

Rho GTPases regulate many essential processes during development, yet the full impact of their upstream regulation through guanine nucleotide exchange factors (GEFs) is only beginning to be appreciated. In this review, Laurin and Côté focus on emerging biological functions of the mammalian Dock family of GEFs in development and disease and discuss how recent discoveries might be exploited for novel therapeutic strategies.
Keywords: mouse models, Dock180, Elmo, neurogenesis, myoblast fusion, migration
Abstract
Rho GTPases play key regulatory roles in many aspects of embryonic development, regulating processes such as differentiation, proliferation, morphogenesis, and migration. Two families of guanine nucleotide exchange factors (GEFs) found in metazoans, Dbl and Dock, are responsible for the spatiotemporal activation of Rac and Cdc42 proteins and their downstream signaling pathways. This review focuses on the emerging roles of the mammalian DOCK family in development and disease. We also discuss, when possible, how recent discoveries concerning the biological functions of these GEFs might be exploited for the development of novel therapeutic strategies.
Regulation and signaling of Rho GTPases
Twenty-one genes in the Ras superfamily encode for Rho GTPases. By orchestrating remodeling of the cytoskeleton, these molecular switches regulate numerous processes throughout embryonic development, and their abnormal regulation is associated with various diseases (Bryan et al. 2005; Cancelas and Williams 2009; Hall and Lalli 2010; Alan and Lundquist 2013). Rho proteins switch between inactive (GDP-bound) and active (GTP-bound) conformations. Three groups of proteins coordinate their cycling states: GTPase-activating proteins (GAPs) and Rho guanine nucleotide dissociation inhibitors act as negative regulators by promoting the intrinsic GTPase activity of Rho proteins or sequestering them in the cytoplasm, respectively. The guanine nucleotide exchange factors (GEFs) are direct positive regulators, mediating GDP/GTP exchange to ultimately promote the binding of Rho proteins to specific effectors (Lazer and Katzav 2011). The first GEF to be discovered was initially characterized in yeast and found to be analogous to the proto-oncogene Dbl in mammals (Hart et al. 1991; Ron et al. 1991). The Dbl family of GEFs, with >70 members, is characterized by the presence of the Dbl domain, which is critical for the GDP/GTP exchange activity (Rossman et al. 2005). The Dock GEFs were discovered later and form an 11-member family, classified into four subgroups, characterized by the presence of two evolutionarily conserved domains: the lipid-binding Dock homology region-1 (DHR-1) and the GEF DHR-2 modules (Fig. 1). Because Dock GEFs lack a Dbl domain, they are often referred to as “atypical GEFs.” A distinctive feature of the Dock GEFs is the specificity of individual family members to activate Rac and/or Cdc42 but not RhoA or other members of the Rho family (Cote and Vuori 2002, 2006). It remains unclear why two subfamilies of GEFs exist and whether they might cosignal in certain conditions. While a recent review discussed the precise roles of DOCK2 and DOCK8 in the immune system (Nishikimi et al. 2013), this review focuses on emerging biological functions of Dock family members in development and disease.
Figure 1.

Dock GEF family. Dock proteins, subdivided into four subfamilies, are characterized by the evolutionarily conserved DHR-1, mediating binding to PIP3, and DHR-2, encompassing the GEF activity toward Rac/Cdc42 GTPases. The N terminus of Dock-A/B GEFs, including a SH3 domain, mediates their interaction with Elmo scaffolding proteins, while the C-terminal PxxP region coordinates interactions with SH3-containing adaptor proteins, such as Crk and Grb2. Dock-D members have a N-terminal-localized PH domain involved in phosphoinositide binding for membrane translocation. Several studies have identified that Dock GEFs are post-translationally modified by kinases and phosphatases. Of in vivo relevance, phosphorylation of DOCK1 (D1) on Y722, Y1811, or S1250 increases its GEF activity toward RAC and is elevated in brain cancers. Akt1 binds to Dock6 (D6) and phosphorylates its S1194 to inhibit its GEF activity; binding of Dock6 to the phosphatase Ppp2ca counteracts this inhibition through dephosphorylation.
Lipids and scaffolds: spatiotemporal activation of Docks
A new twist in Dock1/2 localization
Defining how Dock GEFs reach the membrane for GTPase activation is key to understanding how these proteins signal. The DHR-1 domain of Dock GEFs facilitates their recruitment to the membrane following PI3-kinase activation by directly binding to PIP3. A polybasic region (PBR) (see Fig. 1) in Dock1 and Dock2 was initially thought to bind PIP3, but more recent data suggest that it binds the signaling lipid phosphatidic acid (PA) (Kobayashi et al. 2001; Nishikimi et al. 2009; Sanematsu et al. 2013). Insight into the biological role of this second lipid-binding activity came from analyzing the cytokine-induced temporal production of these lipids in neutrophils; PIP3 is produced early, and PA production peaks soon after (Fig. 2A). Dock2 is critical for migration in these cells, and its rapid and broad recruitment at the site of cytokine receptor activation on the membrane is mediated by a direct DHR-1/PIP3 interaction. The ensuing cell polarization is dependent on phospholipase D-dependent production of PA, which serves to focus the recruitment of Dock2 to the pseudopod via its PBR (Nishikimi et al. 2009). Through such sequential lipid-binding events, Dock2 is optimally positioned to promote polarized neutrophil migration.
Figure 2.

Structural basis of Dock GEF localization and signaling. (A) When neutrophils sense a chemokine gradient, PIP3 is produced early following stimulation of the GPCR, and the DHR-1 domain of Dock2 facilitates its recruitment at the membrane. Neutrophil polarization is next dependent on phospholipase D-dependent production of PA, which serves to narrow the recruitment of Dock2 to the pseudopod via a PBR. (B) In fibroblasts, Pdgf treatment promotes a rapid PIP3-dependant recruitment of both Dock1 and Dock5 at the membrane. PA, produced sequentially to PIP3, narrows the localization of Dock1 to generate characteristic Pdgf-induced dorsal ruffles via Rac activation.
In fibroblasts, activation of the Pdgf receptor promotes two types of Rac-dependent membrane ruffles: Peripheral membrane ruffles are linked to cell migration, while dorsal ruffles are involved in invasion in a three-dimensional (3D) environment. Interestingly, generation of Rac-induced dorsal ruffles is dependent on the production of PA (Fig. 2B). Pdgf treatment of fibroblasts promotes a rapid DHR-1-dependent and PIP3-dependent recruitment of both Dock1 and Dock5 at the membrane, resulting in the activation of Rac1 to form peripheral ruffles. As in neutrophils, PA production lags behind that of PIP3 and acts to focus the localization of Dock1 (but not Dock5, as it lacks a PBR) at the membrane to generate the characteristic Pdgf-induced dorsal ruffles (Sanematsu et al. 2013). These data highlight the importance of localizing individual Dock family members for specific biological functions.
Elmo scaffolds orchestrate Dock-mediated Rac activation
Elmo proteins are binding partners of Dock-A/B, and formation of this complex is mandatory to achieve Rac-dependent cytoskeleton remodeling (Komander et al. 2008). Elmo positions Dock1 to discrete areas of cells to allow for polarized Rac activation. In the basal state, Elmo and Dock1 both exist in closed conformations even though they are physically associated (Fig. 3A, i; Lu et al. 2005; Patel et al. 2010, 2011b). Extracellular cues may sequentially release the autoinhibition constraints on Elmo followed by activation of Dock1 and Rac signaling (Fig. 3B; Patel et al. 2011b). It is not known whether other Dock-A/B GEFs are prebound to Elmo in the basal state. Recent data suggest that Elmo and Dock2 may exist as individual autoinhibited proteins that form a complex following stimulation to relieve both proteins from their inhibited state (Fig. 3A [ii], B; Hanawa-Suetsugu et al. 2012). The recruitment of the Elmo/Dock1 complex to the membrane is also guided by Elmo-interacting proteins (Fig. 3B). Activated GTPases of the Rho and Arf families, Rhog and Arl4a, use Elmo proteins as effectors by binding to the Ras-binding domain (RBD), and this contributes to both relieve Elmo autoinhibition and position the Elmo/Dock1 complex at the membrane for optimal Rac activation (Patel et al. 2010, 2011a). Elmo also directly interacts with the microtubule- and actin-binding spectraplakin Macf1 (also known as Acf7) (Margaron et al. 2013). Upon integrin (Itg) stimulation, cells expressing Elmo and Macf1 form long and persistent membrane protrusions. Recruitment of Elmo at the membrane can also occur following activation of the G-protein-coupled receptor (GPCR) Cxcr4, where the G protein Gαi2 promotes Rac activation and cell invasion in an Elmo/Dock1-dependent manner (Li et al. 2013).
Figure 3.

Elmo: a regulator of the spatiotemporal localization of Dock proteins. Elmo proteins bind Dock-A/B members. Recent studies highlight the key role played by Elmo in positioning Dock1 after cell stimulation. (A, i) At the basal state, Elmo and Dock1 are found in complex and are proposed to be autoinhibited by intramolecular interactions. (ii) Uncomplexed Elmo and Dock2 are also proposed to be autoinhibited, which is suggested to be released upon their interaction. (B) Upon cell stimulation, the recruitment of the Elmo/Dock1/2 complex at the membrane can be guided by Elmo's repertoire of interacting proteins. See the text for further details.
Dock GEFs in development
Rho GTPases regulate many essential processes during development, yet the full impact of their upstream regulation through GEFs is only starting to be appreciated (Heasman and Ridley 2008). Genetic screens in Caenorhabditis elegans and Drosophila have provided clues to the potential developmental functions of Dock GEFs in mammals through their regulation of cell migration, myogenesis, and clearance of apoptotic cells (for review, see Cote and Vuori 2007). Recent breakthroughs, summarized in Table 1, have resulted from the use of various vertebrate in vivo models to explore the biological functions of Dock GEFs in development and are discussed here.
Table 1.
In vivo models of Dock GEFs
Endothelial cells on the move: Dock1 and Elmo1 in cardiovascular development
The chemokine Cxcl12 and its receptor, Cxcr4, provide homing signals for at least two types of endothelial progenitors, but the effector pathways involved in this system have remained elusive (Tachibana et al. 1998; Sierro et al. 2007). Cxcl12 and Cxcr4 are expressed in developing endothelial cells, where they promote the cell migration essential for the establishment of cardiac valves and septa and the vascularization of the developing gastrointestinal tract (Tachibana et al. 1998; Sierro et al. 2007). In agreement with their expression patterns, genetic inactivation of either Cxcr4 or Cxcl12 in mice leads to cardiovascular defects characterized by aberrantly formed heart chambers and defective vascularization of the digestive tract. The characterization of two independent mutant mouse lines revealed an essential role for Dock1 downstream from Cxcr4 in endothelial cell migration (Table 1; Sanematsu et al. 2010). Inactivation of Dock1 is lethal at birth, and mice display severe edema as a consequence of ventricular septal defects, similar to what is observed in Cxcr4 mutants (Tachibana et al. 1998; Sanematsu et al. 2010). Endocardial cells derived from the explanted hearts of embryonic day 8.5 (E8.5) Dock1 mutant mice fail to invade Matrigel despite their ability to undergo morphological changes reminiscent of epithelial-to-mesenchymal transition, consistent with a central role for Dock1 in Cxcr4-dependant endocardial cell migration in vivo. Dock1 mutant animals also display the abnormal vascularization of the gastrointestinal system, as seen in Cxcr4 mutants. Rac1 activation, cytoskeletal changes, and cell migration are all impaired in cells explanted from Dock1 mutant mice following treatment with Cxcl12, while cell migration in response to Vegf remains unaffected, demonstrating a central signaling role for Dock1 downstream from Cxcr4 (Sanematsu et al. 2010). While a role for Dock1 orthologs in cell migration is well established in lower organisms, these studies are the first to uncover an in vivo contribution of this GEF to cell migration in mammals. It will be important to address the molecular mechanism that connects Dock1 to Cxcr4 receptor activation (Fig. 3B). Additional pathways provide guidance during vascular development, and among them, Netrin and the receptor Unc5b ensure the proper migration of endothelial cell progenitors, although the exact molecular connections in this pathway remain unresolved (Lu et al. 2004; Castets et al. 2009; Larrivee et al. 2009). Interestingly, silencing dock1 or elmo1 in zebrafish profoundly impairs vascular development, and a model in which Netrin acts as a chemoattractant through unc5b by dock1/elmo1-mediated activation of rac is suggested (Epting et al. 2010). These data challenge genetic studies defining unc5b as a repellant receptor in endothelial cells (Lu et al. 2004). Since Rac1 is critical for vascular development in mice, investigating the function of Elmo1 and Dock1 in Netrin-induced cardiac progenitor migration may reveal new pathways regulating this process. Collectively, these studies reveal a prominent role for Elmo1/Dock1 signaling downstream from different promigratory receptors in the modulation of Rac1 activation and migration in endothelial cells.
From attraction to retraction: Dock1 regulates Rac in neuronal axon guidance
Axon guidance is critical in brain development, and deregulation of this process may be implicated in certain mental diseases (Nugent et al. 2012). The growth cone is a specialized neuron compartment that integrates guidance signals (Vitriol and Zheng 2012). Netrin, a classical guidance cue, acts via the receptor Dcc to induce Rac- and Cdc42-dependent attraction of commissural axons toward the neural tube floor plate (Round and Stein 2007; Li et al. 2008). Dock1 is reported to colocalize with Dcc in the growth cone following treatment with Netrin in vitro (Fig. 4A). Loss of function of Dock1 leads to impaired axon outgrowth and abnormal commissural axon reorientation toward the guidance factor, demonstrating the important role of this GEF in Netrin-induced Rac activation (Li et al. 2008). These cell-based experiments also hold true in vivo, as electroporation of a siRNA targeting DOCK1 in the developing chicken neural tube causes misguidance of commissural axons (Table 1). These data provide a long-sought mechanism that connects Dcc activation to Rac-mediated axon outgrowth and attraction toward Netrin. However, axon guidance is not sufficient to ensure appropriate neuronal connections, and thus mechanisms to remove unwanted contacts, such as axon pruning, must also take place (Luo and O’Leary 2005). During development of the hippocampus, axonal remodeling occurs, and defects in this process are linked to behavioral diseases (Pittenger and Duman 2008; Ransome et al. 2012). In contrast to Netrin/Dcc-mediated attraction, ephrin B3 reverse signaling favors hippocampal axonal retraction-like pruning (Xu and Henkemeyer 2009). In this case, the adaptor protein Nck2 connects ephrin B3 to Dock1 and activation of Rac and its effector, Pak, to induce axon pruning (Fig. 4B). While this study sheds light on the first intracellular regulators of pruning, it is not clear why Dock1-mediated Rac activation would be required for axonal retraction. A recent study suggests that Sema3F, another receptor involved in hippocampal axonal pruning, also promotes Rac inactivation via the RacGAP β2-Chimaerin (Riccomagno et al. 2012). Understanding the spatial activation of Rac during pruning may clarify the role of the different pools of this GTPase in pruning. Nevertheless, while these studies exploited in vivo and in vitro models, it remains unknown whether commissural and hippocampal axons are misguided in Dock1-null mice. Other proteins controlling Rac signaling, the GAP Oligophrenin 1 and the effector PAK3, are linked to mental retardation syndromes, most likely through their functions on dendritic spine morphogenesis (Allen et al. 1998; Bergmann et al. 2003). Could abnormal DOCK1 and RAC signaling be at the root of some neuronal defects and mental disorders? Future behavioral studies in mice will certainly shed light on the importance of this pathway in more subtle aspects of brain development.
Figure 4.

Docks in neurogenesis. (A) In commissural neurons, Dock1 mediates Rac activation and promotes reorientation of the growth cone following activation of Dcc by the presence of a Netrin-1 gradient at the floor plate. (B) In the hippocampus, ephrin B3 mediates axon pruning. After their engagement to their receptor, ephrin B3 molecules become phosphorylated and recruit the adaptor protein Nck2, which is essential to relocalize Dock1 to the membrane and induce Rac activation to promote repulsion of the axon. (C) In hippocampal neurons, Ntrk2 RTK activation by Bdnf recruits Dock3 to the membrane in a Fyn kinase-dependent manner. Recruitment of Dock3 to the receptor is essential to promote Rac-dependent Bdnf-induced axon outgrowth. Dock3 also directly binds Wasf1 (also known as Wave1) to induce actin remodeling. Dock3 regulates the microtubule network through its interaction with Gsk3b and facilitates the phosphorylation of the kinase on an inhibitory site. Inhibition of Gsk3b leads to the dephosphorylation of Dpysl2 (also known as Crmp-2) and allows for microtubule elongation and axonal growth. (D) At the early developmental stages, Dock6 is found in complex with Ppp2ca. This interaction prevents phosphorylation of Dock6 on Ser1194 by Akt and allows axon growth. At the later developmental stages, the abundance of Akt increases. Akt interacts with Dock6 and phosphorylates Ser1194 to prevent axon growth. The phosphorylation of Dock6 downstream from Ngf activation of Ntrk1 requires PI3-kinase signaling. (E) During cell cycle progression, the interkinetic nuclear migration (INM) of the radial glial progenitors (RGCs) along the apical-to-basal axis contributes to cell fate decision after division. (Left) Dock7, via its binding to Tacc3, regulates the speed of INM. (Right) Down-regulation of Dock7 expression accelerates the basolateral-to-apical INM and leads to the proliferation of the RGC pool at the expense of a reduction in the number of basal progenitors.
Dock3 integrates actin and microtubule dynamics to promote axon outgrowth
Expression of Dock3 is largely restricted to neuronal tissues during embryogenesis, suggesting a role for this GEF in brain development (Kashiwa et al. 2000). Overexpression experiments using primary explanted hippocampal neurons suggest that Dock3 promotes axonal outgrowth by activating Rac1 (Namekata et al. 2010). Transgenic animals overexpressing Dock3 have been generated to test whether this also occurs in vivo (Table 1; Namekata et al. 2010). Global Dock3 overexpression (presumably at high levels in ganglion cells of the retina) stimulates some axonal outgrowth after optic nerve crushing, suggesting that Dock3 might be a useful target to stimulate nerve regeneration, although further experiments will be required to fully test this possibility. In hippocampal neurons, stimulation of axonal growth by activation of Ntrk2 receptor tyrosine kinase (RTK) with brain-derived neurotrophic factor (Bdnf) promotes Rac activation via the Fyn kinase-dependent recruitment of Dock3 to the membrane (Fig. 4C; Namekata et al. 2010). Dock3 also directly regulates actin remodeling following Bdnf stimulation by recruiting the actin nucleation-promoting factor Wasf1 through its DHR-1 domain (Fig. 4C). Whether the sole function of the DHR-1 of Dock3 is to bind Wasf1 or it also serves a membrane recruitment function through PIP3 binding has not been investigated. Dock3 also affects the microtubule network through an interaction with Gsk3b, a broad action kinase important in microtubule dynamics, which facilitates the phosphorylation of the kinase at an inhibitory site (Namekata et al. 2012). Consequently, this sequestering and inhibition of Gsk3b upon Bdnf treatment lead to the dephosphorylation of Dpysl2 (also known as Crmp-2) and allows this protein to promote microtubule elongation and axonal growth (Fig. 4C). While overexpression of Dock3 in mice is neuroprotective, knockout animals display axonal degeneration and impaired sensorimotor functions (Table 1; Chen et al. 2009). In vivo, Dock3 controls the activation of LIM domain-containing protein kinase (Limk1) and therefore the phosphorylation of its target, cofilin. Reduced activation of Limk1 has been proposed to mediate axonal dystrophy characterized by the accumulation of organelles, autophagic vacuoles, and disorganized cytoskeletons (Chen et al. 2009). Pharmacological treatments aimed to increase DOCK3 signaling could therefore be of therapeutic benefit for patients afflicted with spinal chord injuries that require axonal regeneration.
Tug of war between Akt and Ppp2ca: Dock6 is controlled by phosphorylation
The contribution of Dock7 to the differentiation of the neurites of hippocampal neurons into axons (Watabe-Uchida et al. 2006; Cote and Vuori 2007) raises the question of whether the closely related family member Dock6 could also be involved in axon specification. The additional finding that Dock6 is highly expressed in dorsal root ganglion neurons in vivo has prompted studies on its potential role in axon extension (Miyamoto et al. 2013). Transgenic mice expressing shRNAs specific to Dock6 show a reduction in the length of peripheral axons at E11, and these mice also fail to form neuronal fiber extension in an injury model in vivo (Table 1; Miyamoto et al. 2013). In agreement with in vivo observations, down-regulation of Dock6 expression in explanted dorsal root ganglion neurons impairs axon outgrowth and the extent of side branching. Likewise, knockdown of Rac1 decreases both axon length and the number of branch points, while Cdc42 knockdown has only a modest effect on axon outgrowth. These finding are in agreement with biochemical data suggesting that Dock6 is a functional Rac GEF in this system. Concomitant with developmental axon growth, Dock6 gradually becomes phosphorylated on Ser1194, and biochemical studies suggest a GEF inhibitory function for this modification. In a search for regulators of Dock6 phosphorylation, Akt1 was identified as a binding partner of the DHR-1 and was shown to directly phosphorylate Ser1194; in contrast, the phosphatase Ppp2ca has been shown to interact with and dephosphorylate the DHR-2 (Fig. 4D). Accordingly, at early developmental stages, when low levels of Akt1 are present in dorsal root ganglion neurons, Dock6 is in a complex with Ppp2ca. However, at later time points, when the axons have completed their migration, high Akt1 expression and binding to Dock6 correlate with Dock6 phosphorylation.
A GEF-less role: Dock7 controls interkinetic nuclear migration (INM) in neurogenesis
An important question in neurogenesis is what drives progenitor self-renewal versus differentiation. Dock7 is expressed in apical progenitors of the ventricular zone, and elegant experiments using in utero electroporation of these cells in mice have addressed its role in neurogenesis (Table 1; Yang et al. 2012b). Knockdown of Dock7 leads to an expansion of the radial glial progenitor (RGC) pool at the expense of basal progenitors and neurons (Fig. 4E). During cell cycle progression, RGC nuclei move along the apical-to-basal axis by a process termed INM. INM has been suggested to influence the balance between neurogenesis and progenitor pool maintenance by controlling the exposure time of RGC nuclei to neurogenic versus proliferative signals along the basal–apical axis (Taverna and Huttner 2010). In agreement with the phenotypes described above, down-regulation of Dock7 accelerates the basal-to-apical INM of RGCs, resulting in extended apical residency of RGC nuclei and apical mitoses. Conversely, ectopic Dock7 expression impedes basal-to-apical INM of RGCs, leading to extended residence of RGC nuclei at basal locations and mitoses at ectopic sites away from the ventricular surface. Surprisingly, structure/function assays suggest that Dock7 operates in a Rac-independent manner in this context. Instead, Dock7 directly interacts with Tacc3 to promote microtubule growth between the centrosome and the nucleus, providing a molecular explanation for the regulation of INM (Fig. 4E). By influencing RGC INM, Dock7 could be a key protein involved in cell fate decision during neurogenesis.
Unraveling the role of the Dock1 pathway in myoblast fusion
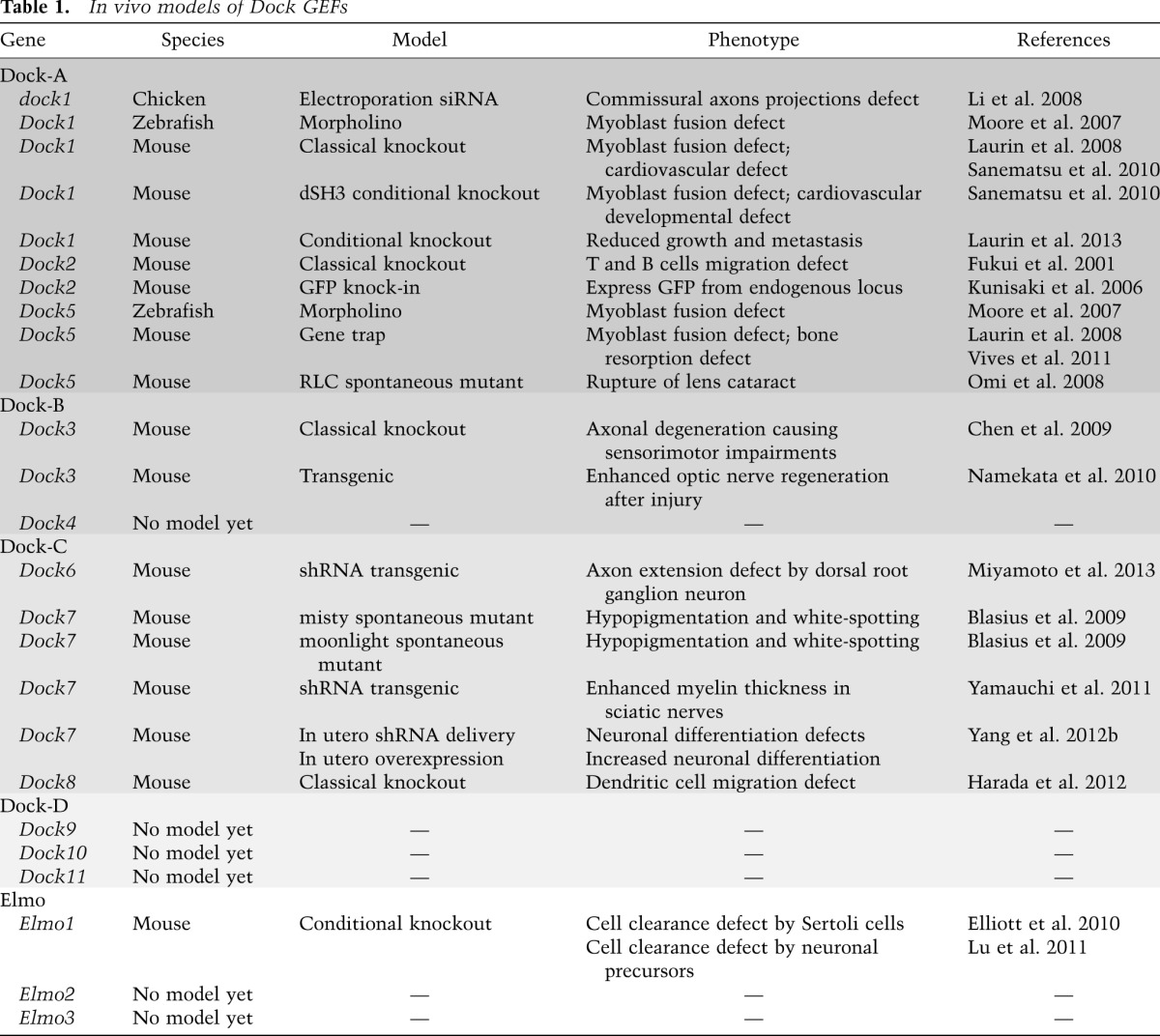
Successive rounds of myoblast fusion govern the formation of primary muscle fibers, yet this process is poorly understood at the molecular level in vertebrates (Abmayr and Pavlath 2012). Genetic screens in Drosophila uncovered cytoskeleton regulators, including myoblast city (mbc, ortholog of Dock1) and Rac, which specifically control the myoblast fusion step (Abmayr and Pavlath 2012). Mice with mutated Dock1 and Rac1 were generated to address whether this pathway plays a universal role in myoblast fusion. Dock1 mutants die at birth and are characterized by a strong block in primary myoblast fusion both in vivo and ex vivo (Table 1; Laurin et al. 2008). Likewise, muscle-specific inactivation of Rac1 severely impairs myoblast fusion (Vasyutina et al. 2009). Although the cell surface proteins promoting myoblast fusion in vertebrates are poorly characterized, two transmembrane proteins belonging to the GPCR family—Bai1 and Bai3, which are not found in Drosophila—promote myoblast fusion by engaging the Dock1/Rac pathway through a direct interaction with Elmo (Fig. 5A,B; Hochreiter-Hufford et al. 2013; Hamoud et al. 2014). The long intracellular tails of these GPCRs have a well-conserved motif that mediates Elmo binding by a mechanism that does not involve heterotrimeric G proteins (Park et al. 2007; Hochreiter-Hufford et al. 2013; Hamoud et al. 2014). Down-regulation of Bai3 in a myoblast cell line, C2C12, completely blocks myoblast fusion, and this phenotype can be rescued by re-expression of wild-type Bai3 but not by mutants unable to engage Elmo. In agreement with these data, uncoupling Bai3–Elmo interactions in vivo in muscle progenitors of chicken embryos also prevents myoblast fusion (Hamoud et al. 2014), and Bai1 overexpression increases fusion in C2C12 myoblasts in an Elmo-dependent manner (Hochreiter-Hufford et al. 2013). Mice lacking Bai1 have smaller muscle fibers and are less efficient at repairing injured muscle tissue than controls. Apoptotic cells themselves have been shown to act as ligands for Bai1 and indirectly promote cell fusion, thus providing a unique mechanism where tissue damage sensing and repair activities are coupled (Fig. 5B; Hochreiter-Hufford et al. 2013). These findings reveal the long sought-after vertebrate transmembrane proteins that engage the Dock1–Elmo–Rac pathway in primary myoblast fusion and regeneration. It remains to be determined whether heterotrimeric G proteins bound to Bai GPCRs have a role in engulfment and myoblast fusion. It has been reported that inhibiting Gαi with pertussis toxin does not prevent the myoblast fusion-promoting activity of Bai1 (Hochreiter-Hufford et al. 2013), but further studies are required to test whether other signaling subunits could be involved.
Figure 5.
The role of Dock GEFs in development. (A) Bai3 is expressed by myoblasts and is essential for myoblast fusion. Activation of Bai3 through an as yet to be determined mechanism and its interaction with Elmo are required for myoblast fusion. (B) Bai1’s ability to recognize phosphatidylserine exposed on apoptotic cells and interact with Elmo is proposed to trigger signaling that indirectly favors the fusion of myoblasts in order to help regenerate injured muscle tissue. (C) In osteoclasts, Dock5 is proposed to be essential downstream from Itg avb3 signaling to promote Bcar1 (also known as p130Cas) phosphorylation and Rac activation, which is essential for the formation and maturation of the sealing zone.
Bone resorption: Dock5 promotes osteoclast adhesion
Regulation of bone mass is controlled by the balanced activities of osteoblasts and osteoclasts (Teitelbaum and Ross 2003). Osteoclasts become multinucleated as a result of cell fusion and tightly adhere to bones to promote their resorption through the secretion of digestive enzymes (Teitelbaum and Ross 2003). The bone remodeling activity of osteoclasts is dependent on the proper assembly and disassembly of the sealing zone, an actin-rich ring structure generated by the association of multiple podosomes (Jurdic et al. 2006). In the absence of Dock5 expression, the sealing zone is poorly established, and the resorbing activity of Dock5-null osteoclasts is impaired (Vives et al. 2011). The phosphorylation of Bcar1 (also known as p130Cas), a central molecule in Itg signaling, is reduced in osteoclasts in the absence of Dock5 expression (Vives et al. 2011). One possibility is that Dock5 contributes to the formation of the sealing zone in osteoclasts by promoting Itgαvβ3 signaling via Bcar1 (p130Cas) phosphorylation and Rac activity (Fig. 5C; Nakamura et al. 1998, 2003; Elsegood et al. 2006; Vives et al. 2011). Interestingly, in osteoclasts lacking Bcar1, Dock5 fails to interact with Src kinases and Ptk2b (also known as Pyk2, a Fak family kinase), leading to defects in Rac activation (Nagai et al. 2013). These results suggest that Bcar1 is an important orchestrator of Rac signaling via Dock5 in osteoclasts. In vivo, Dock5 mutant mice display an increased trabecular bone mass, a symptom of improper bone resorption (Table 1). Pharmacological targeting of the Itgαvβ3/BCAR1/DOCK5 pathway could represent a novel avenue to counteract the osteoclastic activity in osteoporosis patients.
DOCK family in diseases
Through their regulation of the cell cytoskeletons, RHO GTPases orchestrate the ability of cancer cells to invade tissues and establish metastases (Alan and Lundquist 2013). During Drosophila oogenesis, border cells migrate collectively across the egg chamber and have been used as model to identify genes that possibly promote invasion (Montell et al. 2012). Such studies have identified the Drosophila PDGF/VEGF RTK, signaling through the Ced-12/mbc complex, as a promoter of border cell migration (Duchek et al. 2001). Based on these findings, several groups set out to test whether DOCK1 might play a similar role downstream from RTKs that have been amplified or mutated in human cancer. In this section, we only discuss the roles of DOCK1 in glioblastoma progression and breast cancer metastasis where the levels/activity of unmutated DOCK1 appear to increase to promote tumorigenesis. Recently, however, driver mutations in DOCK2 and ELMO1 have been reported in esophageal cancer, and in vitro studies suggest that these molecular lesions directly support cell migration/invasion (Dulak et al. 2013). Likewise, activating mutations in RAC1 have been identified and shown to drive some cancers, in particular melanocyte proliferation and invasion (Krauthammer et al. 2012; Kawazu et al. 2013). Clearly, with emerging genomic techniques, evidence of mutations in DOCK/ELMO/RAC proteins will continue to accumulate and will require careful investigation to understand their functional impact. Because they provide specificity in effector pathway activation, GEFs represent attractive therapeutic targets to block uncontrolled activity of RHO GTPases. In this section, we focus on emerging functions of DOCK GEFs in diseases.
The Adams-Oliver syndrome (AOS) and misregulation of CDC42/RAC: genetic mutations in DOCK6 and ARHGAP31
The AOS is an inherited heterogeneous disorder where patients are afflicted with a range of aplasia cutis congenita, terminal transverse limb defects, and other varying malformations (Whitley and Gorlin 1991). Recent exome sequencing of three unrelated AOS patients identified two mutations in DOCK6 and a truncating mutation in ARHGAP31 that may explain the molecular basis of this disease (Shaheen et al. 2011; Southgate et al. 2011). Homozygous mutations in DOCK6 correspond to a 4-base-pair (bp) deletion and a 1-bp duplication that create stop codons located upstream of the DHR-1 that would presumably result in truncated and GEF-dead proteins in patients. In addition to abnormal hands and feet, patients with DOCK6 mutations also exhibit microencephaly, supporting a possible function of DOCK6 during brain development. Fibroblasts isolated from these patients display severe cytoskeletal defects, with more cells being round and abnormally elongated while lacking lamellipodia. Interestingly, the mutation found in ARHGAP31 is proposed to truncate an autoinhibition fragment, thereby generating a constitutively active GAP for RAC/CDC42. These studies suggest that a reduction in RAC and/or CDC42 activities via either inactivation of a GEF or hyperactivation of a GAP might lead to the development of this disease. Further studies are required to determine whether abnormal cell migration in the absence of functional DOCK6 or activated ARHGAP31 is the primary cause of the OAS.
Amoeboid or mesenchymal movement? Dock GEFs tip the balance
Although metastatic cancer cells acquire the ability to become motile and escape from the primary tumor, individual cancer cells can migrate via either a mesenchymal or an amoeboid mode of migration (Friedl and Wolf 2003). During mesenchymal movement, cells adopt typical Rac phenotypes, including a polarized morphology and a leading edge with active membrane ruffles (Friedl and Wolf 2003). In contrast, amoeboid migration is characterized by round cells with plasma membrane blebbing that is dependent on RhoA signaling (Friedl and Wolf 2003). These modes are interconvertible and are exploited by cancer cells to adapt to their microenvironment. While Rho GTPases are important players in the choice between these types of movement, the GEFs that orchestrate their signaling have remained elusive. Elegant RNAi screens in melanoma cells have implicated two Dock GEFs, Dock3 and Dock10, as central regulators of mesenchymal and amoeboid migration (Fig. 6; Gadea et al. 2008; Sanz-Moreno et al. 2008). The melanoma cell line A375M2 can switch between mesenchymal and amoeboid morphologies when grown in 3D matrices. Silencing Dock3 in these cells enriches for a population that migrates in an amoeboid manner. During mesenchymal movement, the formation of a complex between the scaffold protein Nedd9 (also known as Hef1) and Dock3 is required to orchestrate Rac activation and signaling to the effector protein Wasf2 (also known as Wave2), which negatively regulates the levels of phospho-myosin light chain-2 (p-MLC2), therefore decreasing actomyosin contractility (Fig. 6). In contrast, Rock signaling, which promotes amoeboid migration, represses mesenchymal migration by promoting the inactivation of Rac1 via the Rac-specific Arhgap22 protein (Gadea et al. 2008; Sanz-Moreno et al. 2008).
Figure 6.
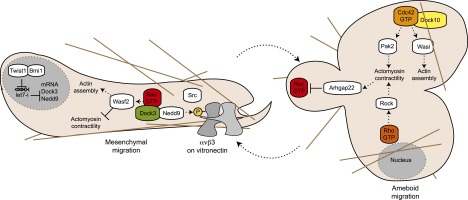
Dock's and cancer cell’s migrating mode. During mesenchymal movement, Nedd9 (also known as Hef1) and Dock3 orchestrate Rac activation and signaling to the effector protein Wasf2 (also known as Wave2), which negatively regulates the levels of p-MLC. Twist1 cooperates with Bmi1 to repress the expression of let-7i, a miRNA targeting Nedd9 and Dock3, and thereby regulates their level of expression. In contrast, Rock signaling promotes amoeboid migration by promoting the inactivation of Rac1 via the RacGAP Arhgap22. Moreover, Dock10 promotes amoeboid migration by promoting the activation of Cdc42 and its downstream effectors, Wasl (also known as N-Wasp) and Pak2.
In a cellular model of head and neck cancer, a master regulator of epithelial-to-mesenchymal transition, Twist1, regulates the level of Nedd9 and Dock3 expression to promote Rac-dependent mesenchymal migration (Yang et al. 2012a). Twist1 cooperates with Bmi1, a polycomb group family member, to repress the expression of let-7i, an miRNA targeting Nedd9 and Dock3, therefore facilitating the epithelial-to-mesenchymal transition. Silencing of Dock10 in melanoma cells grown in a 3D matrix reduces the levels of activated Cdc42 and p-MLC and converts round cells into mesenchymal cells (Fig. 6; Gadea et al. 2008). Consistent with a role of Dock10 in promoting amoeboid migration, overexpression of the Dock10 DHR-2 domain leads to an increase in the amount of round cells, while silencing two of the Cdc42 effectors, Wasl (also known as N-Wasp) and Pak2, favors elongated cells. Overall, these studies highlight the existence of signaling pathways that promote one type of migration while at the same time supporting a repression of the alternative mode.
DOCK1 regulates growth and invasion of glioblastomas
Amplification of the PDGFRA or EGFR locus defines subclasses of glioblastomas that correlate with poor prognosis (Van Meir et al. 2010). In experiments aimed at identifying the molecular pathways that mediate tumor spreading, high expression of DOCK1 and ELMO1 proteins has been observed in invasive areas of glioblastoma tissue sections (Jarzynka et al. 2007). In glioblastoma cell lines overexpressing PDGFRA ligand or EGFRvIII, the suppression of DOCK1 expression prevents cell migration and AKT, ERK1/2, and RAC1 activation (Feng et al. 2011, 2012). When PDGFRA ligand-expressing cells are injected in vivo, they exhibit a deficit in proliferation and invasion of the brain when DOCK1 expression is abrogated, suggesting that DOCK1 is an important downstream effector of this RTK (Feng et al. 2011). Both oncogenic kinases activate DOCK1 by inducing its tyrosine phosphorylation. PDGFRA promotes SRC kinase-dependent phosphorylation of DOCK1 on Y1811 to increase its affinity for RAC and promote GTP loading (Fig. 7A). In vivo, glioblastoma cells expressing PDGFRA ligand fail to invade when a DOCK1Y1811F mutant replaces endogenous DOCK1 (Feng et al. 2011). Patient survival has also been reported to decrease significantly when tumors are positive for both pDOCK1Y1811 and PDGFRA. Similarly, oncogenic EGFRvIII promotes the phosphorylation of DOCK1 on two different sites: Y722 through SFK kinases and S1250 via protein kinase A (Fig. 7B; Feng et al. 2012, 2013). Phosphorylation of both of these sites is required for function, since expression of either DOCK1Y722F or DOCK1S1250A prevents growth and invasion of EGFRvIII-overexpressing glioblastoma cells when injected in the brains of mice (Feng et al. 2012, 2013). Interestingly, down-regulation of DOCK1 in glioblastoma cells significantly impairs their ability to proliferate when injected in the brain, suggesting that this might be an ideal model to dissect the contribution of DOCK1 and RAC1 to tumor growth. Blocking this pathway in patients could also be of interest, as it could limit both growth and dissemination.
Figure 7.

DOCK1 in cancer. Several studies have showed that DOCK1 is phosphorylated downstream from oncogenic RTKs. (A) In glioblastoma cells, activation of the PDGFRA by its ligand promotes the phosphorylation of DOCK1 at Y1811 through SRC family kinases. Phosphorylation of DOCK1 is critical for its interaction with CRK and BCAR1 (also known as p130CAS) and to induce RAC1 activation. In these cells, DOCK1 is also essential for AKT and ERK1/2 activation mediated by PDGFRA. (B) In glioblastoma cells, overexpression EGFRvIII induces the phosphorylation of DOCK1 at Y722 via SRC family kinases and at S1250 via PKA. DOCK1 is essential to promote RAC1, AKT, and ERK1/2 activation mediated by oncogenic EGFRvIII. (C) HER2 activation or overexpression of its oncogenic form induces phosphorylation of DOCK1 at the Y1811 through the SRC kinase in breast cancer cells.
DOCK1 regulates breast cancer metastasis
Mining of genomic data reveals a correlation between high levels of DOCK1 mRNA expression and poor prognosis for cancer patients afflicted with either HER2-positive or basal breast cancers, the two most invasive subtypes of this disease (Laurin et al. 2013). The RTK HER2 is amplified or overexpressed in 20%–30% of breast cancers (Slamon et al. 1987). In breast cancer cell lines, both oncogenic HER2 and activation of endogenous HER2-containing heterodimers by the ligand Heregulin β1 promote DOCK1 phosphorylation on its positive regulatory site, Y1811 (Fig. 7C; Laurin et al. 2013). Either pharmacological inhibition or knockdown of DOCK1 expression in breast cancer cell lines reveals an essential role for this GEF in HER2-mediated RAC activation and migration. The effector pathways of HER2 that promote metastasis in vivo, however, remain incompletely defined. To address whether DOCK1 is a mediator of invasion in vivo, Dock1 was conditionally deleted in mammary epithelial cells in a murine Her2 breast cancer model that mimics the human disease, including metastasis (Table 1; Ursini-Siegel et al. 2007, 2008). Genetic ablation of Dock1 in Her2 transgenic mammary glands reduces the total tumor burden per animal and protects mice from developing lung metastases. Gene expression profiling of Her2 mammary tumors identifies a gene signature under the control of Dock1 that is enriched in genes responding to type I interferon. As some of these genes correlate with poor survival in HER2+ patients, further analysis will be required to understand their contribution to cancer progression. These findings demonstrate that HER2 exploits the DOCK1/RAC module in breast cancer progression to metastasis and further emphasize the central role played by this signaling pathway downstream from RTKs in various forms of tumor progression and metastasis. These studies also suggest that inhibition of DOCK1 signaling could be a promising avenue for adjunct therapy in invasive cancers.
Future directions
In this review, we highlighted novel biological functions carried out by Dock family members. Several Dock GEFs remain to be fully characterized at the biochemical and cell biology levels, and the generation of new in vivo mutant models will be essential to clarify their roles in development and diseases. In particular, the biochemical activities of Dock-C/D GEFs are less well characterized and appear to preferentially activate prenylated and membrane-localized Rac or Cdc42 (Meller et al. 2002; Zhou et al. 2013). These GEFs may also generate a positive feedback loop whereby their activated target (for example, Cdc42-GTP) can bind elsewhere on the Dock protein to sustain its activity (Lin et al. 2006; Zhou et al. 2013). Investigating the structure of these proteins might help to explain how such regulation is occurring. The biological functions of the Dock-C/D family members are also poorly defined in mammals and other model organisms. Recent studies have emphasized the central role played by Dock GEFs in the control of cytoskeletal dynamics. Several Dock proteins, including Dock1, Dock3, and Dock7, are positioned at the interface between the actin and microtubule networks by interacting with various types of microtubule regulators, and a more complete map of these interactomes is needed to clarify this complex cross-talk.
The role of Dock GEFs in migration in vivo is also poorly explored. Two independent spontaneous mouse mutant lines, misty and moonlight (Table 1), were identified due to a hypopigmented and white-spotted phenotype resulting from genomic deletions in the Dock7 locus (Blasius et al. 2009). Surprisingly, the cellular and molecular bases for the hypopigmentation have not yet been addressed in these models; an attractive hypothesis would be that Dock7 is required for melanocyte progenitor differentiation or their migration from the neural crest to the ectoderm (Cichorek et al. 2013).
For more than a decade, Dock1, Dock2, and Dock5 have been viewed as central signaling intermediates promoting Rac activation downstream from Itgs. While Crk adaptors are considered central to the recruitment of Dock1 to Itg signaling complexes, the exact mechanisms controlling Dock1 signaling downstream from these adhesion receptors remain poorly understood. Are Elmo scaffolds needed for Itg-mediated Rac activation? Given the central role of Itg signaling in motility and invasion, understanding how Dock proteins connect to these receptors will be needed to assess the importance of this molecular connection in tumorigenesis.
Recent studies have also expanded the repertoire of membrane receptors acting upstream of Dock1. Notably, several RTKs and GPCRs take advantage of Dock1 to promote migration, invasion, phagocytosis, and myoblast fusion. In contrast to the Itg signaling pathways, the molecular connections between Dock1 and these receptors are being characterized in detail. While Dock1 acts downstream from the GPCR Cxcr4 during endothelial cell migration, this receptor is also implicated in an abundance of developmental processes and diseases. Could Dock1 be contributing to other aspects of Cxcr4 signaling? CXCR4 regulates the homing of cancer cells during metastasis, and DOCK1 acts downstream from oncogenic RTKs during cancer invasion. An attractive hypothesis would be that DOCK1 integrates signaling by these two families of receptors, and probing in vivo models of cancer where DOCK1 is uncoupled from Gαi2 would be a powerful means to investigate the importance of the CXCR4/DOCK1 axis in cancer progression.
Another emerging theme is the complex regulation of Dock family members by phosphorylation. As observed for the control of Dock6 activity, the complicity between a kinase and a phosphatase in the regulation of Dock proteins provides a mechanism for spatiotemporal control of their functions. It will be essential to better understand the role of this post-translational modification in in vivo models. Does it modulate the ability of Dock proteins to interact with other protein partners, or does it instead affect its binding to GTPases by inducing conformational changes?
Finally, Dock GEFs are large proteins in which only the DHR-1 and DHR-2 domains have been thoroughly characterized, and it seems very likely that other regions in these GEFs will provide scaffolding or regulatory functions during signaling. As we highlighted in this review, Dock7 can regulate neuronal differentiation in a GEF-independent manner, and, as such, it is possible that other Dock family members also play important biological functions that are independent of their GEF activity. Mapping the interactome of Dock family proteins may therefore reveal novel pathways regulated by these GEFs.
Acknowledgments
We thank Dr. Michel Cayouette and Dr. Manishha Patel for critical reading of the manuscript. We apologize to colleagues whose work could not be cited due to space constraints. The work in J.F.C’s laboratory is funded by the following agencies: Canadian Institute of Health Research, Canadian Cancer Society, Quebec Breast Cancer Foundation, and the Cancer Research Society. J.-F.C holds a Senior Investigator Award from the Fonds de Recherche du Québec-Santé (FRQ-S).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.236349.113.
References
- Abmayr SM, Pavlath GK 2012. Myoblast fusion: lessons from flies and mice. Development 139: 641–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alan JK, Lundquist EA 2013. Mutationally activated Rho GTPases in cancer. Small GTPases 4: 159–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA 1998. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet 20: 25–30 [DOI] [PubMed] [Google Scholar]
- Bergmann C, Zerres K, Senderek J, Rudnik-Schoneborn S, Eggermann T, Hausler M, Mull M, Ramaekers VT 2003. Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain 126: 1537–1544 [DOI] [PubMed] [Google Scholar]
- Blasius AL, Brandl K, Crozat K, Xia Y, Khovananth K, Krebs P, Smart NG, Zampolli A, Ruggeri ZM, Beutler BA 2009. Mice with mutations of Dock7 have generalized hypopigmentation and white-spotting but show normal neurological function. Proc Natl Acad Sci 106: 2706–2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan BA, Li D, Wu X, Liu M 2005. The Rho family of small GTPases: crucial regulators of skeletal myogenesis. Cell Mol Life Sci 62: 1547–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancelas JA, Williams DA 2009. Rho GTPases in hematopoietic stem cell functions. Curr Opin Hematol 16: 249–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castets M, Coissieux MM, Delloye-Bourgeois C, Bernard L, Delcros JG, Bernet A, Laudet V, Mehlen P 2009. Inhibition of endothelial cell apoptosis by netrin-1 during angiogenesis. Dev Cell 16: 614–620 [DOI] [PubMed] [Google Scholar]
- Chen Q, Peto CA, Shelton GD, Mizisin A, Sawchenko PE, Schubert D 2009. Loss of modifier of cell adhesion reveals a pathway leading to axonal degeneration. J Neurosci 29: 118–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichorek M, Wachulska M, Stasiewicz A, Tyminska A 2013. Skin melanocytes: biology and development. Postepy Dermatol Alergol 30: 30–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote JF, Vuori K 2002. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J Cell Sci 115: 4901–4913 [DOI] [PubMed] [Google Scholar]
- Cote JF, Vuori K 2006. In vitro guanine nucleotide exchange activity of DHR-2/DOCKER/CZH2 domains. Methods Enzymol 406: 41–57 [DOI] [PubMed] [Google Scholar]
- Cote JF, Vuori K 2007. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol 17: 383–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P 2001. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell 107: 17–26 [DOI] [PubMed] [Google Scholar]
- Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, et al. 2013. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 45: 478–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, Zheng S, Park D, Woodson RI, Reardon MA, Juncadella IJ, Kinchen JM, Zhang J, Lysiak JJ, Ravichandran KS 2010. Unexpected requirement for ELMO1 in clearance of apoptotic germ cells in vivo. Nature 467: 333–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsegood CL, Zhuo Y, Wesolowski GA, Hamilton JA, Rodan GA, Duong LT 2006. M-CSF induces the stable interaction of cFms with αVβ3 integrin in osteoclasts. Int J Biochem Cell Biol 38: 1518–1529 [DOI] [PubMed] [Google Scholar]
- Epting D, Wendik B, Bennewitz K, Dietz CT, Driever W, Kroll J 2010. The Rac1 regulator ELMO1 controls vascular morphogenesis in zebrafish. Circ Res 107: 45–55 [DOI] [PubMed] [Google Scholar]
- Feng H, Hu B, Liu KW, Li Y, Lu X, Cheng T, Yiin JJ, Lu S, Keezer S, Fenton T, et al. 2011. Activation of Rac1 by Src-dependent phosphorylation of Dock180(Y1811) mediates PDGFRα-stimulated glioma tumorigenesis in mice and humans. J Clin Invest 121: 4670–4684 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Feng H, Hu B, Jarzynka MJ, Li Y, Keezer S, Johns TG, Tang CK, Hamilton RL, Vuori K, Nishikawa R, et al. 2012. Phosphorylation of dedicator of cytokinesis 1 (Dock180) at tyrosine residue Y722 by Src family kinases mediates EGFRvIII-driven glioblastoma tumorigenesis. Proc Natl Acad Sci 109: 3018–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Hu B, Vuori K, Sarkaria JN, Furnari FB, Cavenee WK, Cheng SY 2013. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene 10.1038/onc.2013.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl P, Wolf K 2003. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3: 362–374 [DOI] [PubMed] [Google Scholar]
- Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, Inayoshi A, Noda M, Oike M, Shirai T, et al. 2001. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature 412: 826–831 [DOI] [PubMed] [Google Scholar]
- Gadea G, Sanz-Moreno V, Self A, Godi A, Marshall CJ 2008. DOCK10-mediated Cdc42 activation is necessary for amoeboid invasion of melanoma cells. Curr Biol 18: 1456–1465 [DOI] [PubMed] [Google Scholar]
- Hall A, Lalli G 2010. Rho and Ras GTPases in axon growth, guidance, and branching. Cold Spring Harb Perspect Biol 2: a001818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamoud N, Tran V, Croteau L-P, Kania A, Côté J-F 2014. G-protein coupled receptor BAI3 controls myoblast fusion in vertebrates. Proc Natl Acad Sci 10.1073/pnas.1313886111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawa-Suetsugu K, Kukimoto-Niino M, Mishima-Tsumagari C, Akasaka R, Ohsawa N, Sekine S, Ito T, Tochio N, Koshiba S, Kigawa T, et al. 2012. Structural basis for mutual relief of the Rac guanine nucleotide exchange factor DOCK2 and its partner ELMO1 from their autoinhibited forms. Proc Natl Acad Sci 109: 3305–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada Y, Tanaka Y, Terasawa M, Pieczyk M, Habiro K, Katakai T, Hanawa-Suetsugu K, Kukimoto-Niino M, Nishizaki T, Shirouzy M, et al. 2012. DOCK8 is a Cdc42 activator critical for interstitial dendritic cell migration during immune responses. Blood 119: 4451–4461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MJ, Eva A, Evans T, Aaronson SA, Cerione RA 1991. Catalysis of guanine nucleotide exchange on the CDC42Hs protein by the dbl oncogene product. Nature 354: 311–314 [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701 [DOI] [PubMed] [Google Scholar]
- Hochreiter-Hufford AE, Lee CS, Kinchen JM, Sokolowski JD, Arandjelovic S, Call JA, Klibanov AL, Yan Z, Mandell JW, Ravichandran KS 2013. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature 497: 263–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarzynka MJ, Hu B, Hui KM, Bar-Joseph I, Gu W, Hirose T, Haney LB, Ravichandran KS, Nishikawa R, Cheng SY 2007. ELMO1 and Dock180, a bipartite Rac1 guanine nucleotide exchange factor, promote human glioma cell invasion. Cancer Res 67: 7203–7211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurdic P, Saltel F, Chabadel A, Destaing O 2006. Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol 85: 195–202 [DOI] [PubMed] [Google Scholar]
- Kashiwa A, Yoshida H, Lee S, Paladino T, Liu Y, Chen Q, Dargusch R, Schubert D, Kimura H 2000. Isolation and characterization of novel presenilin binding protein. J Neurochem 75: 109–116 [DOI] [PubMed] [Google Scholar]
- Kawazu M, Ueno T, Kontani K, Ogita Y, Ando M, Fukumura K, Yamato A, Soda M, Takeuchi K, Miki Y, et al. 2013. Transforming mutations of RAC guanosine triphosphatases in human cancers. Proc Natl Acad Sci 110: 3029–3034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Shirai T, Kiyokawa E, Mochizuki N, Matsuda M, Fukui Y 2001. Membrane recruitment of DOCK180 by binding to PtdIns(3,4,5)P3. Biochem J 354: 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Patel M, Laurin M, Fradet N, Pelletier A, Barford D, Cote JF 2008. An α-helical extension of the ELMO1 pleckstrin homology domain mediates direct interaction to DOCK180 and is critical in Rac signaling. Mol Biol Cell 19: 4837–4851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. 2012. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44: 1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisaki Y, Nishikimi A, Tanaka Y, Takii R, Noda M, Inayoshi A, Watanabe K, Sanematsu F, Sasazuki T, Sasaki T, et al. 2006. DOCK2 is a Rac activator that regulates motility and polarity during neutrophil chemotaxis. J Cell Biol 174: 647–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrivee B, Freitas C, Suchting S, Brunet I, Eichmann A 2009. Guidance of vascular development: lessons from the nervous system. Circ Res 104: 428–441 [DOI] [PubMed] [Google Scholar]
- Laurin M, Fradet N, Blangy A, Hall A, Vuori K, Cote JF 2008. The atypical Rac activator Dock180 (Dock1) regulates myoblast fusion in vivo. Proc Natl Acad Sci 105: 15446–15451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin M, Huber J, Pelletier A, Houalla T, Park M, Fukui Y, Haibe-Kains B, Muller WJ, Cote JF 2013. Rac-specific guanine nucleotide exchange factor DOCK1 is a critical regulator of HER2-mediated breast cancer metastasis. Proc Natl Acad Sci 110: 7434–7439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazer G, Katzav S 2011. Guanine nucleotide exchange factors for RhoGTPases: good therapeutic targets for cancer therapy? Cell Signal 23: 969–979 [DOI] [PubMed] [Google Scholar]
- Li X, Gao X, Liu G, Xiong W, Wu J, Rao Y 2008. Netrin signal transduction and the guanine nucleotide exchange factor DOCK180 in attractive signaling. Nat Neurosci 11: 28–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yang L, Fu H, Yan J, Wang Y, Guo H, Hao X, Xu X, Jin T, Zhang N 2013. Association between Gαi2 and ELMO1/Dock180 connects chemokine signalling with Rac activation and metastasis. Nat Commun 4: 1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q, Yang W, Baird D, Feng Q, Cerione RA 2006. Identification of a DOCK180-related guanine nucleotide exchange factor that is capable of mediating a positive feedback activation of Cdc42. J Biol Chem 281: 35253–35262 [DOI] [PubMed] [Google Scholar]
- Lu X, Le Noble F, Yuan L, Jiang Q, De Lafarge B, Sugiyama D, Breant C, Claes F, De Smet F, Thomas JL, et al. 2004. The netrin receptor UNC5B mediates guidance events controlling morphogenesis of the vascular system. Nature 432: 179–186 [DOI] [PubMed] [Google Scholar]
- Lu M, Kinchen JM, Rossman KL, Grimsley C, Hall M, Sondek J, Hengartner MO, Yajnik V, Ravichandran KS 2005. A Steric-inhibition model for regulation of nucleotide exchange via the Dock180 family of GEFs. Curr Biol 15: 371–377 [DOI] [PubMed] [Google Scholar]
- Lu Z, Elliott MR, Chen Y, Walsh JT, Klibanov AL, Ravichandran KS, Kipnis J 2011. Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat Cell Biol 13: 1076–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, O’Leary DD 2005. Axon retraction and degeneration in development and disease. Annu Rev Neurosci 28: 127–156 [DOI] [PubMed] [Google Scholar]
- Margaron Y, Fradet N, Cote JF 2013. ELMO recruits actin cross-linking family 7 (ACF7) at the cell membrane for microtubule capture and stabilization of cellular protrusions. J Biol Chem 288: 1184–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller N, Irani-Tehrani M, Kiosses WB, Del Pozo MA, Schwartz MA 2002. Zizimin1, a novel Cdc42 activator, reveals a new GEF domain for Rho proteins. Nat Cell Biol 4: 639–647 [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Torii T, Yamamori N, Ogata T, Tanoue A, Yamauchi J 2013. Akt and PP2A reciprocally regulate the guanine nucleotide exchange factor Dock6 to control axon growth of sensory neurons. Sci Signal 6: ra15. [DOI] [PubMed] [Google Scholar]
- Montell DJ, Yoon WH, Starz-Gaiano M 2012. Group choreography: mechanisms orchestrating the collective movement of border cells. Nat Rev Mol Cell Biol 13: 631–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CA, Parkin CA, Bidet Y, Ingham PW 2007. A role for the Myoblast city homologues Dock1and Dock5 and the adaptor proteins Crk and Crk-like in zebrafish myoblast fusion. Development 134: 3145–3153 [DOI] [PubMed] [Google Scholar]
- Nagai Y, Osawa K, Fukushima H, Tamura Y, Aoki K, Ohya K, Yasuda H, Hikiji H, Takahashi M, Seta Y, et al. 2013. p130Cas plays important roles in osteoclastic bone resorption. J Bone Miner Res 28: 2449–2462 [DOI] [PubMed] [Google Scholar]
- Nakamura I, Jimi E, Duong LT, Sasaki T, Takahashi N, Rodan GA, Suda T 1998. Tyrosine phosphorylation of p130Cas is involved in actin organization in osteoclasts. J Biol Chem 273: 11144–11149 [DOI] [PubMed] [Google Scholar]
- Nakamura I, Rodan GA, Duong LT 2003. Distinct roles of p130Cas and c-Cbl in adhesion-induced or macrophage colony-stimulating factor-mediated signaling pathways in prefusion osteoclasts. Endocrinology 144: 4739–4741 [DOI] [PubMed] [Google Scholar]
- Namekata K, Harada C, Taya C, Guo X, Kimura H, Parada LF, Harada T 2010. Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc Natl Acad Sci 107: 7586–7591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekata K, Harada C, Guo X, Kimura A, Kittaka D, Watanabe H, Harada T 2012. Dock3 stimulates axonal outgrowth via GSK-3β-mediated microtubule assembly. J Neurosci 32: 264–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikimi A, Fukuhara H, Su W, Hongu T, Takasuga S, Mihara H, Cao Q, Sanematsu F, Kanai M, Hasegawa H, et al. 2009. Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science 324: 384–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikimi A, Kukimoto-Niino M, Yokoyama S, Fukui Y 2013. Immune regulatory functions of DOCK family proteins in health and disease. Exp Cell Res 319: 2343–2349 [DOI] [PubMed] [Google Scholar]
- Nugent AA, Kolpak AL, Engle EC 2012. Human disorders of axon guidance. Curr Opin Neurobiol 22: 837–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omi N, Kiyokawa E, Matsuda M, Kinoshita K, Yamada S, Yamada K, Matsushima Y, Wang Y, Kawai J, Suzuki M, et al. 2008. Mutation of Dock5, a member of the guanine exchange factor Dock180 superfamily, in the rupture of lens cataract mouse. Exp Eye Res 86: 828–834 [DOI] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS 2007. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450: 430–434 [DOI] [PubMed] [Google Scholar]
- Patel M, Margaron Y, Fradet N, Yang Q, Wilkes B, Bouvier M, Hofmann K, Cote JF 2010. An evolutionarily conserved autoinhibitory molecular switch in ELMO proteins regulates Rac signaling. Curr Biol 20: 2021–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Chiang TC, Tran V, Lee FJ, Cote JF 2011a. The Arf family GTPase Arl4A complexes with ELMO proteins to promote actin cytoskeleton remodeling and reveals a versatile Ras-binding domain in the ELMO proteins family. J Biol Chem 286: 38969–38979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, Pelletier A, Cote JF 2011b. Opening up on ELMO regulation: new insights into the control of Rac signaling by the DOCK180/ELMO complex. Small GTPases 2: 268–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Duman RS 2008. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology 33: 88–109 [DOI] [PubMed] [Google Scholar]
- Ransome MI, Renoir T, Hannan AJ 2012. Hippocampal neurogenesis, cognitive deficits and affective disorder in Huntington’s disease. Neural Plast 2012: 874387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccomagno MM, Hurtado A, Wang H, Macopson JG, Griner EM, Betz A, Brose N, Kazanietz MG, Kolodkin AL 2012. The RacGAP β2-Chimaerin selectively mediates axonal pruning in the hippocampus. Cell 149: 1594–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Zannini M, Lewis M, Wickner RB, Hunt LT, Graziani G, Tronick SR, Aaronson SA, Eva A 1991. A region of proto-dbl essential for its transforming activity shows sequence similarity to a yeast cell cycle gene, CDC24, and the human breakpoint cluster gene, bcr. New Biol 3: 372–379 [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J 2005. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6: 167–180 [DOI] [PubMed] [Google Scholar]
- Round J, Stein E 2007. Netrin signaling leading to directed growth cone steering. Curr Opin Neurobiol 17: 15–21 [DOI] [PubMed] [Google Scholar]
- Sanematsu F, Hirashima M, Laurin M, Takii R, Nishikimi A, Kitajima K, Ding G, Noda M, Murata Y, Tanaka Y, et al. 2010. DOCK180 is a Rac activator that regulates cardiovascular development by acting downstream of CXCR4. Circ Res 107: 1102–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanematsu F, Nishikimi A, Watanabe M, Hongu T, Tanaka Y, Kanaho Y, Cote JF, Fukui Y 2013. Phosphatidic acid-dependent recruitment and function of the Rac activator DOCK1 during dorsal ruffle formation. J Biol Chem 288: 8092–8100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ 2008. Rac activation and inactivation control plasticity of tumor cell movement. Cell 135: 510–523 [DOI] [PubMed] [Google Scholar]
- Shaheen R, Faqeih E, Sunker A, Morsy H, Al-Sheddi T, Shamseldin HE, Adly N, Hashem M, Alkuraya FS 2011. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am J Hum Genet 89: 328–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H, Groom J, Batten M, et al. 2007. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci 104: 14759–14764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL 1987. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235: 177–182 [DOI] [PubMed] [Google Scholar]
- Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, Branney PA, Fisher M, Lee GJ, Simpson MA, et al. 2011. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet 88: 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y, Kitamura Y, Matsushima K, Yoshida N, Nishikawa S, et al. 1998. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393: 591–594 [DOI] [PubMed] [Google Scholar]
- Taverna E, Huttner WB 2010. Neural progenitor nuclei IN motion. Neuron 67: 906–914 [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP 2003. Genetic regulation of osteoclast development and function. Nat Rev Genet 4: 638–649 [DOI] [PubMed] [Google Scholar]
- Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ 2007. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer 7: 389–397 [DOI] [PubMed] [Google Scholar]
- Ursini-Siegel J, Hardy WR, Zuo D, Lam SH, Sanguin-Gendreau V, Cardiff RD, Pawson T, Muller WJ 2008. ShcA signalling is essential for tumour progression in mouse models of human breast cancer. EMBO J 27: 910–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ 2010. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 60: 166–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasyutina E, Martarelli B, Brakebusch C, Wende H, Birchmeier C 2009. The small G-proteins Rac1 and Cdc42 are essential for myoblast fusion in the mouse. Proc Natl Acad Sci 106: 8935–8940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitriol EA, Zheng JQ 2012. Growth cone travel in space and time: the cellular ensemble of cytoskeleton, adhesion, and membrane. Neuron 73: 1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vives V, Laurin M, Cres G, Larrousse P, Morichaud Z, Noel D, Cote JF, Blangy A 2011. The Rac1 exchange factor Dock5 is essential for bone resorption by osteoclasts. J Bone Miner Res 26: 1099–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, John KA, Janas JA, Newey SE, Van Aelst L 2006. The Rac activator DOCK7 regulates neuronal polarity through local phosphorylation of stathmin/Op18. Neuron 51: 727–739 [DOI] [PubMed] [Google Scholar]
- Whitley CB, Gorlin RJ 1991. Adams-Oliver syndrome revisited. Am J Med Genet 40: 319–326 [DOI] [PubMed] [Google Scholar]
- Xu NJ, Henkemeyer M 2009. Ephrin-B3 reverse signaling through Grb4 and cytoskeletal regulators mediates axon pruning. Nat Neurosci 12: 268–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi J, Miyamoto Y, Hamasaki H, Sanbe A, Kusakawa S, Nakamura A, Tsumura H, Maeda M, Nemoto N, Kawahara K, et al. 2011. The atypical guanine-nucleotide exchange factor, dock7, negatively regulates schwann cell differentiation and myelination. J Neurosci 31: 12579–12592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WH, Lan HY, Huang CH, Tai SK, Tzeng CH, Kao SY, Wu KJ, Hung MC, Yang MH 2012a. RAC1 activation mediates Twist1-induced cancer cell migration. Nat Cell Biol 14: 366–374 [DOI] [PubMed] [Google Scholar]
- Yang YT, Wang CL, Van Aelst L 2012b. DOCK7 interacts with TACC3 to regulate interkinetic nuclear migration and cortical neurogenesis. Nat Neurosci 15: 1201–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Johnson JL, Cerione RA, Erickson JW 2013. Prenylation and membrane localization of Cdc42 are essential for activation by DOCK7. Biochemistry 52: 4354–4363 [DOI] [PMC free article] [PubMed] [Google Scholar]