

Abstract
Cancers can exhibit dramatic tumor regression following oncogene inhibition through the phenomenon of “oncogene addiction”. The ability to predict when a tumor will exhibit oncogene addiction would be useful in the development of targeted therapeutics. Oncogene addiction is likely the consequence of many cellular programs. However, we reasoned that many of these inputs may converge on aggregate survival and death signals. To test this, we measured the sequence of changes that occur upon oncogene inactivation in conditional genetically engineered mouse models of K-rasG12D- or MYC-induced lung tumors and lymphoma. We combined quantitative imaging with an in situ analysis of biomarkers of proliferation and apoptosis. Indeed, oncogene addiction could be modeled as differential changes in intracellular survival and death signals following oncogene inactivation. Our model used different imaging methods (CT and bioluminescence imaging) and histochemical markers of proliferation and apoptosis (Ki-67 and caspase 3) to blindly predict the differential in dynamics of several pro-survival and pro-death signaling factors (phosphorylated Erk1/2, Akt1, Stat3/5 and p38) that contribute to the aggregate survival and death signals. The model was predictive of different oncogenes (K-rasG12D and MYC) in multiple tumor types (lung and lymphoma). Furthermore, we could predict the influence of specific genetic lesions (p53-/-, Stat3-d358L and myr-Akt1) on tumor regression upon oncogene inactivation. Finally, our model could utilize quantitative imaging data to predict both EGFR genotype and progression-free survival in human patients with lung cancer shortly after the initiation of treatment with the targeted therapy erlotinib. Hence, the consequences of oncogene inactivation can be accurately modeled based on a relatively small number of parameters that may predict when targeted therapeutics will elicit oncogene addiction.
Introduction
Cancer is largely caused by activation of oncogenes or inactivation of tumor suppressor genes resulting in the pathological disruption of a multitude of cellular signaling programs including cellular proliferation, apoptosis, self-renewal/differentiation as well as host programs of angiogenesis and immune surveillance (1, 2). Surprisingly, the inactivation of even a single oncogene can induce dramatic tumor regression revealing that some tumors can be highly dependent or “addicted” to specific oncogenes (3, 4). Recently, it has been suggested based upon in vitro studies in tumor-derived cell lines that oncogene addiction may be defined by the interaction between survival and death programs (3, 5, 6). However, these experiments were performed in vitro and it is not clear a priori what factors determine oncogene addiction. Indeed, several qualitative models for oncogene addiction have been proposed (3, 7, 8).
Evidence for oncogene addiction comes from two sets of observations: work in experimental mouse model systems and in the clinic using targeted therapeutics. Through using conditional transgenic mouse models, it has been demonstrated that many different tumors exhibit oncogene addiction associated with proliferative arrest, apoptosis and/or differentiation/senescence (9-17). More recently, it has been suggested that other tumor intrinsic mechanisms such as the induction of cellular senescence (18) as well as host-dependent programs, including the shut down of angiogenesis (19) may also be important. The relative contribution of these different cellular programs to the mechanism of oncogene addiction remains to be determined. One possibility is that all of these different cellular programs converge on influencing survival and death signals in a cell (3, 5, 6).
The clinical relevance of oncogene addiction became apparent through the discovery of effective clinical therapies that target specific oncogenes including: epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib for non–small cell lung cancer (NSCLC) (20, 21) and imatinib treatment for chronic myeloid leukemia (22) and gastrointestinal stromal tumors (GIST) (23, 24). Drugs that target EGFR improve the progression free survival of patients with advanced NSCLC (20, 25). Notably, patients with EGFR mutations are the most responsive to these therapies (26-28). The EGFR genotype best predicts the progression free survival in patients treated with EGFR TKIs (20, 29).
Methods that could predict the efficacy of targeted therapeutics would be highly useful in the development of novel therapies. In addition, since targeted therapies are very expensive (30) and are usually only effective to treat a very specific subpopulation of cancer patients, it is important to develop strategies to rapidly discriminate when these agents are effective to help a particular patient. The ideal predictive method would be non-invasive, generate reproducible measurements, and be widely available utilizing technology and clinical skills generally available to most hospitals. Previous efforts to utilize clinical imaging approaches to predict response to therapy, such as dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI), daily mega-voltage computed tomography (MVCT), or functional PET and SPECT are limited (31-37). It is still not possible to predict with a high degree of certainty by using imaging alone, which patients will respond to any particular treatment.
To gain insight into the mechanism of oncogene addiction and to develop methods that predict when targeted therapies will elicit oncogene addiction, we have combined quantitative in vivo imaging with detailed immunohistochemical analysis of conditional transgenic mouse models of lung cancer (14, 38) and lymphoma (9) (overview see Fig. 1). To analyze our data, we used two complementary mathematical approaches: modeling of the dynamics of cellular survival and death signaling based on ordinary differential equations (ODE) and prediction of genotype based on a support vector machine (SVM) classifier.
Fig. 1.
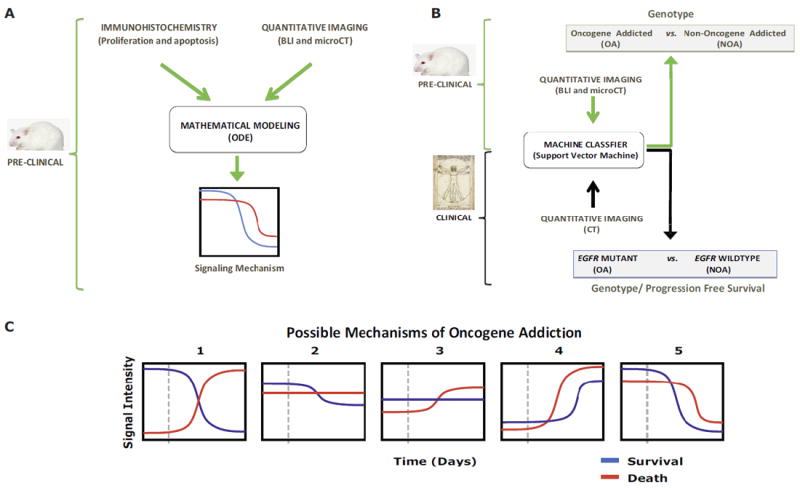
Multiscale mathematical modeling of oncogene addiction. Two general datasets were considered: preclinical conditional mouse tumor models (green arrows and brackets) and human clinical data (black arrows and brackets). Two complementary mathematical approaches were employed (black outlined boxes): (A) ordinary differential equations (ODE) and (B) support vector machine (SVM) classifiers. (A) ODE modeling used immunohistochemistry (IHC) and quantitative imaging data from mice to describe the relationship between aggregate survival and death signaling pathways (“Signaling Mechanism”) following oncogene inactivation. (B) We utilized quantitative imaging from mice and humans to predict shortly after oncogene inactivation whether tumors would exhibit oncogene addiction. For humans we could predict from imaging data alone EGFR genotype and longer progression free survival. (C) Aggregate survival and death signaling behavior following oncogene inactivation. Oncogene addicted tumor cells may display one of the scenarios upon oncogene inactivation: 1) a reduction in survival signals and an increase in death signals; 2) a reduction in survival signals only without change in death signals; 3) an increase in death signals only without change in survival signals; 4) an increase in survival and death signaling but the latter increasing more; or 5) a decrease in survival and death signaling but with the former decreasing more. The vertical dashed line indicates time at oncogene inactivation.
We found oncogene addiction could be accurately modeled as the result of differential changes in aggregate survival and death intracellular signals (Fig. 1A & 1C) (2, 5). Our model generalized to different imaging methods (CT and bioluminescence imaging), different oncogenes (K-rasG12D and MYC), multiple types of tumors (lung tumors and lymphoma) and predicted the effects of other genetic lesions (p53-/-, Stat3-d358L and myr-Akt1). Lastly, we found our models could predict when oncogene inactivation would result in oncogene addicted tumor regression (Fig. 1B). Moreover, we could predict in human patients both EGFR genotype and more importantly their clinical response to EGFR inhibition.
Results
Interrogating Oncogene Addiction through Quantitative Imaging and in situ Analysis
Cancers that display an oncogene addicted phenotype are complex and have disruption of many cellular programs including cellular proliferation, apoptosis, differentiation, self-renewal, as well as host programs of angiogenesis and immune surveillance (39). We reasoned that these complex programs determine tumor formation as well as predict tumor regression as a consequence of changes in cell survival and cell death signaling (2). If this were the case, then oncogene addiction could be described by the interplay between survival and death signaling in five possible ways: (Fig. 1C): 1) a reduction in survival signals and an increase in death signals occurs; 2) a reduction in survival signals only without change in death signals; 3) an increase in death signals only without change in survival signals; 4) an increase in both survival and death signaling but the latter increasing more quickly; or 5) a decrease in both survival and death signaling but with the former decreasing more quickly. To distinguish amongst these possibilities we examined upon oncogene inactivation changes in pro-survival and pro-death signaling molecules (Fig. 3) and their net effect on cells’ decision to proliferate or undergo apoptosis (Table 1) by immunohistochemistry (IHC) (Table 1).
Fig. 3.
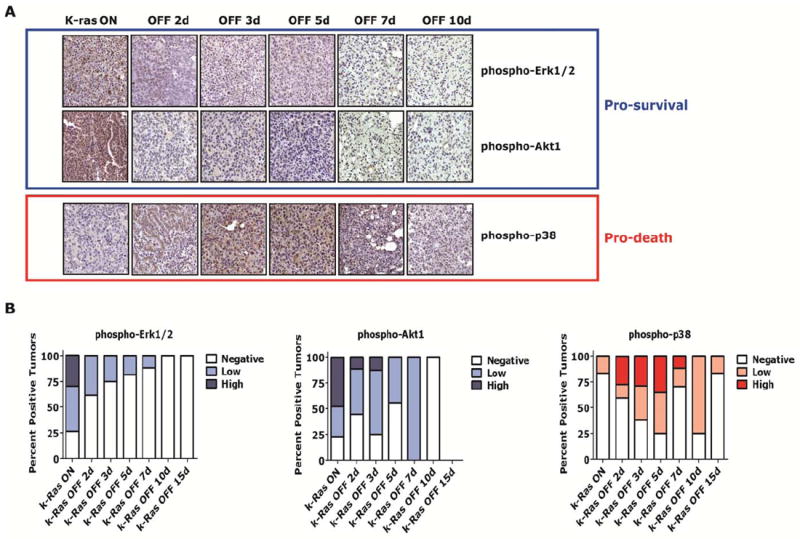
Differential attenuation of pro-survival and pro-death signaling factors during regression of K-rasG12D –induced lung tumors in vivo. (A) CR lung tumors with K-rasG12D oncogene activated (or “ON”) and at time points following oncogene inactivation (or “OFF”) were evaluated for phosphorylation of signaling pathway mediators by IHC. Representative examples show pro-survival pathways mediated by Erk1/2 and Akt1 (blue box) are phosphorylated in lung tumors when K-rasG12D is “ON,” but are de-phosphorylated after turning K-rasG12D “OFF” (day 2-3). Pro-death pathway (red box) is de-phosphorylated with delayed kinetics (day 10-15). (B) IHC was scored as negative, low (<50% positive cells) or high (≥50% positive cells) for tumors.
Table 1.
Multiscale data used to optimize the mathematical model.
| Data Type | K-rasG12D | MYC Lung | MYC Lymphoma |
|---|---|---|---|
| Tumor number scored | 20 | 8 | 21 |
| Cleaved caspase 3 IHC panels scored | 24 | 27 | ND |
| Ki-67 IF panels scored | 19 | 35 | ND |
| microCT volumes scored | 202 | 87 | NA |
| Bioluminescence images scored | NA | NA | 70 |
NA - not applicable
ND - not done
We utilized a transgenic mouse model of K-rasG1D induced lung adenocarcinoma that been shown to exhibit oncogene addiction. The K-rasG12D oncogene has been implicated in the pathogenesis of many human cancers, including lung cancer, through its pleiotropic effects on cell autonomous functions such as proliferation, apoptosis and cellular differentiation that can drive tumorigenesis (40). The conditional transgenic model system of K-rasG12D induced lung cancer utilizes the tetracycline-regulatory ON system (Tet ON), that allows control of target gene expression by the addition or withdrawal of doxycycline in the drinking water of transgenic mice (Fig. 2A) (14). Lung specific expression of K-rasG12D caused multiple primary lung tumors to develop spontaneously in a stochastic manner with moderate latency (Fig. 2B-E, median latency 26 weeks). By using the Tet ON system, we could simulate targeted therapy against an oncogene by simply withdrawing doxycycline to turn the oncogene “OFF” (Fig. 2F-I) (14, 41).
Fig. 2.
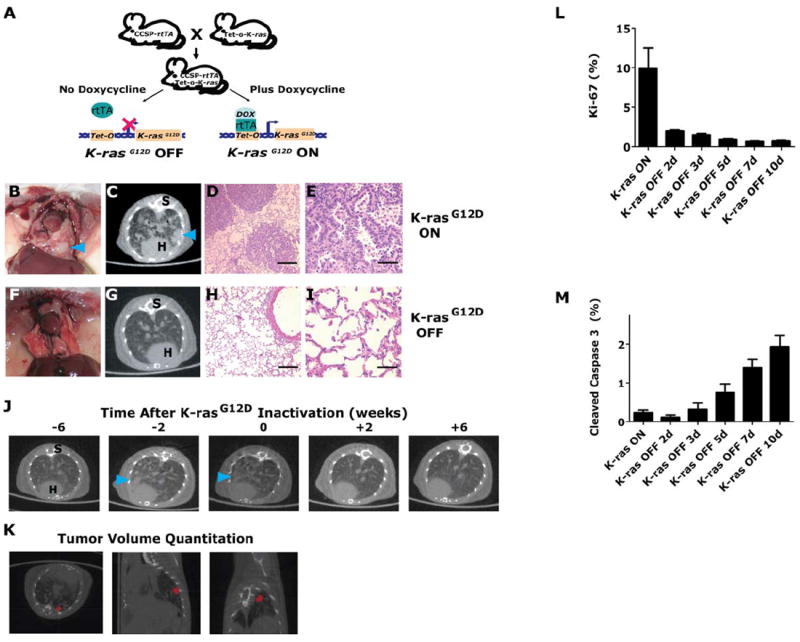
Quantitative analysis of K-rasG12D -induced lung tumor regression. (A) A mouse line containing the Clara cell secretory protein (CCSP) promoter driving the reverse tetracycline transactivating protein (rtTA) is crossed with a line containing K-rasG12D under the control of the tetracycline-responsive promoter (Tet-op). Addition or withdrawal of doxycycline in CCSP-rtTA/Tet-op- K-rasG12D (or CR) enables or prevents, respectively, the rtTA protein from binding and activating the Tet-op promoter. (B) K-rasG12D expression in the lung results in lung tumors (C) radiographically and (D & E) microscopically. K-rasG12D -induced tumors after 6 weeks of K-rasG12D -inactivation are absent on (F) gross examination, (G) radiographically and (H & I) microscopically (n>=11). Serial microCT representative axial images are shown (C & G). S – spine, H – heart and tumors marked by blue arrowheads. H&E histology sections show lung tumors or normal lung epithelium (D, E, H and I). (J & K) Tumors were tracked prospectively using serial microCT. Tumor progression and tumor regression following oncogene inactivation were quantified by semiautomated segmentation of lung nodules (red pixels) in three-dimensions. (L & M) Proliferative and apoptotic indices in CR lung tumors after turning K-rasG12D “OFF” as analyzed by quantization of Ki-67 and cleaved caspase 3 by IHC, respectively.
To determine the behavior of pro-survival and pro-death signaling factors following oncogene inactivation in vivo in this transgenic model (see Fig. 1A), we measured over time in lung tumor cells the phosphorylation status of specific pro-survival and pro-death effector molecules (Fig. 1A and Fig. 3). The primary lung tumor cells demonstrated high steady state levels of phosphorylated Erk1/2, Akt1, Stat3 and Stat5 pro-survival effectors prior to K-rasG12D oncogene inactivation (Fig 3A-B, Fig. S1A-B; blue outlines or blue bars), as measured by IHC. However, as early as 2 days following oncogene inactivation in the primary lung tumors, these pro-survival molecules were largely inactive or no longer phosphorylated and remained inactive during the entire course of the oncogene targeted treatment, day 15 (Fig 3A-B, Fig. S1A-B). Conversely, the pro-death effector p38 (42) (Fig. 3, red outline or red bars) was not phosphorylated initially during the K-rasG12D oncogene inhibition time course (day 0), but phospho-p38 accumulated at day 2, peaked at days 5-7 and then eventually decreased in a delayed fashion to a low basal level by days 10-15 by IHC (Fig. 3A-B). Thus, data from primary lung tumor studies in situ suggested that following K-rasG12D oncogene inactivation there was a rapid attenuation of pro-survival signaling molecules followed by a burst of pro-death signaling and then a lag or differential attenuation in pro-death mediators.
Mathematical Modeling of Oncogene Addiction in Lung Cancer
Among the possible interactions between aggregate survival and death signaling upon oncogene inactivation (Fig. 1C), our results appeared to be most consistent with two signaling scenarios that may elicit oncogene addiction and thereby tumor regression in vivo (see Fig. 1C: panel 1 and 5 for two signaling scenarios). To distinguish further which signaling scenario between aggregate survival and death signaling was likely to elicit oncogene addiction, we generated an ordinary differential equation (ODE) model, based on the statistical representation of the heterogeneous intracellular signals within different cells across the entire tumor. We modeled tumor growth and regression as being primarily dependent upon a balance of aggregate survival and death signals that could account for temporal patterns in tumor growth before and after K-rasG12D oncogene inactivation (2).
Thus, tumor growth is governed by the balance between aggregate survival (S(t)) and death (D(t)) signals. At each time point, cell death and proliferation were simultaneously observed within a tumor and thus, a deterministic response to a single systemic balance of survival and death signals was insufficient to model the observed response. This heterogeneity of cellular responses was modeled with a normal distribution (log-normal distribution produced virtually identical results) on the difference between survival and death signals, leading to one of three mutually exclusive cellular states: cell proliferation (P), cell death (D), or homeostasis/quiescent (H) (Fig. 4A-B). Two threshold values on the signal difference determined which of the three separate responses was chosen. The homeostatic/quiescent population (H) was defined as non-cycling cells and thus may contain cells in G0, differentiated, senescent cells, dormant cells and/or cancer stem cells. Then, we further developed an ODE to model the change in cell number due to proliferation and apoptosis (Fig. 4C) using logistic functions to represent the time course of aggregate survival and death signals resulting from oncogene inactivation (Fig. 4E).
Fig. 4.
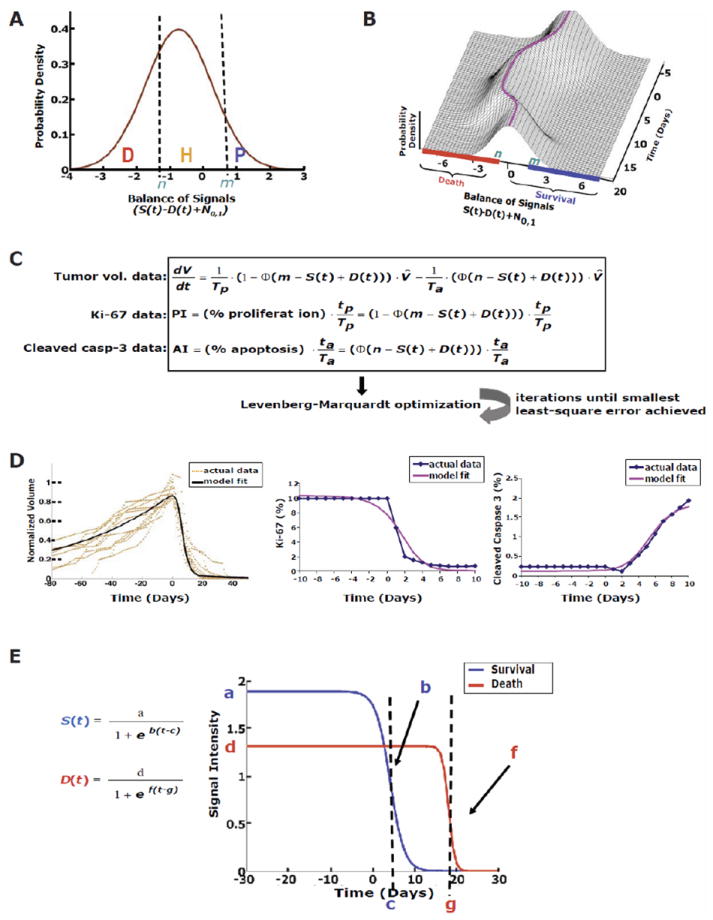
Mathematical modeling of K-rasG12D -induced lung tumors reveals differential attenuation of survival and death signals. (A) Distribution between three cellular decisions directed by the balance of aggregate survival (S(t)) and death signals (D(t)). The percentage of cells in each of these states [death (D), homeostasis (H), and proliferation (P)] is determined by thresholds, n and m, and stochastic variability represented by N0,1 (standard normal distribution). (B) The distribution changes over time with its mean (purple line) centered at S(t)-D(t), shifting the percentage of cells in the three states. (C) The first ordinary differential equation represents changes in tumor cell number due to the increase in cells to the right of threshold m and the decrease in cells to the left of threshold n. Φ is the standard normal CDF. The second and third equations represent the proliferation and apoptosis indices measured by IHC as a balance of input signals. Tp and Ta are the duration of cell proliferation and apoptosis, respectively. tp and ta are duration of detectability of cell proliferation and apoptosis by IHC, respectively. The model fit well to the original (D) tumor volume data and IHC for proliferation and apoptotic indices although there were some points of discordance such as the model fit cleaved caspase 3 appears to be approaching a plateau at day 10. (E) The logistic functions, S(t) and D(t), and optimized parameters showed that survival signals were short-lived following oncogene inactivation compared to the death signals.
To precisely measure the kinetics of tumor formation and regression in vivo, transgenic mice were serially monitored weekly by microCT imaging, following activation and then inactivation of K-rasG12D. In this manner, we interrogated both oncogene-induced tumorigenesis as well as oncogene inactivation induced tumor regression. Following oncogene inactivation in this model system, K-rasG12D induced lung tumors regress within 4 weeks, both radiographically and histologically, as previously described (Fig.2F-J and Video S1) (14, 40). MicroCT imaging has been shown to correlate with the number and volume of murine lung tumors found on necropsy (42). Individual tumor nodule volumes (calculated from segmented microCT images) were quantified at each time point by the ITK-Snap program (Fig. 2K and Fig. S2A-B, Supporting Materials and Methods). Hence, we were able to quantitatively monitor individual tumor sizes in each mouse over time. Importantly, we focused our subsequent analysis on macroscopic tumors that were growing exponentially and therefore by definition are adenocarcinomas (14, 38).
Next, proliferation and apoptosis upon oncogene activation and inactivation were characterized by proliferative index (PI) and apoptotic index (AI) via Ki-67 and cleaved caspase 3 IHC. Measurements were made from in situ lung tumor samples at various time points during the first 10 days following oncogene inactivation (Fig. 2L-M and Fig. S2C-E). Note, this time period was chosen because it corresponded to the most rapid tumor regression by volumetric analysis from microCT (Fig. S2A-B).
These radiologic and biologic data described above (Fig. 2K-M and Fig. S2) were then used to estimate parameter values for the ODE model including parameters of the kinetics of aggregate survival and death signaling, which were not observed directly. The resultant optimization showed an excellent fit with an average error within 6.2% [(root mean square error (RMSE)] between the model fit and the real data (Fig. 4D and Table 1). Given the limited number of parameters in the model (12 total: 3 survival, 3 death, 4 cell cycle duration and 2 thresholds), the risk of overfitting the model to the data was very limited. This was confirmed by statistical analyses of robustness and reproducibility described below.
To examine whether our ODE model of aggregate survival and death signals could accurately describe the dynamics observed between intracellular pro-survival and pro-death factors observed in vivo from IHC (Fig. 3), we solved for survival (S(t)) and death (D(t)) signal intensity and then plotted over time (Fig. 4E). This revealed three striking findings. First, K-rasG12D oncogene addiction could be explained almost entirely by the balance between aggregate survival and death signals as demonstrated by the excellent agreement between the actual radiologic-biologic data (tumor volumes, PI and AI) and the model fitted function (Fig. 4D). Secondly, it surprisingly predicted the almost complete reduction of both aggregate survival and death signaling following oncogene inactivation. Lastly, there was a notable delay for the final degradation of the death signal compared to the survival signal (Fig. 4E and similar to Fig. 1C: panel 5).
Although the aggregate survival and death signals have yet to be linked with single individual markers, we examined a variety of known pro-survival and pro-death factors (Fig. 3). The empirical data from our IHC time course for pro-survival factors agreed well with the mathematical model. The model predicted the initial decline of the aggregate survival signal (S(t)) to occur at day 4 following K-rasG12D inactivation (Fig. 4E), similar to what was experimentally observed by day 2-3 with the pro-survival factors (Fig. 3A-B and Fig. S1). Similarly for the death signal (D(t)), the model predicted attenuation of the death signal following K-rasG12D inactivation to begin at day 13, while in situ tumor data by IHC showed decline of the pro-death factor to occur between days 10-15. Hence, an important implication of the results from our ODE model is that they support the contention that oncogene addiction is a direct consequence of the differential attenuation of aggregate survival and death signals that we observed in vivo (Fig. 1C: panel 5).
Validation of the ODE Model
The robustness of the ODE model to different sets of data was first confirmed by a hold-out analysis of the model by rotating the removal of one contributing data set from the optimization (tumor volumes, PI or AI) and then predicting the aggregate survival and death signals, which yielded essentially identical results (Fig. S3A-C). Next, to validate the reproducibility of the ODE model, we performed both bootstrapping (Fig. S3D) as well as sensitivity analysis (Fig. S3E-I), which yielded highly consistent results. Bootstrapping was chosen over other similar resampling methods such as jackknife and cross-validation due to its ability to better sample distributions of the estimated values and since computational power was not a limitation. Despite the number of parameters modeled by our ODE, although modest, (Fig. 2C) we had sufficiently large numbers of data points (Table 1). All of the statistical validation suggested that the results from our mathematical ODE model were both robust and reproducible.
Next, we tested if our ODE model could generalize to a different transgenic mouse tumor model system and make similar predictions regarding the dynamics of survival and death signals following oncogene inactivation. For this purpose, data was used that involved a different imaging modality, bioluminescence imaging (BLI), the examination of a different type of cancer, lymphoma, and measured the response to the inactivation of a different oncogene, MYC (19). Using the ODE model, as described above (Fig. 4C) modified for optimization with BLI data (Fig. S4C-D; RMSE 7.0-12.6%), we predicted an identical relationship between aggregate survival and death signals between the two different datasets from the transgenic mouse models of lung cancer and lymphoma (compare Fig. 4E and Fig. 5D-F). Thus, our ODE model can be generalized to multiple imaging modalities, the inactivation of other oncogenes and the examination of different types of cancer.
Fig. 5.
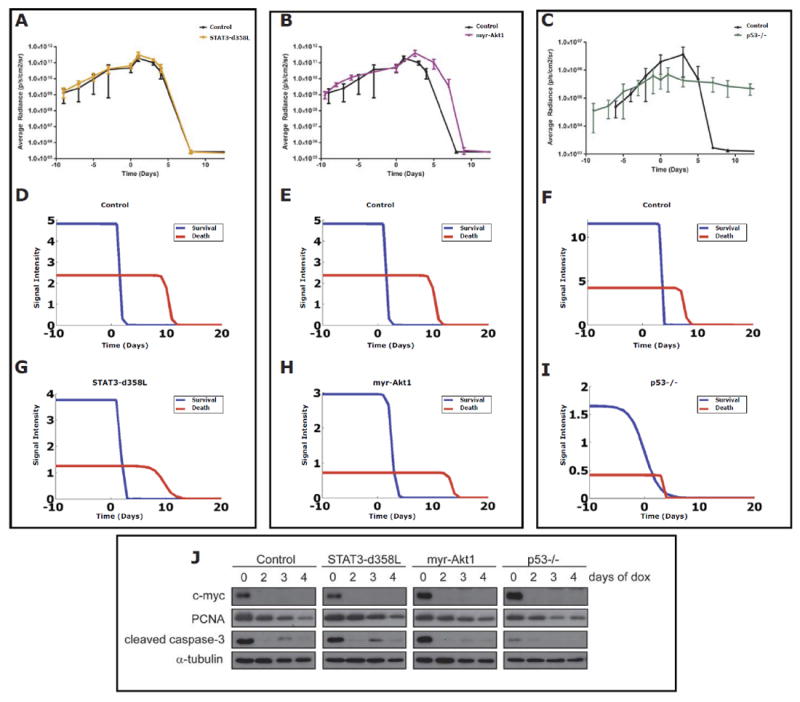
Mathematical modeling of MYC-induced lymphomas with bioluminescence imaging (BLI) shows that genetic perturbations of aggregate survival and death pathways can impede tumor regression following MYC inactivation in vivo. MYC-induced lymphomas were used following transduction with (A & B) control constructs; mutant active forms of (A) Stat3-d358L or (B) myr-Akt1; and (C) a MYC-induced lymphoma derived from a p53-/- background. These MYC-induced lymphoma cells lines were transplanted into immunocompetent mice and tumors tracked using BLI for tumor growth and than regression following MYC inactivation (day 0) in vivo (A-C). Resultant BLI data were used to optimize the mathematical model as in Fig. 4A-C modified for use with BLI (Fig. S4). The survival and death signal plots were then generated from the optimizations for (D-F) Control cell lines, (G) Stat3-d358L, (H) mry-Akt1 and (I) p53-/- derivative lymphomas. The 6780 MYC-induced lymphoma line was used as a control for comparison to the p53-/- derivative for BLI as they had similar basal luciferase activities in vivo. (J) MYC-induced lymphomas transduced with either vector control, Stat3-d358L, or myr-Akt1, or isolated from a p53-/- background, were injected into recipient immunocompetent mice. Tumors were left untreated or treated with doxycycline for 2,3 or 4 days. Samples were harvested at each timepoint and lysates were blotted for expression of c-Myc, PCNA, cleaved caspase-3, and α-tubulin (loading control).
Genetic Changes in Survival and Death Signals Effects Oncogene Addiction
Our ODE model predicted that the modulation of aggregate survival and/or death signaling had predictable effects on tumor regression upon oncogene inactivation. To directly test this in our transgenic mouse model of MYC-induced lymphoma, we perturbed the pathways by introducing the constitutively activated forms of Stat3 (Stat3-d358L) (44) and Akt1 (myr-Akt1) (45), and also by looking at the effect of loss of p53 (p53-/-) (19), in our model of MYC-induced lymphomagenesis (Fig. 5 and Fig. S4). As a negative control, an empty vector alone was introduced into the lymphoma cells (Fig. 5 and Fig. S4). As predicted, the introduction of these mutations into MYC-induced lymphomas had differential effects on apoptosis following MYC inactivation in vitro (Fig. S4B).
Next, the in vivo effects of these mutations on the ability of MYC inactivation to induce lymphoma regression was measured. As predicted from our ODE model, the magnitude of the effect of pro-survival (Stat3-d358L and myr-Akt1) and pro-death (p53-/-) pathways on cell death correlated with how quickly tumors regressed following MYC inactivation (Fig. 5). Specifically, Stat3-d358L had no effect on apoptosis (83% apoptotic cells versus 81% for control, p=0.17; Fig. 5J and Fig. S4B) and no effect on tumor regression (Fig. 5A). Conversely, myr-Akt1 reduced apoptosis modestly (63% apoptotic cells versus 81% for control, p=0.008; Fig. 5J and Fig. S4B) and caused a modest delay in tumor regression (Fig. 5B). Finally, the loss of p53 produced the most dramatic effect on apoptosis (24% apoptotic cells for p53-/- versus 81% for control, p=0.0005; Fig. 5J and Fig. S4B) and abrogated tumor regression so dramatically that tumors no longer appeared oncogene addicted (Fig. 5C and J).
Then, we examined how these mutations resulted in changes in aggregate survival and death signals as predicted by our ODE model (as for Fig. 4). We solved for survival (S(t)) and death (D(t)) signal intensities over time (Fig 5G-I and Fig. S4C-G). As predicted, Stat3d358L and myr-Akt1 containing lymphomas that behaved in an oncogene addicted fashion still demonstrated a continuous excess of death signal compared to survival signal during tumor regression (Fig. 5G & H). Furthermore, for myr-Akt1 tumors (Fig. 5J and Fig. S4B), the predicted survival signal took longer to decay than control tumors or Stat3-d358L tumors (3 days versus 2 days; Fig. 5D-H). Lastly, the loss of p53 had the most dramatic effect on the inhibition of apoptosis (Fig. 5J and Fig. S4B), resulting in the absence of tumor regression (Fig. 5C) and the loss of the differential decay between aggregate survival and death signals such that the two signals crossed over each other (Fig. 5I). Hence, our ODE model, could predict how changes in survival and death pathways influenced the ability of oncogene inactivation to elicit oncogene addiction.
Recently, MYC-inactivation in lung cancer has been shown by us to fail to induce complete tumor regression (41) (Fig. S5). We fitted our mathematical model using data from the MYC-induced lung tumors as had been performed for the K-rasG12D induced lung tumors (Fig. 6A and Table 1). Interestingly, when we solved for aggregate survival (S(t)) and death (D(t)) signals over time the model fit was very poor (Fig. 6A) thereby demonstrating that our ODE model worked well as predicted on oncogene addicted tumors but did not show overfitting to the case of non-oncogene addicted tumors. Thus, when oncogene inactivation is not associated with oncogene addiction, tumor growth kinetics are unable to be explained by differential attenuation of aggregate survival and death signals, as we also observed for the loss of p53 in our model of MYC-induced lymphoma (Fig. 5I). Thus, the consequences of oncogene inactivation largely depend on how genetic and cellular context influence survival and death signaling (9, 11, 12, 19, 46-49).
Fig. 6.
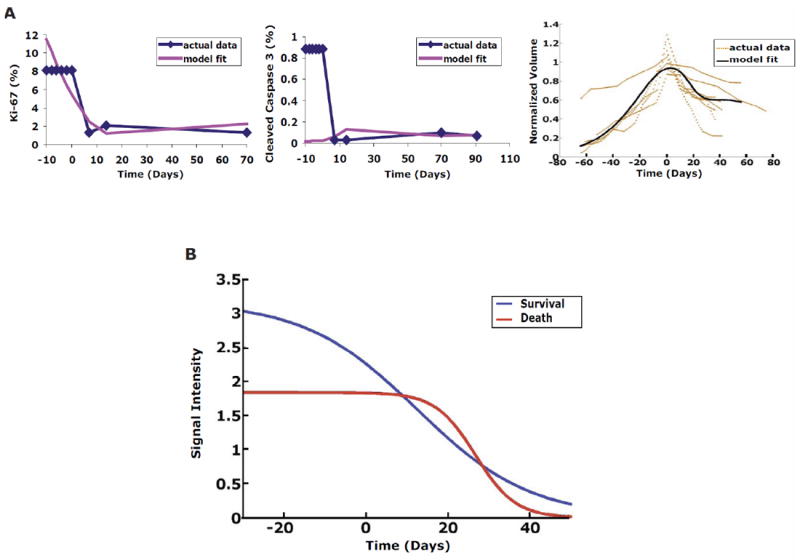
Mathematical model of differential attenuation is not applicable to non-oncogene addicted MYC-induced lung tumors. Model overfitting does not occur when applied to the non-oncogene addicted MYC-induced lung tumor model where differential attenuation is not expected to explain growth kinetics. Model fitting was performed for the (A) volume data (right panel), proliferation (Ki-67; left panel) and apoptosis (cleaved caspase 3; middle panel). Note, these data showed a poor fit using the same mathematical equation for oncogene addicted or K-rasG12D -induced lung tumors. The averaged RMSE was 31%: 8% for volume fit, 18% error for PI fit (Ki-67), and 67% for AI (cleaved caspase3) fit. (B) MYC inactivation in lung tumors is not associated with differential attenuation of aggregate survival and death signals. The plots shown were generated using the same mathematical model as described in detail in Fig. 4 using the data (V, PI and AI) from the MYC-induced lung tumors following oncogene inactivation.
Prediction of Oncogene Addiction
The ability to anticipate whether oncogene inactivation would result in tumor regression would be highly valuable. To address this possibility, we compared the K-rasG12D –induced lung tumor system (oncogene addicted) (14) to our MYC-induced lung tumor system (non-oncogene addicted) where tumor signaling dynamics have a complexity beyond the ODE differential attenuation model above. Thus, we employed a machine learning approach, support vector machine (SVM), to determine whether the early tumor growth and regression kinetics alone contain enough information to accurately classify the oncogene addicted genotype versus non-addicted genotype. The K-rasG12D (Fig. S2A-B) and MYC (Fig. S5) microCT based kinetic regression curves were used to train a support vector machine (SVM) algorithm, which is a supervised learning method for classification of datasets (Fig. 7A) (50, 51). After training the SVM on the difference between these genotype classes following simulated treatment (Fig. 7A-B and Fig. S6A), the SVM was 100% accurate at classifying an oncogene addicted genotype (K-rasG12D) using only the first three serial weekly microCT scans following a simulated oncogene targeted therapy out of 6-10 weeks total to train the SVM (Fig. S6B-C).
Fig. 7.
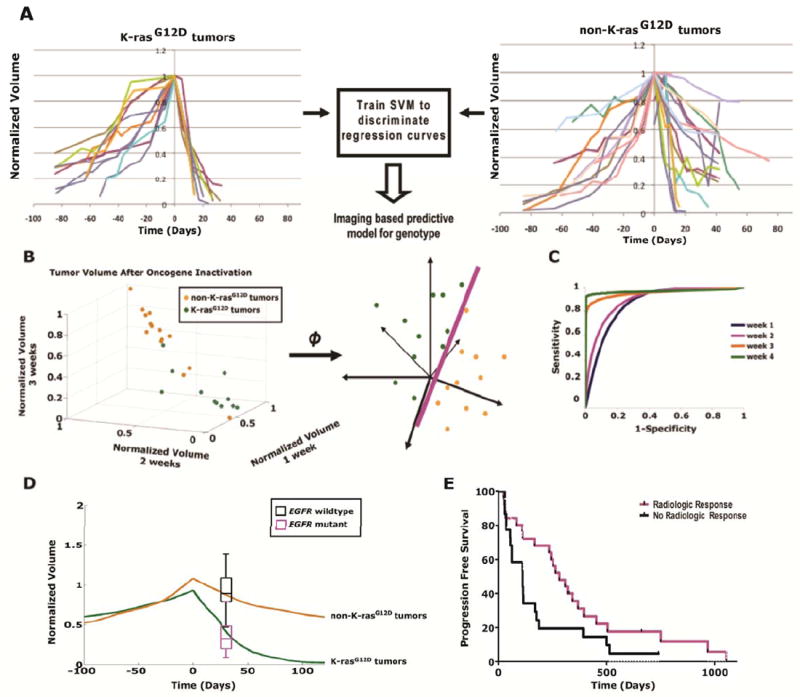
Modeling imaging data from the regression of human lung tumors treated with targeted therapy can be used to classify genotype. (A) Quantitative imaging data following simulated oncogene therapy for K-rasG12D -and non-K-rasG12D -induced lung tumors are used to train a support vector machine (SVM) algorithm. (B) An illustration of SVM mapping the original dataset in a higher dimensional space where a maximal separating hyperplane is constructed that best separates the data points between two different genotypes, K-rasG12D and non-K-rasG12D, for classification. (C) Receiver operating characteristics (ROC) curves show the accuracy of the SVM technique in predicting the oncogene dependent genotypes based on tumor volumes obtained from different length of time after oncogene targeted therapy. (D) K-rasG12D and non-K-rasG12D tumor volumes over time in orange and green, respectively, scaled for differences between mouse and human tumor doubling times (oncogene inactivation at day 0). The black and cyan box plots with error bars represent tumor responses from patients with EGFR mutations (cyan) and wildtype EGFR (black) measured 4 weeks following targeted therapy with erlotinib. Mouse K-rasG12D and non-K-rasG12D tumors behave similarly to human tumors with EGFR mutations and wildtype EGFR, respectively, following targeted therapy. This model had an 80% (12/15) sensitivity, 100% (28/28) specificity, 100% (12/12) positive predictive value and 90% (28/31) negative predictive value for assigning EGFR mutation status. (E) Kaplan-Meier plots of lung cancer patients based on quantitative imaging response at 4 weeks predicted improved progression free survival (p=0.046).
Then, by using imaging data from only the first 2 weekly scans following oncogene targeted therapy, the SVM could classify K-rasG12D and MYC genotypes with 100% sensitivity and 87.5% specificity (Fig. S6B-C). Even after training the SVM with a more heterogeneous data set where MYC- and double MYC/ K-rasG12D –induced tumors were grouped together (non-K-rasG12D) against K-rasG12D–induced tumors (Fig. 7B), the SVM was still able to accurately differentiate between K-rasG12D and non- K-rasG12D genotypes (Fig. 7C and Fig. S6D). After only 2 weeks of oncogene inactivation, the SVM could classify K-rasG12D versus non- K-rasG12D tumors with 95% sensitivity and 86% specificity. The sensitivity improved to 100% and specificity improved to 93% for K-rasG12D versus non- K-rasG12D tumors, respectively, after 4 weeks of imaging data (Fig. S6D). Thus, an SVM classifier tool given quantitative CT mouse imaging data is able to classify oncogene addicted versus non-addicted genotypes and thus predict the tumor response to oncogene inactivation.
Predicting Oncogene Addiction in Human Lung Cancers
We considered that our SVM approach could be useful to predict in human patients with lung cancer if they will clinically respond to a targeted therapy. To evaluate this possibility, we analyzed patients from a prospective clinical trial who had received the EGFR inhibitor, erlotinib, for the treatment of lung adenocarcinoma (52). EGFR mutant lung tumors are reported to behave in an oncogene addicted fashion following EGFR targeted therapy and EGFR mutation status is now considered to be the best surrogate for prediction of tumor response and progression free survival for lung cancer patients undergoing EGFR TKI treatment (20, 25, 29).
We quantitatively analyzed CT scan imaging data from the patients for whom the status of EGFR mutations was known and for whom paired tumor measurements were available before initiation of erlotinib and then again at the early time point of 4 weeks (52). Then, using the SVM classifier generated from the analysis of our transgenic mice as a reference, we correctly assigned the EGFR genotype and thus clinical response of 93% of the patients with a positive predictive value of 100% and a negative predictive value of 91% after only 4 weeks of targeted therapy (Fig. 7D). Most importantly, we could predict longer progression free survival (PFS) of patients directly from the CT data alone (Fig. 7E, log rank analysis p=0.05). Note that from the mouse SVM data we could predict genotype as early as 2 weeks after targeted therapy (Fig. 7B-C). We believe that by using our SVM model we will be able to predict both EGFR genotype and PFS earlier than 4 weeks after erlotinib treatment.
Discussion
We have combined direct in vivo biological analysis of conditional transgenic tumor mouse models and highly quantitative imaging methods to demonstrate that aggregate survival and death signals interact and can predict oncogene addiction at the level of known pro-survival and pro-death effector molecules and as well as clinical response (Figure 1A). A priori, there were several ways in which aggregate survival and death signaling pathways could behave following oncogene addicted tumor response (Figure 1C): 1) decrease in survival signals and an increase in death signals; 2) decrease in survival signals only 3) an increase in death signals only; 4) an increase in survival and quicker increase in death signals; or 5) a decrease in survival and delayed decrease in death signals. We directly measured pro-survival and pro-death signaling molecules in vivo (Figure 3). We then developed a quantitative ODE model that could be used to define and predict the behavior of tumor cells upon oncogene inactivation (Figure 4). Using additional quantitative methods that included serial imaging and in situ measurements of proliferation and apoptosis (Figure 2) our ODE model predicted that intracellular aggregate survival signals decay rapidly at a much earlier time point than death signals following targeted therapy (Figure 4). Then, using an SVM machine classifier we could distinguish oncogene addicted versus non-addicted mouse lung tumors (Figure 7A-C and Figure S6). Using a similar approach we were able to predict genotype and thereby clinical outcome in human patients with lung cancer treated with erlotinib (Figure 7D-E).
Our model provides a description of the aggregate survival and death signals that would not necessarily be expected by examination of individual signaling molecules. Thus, as expected there are some discrepancies between our ODE model and changes in activation of individual signaling proteins. Initially after oncogene inactivation there was no apparent phospho-p38, which was followed by a burst of phospho-p38 (Fig. 3A). It is known that K-Ras regulates the pro-survival effector molecules Erk1/2 and Akt1 that in turn negatively regulate p38 phosphorylation status and thus we expected low initial phospho-p38 levels (53-55).
Our results illustrate that oncogene addiction is associated with a decrease in both aggregate survival and death signaling, but that death signals extinguish more gradually (Fig. S7). Our results are consistent but not identical with previous reports based upon qualitative in vitro observations in tumor-derived cell lines, that have argued that “differential” attenuation of survival and death signals could account for oncogene addiction (5, 6). There are several possible reasons why our results differ slightly from these experiments. The most obvious is that in contrast to our in vivo study, these previous studies were conducted entirely using cell lines in vitro. However, our modeling is not necessarily mutually exclusive from other proposed models of oncogene addiction (3, 7, 8), that propose mechanisms involving dysregulation of the intracellular signaling or “wiring” in cancer cells (3), oncoprotein specific negative feedback loops (56) and/or the reconstitution of normal cellular checkpoint programs (7).
We acknowledge that neither our ODE model nor our statistical SVM classifier model incorporates cellular or host programs that are also presumed to be important for tumorigenesis including: angiogenesis, genetic instability or immune function (1). Indeed, our own previous work has specifically identified that angiogenesis and cellular senescence are important mechanisms of oncogene addiction (19, 47). How can we reconcile that a model that incorporates survival and death signals but does not directly include many of these other processes is still capable of describing oncogene addiction? We infer that aggregate survival and death signals are an integration of many of these other biological programs that ultimately impact proliferation versus cell death, as has been suggested (2, 57). Because our ODE model was generated by analyzing in vivo data, it necessarily incorporates these other biological programs via their effects on survival and death signaling. Also, our ODE model does explicitly consider as a possibility that upon oncogene inactivation tumor cells can become homeostatic that would include cells that differentiate, senescence or become dormant. Indeed, precisely because our analysis was performed using data that was obtained in vivo our measurements necessarily incorporated the effects of the tumor microenvironment and accounted for the resulting heterogeneity. We recognize that the incorporation of other biological variables is likely to further improve and refine our ODE and SVM classifier models and their predictive potential.
A potential limitation of our study is that we used mouse tumor models that can not completely recapitulate human disease. However, we have gone to significant efforts to utilize mouse models that have been shown to approximate closely human tumors. The lung tumor model used in our study has been accepted as representative of human lung tumors that arise stochastically as illustrated by a range in latency of tumor onset and the kinetics of tumor growth; and the tumors are histologically consistent with lung adenocarcinoma (38). Moreover, the mouse K-rasG12D–induced lung tumors have been shown to behave very similarly following oncogene inactivation to EGFR mutated human lung adenocarcinomas following erlotinib treatment (58). Similarly, our transgenic model of lymphoma (9) has been widely utilized as representative of human MYC-induced T-ALL. Therefore, our mouse models appear to be useful to extrapolate tumor behavior that occurs in humans.
Our general approach of using quantitative imaging may be useful to develop new therapeutics as well as to assist therapeutic decisions for cancer. Even though EGFR mutation status is a strong predictor of therapeutic response and progression free survival (20), it is not always possible to sequence for EGFR mutations (59). In contrast, a non-invasive quantitative imaging based method potentially could be widely applied to different types of cancers and could easily be translated into a clinical setting. For most types of cancer, biomarkers for a clinical response to a specific targeted therapeutic are not available. Hence, during the development of targeted therapeutics for lung cancer as well as other types of cancer, it would be highly useful to have noninvasive and robust methods to predict clinical response both in pre-clinical models as well in clinical studies. Our results using an SVM classifier (Fig. 7 and Fig. S6) provide a proof of principle that imaging based approaches may provide a strategy that will accelerate the validation of targeted therapeutics by serving as a surrogate endpoint in early phase clinical research.
Our work demonstrates how transgenic mouse models may be useful to gain insight into the mechanism of oncogene addiction so that it may be possible to more rapidly predict when targeted therapeutics will be effective. The general availability of CT-scanners and the non-invasive nature of our general strategy is ideal for translation to the clinical setting (25, 29), in particular, for the evaluation of patients with lung cancer treated with erlotinib or gefitinib (25, 29). We recognize that the incorporation of additional biological variables, and the utilization of other modalities of quantitative molecular imaging as a biomarker would likely further refine our model and its clinical utility.
Materials and Methods
Transgenic mice
The TRE-MYC transgenic line generated for these experiments was described previously (9). The CCSP-rtTA and Tet-op-K-rasG12D transgenic lines were kindly provided by J. A. Whitsett (Cincinnati Children’s Hospital) and H. Varmus (Memorial Sloan-Kettering Cancer Center, New York), respectively. Mice were mated and screened by PCR as below. CCSP-rtTA/TRE-MYC (termed MYC or CM), CCSP-rtTA/Tet-op-K-rasG12D (termed K-rasG12D or CR) and CCSP-rtTA/TRE-MYC/Tet-op-K-rasG12D (termed CMR) expression was activated in the lung lines by administering doxycycline (Sigma) in the drinking water weekly [2 mg/mL] starting at the age of 3-4 weeks. All procedures were performed in accordance with APLAC protocols and animals were housed in a pathogen-free environment.
Oncogene Inactivation
Transgenic mice were followed by weekly computed tomography scans for a total of >16 weeks. Oncogenes were inactivated in the CM, CR and CMR cohorts by removing doxycycline from the animals’ drinking water before mice became moribund with tumor or when tumors were visualized by microCT.
PCR genotyping
DNA was isolated from mouse tails using the Qiaprep DNeasy kit (Qiagen) in accordance with the manufacturer’s directions. The CCSP-rtTA segment was detected using the following primers: CCSP-F 5’-ACTGCCCATTGCCCAAACAC-3’ and CCSP-R 5’-AAAATCTTGCCAGCTTTCCCC-3’ (yielding a 440 bp product). The TRE-Myc construct was detected with the following primers: Myc-F 5’-TAGTGAACCGTCAGATCGCCTG-3’ and Myc-R 5’-TTTGATGAAGGTCTCGTCGTCC-3’ (yielding a 450 bp product). Tet-op-K-Ras was screened as described previously (Fisher et. al., 2001). DNA was amplified using the following PCR protocol: 94°C denaturation for 2 minutes followed by 35 cycles of 94°C for 15 seconds, 59°C annealing for 30 seconds, and 72°C for 30 seconds, followed by a 5 minute extension at 72°C. PCR products were resolved on a 1.5% gel.
Western blot analysis
Western analysis was performed using conventional techniques. Tissues or cells were disrupted and protein was isolated using a tube homogenizer in RIPA lysis buffer. Equal protein was loaded in each lane, as quantified by the Bicinchoninic Acid (BCA) Protein Assay (Pierce, Rockford, Illinois, United States). Proteins were electrophoresed on 8-10% Tris-HCl polyacrylamide gels at 100 V for 60 min and transferred on PVDF membranes at 100 V for 60 min. Blotting was then performed as directed by the antibody manufacturer. The following antibodies were used: Anti-Stat3 pTyr705 (Cell Signaling); Anti-Stat3 (Cell Signaling); Anti-alpha-tubulin (Sigma); Anti-Akt1 pSer473 (Cell Signaling); Anti-Akt1 (Cell Signaling); and Anti-Erk1/2 pT202/Y204 (Cell Signaling).
Apoptosis and Proliferation assays
Cells were centrifuged, washed twice with PBS and fixed in cold 70% ethanol and stained with propidium iodide and then analyzed with a FACScan flow cytometer for sub-G1 and S-phase DNA content for apoptotic and proliferative growth fractions, respectively. Alternatively, cells were stained with Annexin V-PE (BD Pharmingen) and apoptosis was measured by flow cytometry. Data was analyzed using FlowJo (Tree Star, Inc).
Histology and Immunohistochemistry
Tissues were fixed in 10% buffered formalin for 24 h and then transferred to 70% ethanol until embedded in paraffin. Tissue sections 5 μm thick were cut from paraffin embedded blocks, placed on glass slides and hematoxylin and eosin (H&E) staining was performed using standard procedures (Stanford Histology Core). We measured Ki67 and TUNEL-staining as described previously (Beer et. al., 2004). Antibodies used in our study: c-Myc (C19) (Santa Cruz Biotech.), cleaved caspase 3 (Cell Signaling Tech.), phospho-AKT-S497 (Cell Signaling Tech.), phospho-Erk1/2-T202/Y204 (Cell Signaling Tech.), phospho-Stat3-Y705 (Cell Signaling Tech.), phospho-p38-T180/Y182 (Cell Signaling Tech.) and phospho-Stat5-Y694 (Cell Signaling Tech.). Samples were dewaxed in xylene and rehydrated in a graded series of ethanols. Antigen retrieval for c-Myc, cleaved caspase 3 and phospho-AKT were performed by 14 min microwave irradiation in citrate-based Antigen Unmasking Solution (Vector Laboratories, Burlingame, CA, USA). Antigen retrieval for phospho-Stat3 and -Stat5 were performed by 14 min microwave irradiation in EDTA, pH 8.0, and antigen retrieval for phospho-Erk1/2 and phospho-p38 was performed by10 min incubation in Pronase (Roche, Basel, Switzerland). Endogenous peroxidases were blocked in either 3% hydrogen peroxide in deionized water (phospho-AKT, -pErk, -p38 and -pStat3/5) or 0.3% hydrogen peroxide in methanol (c-Myc and cleaved caspase 3) for 10-20 minutes. Non-specific binding was blocked with 5 -10% goat serum for 60 minutes. Primary antibodies were used at appropriate dilutions (c-Myc, phospho-AKT, -pErk and -p38 at 1:100; cleaved caspase 3 at 1:150; phospho-Stat5 at 1:200; and phospho-Stat3 at 1:50) and sections incubated overnight at 4 degrees Celsius. Detection was conducted using the Vector Elite ABC detection kit (Vector Laboratories) with 3,3’-diaminobenzidine tetrahydrochloride as the chromogen. Sections were counterstained with Gill’s hematoxylin (Vector Laboratories).
Computed Tomography
Micro-computed tomography (μCT) scans were performed on a custom GEHC (London, Ontario) eXplore RS150 cone-beam scanner, which uses a fixed anode with tungsten target source. Animals were anesthetized with 2% isofluorane in a nitrogen/oxygen mixture. Scans were performed at 97μm resolution, using a 70 kV (40 mA) beam to acquire images at 286 radial views over 200 degrees around the subject. Four frames were exposed and averaged in each position. Data were corrected using the GEHC reconstruction utility and volumes generated using the same application, which were viewed using the GEHC Microview software. Mice were exposed to 19.4 rads per weekly microCT scan.
Sample Population
Weekly microCT scans were collected on twenty K-rasG12D-induced mouse lung tumors, eight MYC-induced mouse lung tumors and four CMR-induced lung tumors. Nodular opacities were designated tumors after retrospective review from serial microCT. For the purpose of data analysis, dates of the scans are referred to relative to oncogene inactivation at day 0.
Tumor Segmentation
A semi-automated open source image analysis application, ITK-Snap (60), was employed for segmentation of the lung tumors from the microCT images. The tool uses a level set algorithm to semi-automatically delineate tumor from background by expanding a level set from a user-defined seed point. We viewed 2D images in sagittal, coronal and transverse views to detect tumors and track them over time. The segmentation algorithm was seeded manually and stopped once the tumor boundary was detected. The postprocessing of the segmented data provided the voxel counts, the volume (in cubic millimeters) and displayed the shape of the segmented structure. Matching tumors across time points was performed manually by simultaneous viewing of the serial data. This volume information was then used for analysis of the temporal changes in lung tumor nodules.
Normalization
Peak tumor volumes at the time of oncogene inactivation had significant variability due to the subjective determination of when the oncogene was inactivated and also due to the variability in tumor sizes in the same mouse. However, simple normalization by peak tumor volume was artificially biased toward a single time point due to zero variance at time zero (peak) and exaggerated variance at large negative and positive times. Instead, we weighted each time point equally in the normalization process. Due to the exponential-like growth and regression pattern observed in volume data, two straight lines were fitted by least squares to tumor volume in semi-log space, one for the growth phase and one for the regression phase. The value at the intersection of the two straight lines was then used to normalize the volume for each tumor time series (Fig. S2A). Normalized tumor volumes at each scan time were averaged across all tumors to obtain a general trend of the growth and regression patterns leading to much more consistent variance in tumor volume measurements across time.
MYC-induced Lymphoma Mouse System
MYC-induced lymphoma cells expressing luciferase were transduced using retrovirus carrying either pBabe-puro, pMx-Stat3d358L-puro, or pBabe-MyrAktHA-puro. Infected cells were then selected with 1 μg/ml puromycin. Ten million MYC-induced lymphoma cells were injected subcutaneously and then were scanned every 2-5 days prior to oncogene inactivation and 20 days after oncogene inactivation using BLI imaging (19). Average radiance (p/s/cm2/sr) of each scan was collected and treated similarly as volumetric measurements for the K-rasG12D–induced lung tumors and was used to study tumor kinetics. A similar normalization method was used on the BLI measurements. We fitted the BLI data to equation (2) to obtain the necessary parameters for estimation of S(t) and D(t) behaviors.
Support Vector Machine (SVM) Classification
v-SVM and leave-one-out techniques were used for the classification and prediction of tumor genotype based on the first few serial weekly scans after oncogene inactivation. Two classes of data points were constructed: (1) 20 K-rasG12D oncogene-induced tumors, and (2) 8 non-oncogene addicted MYC-induced mouse tumors also following simulated MYC oncogene targeted treatment. v-SVM algorithm can be summarized as follows. Given a dataset D of n samples, (S1,L1),⋯,(Sn,Ln), where Si = <s1,s2,⋯,sm> was a set of feature vectors which in our case were the tumor volumes from weekly scan, and Li ε {-1,+1} represented the binary class membership, SVMs mapped the input data into a high-dimensional feature space, and constructed a maximum margin hyperplane to separate the two classes in order to predict the label of a new sample. In this work, we used the Gaussian kernel k (S, S′) = exp(−d (S, S′)2) /σ2) to map the input data to the high-dimensional feature space, where S, S’ were two feature vectors, d was the Euclidean distance, and σ the width parameter of the kernel. We selected the width σ of the kernel by minimizing the leave-one-out error estimate of the classification error between the true label and the predicted value of the v-SVM classification function for each σ. In our experiments simulating a more heterogeneous population, we included 20 oncogene addicted K-rasG12D tumors, 8 non-oncogene addicted MYC tumors and 6 double mutant, MYC/ K-rasG12D, tumors for training and classification. Training data was derived from normalized tumors volumes.
Mouse and Human Lung Tumor Regression Analysis
Human lung tumor measurements from CT scan data were performed according to the World Health Organization (WHO) standards. Bi-dimensional tumor measurements were performed before and 4 weeks following erlotinib therapy by obtaining the longest diameter of the tumor in the axial plane and the greatest perpendicular to it. Tumor volume was calculated as a sphere and percent change at follow up was calculated as a percentage of baseline value. The median doubling time of human lung tumors is 144 days (61-63) while the conditional transgenic mouse lung tumors in our study double every 37.04 days. To correct for different growth kinetics between species, mouse tumor kinetics were expanded by a factor of 3.89 (human tumor doubling time divided by mouse tumor doubling time). Using this analysis, at one month after oncogene inactivation, the average change in volume of oncogene addicted mouse lung tumors (K-rasG12D) was 58.5% while patients with EGFR mutations had average change in tumor volume of 60.9%. Similarly, the average change in volume of non-oncogene addicted mouse lung tumors (non-K-rasG12D) was 12.5% while patients with wildtype EGFR had an average change of 5.7%.
Supplementary Material
Figure S1. Attenuation of pro-survival and pro-death signaling pathway mediators, during regression of K-rasG12D –induced lung tumors in vivo.
Figure S2. K-rasG12D induced murine lung tumors are oncogene-dependent.
Figure S3. Validation of the ODE mathematical model for oncogene addicted tumor behavior.
Figure S4. Genetic perturbation of pro-survival and pro-death pathways in MYC-induced lymphomas can impede tumor regression following MYC inactivation in vivo.
Figure S5. MYC-induced lung tumors are not oncogene addicted.
Figure S6. Support vector machine (SVM) trained with quantified imaging data can be used to distinguish K-rasG12D and MYC-induced lung tumors.
Figure S7. Schematic integrating the temporal phospho-IHC in relation to K-rasG12D activation state.
Supplementary Videos: Regression of K-rasG12D -induced lung tumors imaged serially using microCT.
Acknowledgments
We thank members of the Felsher and Paik laboratories for helpful discussions and a critical reading of the manuscript. We thank Qiwei Yang for her assistance in the generation and characterization of transgenic mice and Dr’s Howard Chang, Sam Gambhir, Ted Graves, Paul Khavari, Sylvia Plevritis, and Charles Rudin for their helpful comments. Additionally, we appreciate the support of Tim Doyle and the staff at the Stanford Small Animal Imaging Facility (SAIF) during the conduction of these experiments. This work was supported by an RSNA Fellow Research grant, a Parker B. Francis Fellowship and the Henry S. Kaplan Research Fellow award (to P.T.T.); the Howard Hughes Medical Institute Medical Student Research Fellowship (to P.K.B.); the Stanford Molecular Imaging Scholars program (to H.J.L); and National Cancer Institute (NCI) Grants; National Institutes of Health (NIH)_NCI In Vivo Cellular and Molecular Imaging Center Grant and NCI Clinical Center for Systems Biology Grant; the Leukemia and Lymphoma Society; the Burroughs Wellcome Fund; and the Damon Runyon Lilly Clinical Investigator Award (to D.W.F.).
Footnotes
The authors’ declare no competing interests
References
- 1.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–37. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432(7015):307–15. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 3.Weinstein IB, Joe A. Oncogene addiction. Cancer Res. 2008;68(9):3077–80. doi: 10.1158/0008-5472.CAN-07-3293. discussion 3080. [DOI] [PubMed] [Google Scholar]
- 4.Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297(5578):63–4. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- 5.Sharma SV, Gajowniczek P, Way IP, Lee DY, Jiang J, Yuza Y, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10(5):425–35. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–98. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 7.Felsher DW. Oncogene addiction versus oncogene amnesia: perhaps more than just a bad habit? Cancer Res. 2008;68(9):3081–6. doi: 10.1158/0008-5472.CAN-07-5832. discussion 3086. [DOI] [PubMed] [Google Scholar]
- 8.Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 2007;21(24):3214–31. doi: 10.1101/gad.1609907. [DOI] [PubMed] [Google Scholar]
- 9.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4(2):199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 10.Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science. 2002;297(5578):102–4. doi: 10.1126/science.1071489. [DOI] [PubMed] [Google Scholar]
- 11.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400(6743):468–72. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 12.D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7(2):235–9. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 13.Pelengaris S, Littlewood T, Khan M, Elia G, Evan G. Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell. 1999;3(5):565–77. doi: 10.1016/s1097-2765(00)80350-0. [DOI] [PubMed] [Google Scholar]
- 14.Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15(24):3249–62. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perera SA, Li D, Shimamura T, Raso MG, Ji H, Chen L, et al. HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci U S A. 2009;106(2):474–9. doi: 10.1073/pnas.0808930106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji H, Wang Z, Perera SA, Li D, Liang MC, Zaghlul S, et al. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007;67(10):4933–9. doi: 10.1158/0008-5472.CAN-06-4592. [DOI] [PubMed] [Google Scholar]
- 17.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu CH, Sahoo D, Arvanitis C, Bradon N, Dill DL, Felsher DW. Combined analysis of murine and human microarrays and ChIP analysis reveals genes associated with the ability of MYC to maintain tumorigenesis. PLoS Genet. 2008;4(6):e1000090. doi: 10.1371/journal.pgen.1000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giuriato S, Ryeom S, Fan AC, Bachireddy P, Lynch RC, Rioth MJ, et al. Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci U S A. 2006;103(44):16266–71. doi: 10.1073/pnas.0608017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Govindan R. INTERESTing Biomarker to Select IDEAL Patients for Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors: Yes, for EGFR Mutation Analysis, Others, I PASS. J Clin Oncol. 2010;28(5):713–5. doi: 10.1200/JCO.2009.25.1637. [DOI] [PubMed] [Google Scholar]
- 21.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–57. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 22.Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–17. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 23.Heinrich MC, Blanke CD, Druker BJ, Corless CL. Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol. 2002;20(6):1692–703. doi: 10.1200/JCO.2002.20.6.1692. [DOI] [PubMed] [Google Scholar]
- 24.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342–9. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 25.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–74. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 26.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 28.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 29.Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25(5):587–95. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 30.Uyl-de Groot CA, Giaccone G. Health economics: can we afford an unrestricted use of new biological agents in gastrointestinal oncology? Curr Opin Oncol. 2005;17(4):392–6. doi: 10.1097/01.cco.0000168535.25330.6a. [DOI] [PubMed] [Google Scholar]
- 31.Kartachova M, van Zandwijk N, Burgers S, van Tinteren H, Verheij M, Valdes Olmos RA. Prognostic significance of 99mTc Hynic-rh-annexin V scintigraphy during platinum-based chemotherapy in advanced lung cancer. J Clin Oncol. 2007;25(18):2534–9. doi: 10.1200/JCO.2006.10.1337. [DOI] [PubMed] [Google Scholar]
- 32.Zahra MA, Hollingsworth KG, Sala E, Lomas DJ, Tan LT. Dynamic contrast-enhanced MRI as a predictor of tumour response to radiotherapy. Lancet Oncol. 2007;8(1):63–74. doi: 10.1016/S1470-2045(06)71012-9. [DOI] [PubMed] [Google Scholar]
- 33.Schelling M, Avril N, Nahrig J, Kuhn W, Romer W, Sattler D, et al. Positron emission tomography using [(18)F]Fluorodeoxyglucose for monitoring primary chemotherapy in breast cancer. J Clin Oncol. 2000;18(8):1689–95. doi: 10.1200/JCO.2000.18.8.1689. [DOI] [PubMed] [Google Scholar]
- 34.Smith IC, Welch AE, Hutcheon AW, Miller ID, Payne S, Chilcott F, et al. Positron emission tomography using [(18)F]-fluorodeoxy-D-glucose to predict the pathologic response of breast cancer to primary chemotherapy. J Clin Oncol. 2000;18(8):1676–88. doi: 10.1200/JCO.2000.18.8.1676. [DOI] [PubMed] [Google Scholar]
- 35.Advani R, Maeda L, Lavori P, Quon A, Hoppe R, Breslin S, et al. Impact of Positive Positron Emission Tomography on Prediction of Freedom From Progression After Stanford V Chemotherapy in Hodgkin’s Disease. J Clin Oncol. 2007;25(25):3902–3907. doi: 10.1200/JCO.2007.11.9867. [DOI] [PubMed] [Google Scholar]
- 36.Kong F-MS, Frey KA, Quint LE, Haken RKT, Hayman JA, Kessler M, et al. A Pilot Study of [18F]Fluorodeoxyglucose Positron Emission Tomography Scans During and After Radiation-Based Therapy in Patients With Non Small-Cell Lung Cancer. J Clin Oncol. 2007;25(21):3116–3123. doi: 10.1200/JCO.2006.10.3747. [DOI] [PubMed] [Google Scholar]
- 37.Nahmias C, Hanna WT, Wahl LM, Long MJ, Hubner KF, Townsend DW. Time Course of Early Response to Chemotherapy in Non-Small Cell Lung Cancer Patients with 18F-FDG PET/CT. J Nucl Med. 2007;48(5):744–51. doi: 10.2967/jnumed.106.038513. [DOI] [PubMed] [Google Scholar]
- 38.Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, et al. Classification of Proliferative Pulmonary Lesions of the Mouse: Recommendations of the Mouse Models of Human Cancers Consortium. Cancer Res. 2004;64(7):2307–2316. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 39.Felsher DW. MYC Inactivation Elicits Oncogene Addiction through Both Tumor Cell-Intrinsic and Host-Dependent Mechanisms. Genes Cancer. 1(6):597–604. doi: 10.1177/1947601910377798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 41.Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J, et al. Combined Inactivation of MYC and K-Ras Oncogenes Reverses Tumorigenesis in Lung Adenocarcinomas and Lymphomas. PLoS ONE. 2008;3(5):e2125. doi: 10.1371/journal.pone.0002125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bradham C, McClay DR. p38 MAPK in development and cancer. Cell Cycle. 2006;5(8):824–8. doi: 10.4161/cc.5.8.2685. [DOI] [PubMed] [Google Scholar]
- 43.Cody DD, Nelson CL, Bradley WM, Wislez M, Juroske D, Price RE, et al. Murine lung tumor measurement using respiratory-gated micro-computed tomography. Invest Radiol. 2005;40(5):263–9. doi: 10.1097/01.rli.0000160070.67270.05. [DOI] [PubMed] [Google Scholar]
- 44.Kawashima T, Bao YC, Minoshima Y, Nomura Y, Hatori T, Hori T, et al. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol. 2009;29(7):1796–813. doi: 10.1128/MCB.01423-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kulik G, Klippel A, Weber MJ. Antiapoptotic signalling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol. 1997;17(3):1595–606. doi: 10.1128/mcb.17.3.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell. 2002;109(3):321–34. doi: 10.1016/s0092-8674(02)00738-9. [DOI] [PubMed] [Google Scholar]
- 47.Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shachaf CM, Gentles AJ, Elchuri S, Sahoo D, Soen Y, Sharpe O, et al. Genomic and proteomic analysis reveals a threshold level of MYC required for tumor maintenance. Cancer Res. 2008;68(13):5132–42. doi: 10.1158/0008-5472.CAN-07-6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Michor F, Hughes TP, Iwasa Y, Branford S, Shah NP, Sawyers CL, et al. Dynamics of chronic myeloid leukaemia. Nature. 2005;435(7046):1267–70. doi: 10.1038/nature03669. [DOI] [PubMed] [Google Scholar]
- 50.Noble WS. What is a support vector machine? Nat Biotechnol. 2006;24(12):1565–7. doi: 10.1038/nbt1206-1565. [DOI] [PubMed] [Google Scholar]
- 51.Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, et al. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A. 2000;97(1):262–7. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller VA, Riely GJ, Zakowski MF, Li AR, Patel JD, Heelan RT, et al. Molecular characteristics of bronchioloalveolar carcinoma and adenocarcinoma, bronchioloalveolar carcinoma subtype, predict response to erlotinib. J Clin Oncol. 2008;26(9):1472–8. doi: 10.1200/JCO.2007.13.0062. [DOI] [PubMed] [Google Scholar]
- 53.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270(5240):1326–31. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 54.Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. 1997;275(5296):90–4. doi: 10.1126/science.275.5296.90. [DOI] [PubMed] [Google Scholar]
- 55.Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21(3):893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A. 2009;106(11):4519–24. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma SV, Fischbach MA, Haber DA, Settleman J. “Oncogenic shock”: explaining oncogene addiction through differential signal attenuation. Clin Cancer Res. 2006;12(14 Pt 2):4392s–4395s. doi: 10.1158/1078-0432.CCR-06-0096. [DOI] [PubMed] [Google Scholar]
- 58.Milton DT, Riely GJ, Pao W, Miller VA, Kris MG, Heelan RT. Molecular on/off switch. J Clin Oncol. 2006;24(30):4940–2. doi: 10.1200/JCO.2006.06.4410. [DOI] [PubMed] [Google Scholar]
- 59.Sequist LV, Engelman JA, Lynch TJ. Toward noninvasive genomic screening of lung cancer patients. J Clin Oncol. 2009;27(16):2589–91. doi: 10.1200/JCO.2008.20.4875. [DOI] [PubMed] [Google Scholar]
- 60.Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31(3):1116–28. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 61.Yankelevitz DF, Kostis WJ, Henschke CI, Heelan RT, Libby DM, Pasmantier MW, et al. Overdiagnosis in chest radiographic screening for lung carcinoma: frequency. Cancer. 2003;97(5):1271–5. doi: 10.1002/cncr.11185. [DOI] [PubMed] [Google Scholar]
- 62.Lindell RM, Hartman TE, Swensen SJ, Jett JR, Midthun DE, Tazelaar HD, et al. Five-year lung cancer screening experience: CT appearance, growth rate, location, and histologic features of 61 lung cancers. Radiology. 2007;242(2):555–62. doi: 10.1148/radiol.2422052090. [DOI] [PubMed] [Google Scholar]
- 63.El Sharouni SY, Kal HB, Battermann JJ. Accelerated regrowth of non-small-cell lung tumours after induction chemotherapy. Br J Cancer. 2003;89(12):2184–9. doi: 10.1038/sj.bjc.6601418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Attenuation of pro-survival and pro-death signaling pathway mediators, during regression of K-rasG12D –induced lung tumors in vivo.
Figure S2. K-rasG12D induced murine lung tumors are oncogene-dependent.
Figure S3. Validation of the ODE mathematical model for oncogene addicted tumor behavior.
Figure S4. Genetic perturbation of pro-survival and pro-death pathways in MYC-induced lymphomas can impede tumor regression following MYC inactivation in vivo.
Figure S5. MYC-induced lung tumors are not oncogene addicted.
Figure S6. Support vector machine (SVM) trained with quantified imaging data can be used to distinguish K-rasG12D and MYC-induced lung tumors.
Figure S7. Schematic integrating the temporal phospho-IHC in relation to K-rasG12D activation state.
Supplementary Videos: Regression of K-rasG12D -induced lung tumors imaged serially using microCT.