

Abstract
The Rpd3-like members of the class I lysine deacetylase family are important regulators of chromatin structure and gene expression and have pivotal functions in the control of proliferation, differentiation and development. The highly related class I deacetylases HDAC1 and HDAC2 have partially overlapping but also isoform-specific roles in diverse biological processes, whereas HDAC3 and HDAC8 have unique functions. This review describes the role of class I KDACs in the regulation of transcription as well as their non-transcriptional functions, in particular their contributions to splicing, mitosis/meiosis, replication and DNA repair. During the past years, a number of mouse loss-of-function studies provided new insights into the individual roles of class I deacetylases in cell cycle control, differentiation and tumorigenesis. Simultaneous ablation of HDAC1 and HDAC2 or single deletion of Hdac3 severely impairs cell cycle progression in all proliferating cell types indicating that these class I deacetylases are promising targets for small molecule inhibitors as anti-tumor drugs.
Keywords: Histone deacetylases, Histone acetylation, Chromatin modifications, Epigenetics, Development, Tumor drugs
Introduction
In mammalian cells, a considerable part of the proteome is subject to reversible acetylation of lysine residues (Choudhary et al. 2009). Acetylation of the epsilon amino group of lysine side chains results in loss of the positive charge and affects several properties such as protein stability, protein–protein interaction and association of proteins with DNA. Moreover, acetylated lysine residues are recognized and bound by bromodomain-containing factors. Thus reversible lysine acetylation has an impact on a diverse range of important biological processes in the cell (Yang and Seto 2007). The best-studied substrates for this post-translational modification are the basic histone proteins. Acetylation of the N-terminal histone tails increases the accessibility of DNA for the RNA polymerase machinery and sets the stage for transcription. The dynamic process of histone acetylation is controlled by two antagonistic enzyme families, the histone acetyltransferases (HATs) and the histone deacetylases (HDACs) (reviewed by Brunmeir et al. 2009). Transcriptionally permissive chromatin is usually associated with histone acetylation, conducted by the HATs, whereas HDACs remove the acetyl groups from the histone tail and are generally considered as transcriptional co-repressors (Grunstein 1997). While histones were the first proteins found to be deacetylated by HDACs, a plethora of non-histone substrates has been identified during the last decade (Patel et al. 2011; Wolffe 1996). Therefore HDACs are also called lysine (K) deacetylases (KDACs). However, to better link the discussed topics to published data we will refer to the individual enzymes as HDACs. Based on the homologies to yeast enzymes, 18 KDACs have been identified in mammals. The KDAC family encompasses the following deacetylases: Rpd3-like class I: HDAC1, 2, 3, 8; Hda1-like class II: HDAC4, 5, 6, 7, 9 and 10; class IV: HDAC11 and the mechanistically unrelated Sir2-like class II: sirtuins SIRT1-7 (Witt et al. 2009).
The first protein shown to have histone deacetylase activity was HDAC1, a mammalian homolog of the yeast pleiotropic transcriptional regulator Rpd3 (reduced potassium dependency 3) (Taunton et al. 1996). Yeast Rpd3 was initially identified in genetic screens as regulatory factor “required to achieve maximum positive and negative transcriptional states” (Stillman et al. 1994; Vidal and Gaber 1991). Three additional Rpd3-like deacetylases —HDAC2, HDAC3 and HDAC8 — have been subsequently found in mammalian cells and constitute together with HDAC1 the class I KDAC subfamily (Dangond et al. 1998; Emiliani et al. 1998; Hu et al. 2000; Yang et al. 1996, 1997).
Structure and localization of class I KDACs
The structures of the genes for Hdac1 and Hdac2 are nearly identical indicating that the two genes most probably arose from gene duplication of a common ancestor (Khier et al. 1999; Zeng et al. 1998). In contrast, the Hdac3 and Hdac8 genes have different exon/intron structures (Mahlknecht et al. 1999). Accordingly, HDAC1 and HDAC2 proteins are the most related among the class I HDACs, exhibiting 86 % of amino acid sequence identity in mice and men, suggesting that they have undergone only little functional divergence from each other (Gregoretti et al. 2004). The catalytic domain is positioned at the N-terminus of HDAC1 and HDAC2 forming the major part of the protein. The N-terminus of HDAC1 also harbors the HDAC association domain (HAD) important for homo-dimerization, whereas the C-terminal part contains a nuclear localization domain (NLS) (Taplick et al. 2001). A coiled-coil domain which presumably serves as protein–protein interaction domain is present within the C-terminus of HDAC2 (Gregoretti et al. 2004). Both enzymes are usually localized to the nucleus. As an exception HDAC1 was reported to show cytosolic localization in the axons of human and murine neurons under pathological conditions (Kim et al. 2010).
HDAC3 shares 63/62 % identical amino acids with HDAC1/HDAC2 and has 43 % sequence identity to HDAC8. In addition to the NLS on the C terminus, HDAC3 has a nuclear export signal (NES), consistent with its ability to localize both to the nucleus as well as to the cytoplasm (Takami and Nakayama 2000; Yang et al. 2002). HDAC3 forms homo-oligomers but can also associate with class II KDACs (Fischle et al. 2001, 2002; Yang et al. 2002). HDAC8, the most recently identified class I KDAC comprises the NLS in the center of the catalytic domain and locates to the nucleus upon overexpression in human cells (Hu et al. 2000; Van den Wyngaert et al. 2000). Another report has described a cytosolic localization of HDAC8 in smooth muscle cells (Waltregny et al. 2005).
Complexes and modifications of class I KDACs
HDAC1 and HDAC2 can homo- and hetero-dimerize (Hassig et al. 1998; Taplick et al. 2001), while HDAC3 forms homo-oligomers (Gregoretti et al. 2004; Yang et al. 2002) and HDAC8 is found as a dimer (Vannini et al. 2004, 2007). Recombinant HDAC8 catalyzes the deacetylation of specific substrates in the absence of additional proteins (reviewed by Wolfson et al. 2013). In contrast, the other three class I KDACs are enzymatically inactive after purification (Gregoretti et al. 2004; Sengupta and Seto 2004; Yang and Seto 2003). The catalytic activity of HDAC1 and HDAC2 is largely dependent on its incorporation into multiprotein complexes (Alland et al. 2002; Zhang et al. 1999). These complexes provide proteins important for the deacetylase activity, DNA- and chromatin-binding as well as substrate specificity (Grozinger and Schreiber 2002). The predominant HDAC1/HDAC2 complexes in mammalian cells are the Sin3, NuRD and CoREST complexes (Alland et al. 1997; Ballas et al. 2001; Heinzel et al. 1997; Laherty et al. 1997; Zhang et al. 1997). The NODE complex is a specialized HDAC1/HDAC2 complex present in embryonic stem cells and the SHIP complex has a specific function during spermatogenesis (Choi et al. 2008; Liang et al. 2008). MiDAC is a novel mitosis-specific deacetylase complex recently identified in a chemoproteomics approach (Bantscheff et al. 2011). Interestingly, in cardiomyocytes HDAC1 was shown to associate with the class II KDAC HDAC5 during the regulation of sodium/calcium exchanger (Chandrasekaran et al. 2009).
HDAC3 is the catalytic component of the N-CoR/SMRT complex. The enzyme is re-folded by the TCP-1 ring complex before connecting to the SMRT and the N-CoR co-repressors which harbor a deacetylase-activating domain for the stimulation of the enzymatic activity of the HDAC3 protein (Guenther et al. 2001, 2002). In addition, HDAC3 can associate with the class II KDACs HDAC4, HDAC5 and HDAC7 and the enzymatic activity of HDAC7 was shown to be dependent on the interaction with HDAC3 (Fischle et al. 2001; Yang et al. 2002). An overview of the different co-repressor complexes of class I KDACs is given in Fig. 1. Interestingly, it was shown recently that inositol-tetraphosphate (IP4) positively affects class I KDAC co-repressor complex formation and activity (Millard et al. 2013; Watson et al. 2012). Based on these findings, the authors suggested that IP4 acts as intermolecular glue between deacetylases and co-repressor proteins thereby enhancing the deacetylase activity of HDAC3–SMRT and HDAC1–MTA1 complexes.
Fig. 1.
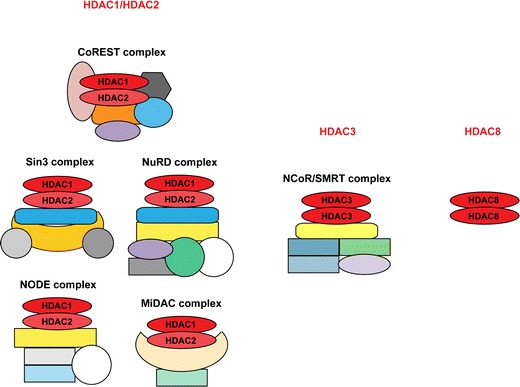
Class I KDACs are deposited in distinct multi-subunit complexes or act on their own. HDAC1/HDAC2 homo- and hetero-dimers are found in the three canonical co-repressor complexes, CoREST, Sin3 and NuRD, and additionally in the ES cell-specific NODE and the mitotic MiDAC complex (left panel). HDAC3 assembles as dimer with the NCoR/SMRT complex (middle panel), while HDAC8 is not dependent on incorporation into complexes for its enzymatic activity (right panel)
Besides their subcellular localization and incorporation into multi-subunit complexes, class I KDACs can also be regulated by post-translational modifications including phosphorylation, acetylation, ubiquitination, SUMOylation, nitrosylation and carbonylation (reviewed by Segre and Chiocca 2011; Wolfson et al. 2013). These modifications modulate their catalytic activity, localization and complex assembly.
HDAC1, HDAC2 and HDAC3 are subjected to phosphorylation by the protein kinase CK2 which can enhance the enzymatic activity as well as the interaction with multi-subunit complex partners (Pflum et al. 2001; Tsai and Seto 2002). Interestingly, CK2-mediated phosphorylation of HDAC1/HDAC2 during mitosis leads to dissociation from each other and thereby to the absence of mitotic HDAC1/2 hetero-dimers (Khan et al. 2013). HDAC8 is phosphorylated in vitro and in vivo by protein kinase A (PKA), which negatively impacts on the catalytic activity (Lee et al. 2004).
Likewise, KDACs are also subject to reversible acetylation. HDAC1 can be acetylated by CBP/p300 resulting in decreased enzymatic activity (Qiu et al. 2006). Deacetylation and concomitant activation of HDAC1 by SIRT1 has been shown to be important for the maintenance of genomic stability in neurons (Dobbin et al. 2013). Despite the fact that five of the six acetylatable lysines are conserved in HDAC2, it cannot be acetylated in vitro due to the lack of lysine 432, which seems to be the crucial residue for acetylation. Accordingly, C-terminal tail-swapping experiments between HDAC1 and HDAC2 showed full HDAC2 acetylation due to the substituted C-terminal region originating from HDAC1 (Luo et al. 2009).
Class I KDACs are also subjected to ubiquitination, which targets them for proteasomal degradation, and sumoylation. For instance, HDAC1 can be sumoylated (Colombo et al. 2002; David et al. 2002), but only SUMO1 and not SUMO2 conjugation to HDAC1 promotes its ubiquitination and degradation (Citro et al. 2013). Thus, specific SUMO paralog conjugation affects HDAC1 protein turnover in mammalian cells.
Cigarette smoke extract-induced tyrosine nitration of HDAC1, HDAC2 and HDAC3 and down-regulation of their protein levels was observed in macrophages (Yang et al. 2006). In neurons cysteine (S)-nitrosylation of HDAC2 was reported to induce chromatin remodeling, triggering the release of HDAC2 from neurotrophin-dependent promoters and thereby stimulating transcription (Nott et al. 2008). In addition, HDAC8 can be S-nitrosylated in vitro, which reversibly inhibits its enzymatic activity (Feng et al. 2011). However, this modification of HDAC8 could not be detected under physiological conditions in vivo.
Moreover, class I deacetylases with the exception of HDAC8 can be carbonylated, which negatively affects their deacetylase activity and hence their transcriptional repressor activity (Doyle and Fitzpatrick 2010).
Non-histone substrates
Genealogical studies indicate that the deacetylation of non-histone substrates was their primary function, since in evolution bacterial KDACs arose ahead of the histone proteins (Gregoretti et al. 2004). Analysis of the acetylome by high-resolution mass spectrometry has revealed the presence of 3,600 lysine acetylation sites on 1,750 human proteins (Choudhary et al. 2009). A subset of these acetylation marks was increased by treatment with the class I KDAC inhibitor MS-275 indicating that class I enzymes recognize many non-histone proteins as substrates. Such non-histone targets comprise transcription factors, proteins involved in chromatin modifications, DNA repair, signal transduction, nuclear import as well as chaperone proteins, structural proteins, tumor suppressors and steroid receptors. Interestingly, several components of KAT and KDAC complexes including HDAC1, HDAC2, and several other proteins of the Sin3 and the NuRD complexes are targets for lysine acetylation (Choudhary et al. 2009). Furthermore, HDAC3 was shown to deacetylate not only the transcription factor MEF2 but also the corresponding acetyltransferases PCAF and p300/CBP (Gregoire et al. 2007). Given that class I KDACs can deacetylate histones, KDAC-recruiting factors, components of the co-repressor complexes and even KATs, it is challenging to dissect the individual contributions of histone and non-histone substrates to the biological function of specific KDACs (see below).
Role of class I KDACs in transcription
According to the textbook view histone acetylation is linked with transcriptional activation, while histone deacetylation is associated with gene repression. Co-repressor complexes are usually recruited by transcription factors, however non-coding RNAs might also play a role in the association of KDAC complexes with chromatin (Yang and Seto 2007). The association of the KDAC complexes with chromatin is further modulated by the affinity of histone interaction domains of specific components of the co-repressor complexes. All major HDAC1/HDAC2 complexes have components with reader domains such as SANT, chromo- and tudor domains, PhD fingers or WD40 repeats, which interact with histones in a modification-dependent manner (Kelly and Cowley 2013). In vitro experiments have shown that HDAC3-containing N-CoR/SMRT complexes bind preferentially to hypoacetylated histones (Vermeulen et al. 2006; Yoon et al. 2005; Yu et al. 2003). A similar feed-forward mechanism based on the preferential association with hypoacetylated histone H3 was also proposed for the HDAC1/HDAC2 co-repressor complex Sin3 (Vermeulen et al. 2006). On the other hand, the presence of active histone marks such as H3K4me3 and H3S10ph reduced the affinity of HDAC1/HDAC2 complexes for the N-terminus of histone H3 in in vitro assays (He et al. 2013; Nishioka et al. 2002). Thus, both the DNA sequence and local chromatin modification patterns control the recruitment of class I KDAC complexes to specific genomic regions.
However, this conventional view of class I KDACs as silencing factors has been challenged during the past years by several studies showing that histone deacetylase activity is important for the activation of certain genes and that HDAC1 and HDAC2 preferentially associate with active genes (Clayton et al. 2006; Kidder and Palmer 2012; Wang et al. 2002, 2009; Zupkovitz et al. 2006). In a genome-wide ChIP-seq analysis in human CD4+ cells a positive correlation between HDAC1/HDAC2 binding, histone acetylation and transcription was discovered (Wang et al. 2009). In this cell system high levels of class I KDACs and KATs associated with transcriptionally active genes. In contrast, silent genes lacked the presence of KATs or KDACs and were not associated with hyperacetylated histones upon KDAC inhibition. According to the model proposed by the authors KDACs (and KATs) are recruited to active genes to dynamically change histone acetylation patterns and to reset the chromatin structure for the next rounds of transcription (Wang et al. 2009).
The situation is further complicated by the fact that class I KDACs not only remove acetyl groups from histones tails but can also deacetylate transcription factors and other chromatin-associated proteins. Indeed, several HDAC1/HDAC2 recruiting transcription factors and components of HDAC1/HDAC2 co-repressor complexes have been also shown to be substrates for acetylation (Choudhary et al. 2009; Patel et al. 2011). In addition, as mentioned above KATs and KDACs are also subject to acetylation (Dobbin et al. 2013; Luo et al. 2009; Qiu et al. 2006). Thus, chromatin-associated class I KDACs can affect gene transcription on several levels (Fig. 2). Recruitment of KDACs as catalytic components of multiprotein complexes results in dynamic changes of histone acetylation patterns. In addition, several of the recruiting transcription factors are themselves targets for reversible acetylation and their protein stability or function can be regulated by class I KDACs. Furthermore, acetylation of specific components of the co-repressor complexes might modulate the activity and stability of these multiprotein complexes. Finally, the acetylation cross-talk between KDACs and KATs by mutual modification can result in modulation of the local activity of the respective chromatin-associated enzymes.
Fig. 2.
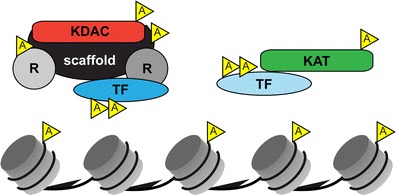
Intricate network of acetylated substrates involved in transcription. In addition to the dynamic and reversible acetylation/deacetylation of histone tails, transcription factors (TF), KATs and KDACs themselves as well as structural (scaffold) and regulatory (labeled as R) components of co-repressor complexes can be reversibly acetylated, potentially leading to changes in local transcriptional activity
Non-transcriptional functions of class I KDACs
In addition to their well-studied role in transcription, other important functions of class I KDACs have emerged during the past years (summarized in Fig. 3).
Fig. 3.
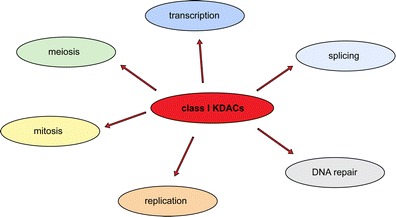
The class I subfamily of lysine deacetylases is involved in several cellular processes. Besides transcription, HDAC1, HDAC2, HDAC3 and/or HDAC8 play crucial roles during splicing, DNA repair, replication, mitosis and meiosis
Splicing
For instance, KDACs have been shown to affect alternative splicing (Hnilicova et al. 2011; Nogues et al. 2002). Analysis of genome-wide nucleosome positioning data had previously revealed increased nucleosomal occupancy at exons (Schwartz et al. 2009). Hu RNA-binding proteins can associate with HDAC2 thereby inhibiting its deacetylation activity and inducing local histone hyperacetylation at the chromatin of alternatively spliced genes (Zhou et al. 2011). KDAC inhibitor treatment of HeLa cells led to altered splicing of nearly 700 human genes. Treatment with sodium butyrate induced histone H4 acetylation and increased RNA Pol II processivity and exon skipping at the fibronectin gene (Hnilicova et al. 2011). This effect could be recapitulated by siRNA-mediated knock-down of HDAC1 but not HDAC2. The alternative splicing defect in HDAC1-depleted cells could be rescued by the expression of the wildtype HDAC1 protein, but not by a catalytically inactive variant, suggesting a specific requirement for HDAC1-associated deacetylase activity for the regulation of alternative splicing. Notably, about 50 % of the proteins building the spliceosome are acetylated, however the relevance of this potential link between KDAC function and splicing remains to be established (Choudhary et al. 2009).
Replication and DNA repair
KDAC inhibition in cancer cells has been shown to slow down replication and to induce DNA damage suggesting an important function of KDACs during DNA synthesis (Conti et al. 2010). Indeed, class I KDACs have been identified as crucial components of the replication and DNA repair machinery.
The simultaneous deletion of Hdac1 and Hdac2 resulted in G1 arrest and severely reduced BrdU incorporation indicating a block in S-phase transition (Wilting et al. 2010; Yamaguchi et al. 2010). Ablation of HDAC1 and HDAC2 was recently reported to cause increased acetylation of H4K16 on nascent chromatin, decreased replication fork velocity and activation of the replication stress response (Bhaskara et al. 2013). HDAC1 and HDAC2 were found to be located at the replication fork and seem to be required for removing specific histone acetylation marks that might otherwise interfere with the activity of ISWI-family chromatin remodelers (Bhaskara et al. 2013). Induction of DNA double-strand breaks in human osteosarcoma cells in the absence of HDAC1/HDAC2 resulted in increased H3K56 acetylation, a modification linked to DNA damage (Miller et al. 2010). In this study, loss of HDAC1 and HDAC2 was found to affect the persistence of NHEJ (non-homologous end-joining) factors at DNA double-strand breaks and to result in hypersensitivity to DNA-damaging agents suggesting a function of these enzymes in the DNA-damage response. In accordance, simultaneous ablation of HDAC1 and HDAC2 in the developing mouse brain led to DNA damage and enhanced H3K56 acetylation (Hagelkruys et al. 2013). Interestingly, HDAC1 was recently found to collaborate with SIRT1 in the maintenance of genomic stability in neurons (Dobbin et al. 2013).
HDAC3-deficient fibroblasts display impaired S phase progression and inefficient DNA repair leading to DNA damage and apoptosis (Bhaskara et al. 2008). Furthermore, ablation of HDAC3 in hematopoietic progenitor cells revealed a requirement of this deacetylase for the passage through S phase (Summers et al. 2013). HDAC3-deficient cells show elevated levels of nucleosome deposition marks such as H4K5ac and H4K12ac accompanied by a loss of heterochromatin, an increase in DNA double-strand breaks and reduced proliferation (Bhaskara et al. 2008, 2010). In summary, HDAC1/HDAC2 and HDAC3 have distinct non-redundant functions during replication and DNA repair.
Mitosis/meiosis
Overexpression of HDAC1 in mouse fibroblasts resulted in a partial G2/M block and aberrant nuclear morphology, whereas knock-down of HDAC1 in human tumor cells also led to impaired mitosis suggesting a requirement for balanced acetylation/deacetylation during mitosis (Bartl et al. 1997; Senese et al. 2007). Along this line, HDAC1/HDAC2-deficient transformed mouse fibroblasts display nuclear bridging, nuclear fragmentation, and mitotic catastrophe (Haberland et al. 2009a). Recently, the multi-zinc-finger protein TRPS1 encoded by the gene mutated in human "Tricho-Rhino-Phalangeal syndrome" was shown to associate with HDAC1 and HDAC4 (Wuelling et al. 2013). Loss of TRPS1 in murine chondrocytes resulted in increased H3 acetylation and impaired chromatin condensation during mitosis. However the underlying molecular mechanisms for the essential function of HDAC1/HDAC2 during mitosis remain to be identified. Remarkably, loss of HDAC2 causes increased H4K16 acetylation and defective chromosome condensation and segregation during oogenesis providing evidence for a critical role of HDAC2 during meiosis (Ma and Schultz 2013).
Several reports describe an essential role for HDAC3 during mitosis. Li et al. have shown that HDAC3 is targeted together with the A-Kinase-Anchoring proteins AKAP95 and HA95 to mitotic chromosomes to generate a hypoacetylated H3 tail as preferred template for Aurora B kinase (Li et al. 2006). Phosphorylation of H3S10 by Aurora B led to the dissociation of HP1 from the neighboring methylated H3K9 residue and is a crucial step during mitosis. Thus, the non-transcriptional function of HDAC3 during mitosis is a prerequisite for the H3S10 kinase activity of Aurora B. Furthermore, HDAC3 was shown to be required for deacetylation of H3K4 at the centromere and sister chromatin cohesion (Eot-Houllier et al. 2008). Finally, a complex formed by HDAC3, N-CoR, TBL1 and TBLR1 is localized at the mitotic spindle and its activity is pivotal for kinetochore-microtubule attachment (Ishii et al. 2008).
Recently, it was reported that HDAC8 deacetylates cohesin and that the enzyme is implicated in Cornelia de Lange Syndrome (CdLS) (Deardorff et al. 2012). Cohesins form a ring structure encircling sister chromatids and have important functions for sister chromatid cohesion as well as for transcription control. Loss of HDAC8 resulted in elevated acetylation of one of the cohesion subunits SMC3 and impaired dissolution of the cohesin complex from chromatin during mitosis. Importantly, Deardorff et al. (2012) identified loss-of-function mutations in the Hdac8 gene in six CdLS probands.
Knock-out studies — full deletions
A lot of effort has been put into the investigation of the specific functions of class I KDACs in development, proliferation and differentiation by loss-of-function studies in mice. Germline deletion of Hdac1 in mice results in embryonic lethality before embryonic day E10.5. These mice show severe developmental and proliferation defects as well as growth retardation (Lagger et al. 2002; Montgomery et al. 2007; Yamaguchi et al. 2010). HDAC1 represses the cyclin-dependent kinase inhibitor p21WAF1/CIP1 and thus positively regulates proliferation. Indeed p21WAF1/CIP1 is up-regulated in HDAC1 knock-out mice and HDAC1-deficient mouse embryonic stem (ES) cells (Lagger et al. 2002). The proliferation defect in ES cells but not the lethal phenotype of HDAC1 knock-out mice could be rescued by the deletion of p21 WAF1/CAF1 (Zupkovitz et al. 2010). In mice and ES cells deficient for HDAC1, the HDAC2 protein is up-regulated but is not fully capable to compensate for the loss of HDAC1 (Lagger et al. 2002; Zupkovitz et al. 2006, 2010). Depending on the knock-out strategy, deletion of Hdac2 in mice results in perinatal lethality (Montgomery et al. 2007), partial perinatal lethality (Guan et al. 2009) or partial lethality within the first months (Trivedi et al. 2007; Zimmermann et al. 2007). These knock-out studies attribute specific functions to the highly homologous enzymes HDAC1 and HDAC2. Knock-out of HDAC3 also results in embryonic lethality before embryonic day E9.5 due to gastrulation defects (Bhaskara et al. 2008; Montgomery et al. 2008). Global loss of HDAC8 in mice results in skull instability and perinatal lethality (Haberland et al. 2009b).
Knock-out studies — conditional deletions
Since global germline ablation of class I KDACs caused early lethality, conditional cre recombinase-mediated deletions were instrumental in revealing the functions of these deacetylases in specific tissues. Loss of either HDAC1 or HDAC2 in cell types including ES cells, fibroblasts, B cells, thymocytes, keratinocytes, cardiomyocytes, neurons, Schwann cells and various cell lines caused no or only mild effects (Chen et al. 2011; Dovey et al. 2010, 2013; Grausenburger et al. 2010; Guan et al. 2009; Jawerka et al. 2010; Jurkin et al. 2011; Lagger et al. 2002; LeBoeuf et al. 2011; Montgomery et al. 2007; Winter et al. 2013; Yamaguchi et al. 2010). In most of these cell types, deletion of Hdac1 led to an increased protein level of HDAC2 and vice versa, most probably compensating for the loss of the respective paralog and thereby masking the knock-out phenotype.
Mild phenotypes were observed for instance upon knock-out of HDAC1 in T cells and ES cells as well as loss of HDAC2 in oocytes and the nervous system. In T cells absence of HDAC1 provoked increased airway inflammation and elevated Th2 cytokine production in an in vivo asthma mouse model, indicating that HDAC1 modulates the inflammatory response (Grausenburger et al. 2010). Similarly deletion of Hdac1 in ES cells resulted in enhanced differentiation of embryoid bodies, highlighting a crucial role of HDAC1 in cell fate determination during differentiation (Dovey et al. 2010). Conversely, loss of HDAC2 in oocytes led to sub-fertile mice due to H4K16 hyperacetylation and affected chromosome segregation (Ma et al. 2012; Ma and Schultz 2013). Deficiency of HDAC2 in adult neural stem cells caused defects in adult neurogenesis (Jawerka et al. 2010), while another report found enhanced synapse number and memory formation upon loss of HDAC2 (Guan et al. 2009). Together these data suggest requirement of HDAC2 during adult neuronal progenitor differentiation and implication of HDAC2 in synaptic plasticity after neuronal maturation. Deletion of either Hdac1 or Hdac2 in the murine epidermis does not evoke an obvious phenotype (LeBoeuf et al. 2011; Winter et al. 2013). Importantly, the ablation of HDAC1, but not of HDAC2 in a genetic skin tumor model results in accelerated tumor development, implicating a tumor suppressor function for HDAC1 in the epidermis (Winter et al. 2013). A potential tumor suppressor function for HDAC1 was also discovered in T cells and B cells (Dovey et al. 2013; Heideman et al. 2013; Santoro et al. 2013). In accordance loss of HDAC1 in teratomas resulted in decreased differentiation and enhanced malignancy due to deregulated SNAIL1 signaling (Lagger et al. 2010).
In contrast to individual conditional deletions of Hdac1 or Hdac2, which mostly do not evoke strong phenotypes, the combined conditional loss of HDAC1 and HDAC2 results in dramatic defects in proliferation, differentiation, survival and transcriptional regulation in most cell types and tissues. Both in fibroblasts and B cells, simultaneous loss of HDAC1 and HDAC2 led to a strong cell cycle block in G1 followed by cell death (Yamaguchi et al. 2010). Similarly, dual ablation of HDAC1 and HDAC2 in neuronal precursor cells resulted in failure to differentiate into mature neurons and apoptosis (Montgomery et al. 2009). Collective loss of HDAC1 and HDAC2 led to impaired development of different cell types due to transcriptional deregulation of important signaling pathways. Combined loss of HDAC1 and HDAC2 was linked to deregulation of the p53/p63 pathway and apoptosis in the epidermis (LeBoeuf et al. 2011), de-repression of Bmp4 and Rb1 in the lung (Wang et al. 2013), inappropriate activation of Wnt/β-catenin signaling in oligodendrocytes (Ye et al. 2009) and impaired T-cell receptor signaling and apoptosis in thymocytes (Dovey et al. 2013; Heideman et al. 2013). Likewise, dual HDAC1/2 ablation resulted in apoptotic megakaryocytes, anemia and thrombocytopenia in the hematopoietic system (Wilting et al. 2010; Yamaguchi et al. 2010), hyperacetylation of TRP53 followed by apoptosis in oocytes (Ma et al. 2012), mitotic failure in proliferating hepatocytes (Xia et al. 2013) or decreased autophagy in skeletal muscles (Moresi et al. 2012). Upon simultaneous HDAC1/2 deletion in Schwann cells, myelin deficiency and massive cell loss were observed and attributed at least in part to hyperacetylation of NF-κB (Chen et al. 2011; Jacob et al. 2011).
Notably, combined loss of HDAC1 and HDAC2 induces DNA damage or chromosomal abnormalities finally resulting in apoptosis in a variety of cell types and organs including T cells (Dovey et al. 2013; Heideman et al. 2013), B cells (Yamaguchi et al. 2010), brain (Hagelkruys et al. 2013) and transformed fibroblasts (Haberland et al. 2009a). Accordingly, proliferating cells cannot tolerate simultaneous lack of HDAC1 and HDAC2, implying that drugs inhibiting both enzymes are promising anti-cancer drugs.
Importantly, deletion of three of the four Hdac1/Hdac2 alleles allowed dissecting the individual functions of HDAC1 and HDAC2 in different cell types and tissues. Haploinsufficency of Hdac1 in the absence of HDAC2 and thereby a more important function of HDAC2 was identified in oocytes, where the enzyme regulates chromosome segregation and kinetochore function (Ma and Schultz 2013). A similar situation was observed in the developing nervous system, where HDAC2 represses protein kinase C delta and thereby inhibits premature differentiation (Hagelkruys et al. 2013).
Interestingly, only the opposite allelic combination, a single Hdac2 allele in the absence of HDAC1, resulted in phenotypes when deleted in keratinocytes and thymocytes. Haploinsufficiency of Hdac2 in the absence of HDAC1 in the epidermis led to strongly impaired epidermal development due to mobilization of epidermal stem cells, hyperproliferation and increased differentiation (Winter et al. 2013). Elevated proliferation in the epidermis was accompanied by post-transcriptional up-regulation of the c-Myc protein, highlighting once more transcription-dependent and independent effects of class I KDACs.
In T cells, a comparable positive effect on cell proliferation by a single Hdac2 allele in the absence of HDAC1 was shown to favor tumor formation (Dovey et al. 2013; Heideman et al. 2013). Furthermore, one allele of Hdac1 in the absence of HDAC2 was sufficient to restore proper B cell development, while the vice versa situation (one allele of Hdac2 in the absence of HDAC1) could not rescue the cell cycle block and apoptosis (Reichert et al. 2012). Interestingly, the different Hdac1/2 allele-specific effects are in some cases dependent on the remaining level of HDAC activity (Dovey et al. 2013; Heideman et al. 2013; Matthias 2013). In contrast, in the developing brain the highly divergent phenotypes of Hdac1+/− Hdac2−/− and Hdac1−/− Hdac2+/− mice could not be explained by differential reduction of HDAC activity, but rather by specific effects of HDAC1 and HDAC2 on co-repressor function. It is tempting to speculate that HDAC1 and HDAC2 have different cell type-specific affinities for co-repressor complexes that are further modulated by various post-translational modifications such as phosphorylation, acetylation and sumoylation. Taken together this reveals overlapping, but specific functions of HDAC1 and HDAC2 in different tissues during mouse development.
Since HDAC1 and HDAC2 are paralogs and show redundancy in many cell types, their individual deletion did not evoke severe phenotypes. On the contrary, loss of HDAC3 and HDAC8 could not be compensated by another deacetylase and caused developmental defects and often lethality. Liver-specific ablation of HDAC3 led to hepatocellular carcinomas due to impaired response to DNA damage and genomic instability in hepatocytes (Bhaskara et al. 2010). Conditional deletion of Hdac3 in osteo-chondroprogenitor cells decreased bone length and caused severe osteopenia due to suppression of the Akt/mTOR pathway (Bradley et al. 2013; Razidlo et al. 2010). Cardiac-specific deletion of Hdac3 resulted in cardiac hypertrophy and aberrant expression of cardiac metabolism genes, highlighting the role of HDAC3 in the maintenance of cardiac function and in the regulation of cardiac energy metabolism (Montgomery et al. 2008). Besides the crucial role of HDAC3 in genomic stability in different cell types, maintenance of cardiac metabolism and formation/maturation of bone, HDAC3 was found to negatively regulate memory formation (McQuown et al. 2011).
Conditional deletion of Hdac8 in neural crest cells phenocopied the global deletion, showing skull instability and perinatal lethality (Haberland et al. 2009b).
Taken together, these findings demonstrate that class I KDACs possess highly specific in vivo functions.
Summary and outlook
By deacetylating histones and a growing number of non-histone substrates, class I KDACs play fundamental roles in transcription. Importantly, lysine deacetylases do not only control dynamic changes in histone acetylation, but also remove acetyl groups from transcription factors, co-repressor complex subunits and KATs and KDACs themselves thereby potentially modulating their stability and function.
During the past years, several non-transcriptional roles of the class I subfamily have emerged and have implicated HDAC1, HDAC2, HDAC3 and HDAC8 in biological processes including replication, DNA repair, splicing and mitosis/meiosis.
Originating from gene duplication the two paralogs HDAC1 and HDAC2 have partially overlapping, but also specific functions in different cell types, while the other class I subfamily members HDAC3 and HDAC8 are more distinct and thereby have non-redundant roles. The redundant functions of HDAC1 and HDAC2 are visible upon simultaneous deletion of both enzymes, which is incompatible with normal cellular proliferation and development. Since the individual contributions of HDAC1 and HDAC2 are partially masked by the up-regulated paralog in single knock-outs, an important step to reveal isoform-specific functions was the deletion of three of the four Hdac1/2 alleles in different cellular systems.
Due to the contribution of class I KDACs to cancer and neurological disorders, KDAC inhibitors are promising drugs in several diseases. Despite the growing number of KDAC knock-out mice, identification of non-histone substrates and increasing knowledge about KDACs in pathological conditions, the individual contribution of particular KDAC members to normal development and disease is not completely understood. The key to fully elucidate the effects of KDAC knock-outs or KDAC inhibitor treatment is to complete the picture by comparing the cell type-specific expression patterns and local chromatin association of KDACs with the ones of the antagonistic KATs.
Acknowledgments
This work was supported by the GEN-AU project "Epigenetic Regulation of Cell Fate Decisions" (Federal Ministry for Education, Science, and Culture) and the Austrian Science Fund (P25807). M.M. and A.H. are fellows of the International Ph.D. program "Molecular Mechanisms of Cell Signaling" supported by the Austrian Science Fund (DK W1220).
Footnotes
M.A. Moser and A. Hagelkruys made equal contributions to this study.
References
- Alland L, David G, Shen-Li H, Potes J, Muhle R, Lee HC, Hou H, Jr, Chen K, DePinho RA. Identification of mammalian Sds3 as an integral component of the Sin3/histone deacetylase corepressor complex. Mol Cell Biol. 2002;22:2743–2750. doi: 10.1128/MCB.22.8.2743-2750.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alland L, Muhle R, Hou H, Jr, Potes J, Chin L, Schreiber-Agus N, DePinho RA. Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, Federoff HJ, Rose DW, Rosenfeld MG, Brehm P, Mandel G. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol. 2011;29:255–265. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Bartl S, Taplick J, Lagger G, Khier H, Kuchler K, Seiser C. Identification of mouse histone deacetylase 1 as a growth factor-inducible gene. Mol Cell Biol. 1997;17:5033–5043. doi: 10.1128/mcb.17.9.5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, Hiebert SW. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30:61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara S, Jacques V, Rusche JR, Olson EN, Cairns BR, Chandrasekharan MB. Histone deacetylases 1 and 2 maintain S-phase chromatin and DNA replication fork progression. Epigenetics Chromatin. 2013;6:27. doi: 10.1186/1756-8935-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, Yenamandra A, Locke K, Yuan JL, Bonine-Summers AR, Wells CE, Kaiser JF, Washington MK, Zhao Z, Wagner FF, Sun ZW, Xia F, Holson EB, Khabele D, Hiebert SW. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–447. doi: 10.1016/j.ccr.2010.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley EW, Carpio LR, Westendorf JJ. Histone deacetylase 3 suppression increases PH domain and leucine-rich repeat phosphatase (Phlpp)1 expression in chondrocytes to suppress Akt signaling and matrix secretion. J Biol Chem. 2013;288:9572–9582. doi: 10.1074/jbc.M112.423723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int J Dev Biol. 2009;53:275–289. doi: 10.1387/ijdb.082649rb. [DOI] [PubMed] [Google Scholar]
- Chandrasekaran S, Peterson RE, Mani SK, Addy B, Buchholz AL, Xu L, Thiyagarajan T, Kasiganesan H, Kern CB, Menick DR. Histone deacetylases facilitate sodium/calcium exchanger up-regulation in adult cardiomyocytes. FASEB J. 2009;23:3851–3864. doi: 10.1096/fj.09-132415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang H, Yoon SO, Xu X, Hottiger MO, Svaren J, Nave KA, Kim HA, Olson EN, Lu QR. HDAC-mediated deacetylation of NF-kappaB is critical for Schwann cell myelination. Nat Neurosci. 2011;14:437–441. doi: 10.1038/nn.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E, Han C, Park I, Lee B, Jin S, Choi H, Kim do H, Park ZY, Eddy EM, Cho C. A novel germ cell-specific protein, SHIP1, forms a complex with chromatin remodeling activity during spermatogenesis. J Biol Chem. 2008;283:35283–35294. doi: 10.1074/jbc.M805590200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Citro S, Jaffray E, Hay RT, Seiser C, Chiocca S (2013) A role for paralog-specific sumoylation in histone deacetylase 1 stability. Journal of molecular cell biology [DOI] [PubMed]
- Clayton AL, Hazzalin CA, Mahadevan LC. Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell. 2006;23:289–296. doi: 10.1016/j.molcel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Colombo R, Boggio R, Seiser C, Draetta GF, Chiocca S. The adenovirus protein Gam1 interferes with sumoylation of histone deacetylase 1. EMBO Rep. 2002;3:1062–1068. doi: 10.1093/embo-reports/kvf213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, Aladjem MI, Pommier Y. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res. 2010;70:4470–4480. doi: 10.1158/0008-5472.CAN-09-3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangond F, Hafler DA, Tong JK, Randall J, Kojima R, Utku N, Gullans SR. Differential display cloning of a novel human histone deacetylase (HDAC3) cDNA from PHA-activated immune cells. Biochem Biophys Res Commun. 1998;242:648–652. doi: 10.1006/bbrc.1997.8033. [DOI] [PubMed] [Google Scholar]
- David G, Neptune MA, DePinho RA. SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. J Biol Chem. 2002;277:23658–23663. doi: 10.1074/jbc.M203690200. [DOI] [PubMed] [Google Scholar]
- Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbin MM, Madabhushi R, Pan L, Chen Y, Kim D, Gao J, Ahanonu B, Pao PC, Qiu Y, Zhao Y, Tsai LH. SIRT1 collaborates with ATM and HDAC1 to maintain genomic stability in neurons. Nat Neurosci. 2013;16:1008–1015. doi: 10.1038/nn.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey OM, Foster CT, Conte N, Edwards SA, Edwards JM, Singh R, Vassiliou G, Bradley A, Cowley SM (2013) Histone deacetylase (HDAC) 1 and 2 are essential for normal T cell development and genomic stability in mice. Blood [DOI] [PMC free article] [PubMed]
- Dovey OM, Foster CT, Cowley SM. Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci U S A. 2010;107:8242–8247. doi: 10.1073/pnas.1000478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle K, Fitzpatrick FA. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem. 2010;285:17417–17424. doi: 10.1074/jbc.M109.089250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emiliani S, Fischle W, Van Lint C, Al Abed Y, Verdin E. Characterization of a human RPD3 ortholog, HDAC3. Proc Natl Acad Sci U S A. 1998;95:2795–2800. doi: 10.1073/pnas.95.6.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eot-Houllier G, Fulcrand G, Watanabe Y, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase 3 is required for centromeric H3K4 deacetylation and sister chromatid cohesion. Genes Dev. 2008;22:2639–2644. doi: 10.1101/gad.484108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng JH, Jing FB, Fang H, Gu LC, Xu WF. Expression, purification, and S-nitrosylation of recombinant histone deacetylase 8 in Escherichia coli. Biosci Trends. 2011;5:17–22. doi: 10.5582/bst.2011.v5.1.17. [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Fillion M, Hendzel MJ, Voelter W, Verdin E. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J Biol Chem. 2001;276:35826–35835. doi: 10.1074/jbc.M104935200. [DOI] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, Zhang Y, Rembold M, Gaisberger M, Hartl A, Epstein MM, Matthias P, Seiser C, Ellmeier W. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol. 2010;185:3489–3497. doi: 10.4049/jimmunol.0903610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, Wong J, Seto E, Yang XJ. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol Cell Biol. 2007;27:1280–1295. doi: 10.1128/MCB.00882-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem Biol. 2002;9:3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol. 2001;21:6091–6101. doi: 10.1128/MCB.21.18.6091-6101.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Yu J, Kao GD, Yen TJ, Lazar MA. Assembly of the SMRT-histone deacetylase 3 repression complex requires the TCP-1 ring complex. Genes Dev. 2002;16:3130–3135. doi: 10.1101/gad.1037502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Johnson A, Mokalled MH, Montgomery RL, Olson EN. Genetic dissection of histone deacetylase requirement in tumor cells. Proc Natl Acad Sci U S A. 2009;106:7751–7755. doi: 10.1073/pnas.0903139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Mokalled MH, Montgomery RL, Olson EN. Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 2009;23:1625–1630. doi: 10.1101/gad.1809209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagelkruys A, Lagger S, Krahmer J, Bedeir A, Artaker M, Zezula J, Weissmann S, Xie Y, Schöfer C, Pusch O, Schlederer M, Brosch G, Matthias P, Lassmann H, Knoblich J, Seiser C (2013) A single allele of Hdac2 but not Hdac1 is sufficient for normal mouse brain development and survival in the absence of its paralog. Development (manuscript in revision) [DOI] [PMC free article] [PubMed]
- Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG, Ayer DE, Schreiber SL. A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc Natl Acad Sci U S A. 1998;95:3519–3524. doi: 10.1073/pnas.95.7.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Khan DH, Winter S, Seiser C, Davie JR. Dynamic distribution of HDAC1 and HDAC2 during mitosis: association with F-actin. J Cell Physiol. 2013;228:1525–1535. doi: 10.1002/jcp.24311. [DOI] [PubMed] [Google Scholar]
- Heideman MR, Wilting RH, Yanover E, Velds A, de Jong J, Kerkhoven RM, Jacobs H, Wessels LF, Dannenberg JH (2013) Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood [DOI] [PMC free article] [PubMed]
- Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression [see comments] Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- Hnilicova J, Hozeifi S, Duskova E, Icha J, Tomankova T, Stanek D. Histone deacetylase activity modulates alternative splicing. PLoS One. 2011;6:e16727. doi: 10.1371/journal.pone.0016727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Chen ZX, Fredrickson T, Zhu Y, Kirkpatrick R, Zhang GF, Johanson K, Sung CM, Liu RG, Winkler J. Cloning and characterization of a novel human. Class I histone deacetylase that functions as a transcription repressor. J Biol Chem. 2000;275(20):15254–15264. doi: 10.1074/jbc.M908988199. [DOI] [PubMed] [Google Scholar]
- Ishii S, Kurasawa Y, Wong J, Yu-Lee LY. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc Natl Acad Sci U S A. 2008;105:4179–4184. doi: 10.1073/pnas.0710140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C, Christen CN, Pereira JA, Somandin C, Baggiolini A, Lotscher P, Ozcelik M, Tricaud N, Meijer D, Yamaguchi T, Matthias P, Suter U. HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat Neurosci. 2011;14:429–436. doi: 10.1038/nn.2762. [DOI] [PubMed] [Google Scholar]
- Jawerka M, Colak D, Dimou L, Spiller C, Lagger S, Montgomery RL, Olson EN, Wurst W, Gottlicher M, Gotz M. The specific role of histone deacetylase 2 in adult neurogenesis. Neuron Glia Biol. 2010;6:93–107. doi: 10.1017/S1740925X10000049. [DOI] [PubMed] [Google Scholar]
- Jurkin J, Zupkovitz G, Lagger S, Grausenburger R, Hagelkruys A, Kenner L, Seiser C. Distinct and redundant functions of histone deacetylases HDAC1 and HDAC2 in proliferation and tumorigenesis. Cell Cycle. 2011;10:406–412. doi: 10.4161/cc.10.3.14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly RD, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41:741–749. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- Khan DH, He S, Yu J, Winter S, Cao W, Seiser C, Davie JR. Protein kinase CK2 regulates the dimerization of histone deacetylase 1 (HDAC1) and HDAC2 during mitosis. J Biol Chem. 2013;288:16518–16528. doi: 10.1074/jbc.M112.440446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khier H, Bartl S, Schuettengruber B, Seiser C. Cloning and characterization of the mouse histone deacetylase 1 gene: integration of a retrovirus in 129SV mice. Biochim Biophys Acta. 1999;1489:365–373. doi: 10.1016/s0167-4781(99)00203-1. [DOI] [PubMed] [Google Scholar]
- Kidder BL, Palmer S. HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res. 2012;40:2925–2939. doi: 10.1093/nar/gkr1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Shen S, Dietz K, He Y, Howell O, Reynolds R, Casaccia P. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci. 2010;13:180–189. doi: 10.1038/nn.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger G, O'Carroll D, Rembold M, Khier H, Tischler J, Weitzer G, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, Seiser C. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. Embo J. 2002;21:2672–2681. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger S, Meunier D, Mikula M, Brunmeir R, Schlederer M, Artaker M, Pusch O, Egger G, Hagelkruys A, Mikulits W, Weitzer G, Muellner EW, Susani M, Kenner L, Seiser C (2010) Crucial function of histone deacetylase 1 for differentiation of teratomas in mice and humans. Embo J 29:3992–4007 [DOI] [PMC free article] [PubMed]
- Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell. 1997;89:349–356. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- LeBoeuf M, Terrell A, Trivedi S, Sinha S, Epstein JA, Olson EN, Morrisey EE, Millar SE. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev Cell. 2011;19:807–818. doi: 10.1016/j.devcel.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Rezai-Zadeh N, Seto E. Negative regulation of histone deacetylase 8 activity by cyclic AMP-dependent protein kinase A. Mol Cell Biol. 2004;24:765–773. doi: 10.1128/MCB.24.2.765-773.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kao GD, Garcia BA, Shabanowitz J, Hunt DF, Qin J, Phelan C, Lazar MA. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 2006;20:2566–2579. doi: 10.1101/gad.1455006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Wan M, Zhang Y, Gu P, Xin H, Jung SY, Qin J, Wong J, Cooney AJ, Liu D, Songyang Z. Nanog and Oct4 associate with unique transcriptional repression complexes in embryonic stem cells. Nat Cell Biol. 2008;10:731–739. doi: 10.1038/ncb1736. [DOI] [PubMed] [Google Scholar]
- Luo Y, Jian W, Stavreva D, Fu X, Hager G, Bungert J, Huang S, Qiu Y. Trans-regulation of histone deacetylase activities through acetylation. J Biol Chem. 2009;284:34901–34910. doi: 10.1074/jbc.M109.038356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma P, Pan H, Montgomery RL, Olson EN, Schultz RM. Compensatory functions of histone deacetylase 1 (HDAC1) and HDAC2 regulate transcription and apoptosis during mouse oocyte development. Proc Natl Acad Sci U S A. 2012;109:E481–E489. doi: 10.1073/pnas.1118403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma P, Schultz RM. Histone deacetylase 2 (HDAC2) regulates chromosome segregation and kinetochore function via H4K16 deacetylation during oocyte maturation in mouse. PLoS Genet. 2013;9:e1003377. doi: 10.1371/journal.pgen.1003377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahlknecht U, Emiliani S, Najfeld V, Young S, Verdin E. Genomic organization and chromosomal localization of the human histone deacetylase 3 gene. Genomics. 1999;56:197–202. doi: 10.1006/geno.1998.5645. [DOI] [PubMed] [Google Scholar]
- Matthias P. Too much or too little, how much HDAC activity is good for you? Blood. 2013;121:1930–1931. doi: 10.1182/blood-2013-02-480400. [DOI] [PubMed] [Google Scholar]
- McQuown SC, Barrett RM, Matheos DP, Post RJ, Rogge GA, Alenghat T, Mullican SE, Jones S, Rusche JR, Lazar MA, Wood MA. HDAC3 is a critical negative regulator of long-term memory formation. J Neurosci. 2011;31:764–774. doi: 10.1523/JNEUROSCI.5052-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L, Schwabe JW. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell. 2013;51:57–67. doi: 10.1016/j.molcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, Jackson SP. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol. 2010;17:1144–1151. doi: 10.1038/nsmb.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, Qi X, Hill JA, Richardson JA, Olson EN. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Hsieh J, Barbosa AC, Richardson JA, Olson EN. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc Natl Acad Sci U S A. 2009;106:7876–7881. doi: 10.1073/pnas.0902750106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, Richardson JA, Bassel-Duby R, Olson EN. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588–3597. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moresi V, Carrer M, Grueter CE, Rifki OF, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc Natl Acad Sci U S A. 2012;109:1649–1654. doi: 10.1073/pnas.1121159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogues G, Kadener S, Cramer P, Bentley D, Kornblihtt AR. Transcriptional activators differ in their abilities to control alternative splicing. J Biol Chem. 2002;277:43110–43114. doi: 10.1074/jbc.M208418200. [DOI] [PubMed] [Google Scholar]
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- Patel J, Pathak RR, Mujtaba S. The biology of lysine acetylation integrates transcriptional programming and metabolism. Nutr Metab (Lond) 2011;8:12. doi: 10.1186/1743-7075-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflum MK, Tong JK, Lane WS, Schreiber SL. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem. 2001;276:47733–47741. doi: 10.1074/jbc.M105590200. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Zhao Y, Becker M, John S, Parekh BS, Huang S, Hendarwanto A, Martinez ED, Chen Y, Lu H, Adkins NL, Stavreva DA, Wiench M, Georgel PT, Schiltz RL, Hager GL. HDAC1 acetylation is linked to progressive modulation of steroid receptor-induced gene transcription. Molecular cell. 2006;22:669–679. doi: 10.1016/j.molcel.2006.04.019. [DOI] [PubMed] [Google Scholar]
- Razidlo DF, Whitney TJ, Casper ME, McGee-Lawrence ME, Stensgard BA, Li X, Secreto FJ, Knutson SK, Hiebert SW, Westendorf JJ. Histone deacetylase 3 depletion in osteo/chondroprogenitor cells decreases bone density and increases marrow fat. PloS One. 2010;5:e11492. doi: 10.1371/journal.pone.0011492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert N, Choukrallah MA, Matthias P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol Life Sci: CMLS. 2012;69:2173–2187. doi: 10.1007/s00018-012-0921-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L, Barozzi I, Senese S, Fornasari L, Moretti S, Altucci L, Pelicci PG, Chiocca S, Johnstone RW, Minucci S. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood. 2013;121:3459–3468. doi: 10.1182/blood-2012-10-461988. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Meshorer E, Ast G. Chromatin organization marks exon–intron structure. Nat Struct Mol Biol. 2009;16:990–995. doi: 10.1038/nsmb.1659. [DOI] [PubMed] [Google Scholar]
- Segre CV, Chiocca S. Regulating the regulators: the post-translational code of class I HDAC1 and HDAC2. J Biomed Biotechnol. 2011;2011:690848. doi: 10.1155/2011/690848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A, Bernard L, Draetta GF, Alcalay M, Seiser C, Chiocca S. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. 2007;27:4784–4795. doi: 10.1128/MCB.00494-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- Stillman DJ, Dorland S, Yu Y. Epistasis analysis of suppressor mutations that allow HO expression in the absence of the yeast SW15 transcriptional activator. Genetics. 1994;136:781–788. doi: 10.1093/genetics/136.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers AR, Fischer MA, Stengel KR, Zhao Y, Kaiser JF, Wells CE, Hunt A, Bhaskara S, Luzwick JW, Sampathi S, Chen X, Thompson MA, Cortez D, Hiebert SW. HDAC3 is essential for DNA replication in hematopoietic progenitor cells. J Clin Invest. 2013;123:3112–3123. doi: 10.1172/JCI60806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami Y, Nakayama T. N-terminal region, C-terminal region, nuclear export signal, and deacetylation activity of histone deacetylase-3 are essential for the viability of the DT40 chicken B cell line. J Biol Chem. 2000;275:16191–16201. doi: 10.1074/jbc.M908066199. [DOI] [PubMed] [Google Scholar]
- Taplick J, Kurtev V, Kroboth K, Posch M, Lechner T, Seiser C. Homo-oligomerisation and nuclear localisation of mouse histone deacetylase 1. J Mol Biol. 2001;308:27–38. doi: 10.1006/jmbi.2001.4569. [DOI] [PubMed] [Google Scholar]
- Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS, Gruber PJ, Epstein JA. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324–331. doi: 10.1038/nm1552. [DOI] [PubMed] [Google Scholar]
- Tsai SC, Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. J Biol Chem. 2002;277:31826–31833. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
- Van den Wyngaert I, de Vries W, Kremer A, Neefs J, Verhasselt P, Luyten WH, Kass SU. Cloning and characterization of human histone deacetylase 8. FEBS Lett. 2000;478:77–83. doi: 10.1016/s0014-5793(00)01813-5. [DOI] [PubMed] [Google Scholar]
- Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini A, Volpari C, Gallinari P, Jones P, Mattu M, Carfi A, De Francesco R, Steinkuhler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Walter W, Le Guezennec X, Kim J, Edayathumangalam RS, Lasonder E, Luger K, Roeder RG, Logie C, Berger SL, Stunnenberg HG. A feed-forward repression mechanism anchors the Sin3/histone deacetylase and N-CoR/SMRT corepressors on chromatin. Mol Cell Biol. 2006;26:5226–5236. doi: 10.1128/MCB.00440-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Gaber RF. RPD3 encodes a second factor required to achieve maximum positive and negative transcriptional states in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:6317–6327. doi: 10.1128/mcb.11.12.6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltregny D, Glenisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–968. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- Wang A, Kurdistani SK, Grunstein M. Requirement of Hos2 histone deacetylase for gene activity in yeast. Science. 2002;298:1412–1414. doi: 10.1126/science.1077790. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tian Y, Morley MP, Lu MM, Demayo FJ, Olson EN, Morrisey EE. Development and regeneration of Sox2+ endoderm progenitors are regulated by a Hdac1/2–Bmp4/Rb1 regulatory pathway. Dev Cell. 2013;24:345–358. doi: 10.1016/j.devcel.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell. 2009;138:1019–1031. doi: 10.1016/j.cell.2009.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PJ, Fairall L, Santos GM, Schwabe JW. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–340. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre J, DePinho RA, Dannenberg JH. Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J. 2010;29:2586–2597. doi: 10.1038/emboj.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M, Moser M, Meunier D, Fischer C, Machat G, Mattes K, Lichtenberger B, Brunmeir R, Weissmann S, Murko C, Brosch G, Matthias P, Sibilia M, Seiser C (2013) Divergent roles of HDAC1 and HDAC2 in the regulation of epidermal development and tumorigenesis. EMBO J (manuscript in revision) [DOI] [PMC free article] [PubMed]
- Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: what are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- Wolffe AP. Histone deacetylase: a regulator of transcription [comment] Science. 1996;272:371–372. doi: 10.1126/science.272.5260.371. [DOI] [PubMed] [Google Scholar]
- Wolfson NA, Ann Pitcairn C, Fierke CA. HDAC8 substrates: histones and beyond. Biopolymers. 2013;99:112–126. [Google Scholar]
- Wuelling M, Pasdziernik M, Moll CN, Thiesen AM, Schneider S, Johannes C, Vortkamp A. The multi zinc-finger protein Trps1 acts as a regulator of histone deacetylation during mitosis. Cell Cycle. 2013;12:2219–2232. doi: 10.4161/cc.25267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia J, Zhou Y, Ji H, Wang Y, Wu Q, Bao J, Ye F, Shi Y, Bu H (2013) Loss of histone deacetylases 1 and 2 in hepatocytes impairs murine liver regeneration through Ki67 depletion. Hepatology [DOI] [PubMed]
- Yamaguchi T, Cubizolles F, Zhang Y, Reichert N, Kohler H, Seiser C, Matthias P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010;24:455–469. doi: 10.1101/gad.552310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46–L57. doi: 10.1152/ajplung.00241.2005. [DOI] [PubMed] [Google Scholar]
- Yang W-M, Inouye C, Zeng Y, Bearss D, Seto E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc Natl Acad Sci U S A. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. Functional domains of histone deacetylase-3. J Biol Chem. 2002;277:9447–9454. doi: 10.1074/jbc.M105993200. [DOI] [PubMed] [Google Scholar]
- Yang WM, Yao YL, Sun JM, Davie JR, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem. 1997;272:28001–28007. doi: 10.1074/jbc.272.44.28001. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13:143–153. doi: 10.1016/s0959-437x(03)00015-7. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Seto E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 2007;26:5310–5318. doi: 10.1038/sj.onc.1210599. [DOI] [PubMed] [Google Scholar]
- Ye F, Chen Y, Hoang T, Montgomery RL, Zhao XH, Bu H, Hu T, Taketo MM, van Es JH, Clevers H, Hsieh J, Bassel-Duby R, Olson EN, Lu QR. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin–TCF interaction. Nat Neurosci. 2009;12:829–838. doi: 10.1038/nn.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HG, Choi Y, Cole PA, Wong J. Reading and function of a histone code involved in targeting corepressor complexes for repression. Mol Cell Biol. 2005;25:324–335. doi: 10.1128/MCB.25.1.324-335.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Li Y, Ishizuka T, Guenther MG, Lazar MA. A SANT motif in the SMRT corepressor interprets the histone code and promotes histone deacetylation. EMBO J. 2003;22:3403–3410. doi: 10.1093/emboj/cdg326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng YY, Tang CM, Yao YL, Yang WM, Seto E. Cloning and characterization of the mouse histone deacetylase-2 gene. J Biol Chem. 1998;273(44):28921–28930. doi: 10.1074/jbc.273.44.28921. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Reinberg D. Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell. 1997;89:357–364. doi: 10.1016/s0092-8674(00)80216-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HL, Hinman MN, Barron VA, Geng C, Zhou G, Luo G, Siegel RE, Lou H. Hu proteins regulate alternative splicing by inducing localized histone hyperacetylation in an RNA-dependent manner. Proc Natl Acad Sci U S A. 2011;108:E627–E635. doi: 10.1073/pnas.1103344108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann S, Kiefer F, Prudenziati M, Spiller C, Hansen J, Floss T, Wurst W, Minucci S, Gottlicher M. Reduced body size and decreased intestinal tumor rates in HDAC2-mutant mice. Cancer Res. 2007;67:9047–9054. doi: 10.1158/0008-5472.CAN-07-0312. [DOI] [PubMed] [Google Scholar]
- Zupkovitz G, Grausenburger R, Brunmeir R, Senese S, Tischler J, Jurkin J, Rembold M, Meunier D, Egger G, Lagger S, Chiocca S, Propst F, Weitzer G, Seiser C. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cellular proliferation. Mol Cell Biol. 2010;30:1171–1181. doi: 10.1128/MCB.01500-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupkovitz G, Tischler J, Posch M, Sadzak I, Ramsauer K, Egger G, Grausenburger R, Schweifer N, Chiocca S, Decker T, Seiser C. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol. 2006;26:7913–7928. doi: 10.1128/MCB.01220-06. [DOI] [PMC free article] [PubMed] [Google Scholar]