector-driven overexpression of iPLA2β in macrophages results in increased expression of Nox4 protein (Figure 4E). Incubation of macrophages in medium supplemented with HG and LDL also resulted in increased expression of Nox2 but not Nox1 protein, but BEL did not affect the Nox2 response (Figure 4F). Moreover, adenoviral vector-driven overexpression of iPLA2β did not affect macrophage Nox2 expression levels (Figure 4G). We also observed Nox activity in macrophages isolated from Nox2 knockout mice. Upon incubation in medium supplemented with HG and LDL, total Nox activity in Nox2 deficient macrophages was lower than that in wild type macrophages incubated under the same conditions, but this effect did not achieve statistical significance. In contrast, total Nox activity was reduced by over 70% by adding either the Nox4 inhibitor GKT13783 or Nox4 siRNA (Online Figure IV). These observations indicate that the increased ROS production observed in macrophages incubated in medium supplemented with HG and LDL is predominantly attributable to increased expression of Nox4 protein rather than to another Nox isozyme and that signals derived from the increased expression of iPLA2β under these conditions are required for the effects on macrophage Nox4 expression and ROS production.
Figure 4. Culture of mouse peritoneal macrophages ex vivo with high concentrations of glucose and/or lipoproteins induces an increase in production of ROS and H2O2 and in Nox4 expression and activity, and these effects require the participation of iPLA2β.
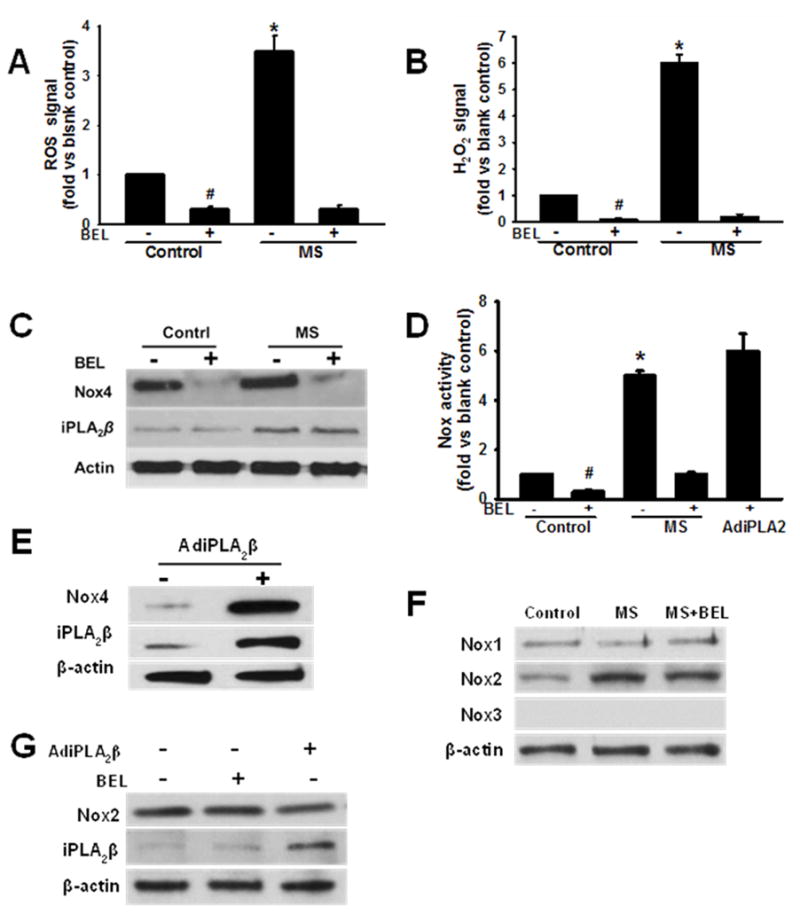
A, Macrphage ROS production. Isolated mouse peritoneal macrophages were incubated (24 hr) in medium containing 5 mM D-glucose (Control) or 30 mM D-glucose and 100 μg/ml LDL (MS) in the presence of absence of BEL, then loaded with DCFH-DA (10 μM, 30 min), and intracellular ROS production reflected by DCF fluorescence was determined by FACS. An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition compared to the other groups, and a hashtag symbol (#) denotes a p value < 0.01 compared to the blank control. B, Intracellular H2O2 production was measured with an Abnova Hydrogen Peroxide Assay kit (Catalog no. KA0801). An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition compared to the other groups, and a hashtag symbol (#) denotes a p value < 0.01 vs. the blank control. C, Immunoblots of Nox4, iPLA2β, and actin in macrophages incubated under control or MS conditions without or with BEL. D, Nox enzymatic activity assays for macrophages incubated under control or MS conditions without or with BEL and effect of adenoviral vector-driven iPLA2β overexpression (AdiPLA2β) to restore increased Nox4 activity to BEL-pretreated cells. An asterisk (*) denotes a p value < 0.01 vs. the control or MS+BEL conditions, and a hashtag symbol (#) denotes a p value < 0.01 vs. the blank control. E, Immunoblot of Nox4 and iPLA2β in macrophages infected with AdiPLA2β or uninfected control macrophages. F, Immunoblots of Nox1, Nox2 and Nox3 isoforms and actin in macrophages cultured under control, MS conditions or in the presence of BEL. G, Immunoblots of Nox2, iPLA2β, and actin in macrophages infected with AdiPLA2β, uninfected control macrophages, or in the presence of BEL.
Nox4 activity regulates macrophage migratory responses to MCP-1
We recently reported that Nox4 is the major source of ROS in macrophages and that Nox4-derived H2O2 participates regulating macrophage migration and in the signaling events that lead to monocyte dysfunction and a hyperchemotactic, proatherogenic macrophage phenotype.10, 11 Here we used the pharmacologic agent GKT137831, which is a potent inhibitor of Nox4 and Nox1 activity, and Nox4 siRNA to examine Nox4 involvement in the macrophage migratory response to MCP-1 (Figure 5). We found that GKT137831 greatly attenuated (by 75%) the enhancement in MCP-1-induced migration that occurred with macrophages that had been incubated in medium supplemented with HG and LDL, that the migratory response was restored by adenoviral vector-driven overexpression of Nox4, and that Nox4 siRNA also suppressed the enhancement of migration with macrophages incubated with HG and LDL (Figure 5A). These findings indicate that Nox4 is a critical component of the machinery that produces enhanced MCP-1-induced migration of macrophages subjected to metabolic stress. To examine further the effect of Nox4-derived H2O2 on cell migration, we performed experiments in the presence of PEG-catalase and PEG-superoxide dismutase (SOD) and measured H2O2 production with an Amplex Red Assay. PEG-catalase pretreatment was found to suppress the enhancement of MCP-1-induced macrophage migration otherwise observed in macrophages that had been incubated in medium supplemented with HG and LDL, and this was associated with a reduction in measured H2O2 levels. This is consistent with catalase-catalyzed decomposition of H2O2 and with the hypothesis that H2O2 is involved in producing the enhanced migratory response. No similar effect of PEG-SOD was observed on cell migration and H2O2 production, which suggests that endogenous SOD activity is sufficient to support maximal H2O2 production so that no additional effect of exogenous SOD is observed (Figure 5B and 5C).
Figure 5. The Nox4 inhibitor GKT137831 and catalase reduce MCP-1 induced macrophage migration.

A, GKT137831 reduces MCP-1 induced migration of both control macrophages and those subjected to metabolic stress by incubation with high concentrations of glucose and low density lipoprotein. Mouse peritoneal macrophages were incubated (16 hr) in medium containing 5 mM D-glucose (“Control” condition) or in medium supplemented with 30 mM D-glucose and 100 μg/mL LDL (“MS” condition). MCP-1-induced migration was measured in a Boyden chamber assay in the presence or absence of the Nox4 inhibitor GKT137831 (10 μM) for macrophages infected with an adenoviral vector driving overexpression of Nox4 (AdNox4), control uninfected cells, or cells treated with Nox4 siRNA. B and C, Pretreatment with PEG-Catalase (500 U/ml) but not with PEG-SOD (500 U/ml) reduces the effect of metabolic stress to enhance MCP-1-induced macrophage migration and H2O2 production, as measured with an Amplex Red assay kit (Invitrogen, Cat# A22188). An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition compared to PEG alone (n = 3).
Lysophosphatidic acid (LPA) derived from iPLA2β action is required for the increased Nox4 expression and ROS production that occurs in macrophages subjected to metabolic stress
We next examined the possibility that products of iPLA2β action are responsible for the increased Nox4 expression and ROS production by macrophages subjected to metabolic stress. Such products include a free fatty acid, e.g., arachidonic acid, and a 2-lysophospholipid, e.g. 2-lysophosphatidylcholine (LPC) generated from phosphatidylcholine or 2-lysophosphatidic acid (LPA) generated directly from phosphatidic acid or indirectly by the action of autotaxin on LPC.24 Generation of LPA and LPC is known to be severely impaired in some circumstances in iPLA2β-knockout mice.25 In addition, arachidonic acid can be further metabolized to biologically active eicosanoids via lipoxygenases (LO), cyclooxygenases (COX), and cytochrome P450-dependent monooxygenases (CYP) that produce a variety of mediators that affect inflammation.26 Incubation of isolated mouse peritoneal macrophages in medium supplemented with arachidonic acid without or with inhibitors of COX (indomethacin), LO (nordihydoguaiaretic acid) or CYP (17-octadecynoic acid) under conditions described in Experimental Procedures did not affect macrophage Nox4 expression or activity or MCP-1-induced migration (not shown). In contrast, adenoviral vector-driven overexpression of iPLA2β or incubation of macrophages in medium supplemented with HG and LDL resulted in a several-fold increase in LPA production, and the response to both conditions together was additive (Figure 6A). Moreover, BEL prevented a rise in LPA production in response to either or both conditions (Figure 6A). That this increased LPA production has functional significance was suggested by the fact that the LPA receptor antagonist VPC32183 prevented the increase in macrophage Nox4 expression (Figure 6B), H2O2 production (Figure 6C), and MCP-1-induced migration (Figure 6D) that otherwise occurred after incubation in medium supplemented with HG and LDL. Moreover, exogenous LPA reversed the BEL-induced suppression of the rises in Nox4 expression (Figure 7A), H2O2 production (Figure 7B), and MCP-1-induced migration (Figure 7C) that occurred with macrophages incubated in medium supplemented with HG and LDL. To complement the findings with the LPA receptor antagonist, we examined effects of reducing LPA receptor expression with siRNAs directed at LPA1 and LPA3 receptors. Knockdown of LPA1 and LPA3 was found to attenuate metabolic stress-induced increases in Nox4 expression, ROS production, and cell migration, and these findings are concordant with those obtained with the LPA receptor antagonist VPC32183 (Online Figure I). These findings indicate that LPA derived from the action of iPLA2β participates in the increased expression of Nox4, production of ROS, and enhanced MCP-1-induced migration that occur in macrophages subjected to metabolic stress ex vivo.
Figure 6. Macrophage production of lysophosphatidic acid (LPA) derived from iPLA2β action increases in cells subjected to metabolic stress by incubation with high concentrations of glucose and lipoprotein, and LPA drives increased Nox4 expression, H2O2 production, and enhanced MCP-1-induced migration.
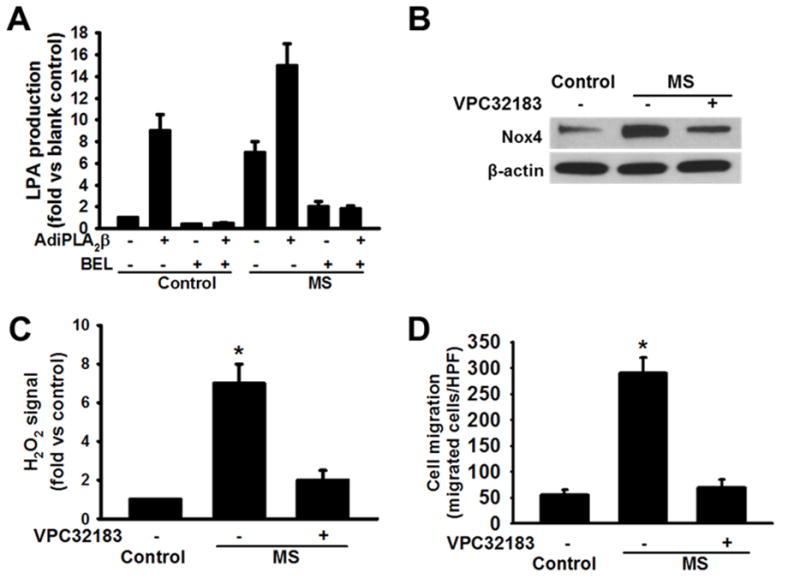
Peritoneal macrophages isolated from C57BL/6J mice were incubated in medium containing 5 mM D-glucose (“Control” condition) or supplemented with 30 mM D-glucose and 100 μg/mL LDL (“MS” condition) without or with BEL and/or infection with an adenovirus driving iPLA2β overexpression (AdiPLA2β). A, Macrophage LPA production. In panels B – D, effects of pretreating macrophages with the LPA1/3 receptor antagonist VPC32183 (5 nM, 30 min) before exposing them to MS conditions (24 hr) were examined, and VPC3218 pretreatment was found to: B. Suppress MS-induced increases in macrophage immunoreactive Nox4 expression in Western blotting experiments; C. Prevent MS-induced increases in macrophage H2O2 production; and D. Prevent MS-induced enhancement of migration in response to MCP-1. An asterisk (*) denotes a p value < 0.01 for the difference between the indicated condition compared to the rest of the groups (n = 3).
Figure 7. Exogenous LPA reverses the effects of the iPLA2β inhibitor BEL to suppress macrophage Nox4 expression, increased production of H2O2, and enhanced migration in response to MCP-1 that otherwise occur in cells subjected to metabolic stress by incubation ex vivo with high concentrations of glucose and lipoprotein.
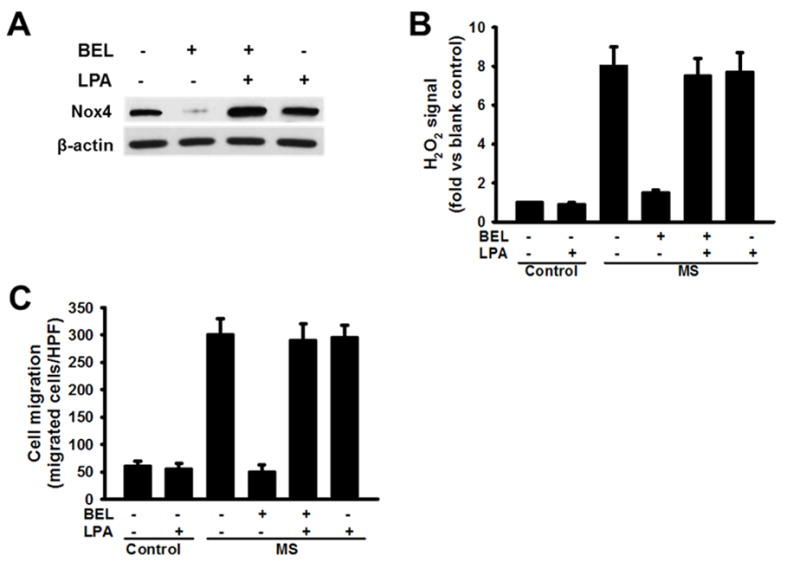
A. Immunoblots of Nox4 and actin in macrophages preincubated (24 hr) under control or MS conditions and then incubated (12 hr) without or with LPA (100 nM) and/or BEL (20 μM). B. H2O2 production by macropahges preincubated under control or MS conditions and then incubated without or with LPA and/or BEL; C. MCP-1-induced migration by macrophages preincubated under control or MS conditions and then incubated without or with LPA and/or BEL. Displayed values represent the means of 3 independent experiments.
LPA derived from iPLA2β action could arise from direct action on phosphatidic acid or indirectly via initial action on phosphatidylcholine to produce lysophosphatidylcholine, followed by conversion of lysophosphatidylcholine to LPA by the action of a phospholipase D (PLD). To examine these possibilities further, we measured PLD activity and performed immunoblotting experiments to determine the phosphorylation state of PLD1 and PLD2 in macrophages incubated under control conditions or in medium supplemented with HG and LDL. Neither an increase in PLD activity nor an increase in phosphorylation of PLD1 or PLD2 was observed in these experiments (data not shown). These observations are consistent with a direct action of iPLA2β on PA to produce LPA but do not definitively exclude a contribution of PLD. To address whether inhibition of PLD would block the production and effects of LPA, we measured LPA production and Nox activity in metabolic stress-challenged macrophages in the presence of PLD1 and PLD2 inhibitors. The results of these experiments are illustrated in Online Figure VII and demonstrated that the PLD2 inhibitor, but not the PLD1 inhibitor, resulted in slight suppression of LPA production and Nox activity, but these effects did achieve statistical significance relative to control cell incubations without inhibitors. These data suggest that PLD activity is not required for the effects or production of the LPA that arises as a consequence of iPLA2β action..
Nox4 activity might be increased by promoting phosphorylation of its binding partner p22phox, and we therefore conducted immunoblotting experiments to examine the phosphorylation state of p22phox under the conditions of our experiments. Knockdown of LPA receptors (LPARs) with siRNA was observed to reduce phosphorylation of p22phox, as illustrated in Online Figure II. This suggests that the LPA/LPAR signaling axis may act to increase p22phox phosphorylation, which could represent one means by which Nox4 activation is achieved.
DISCUSSION
We have demonstrated previously that Nox4 is a major source of ROS in macrophages and participates in the monocyte priming and enhanced macrophage chemotaxis that occur in diabetes mellitus,10, 11 but the processes that regulate Nox4 expression and activity had not been determined. Here we demonstrate that expression of the enzyme iPLA2β increases markedly in macrophages subjected to metabolic stress in vivo in mice with diabetes or ex vivo by incubation in medium supplemented with HG and LDL and that this results in increased production of LPA via iPLA2β action. LPA then triggers a signaling cascade that leads to increased Nox4 expression and activity, increased ROS production, and enhanced MCP-1-induced macrophage migration under conditions of diabetic metabolic stress (DMS).
Of 7 recognized Nox isoenzymes, Nox2 and Nox4 are the predominant ROS producers in macrophages.10 Nox activity in phagocytic cells is known to be regulated phosphorylation/dephosphorylation reactions.27, 28 Phosphorylation of the p47phox subunit stimulates interaction with p67phox, and the resultant complex associates with Nox2 in the plasma membrane to assemble the active form of the oxidase. It has been reported that p41 (NoxO1) and p51 (NoxA1), which are homologues of p47phox and p67phox, respectively, are required for Nox activation in nonphagocytic cells.29, 30 In contrast, Nox4 is constitutively active and is primarily regulated at the transcriptional level, although recent evidence suggests that post-translational regulation also occurs.31, 32 Although Nox2 localizes primarily in the plasma membrane, Nox4 is distributed in intracellular membranous loci that include mitochondria, endoplasmic reticulum, and nuclear membranes.33–35 There is little information about how Nox4 expression and activity are regulated under physiologic or pathologic conditions, although a recent report indicates that Poldip2 associates with p22phox to activate Nox4, which regulates focal adhesion turnover and migration of vascular smooth muscle cell and thereby links ROS production and cytoskeletal remodeling.36 Our studies here are the first of which we are aware to demonstrate relationships among macrophage iPLA2β activity, its product LPA, expression of Nox4, ROS production, and migratory responses to MCP-1.
Formerly, Nox2 was thought to be the predominant source of macrophage ROS, and its roles in the respiratory bust of phagocytes have been extensively examined.8, 9 Nonetheless, participation of Nox2 in macrophage migration and accumulation at inflammatory sites in diabetes and atherosclerosis had not been demonstrated, although a recent report indicates that Nox2 participates in the migration of bone marrow derived macrophages toward a gradient of increasing colony-stinulating factor-1 concentrations.37 Despite this, transfer of Nox2-deficient bone marrow cells into apoE−/− mice had no significant effect atherosclerotic lesion area at 24 weeks even though Nox2-deficient macrophage exhibited a profound reduction in basal and PMA-induced superoxide production.38 This suggests that Nox2 is not essential for monocyte recruitment to sites of vascular inflammation in vivo. We have previously demonstrated that knockdown of Nox4 protected monocytes from metabolic priming, although Nox2 expression was unaffected, and this also argues against a significant role for Nox2 in metabolic stress-induced monocyte dysfunction.10, 11 Here, we demonstrate that incubation of macrophages in medium supplemented with HG and LDL does result in increased Nox2 expression, but neither iPLA2β inhibition with BEL or adenoviral vector-driven overexpression of iPLA2β affected Nox2 expression, although those interventions had profound effects on macrophage Nox4 expression, ROS production, and MCP-1-induced migration. Together, these observations indicate Nox4 expression is regulated by iPLA2β activity and that Nox4-derived ROS play an essential role in accelerated migration of macrophages subjected to diabetic metabolic stress but that Nox2 does not play a similar role in these processes.
The profile ROS derived from Nox4-transfected is characterized by a greater proportion of H2O2 relative to O2•− than is the case for Nox1, 2, or 3 or for Nox5.39–41 The mechanism underlying the preferential production of H2O2 vs O2•− by Nox4 is proposed to be a highly conserved histidine residue in the E-loop that promotes the rapid dismutation of O2•− before it is released from the enzyme.23 Here we demonstrate that increased iPLA2β expression in macrophages incubated with high concentrations of glucose and LDL results in production of ROS with a prominent H2O2 component and that LPA derived from iPLA2β action participates in upregulating macrophage Nox4 expression.
Oxidation of cellular proteins via regulated redox systems is among the post-translational modifications that can modulate enzyme action in physiological and pathological conditions, and ionizable cysteine thiol groups react readily with H2O2.42 For example, reversible S-glutathionylation of actin at Cys374 promotes actin depolymerization,43 and prevention of this modification by site-directed mutagenesis of Cys374 to alanine inhibits cell spreading.44 Nox4 co-localizes with α-actin in smooth muscle cells and is found in actin-rich invadopodia of cancer cells, suggesting that Nox enzymes might be involved in the S-glutathionylation and redox regulation of actin. Continual treadmilling of F-actin filaments via polymerization-depolymerization is the process that underlies formation of retractable pseudopods in mobile macrophages. We have reported previously that Nox4 is recruited to and colocalizes with F-actin filaments in macrophages subjected to diabetic metabolic stress or in which Nox4 overexpression is driven by an adenoviral vector and that this results in increased actin S-glutathionylation and accelerated F-actin turnover.11 Moreover, in preliminary experiments (not shown) we have observed that Nox4 activation affects F-actin assembly by promoting S-glutathionylation and consequent dephosphorylation of cofilin, which is an important actin depolymerizing factor (ADF). These observations suggest that Nox4-derived ROS may regulate S-glutathionylation of actin and/or of other redox regulated proteins, e.g. cofilin. Other signaling complexes essential for macrophage migration might be similarly affected, although the molecular mechanisms by which Nox4 translocates to various intracellular loci and by which Nox4-derived H2O2 selectivity modifies target proteins remain the subjects of ongoing investigation and are incompletely understood at present.
We searched for but failed to find complexes of iPLA2β and Nox4 in immunoprecipitation and immunoblotting experiments (data not shown), which argues against the possibility that iPLA2β regulates Nox4 activity via a physical interaction. This suggests that an iPLA2β reaction product might produce the downstream effects that result from increased iPLA2β expression. One such product is a free fatty acid, such as arachidonic acid. This polyunsaturated fatty acid is distributed primarily in the sn-2 position of phospholipids, which is the site of action of iPLA2β. Arachidonic acid has essential mediator functions that regulate the activities of signaling enzymes, such as phospholipase C and protein kinase C isoforms (PLC-γ, PLC-δ, and PKC-α, - β, and γ). In addition, arachidonic acid is a key inflammatory intermediate in metabolic cascades that result in production of prostaglandins, thromboxane, leukotrienes, epoxyeicosatetraenoic acids, and a variety of other eicosanoids with signaling functions.26 We have previously demonstrated that transgenic overexpression of iPLA2β in vascular smooth muscle cells promotes production of inflammatory cytokines, macrophages infiltration, and proliferation of vascular smooth muscle cells in response to arterial injuries and that these responses are dependent on production of AA metabolites. Here we failed to observe any effects of adding exogenous arachidonic acid without or with various inhibitors of arachidonic acid oxygenases on macrophage Nox4 expression, which argues against the possibility that arachidonic acid or its metabolites represent the product(s) of iPLA2β action that regulate macrophage Nox4 expression and downstream events.
In addition to free fatty acids, such as arachidonic acid, another product of iPLA2β action is a 2-lysophospholipid, such as LPA, and LPA production is greatly reduced in iPLA2β-knockout mice in some circumstances.25 LPA has diverse biological actions that affect Ca2+ mobilization, cAMP accumulation, phospholipase D activity,45 changes in cell shape, cell motility, actin rearrangement, and proliferation in various cells.46, 47 Extracellular LPA has also been implicated in the pathogenesis of atherosclerosis and cancer.48, 49 LPA can be generated in two potential pathways that involve iPLA2β, including direct hydrolysis of phosphatidic acid to yield LPA or via an indirect route that involves initial action on phosphatidylcholine to yield lysophosphatidylcholine followed by the action of the phospholipase D autotaxin to yield LPA (57). Here we have demonstrated that LPA derived from iPLA2β action is required for increased Nox4 expression, H2O2 generation, and MCP-1-induced migration that occurs in macrophages subjected to diabetic metabolic stress. Our data suggest that LPA derived from the action of iPLA2β acts through LPA receptors to increase Nox4 activity and that the mechanism of this effect may involve phosphorylation of p22phox, which is a critical component of superoxide-generating NADH/NADPH oxidase.
In summary, this study identifies iPLA2β as a regulator of macrophage Nox4 expression and activity by the pathway depicted in Figure 8. Subjecting macrophages to metabolic stress in the diabetic state in vivo or by incubation ex vivo in medium supplemented with high concentrations of glucose and LDL results in increased expression of iPLA2β, and LPA derived from its action causes increased macrophage expression of Nox4 and production H2O2 that leads to enhanced migration of macrophages into sites of vascular inflammation in vivo and in response to stimulation with MCP-1 ex vivo. Inhibition of iPLA2β with BEL, blockade of LPA receptors with VPC32183, or inhibition of Nox4 with GKT137831 interrupts this cascade and the downstream events of increased macrophage H2O2 production and enhanced migration. These findings suggest that iPLA2β may be the long-sought link between diabetes and accelerated atherosclerosis (and perhaps other complications of diabetes) that results from increased inflammation and that iPLA2β might thus be a suitable target for therapeutically beneficial interventions on these processes. A combinatorial approach directed at inhibiting iPLA2β, blocking LPA receptors, and inhibiting Nox4 might be particularly useful in that regard.
Figure 8. Model for participation of iPLA2β in the signaling cascade induced by subjecting macrophages to metabolic stress that leads to increased Nox4 expression and enhanced migration.
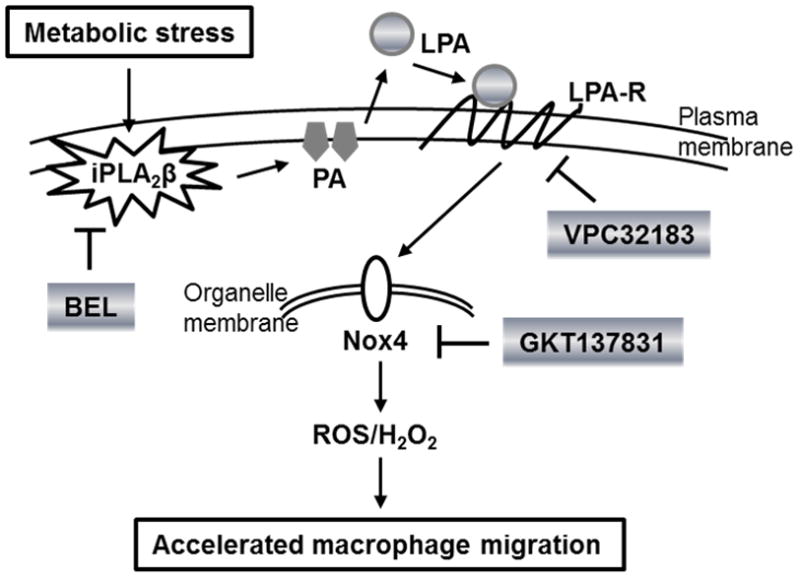
Under conditions of diabetes mellitus (DM) in vivo or upon exposure of macrophages to high concentrations of glucose and lipoproteins ex vivo, iPLA2β expression and activity rise, and LPA production increases directly from hydrolysis of PA or indirectly by hydrolysis of PC to yield LPC that is then metabolized to LPA. Interaction of LPA with its receptor(s) initiates a signaling cascade that results in increased Nox4 expression and activity, which in turn leads to increased production of ROS and H2O2 that enhance the migratory responses of macrophages to signals in vivo that lead to their accumulation at sites of vascular inflammation and to MCP-1 ex vivo. This sequence can be interrupted by inhibiting iPLA2β (with BEL or antisense oligonucleotides or in iPLA2β-KO mice), by blocking LPA receptors (with the competitive antagonist VPC32183), or by inhibiting Nox4 (with GKT137831). In the diagram, arrows indicate activation or production, and T-lines indicate inhibition.
Supplementary Material
SIGNIFICANCE.
Diabetes is associated with increased inflammation and macrophage infiltration into inflamed tissues. We previously demonstrated that NADPH oxidase 4 (Nox4) is a major source of macrophage reactive oxygen species and participates in enhanced macrophage chemotaxis in diabetes, but how Nox4 is regulated is incompletely understood. Here we have identified lysophosphatidic acid (LPA) derived from the action of Group VIA Phospholipase A2 (iPLA2β) as a critical macrophage Nox4 regulator. Our findings suggest that iPLA2β may represent a long-sought link between diabetes and accelerated atherosclerosis that results from increased inflammation and that iPLA2β might thus be a suitable target for therapeutically beneficial interventions in these processes. A combinatorial approach directed at inhibiting iPLA2β, blocking LPA receptors, and inhibiting Nox4 might be particularly useful in that regard.
Acknowledgments
SOURCES OF FUNDING
This work was supported by a grant to QDZ from American Heart Association AHA (11SDG5380002). The laboratory of JT was supported by United States Public Health Service grants R37-DK34388, P41-RR00954, P60-DK20579, and P30-DK56341.
Abbreviations
- BEL
bromoenol lactone
- DMS
diabetic metabolic stress
- HFD
high fat diet
- HG
high glucose
- iPLA2
Ca2+-Independent Phospholipase A2
- LDL
low density lipoprotein
- LPA
lysophosphatidic acid
- Nox
NADPH oxidase
- ROS
reactive oxygen species
- siRNA
small interfering RNA
Footnotes
DISCLOSURES
None
References
- 1.Grundy SM, Howard B, Smith S, Jr, Eckel R, Redberg R, Bonow RO. Prevention conference vi: Diabetes and cardiovascular disease: Executive summary: Conference proceeding for healthcare professionals from a special writing group of the american heart association. Circulation. 2002;105:2231–2239. doi: 10.1161/01.cir.0000013952.86046.dd. [DOI] [PubMed] [Google Scholar]
- 2.Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. The New England journal of medicine. 1998;339:229–234. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- 3.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 4.Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Jr, Broxmeyer HE, Charo IF. Impaired monocyte migration and reduced type 1 (th1) cytokine responses in c-c chemokine receptor 2 knockout mice. The Journal of clinical investigation. 1997;100:2552–2561. doi: 10.1172/JCI119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in ccr2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 6.Rollins BJ. Monocyte chemoattractant protein 1: A potential regulator of monocyte recruitment in inflammatory disease. Molecular medicine today. 1996;2:198–204. doi: 10.1016/1357-4310(96)88772-7. [DOI] [PubMed] [Google Scholar]
- 7.Lambeth JD, Kawahara T, Diebold B. Regulation of nox and duox enzymatic activity and expression. Free radical biology & medicine. 2007;43:319–331. doi: 10.1016/j.freeradbiomed.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segal AW. Absence of both cytochrome b-245 subunits from neutrophils in x-linked chronic granulomatous disease. Nature. 1987;326:88–91. doi: 10.1038/326088a0. [DOI] [PubMed] [Google Scholar]
- 9.Teahan C, Rowe P, Parker P, Totty N, Segal AW. The x-linked chronic granulomatous disease gene codes for the beta-chain of cytochrome b-245. Nature. 1987;327:720–721. doi: 10.1038/327720a0. [DOI] [PubMed] [Google Scholar]
- 10.Lee CF, Qiao M, Schroder K, Zhao Q, Asmis R. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circulation research. 2010;106:1489–1497. doi: 10.1161/CIRCRESAHA.109.215392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ullevig S, Zhao Q, Lee CF, Seok Kim H, Zamora D, Asmis R. Nadph oxidase 4 mediates monocyte priming and accelerated chemotaxis induced by metabolic stress. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:415–426. doi: 10.1161/ATVBAHA.111.238899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsson PK, Claesson HE, Kennedy BP. Multiple splice variants of the human calcium-independent phospholipase a2 and their effect on enzyme activity. The Journal of biological chemistry. 1998;273:207–214. doi: 10.1074/jbc.273.1.207. [DOI] [PubMed] [Google Scholar]
- 13.Moolenaar WH. Lysophosphatidic acid, a multifunctional phospholipid messenger. The Journal of biological chemistry. 1995;270:12949–12952. doi: 10.1074/jbc.270.22.12949. [DOI] [PubMed] [Google Scholar]
- 14.Brash AR. Arachidonic acid as a bioactive molecule. The Journal of clinical investigation. 2001;107:1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, Lin ME, Teo ST, Park KE, Mosley AN, Chun J. Lpa receptors: Subtypes and biological actions. Annual review of pharmacology and toxicology. 2010;50:157–186. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- 16.Mishra RS, Carnevale KA, Cathcart MK. Ipla2beta: Front and center in human monocyte chemotaxis to mcp-1. The Journal of experimental medicine. 2008;205:347–359. doi: 10.1084/jem.20071243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu S, Xie Z, Zhao Q, Pang H, Turk J, Calderon L, Su W, Zhao G, Xu H, Gong MC, Guo Z. Smooth muscle-specific expression of calcium-independent phospholipase a2beta (ipla2beta) participates in the initiation and early progression of vascular inflammation and neointima formation. The Journal of biological chemistry. 2012;287:24739–24753. doi: 10.1074/jbc.M112.340216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao S, Jin C, Zhang S, Turk J, Ma Z, Ramanadham S. Beta-cell calcium-independent group via phospholipase a(2) (ipla(2)beta): Tracking ipla(2)beta movements in response to stimulation with insulin secretagogues in ins-1 cells. Diabetes. 2004;53 (Suppl 1):S186–189. doi: 10.2337/diabetes.53.2007.s186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Z, Gong MC, Su W, Xie D, Turk J, Guo Z. Role of calcium-independent phospholipase a2beta in high glucose-induced activation of rhoa, rho kinase, and cpi-17 in cultured vascular smooth muscle cells and vascular smooth muscle hypercontractility in diabetic animals. The Journal of biological chemistry. 2010;285:8628–8638. doi: 10.1074/jbc.M109.057711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ayilavarapu S, Kantarci A, Fredman G, Turkoglu O, Omori K, Liu H, Iwata T, Yagi M, Hasturk H, Van Dyke TE. Diabetes-induced oxidative stress is mediated by ca2+-independent phospholipase a2 in neutrophils. J Immunol. 2010;184:1507–1515. doi: 10.4049/jimmunol.0901219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of nox1 and nox4 in basal and angiotensin ii-stimulated superoxide and hydrogen peroxide production. Free radical biology & medicine. 2008;45:1340–1351. doi: 10.1016/j.freeradbiomed.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmcke I, Heumuller S, Tikkanen R, Schroder K, Brandes RP. Identification of structural elements in nox1 and nox4 controlling localization and activity. Antioxidants & redox signaling. 2009;11:1279–1287. doi: 10.1089/ars.2008.2383. [DOI] [PubMed] [Google Scholar]
- 23.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The e-loop is involved in hydrogen peroxide formation by the nadph oxidase nox4. The Journal of biological chemistry. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakanaga K, Hama K, Aoki J. Autotaxin--an lpa producing enzyme with diverse functions. Journal of biochemistry. 2010;148:13–24. doi: 10.1093/jb/mvq052. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Zhao Z, Wei G, Yan L, Wang D, Zhang H, Sandusky GE, Turk J, Xu Y. Group via phospholipase a2 in both host and tumor cells is involved in ovarian cancer development. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:4103–4116. doi: 10.1096/fj.10-161356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Needleman P, Turk J, Jakschik BA, Morrison AR, Lefkowith JB. Arachidonic acid metabolism. Annual review of biochemistry. 1986;55:69–102. doi: 10.1146/annurev.bi.55.070186.000441. [DOI] [PubMed] [Google Scholar]
- 27.Bokoch GM, Diebold B, Kim JS, Gianni D. Emerging evidence for the importance of phosphorylation in the regulation of nadph oxidases. Antioxidants & redox signaling. 2009;11:2429–2441. doi: 10.1089/ars.2009.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hordijk PL. Regulation of nadph oxidases: The role of rac proteins. Circulation research. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 29.Lambeth JD. Nox enzymes and the biology of reactive oxygen. Nature reviews. Immunology. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 30.Bedard K, Krause KH. The nox family of ros-generating nadph oxidases: Physiology and pathophysiology. Physiological reviews. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 31.Gorin Y, Block K, Hernandez J, Bhandari B, Wagner B, Barnes JL, Abboud HE. Nox4 nad(p)h oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. The Journal of biological chemistry. 2005;280:39616–39626. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 32.Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, Karwande SV, Stringham JC, Bull DA, Gleich M, Kennedy TP, Hoidal JR. Transforming growth factor-beta1 induces nox4 nad(p)h oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. American journal of physiology. Lung cellular and molecular physiology. 2006;290:L661–L673. doi: 10.1152/ajplung.00269.2005. [DOI] [PubMed] [Google Scholar]
- 33.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circulation research. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. Nadph oxidase 4 (nox4) is a major source of oxidative stress in the failing heart. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Block K, Gorin Y, Abboud HE. Subcellular localization of nox4 and regulation in diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circulation research. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chaubey S, Jones GE, Shah AM, Cave AC, Wells CM. Nox2 is required for macrophage chemotaxis towards csf-1. PloS one. 2013;8:e54869. doi: 10.1371/journal.pone.0054869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirk EA, Dinauer MC, Rosen H, Chait A, Heinecke JW, LeBoeuf RC. Impaired superoxide production due to a deficiency in phagocyte nadph oxidase fails to inhibit atherosclerosis in mice. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:1529–1535. doi: 10.1161/01.atv.20.6.1529. [DOI] [PubMed] [Google Scholar]
- 39.Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of nox4 reveals unique characteristics compared to other nadph oxidases. Cellular signalling. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 40.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. Nox4 activity is determined by mrna levels and reveals a unique pattern of ros generation. The Biochemical journal. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brandes RP, Takac I, Schroder K. No superoxide--no stress?: Nox4, the good nadph oxidase! Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1255–1257. doi: 10.1161/ATVBAHA.111.226894. [DOI] [PubMed] [Google Scholar]
- 42.Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free radical biology & medicine. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Wang J, Boja ES, Tan W, Tekle E, Fales HM, English S, Mieyal JJ, Chock PB. Reversible glutathionylation regulates actin polymerization in a431 cells. The Journal of biological chemistry. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 44.Fiaschi T, Cozzi G, Raugei G, Formigli L, Ramponi G, Chiarugi P. Redox regulation of beta-actin during integrin-mediated cell adhesion. The Journal of biological chemistry. 2006;281:22983–22991. doi: 10.1074/jbc.M603040200. [DOI] [PubMed] [Google Scholar]
- 45.Tou JS, Gill JS. Lysophosphatidic acid increases phosphatidic acid formation, phospholipase d activity and degranulation by human neutrophils. Cellular signalling. 2005;17:77–82. doi: 10.1016/j.cellsig.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Moolenaar WH. Bioactive lysophospholipids and their g protein-coupled receptors. Experimental cell research. 1999;253:230–238. doi: 10.1006/excr.1999.4702. [DOI] [PubMed] [Google Scholar]
- 47.Ye X, Ishii I, Kingsbury MA, Chun J. Lysophosphatidic acid as a novel cell survival/apoptotic factor. Biochimica et biophysica acta. 2002;1585:108–113. doi: 10.1016/s1388-1981(02)00330-x. [DOI] [PubMed] [Google Scholar]
- 48.Siess W, Zangl KJ, Essler M, Bauer M, Brandl R, Corrinth C, Bittman R, Tigyi G, Aepfelbacher M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:6931–6936. doi: 10.1073/pnas.96.12.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maschberger P, Bauer M, Baumann-Siemons J, Zangl KJ, Negrescu EV, Reininger AJ, Siess W. Mildly oxidized low density lipoprotein rapidly stimulates via activation of the lysophosphatidic acid receptor src family and syk tyrosine kinases and ca2+ influx in human platelets. The Journal of biological chemistry. 2000;275:19159–19166. doi: 10.1074/jbc.M910257199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.