

Abstract
The design and synthesis of a series of urea-based non-polycyclic aromatic ligands with alkylaminoanilino side chains as telomeric and genomic G-quadruplex DNA interacting agents is described. Their interactions with quadruplexes have been examined by means of Fluorescent Resonance Energy Transfer melting, circular dichroism and surface plasmon resonance-based assays. These validate the design concept for such urea-based ligands and also show that they have significant selectivity for compared to duplex DNA, as well as for particular G-quadruplexes. The ligand-quadruplex complexes were investigated by computational molecular modeling, providing further information on structure activity relationships. Preliminary biological studies using short-term cell growth inhibition assays show that some of the ligands have cancer cell selectivity, although they appear to have low potency for intracellular telomeric G-quadruplex structures, suggesting that their cellular targets may be other, possibly oncogene-related quadruplexes.
Keywords: rational design, diarylureas, quadruplex DNA
Introduction
Nucleic acids containing repetitive guanine-rich tracts can form higher-order structures, G-quadruplexes, which are held together by the G-quartet motif of four in-plane hydrogen-bonded guanines.1-4 Quadruplex arrangements were first described for eukaryotic telomeric DNA sequences, which comprise long tandem G-tracts of simple repeats such as TTAGGG in mammals.5, 6 The terminal 3′ ends of human and other mammalian telomeres are single-stranded,7, 8 and although these are normally associated with single-stranded binding proteins such as hPOT1 in human telomeres,9-12 they can have enhanced potential to fold up into quadruplex arrangements compared to such sequences within duplex DNA. Telomeres in normal human cells progressively shorten with successive rounds of replication,13 but are maintained in length in many cancer cells by the reverse transcriptase action of the telomerase enzyme complex.14-18 Telomerase activity requires a linear 3′ end telomeric DNA template for transcribing, which is inhibited by folding it into a quadruplex, which can be induced by a wide range of quadruplex-stabilizing ligands.19-36
The potential to form quadruplexes have been established in the human and a number of other genomes, and appears to have an elevated frequency of occurrence in eukaryotic promoter regions37 notably in a number of oncogenes such as c-myc,38-41 c-kit,42-44 bcl-245 and K-ras.46 A quadruplex sequence has also been identified in the 5′-untranslated region of the N-ras oncogene.47 The recent discovery of a functional interaction between the human nuclear protein poly (ADP-ribose) polymerase-1 and several quadruplexes, suggests a further role for these structures.48
Telomere regulation and stability is achieved through a complex capping mechanism,49-51 required to protect the chromosome ends from being recognized as damaged DNA, and is necessary for cellular viability.52 Disruption of the capping status of the telomere results in the up-regulation of the DNA damage response system, telomere end – end fusions and other genomic rearrangements.51-55 It has been demonstrated that several established telomerase inhibitors,26, 56 including the trisubstituted acridine compound BRACO-19 (3,6-bis(3-pyrrolidin-1-ylpropionamido)-9-(4-dimethylaminophenylamino) acridine),32 are able to induce rapid cellular responses of senescence and apoptosis in the absence of the characteristic lag period associated with classical telomerase inhibition.57-60 This more rapid response has been associated with intracellular displacement of the telomeric proteins, a γH2AX/p53/p21 mediated DNA damage response and mitotic abnormalities such as telomere end – end fusions and anaphase bridges.22, 27, 61-67 These observations are indicative that these ligands can have a dual action in cells, disrupting the protective capping function of the telomere, as well as acting as classic telomerase inhibitors as seen by progressive telomere shortening.
Knowledge of the details of telomeric68-72 and genomic38, 39, 43 G-quadruplex molecular structures, as well as those of G-quadruplex-ligand complexes39, 73-75 such as that involving BRACO-19,76 can facilitate the rational design, not only of new ligand analogues, but also of novel G-quadruplex DNA binding scaffolds. Crystallographic studies on several telomeric quadruplex-ligand complexes have shown: 1. that in all structures solved to date the quadruplexes have the all-parallel strand topology, as initially observed in the native structures, and 2. the details of loop and groove geometry are variable, dependent on the nature of the individual ligand.75, 76 We have used the general features of the parallel quadruplex to rationally design a new category of non-polycyclic quadruplex ligand, that incorporates features of the inherently planar 1,3-diphenyl urea functionality, a ‘privileged’ scaffold that has well-established drug-like characteristics as well as optimal dimensions in its more stable trans-trans conformation77, 78 for efficient G-tetrad overlap (Figure 1a). This scaffold is present in a number of clinically-approved and experimental therapeutic agents. We report here the design, synthesis and evaluation of a library of molecules, based around this diarylurea functionality.78 Structure-based design has indicated that incorporation of additional phenylcarbamoyl groups allows all four guanine bases of a G-tetrad to be targeted, with addition of cationic side chains to enhance G-quadruplex potency, selectivity and aid aqueous solubility.79 These groups were systematically altered throughout the ligand series to gain an insight into structure-activity relationships (schemes 1 and 2). Telomeric and a small set of promoter (c-kit and c-myc) G-quadruplex interactions were assessed by biophysical assays. Our ultimate aim is to develop a novel series of ligands with low levels of short-term toxicity, so that cellular and ultimately in vivo administration can be undertaken at concentrations that affect telomere maintenance in cancer cells, in the absence of generalized toxic effects.
Figure 1.
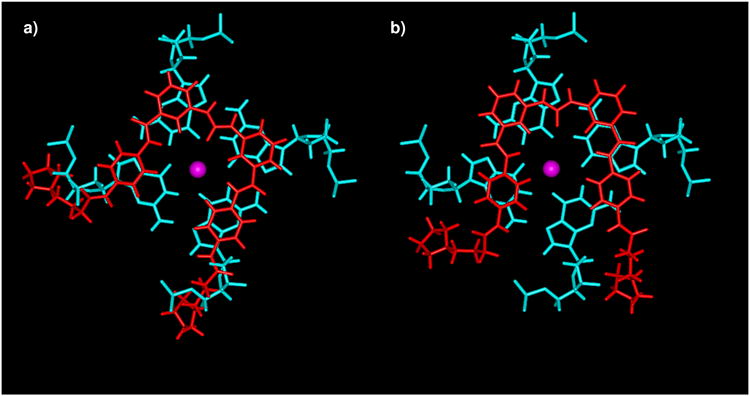
Molecular models showing the overlap of the diarylurea ligand 9c (red) with a terminal G-quartet from the human parallel intermolecular quadruplex (cyan).
a) shows the initial idealized position with maximum overlap between phenyl rings and guanine bases, as used in the initial design concept for the series.
b) shows the position of the same ligand after docking and energy minimization. Note the change in orientation of the ligand, necessary in order to avoid steric clashes between the cationic substituents and quadruplex backbone atoms.
Scheme 1.

The synthetic route to target ligands 8a-f, 9a-i and 10a-f.a
a(i) Cl(CH2)n=1-3COCl, (TEA, THF), rt; (ii) pyrrolidine/dimethylamine/piperidine/morpholine, MeOH/THF, rt; (iii) ammonium formate, 10% Pd/C, MeOH, rt; (iv) tBuOH, DCC, DMAP, DCM/DMF, 0° to rt; (v) H2, 10% Pd/C, MeOH, rt; (vi) CDI, THF, N2, µW; (vii) TFA, rt; viii) PyBOP, DMF, rt.
Scheme 2.

The synthetic route to target ligands 15a-c.a
a(i) Aniline, DCC, HOBt, DMF, rt; (ii) Cl(CH2)n=1-3COCl, TEA, THF, rt; (iii) pyrrolidine, THF, rt; (iv) ammonium formate, 10% Pd/C, MeOH, µW; (v) CDI, THF, N2, µW.
Results
Chemistry
The synthesis of compounds 9a-i and 10a-f was achieved through two key building blocks – the alkylaminoanilino side chains 3a-i, and the urea-bearing di-benzoic acids 7j-l (Scheme 1).
The alkylaminoanilino side chains 3a-i were synthesized from 3- and 4-nitroaniline in overall yields (3 steps) of 19-99%, following methodology adapted from previously reported procedures.30, 31 Acylation was achieved by reaction with the required neat acid chloride, or the acid chloride in the presence of TEA/THF to afford 1a-f in yields of 62-100%. Amination of 1a-b/d-e/g-i was achieved by reaction with the required tertiary amine base (pyrrolidine/dimethylamine/piperidine/morpholine) in the presence of methanol or THF to yield 2a-b/d-e/g-i in 86-100%. 1c/f was reacted with neat pyrrolidine to yield 2c/f in 60-88%. Catalytic hydrogenation afforded the alkylaminoanilino side chains 3a-i in yields of 40-100%.
The synthesis of the diarylurea building blocks 7j-l was achieved from 2-, 3- and 4-nitrobenzoic acid in overall yields (4 steps) of 33-68%. Protection of the carboxylic acid was required to avoid amide bond formation upon treatment with CDI in the urea forming step,80 and was achieved with tbutanol in the presence of DCC and catalytic DMAP81 to afford 4j-l in yields of 72-86%. Catalytic hydrogenation gave the amines 5j-l in yields of 84-100%, which were subsequently coupled into symmetrical ureas 6j-l by reaction with CDI in refluxing anhydrous THF.82-84 6k was isolated following reaction with 0.6 equivalents of CDI in a yield of 93% following workup, whereas 6j/l required an additional 0.6 equivalents of CDI to consume all of the starting amine. This gave 6j/l as crude material which was used without further purification. 7j-l were isolated by suspension of 6j-l in TFA.85 Isolation of the precipitate afforded 7k/l in yields of 95-100%, whereas 7j demonstrated enhanced TFA solubility and was isolated in 46% yield following ether precipitation.
The target ligands 9a-i and 10a-f were synthesized by reaction of the alkylaminoanilino side chains 3a-i with the diarylurea building blocks 7k/l. Several reagents were evaluated in order to achieve this transformation, with PyBOP in DMF86 proving the most efficient without requiring the addition of tertiary amine base due to the basic nature of 3a-i. Basic workup gave the free base of 9a-i and 10a-f in yields of 46-99%. The analogous reaction of the ortho-substituted 7j with the alkylaminoanilino side chains 3a-f led to cyclization of the reactive intermediate, yielding the non-planar quinazoline dione ligands 8a-f (Supplementary Information).
The target ligands 15a-c were synthesized from 3-amino-5-nitrobenzoic acid in overall yields (5 steps) of 18-32% (Scheme 2). Amide bond formation was achieved using DCC/HOBt in DMF87 with an excess of aniline to afford 11 in a yield of 91%. Subsequent acylation to 12a-c in yields of 66-92%, and amination reactions to 13a-c in yields of 77-91% was achieved following the methodology discussed for the synthesis of 2a-i. In the case of the n = 2 side chain, elimination of HCl occurred affording the acrylamide 12b. Catalytic hydrogenation was aided by microwave irradiation on a short time-scale to give 14a-c in yields of 82-92%. Symmetrical urea bond formation was also accomplished by short time-scale microwave irradiation, with 1.0 equivalents of CDI to give the target ligands 15a-c in unoptimized yields of 44-56%.
The free base reaction products 8a-f, 9a-i, 10a-f and 15a-c were purified by semi-preparative HPLC to give the target ligands in suitable analytical purity for biological evaluation.
Circular dichroism studies
The CD spectrum of the native intramolecular human telomeric quadruplex sequence d(TTAGGGTTAGGGTTAGGGTTAGGG), corresponding to four telomeric repeats, shows a small peak at ∼250 and more pronounced peaks at 270 and 288 nm, with a shoulder at (Figure 2). This spectrum is likely to correspond to a mixture of parallel and anti-parallel forms, possibly including hybrid forms as well68-71, 110-112. Five diarylureas were evaluated for their effects on this sequence, compounds 9b, 9d, 15b and two further diarylureas incorporating triazole groups113. Figure 2 shows that the quadruplex topology is retained on binding all of these compounds, with only minor changes in the spectra being apparent, indicating that none of these ligands induce major changes in telomeric quadruplex topology on binding.
Figure 2.

CD spectra of native sequence d(TTAGGGTTAGGGTTAGGGTTAGGG), marked as control, in 100mM potassium chloride/phosphate buffer, and in the presence of five different ligands 9b, 9d, 15b and two triazole-containing diarylureas113.
FRET Assays of Quadruplex Binding and Selectivity
A FRET (fluorescence resonance energy transfer)–based melting assay was used to assess the G-quadruplex stabilization29, 30, 88 and selectivity19, 29, 31, 89 of the ligand library. This showed that all ligands were less effective at quadruplex stabilization than the BRACO-19 molecule.22, 32 However they were significantly more selective for G-quadruplexes.
Ligands 9a-i and 15a-c are superior G-quadruplex DNA stabilizers (Table 1) compared to 8a-f (Supplementary Information) and 10d-f (compounds 10a-c were insoluble under the experimental conditions used). The ΔTm values for 9a-f show that compounds with a n = 1 side chain are less effective quadruplex stabilizers than the n = 2/3 analogues, which show approximately equivalent behavior. This pattern was however not consistent for compounds 15a-c which demonstrate little dependence on length of the side-chains. It is also apparent that the para-side chain substitution pattern of compounds 9a-c generally results in enhanced quadruplex stabilization, except against the F21T (telomeric) G-quadruplex where little substitution pattern dependence was observed. The pyrrolidino basic group (9b) was consistently the most potent end-group, with compounds having piperidino (9h) and morpholino (9i) groups always showing reduced stabilization.
Table 1.
Quadruplex and duplex DNA FRET assay data showing the extent to which the ligand stabilizes DNA sequences against melting. ΔTm1μM is the change in melting temperature (°C) at a 1μM ligand concentration. nd represents values not determined due to insolubility.
| FRET ΔTm at a 1μM ligand concentration (°C) | ||||
|---|---|---|---|---|
| F21T | c-kit1 | c-kit2 | Duplex | |
| BRACO-19 | 25.9 ± 0.2 | 20.1 ± 0.3 | 25.3 ± 0.4 | 11.2 ± 0.6 |
| 9a | 7.9 ± 0.9 | 3.6 ± 1.1 | 11.3 ± 1.4 | 0.0 ± 0.6 |
| 9b | 13.5 ± 0.6 | 10.2 ± 1.1 | 18.7 ± 1.2 | 0.4 ± 0.3 |
| 9c | 12.0 ± 1.3 | 10.1 ± 0.5 | 17.1 ± 1.3 | 0.0 ± 0.6 |
| 9d | 6.8 ± 0.8 | 3.4 ± 0.8 | 10.5 ± 0.6 | 0.3 ± 0.3 |
| 9e | 14.1 ± 0.3 | 6.7 ± 0.7 | 15.3 ± 1.1 | 2.8 ± 0.7 |
| 9f | 13.6 ± 1.3 | 7.8 ± 1.1 | 16.3 ± 0.7 | 0.0 ± 0.3 |
| 9g | 12.3 ± 0.5 | 2.9 ± 1.1 | 13.6 ± 1.0 | 0.1 ± 0.2 |
| 9h | 5.2 ± 0.3 | 7.0 ± 0.2 | 7.1 ± 0.6 | 0.1 ± 0.1 |
| 9i | 1.0 ± 0.2 | 0.0 ± 1.1 | 1.3 ± 0.3 | 0.2 ± 0.3 |
| 10a | nd | nd | nd | nd |
| 10b | nd | nd | nd | nd |
| 10c | nd | nd | nd | nd |
| 10d | 1.8 ± 0.7 | 0.7 ± 0.5 | 2.0 ± 0.4 | 2.5 ± 0.0 |
| 10e | 5.1 ± 0.9 | 0.5 ± 1.0 | 7.5 ± 0.8 | 4.9 ± 0.5 |
| 10f | 3.3 ± 0.6 | 0.0 ± 0.8 | 6.5 ± 1.0 | 5.5 ± 0.6 |
| 15a | 13.6 ± 0.5 | 5.3 ± 0.6 | 8.7 ± 0.3 | 0.0 ± 0.3 |
| 15b | 13.3 ± 0.8 | 4.9 ± 1.2 | 12.9 ± 0.9 | 0.0 ± 0.3 |
| 15c | 12.3 ± 0.6 | 6.3 ± 0.8 | 11.7 ± 0.9 | 0.5 ± 0.0 |
Inter-G-quadruplex selectivity was assessed by the relative ΔTm1µM values measured for the F21T (telomeric),88 and the c-kit143, 44 and c-kit242 G-quadruplexes (Table 1). These show a broad correlation for G-quadruplex stabilization, with the selectivity trend being c-kit2 > F21T > c-kit1 (see Supplementary Information). It should be noted that the c-kit1 G-quadruplex did not produce a simple sigmoidal melting curve in the presence of ligand (i.e. there were multiple phases to the G-quadruplex melting). Hence the experimental melting curves were fitted to idealized sigmoid curves to enable ΔTm values to be obtained by extrapolation (see Supplementary Information).
G-quadruplex vs duplex DNA selectivity was assessed by several FRET-based methods. Low ΔTm1µM stabilization of a FRET-tagged duplex DNA sequence19, 31 indicated high G-quadruplex selectivity (Table 1). Ligands 10d-f however demonstrated comparable G-quadruplex and duplex DNA affinity. Quadruplex selectivity was further probed for the ligands 9a-h and 15a-c by a FRET-based competition assay19, 29, 31, 89 using calf thymus DNA, which demonstrated that all ligands had enhanced G-quadruplex DNA selectivity relative to BRACO-19, with the para-side chain substitution pattern of compounds 9a-c/g/h being optimal (Figure 3).
Figure 3.

FRET-based competition assay data, showing the percentage of retained F21T ΔTm1µM stabilization when an increased ratio of calf thymus duplex DNA competitor is added.
SPR Assays of Quadruplex Binding and Selectivity
The equilibrium binding constants of the potent and highly G-quadruplex selective ligands 9a-c were assessed by surface plasmon resonance (SPR)90, 91 against quadruplex sequences originating from the human telomere (hTel), as well as from the c-kit1/2 and c-myc proto-oncogenes. G-quadruplex selectivity was further assessed using a duplex DNA sequence (Table 2). Figure 4 shows example sensorgrams for ligands 9a-c with c-myc and hTel quadruplexes, and a DNA duplex.
Table 2.
Quadruplex and duplex DNA SPR equilibrium binding constants (KA × 106 M-1) for the strong binding site. na is not available. nd, cannot be determined accurately since KA < 1.0 × 103 M-1.
| SPR Ligand KA (× 106 M-1) for a single-site fit | |||||
|---|---|---|---|---|---|
| hTel | c-kit1 | c-kit2 | c-myc | Duplex | |
| BRACO-1991 | 31.0 | na | na | na | 0.5 |
| 9a | 1.4 | 2.1 | 5.1 | 14.0 | nd |
| 9b | 1.1 | 1.4 | 5.0 | 2.1 | nd |
| 9c | 0.4 | 0.4 | 3.1 | 0.9 | nd |
Figure 4.
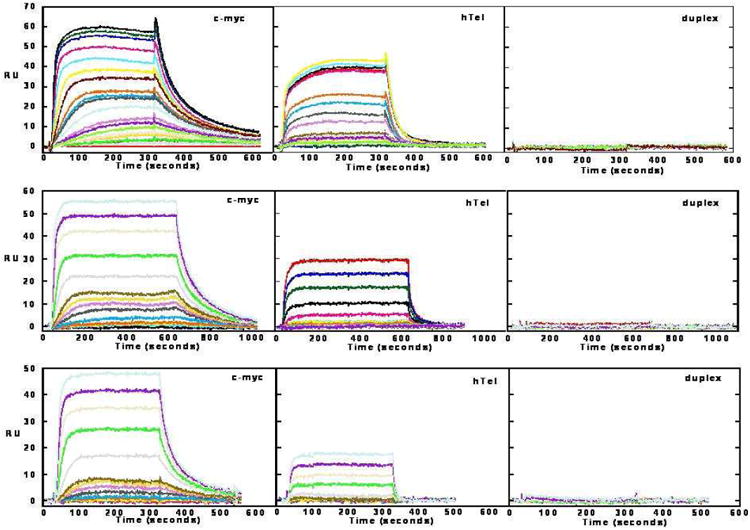
SPR sensorgrams for compounds 9a (top panel), 9b (middle panel) and 9c (bottom panel) with immobilized sequences for the c-myc and hTel quadruplexes, and a DNA hairpin duplex (sequences are given in the Experimental Section).
In terms of G-quadruplex affinity, SPR produced equilibrium binding constants which fit a single strong binding-site model with a significantly weaker secondary binding observed in most cases. In agreement with the FRET ΔTm assay, the SPR results for hTel and c-kit1/2 indicate the strongest binding for each compound is to c-kit2, with c-kit1 and hTel having weaker and more similar affinities (Table 2). The SPR binding constants are inversely proportional to side-chain length, a trend which is not consistent with the results of the FRET assay. There is no FRET data for interactions with the c-myc quadruplex but SPR results indicate inter-G-quadruplex selectivity for the c-myc G-quadruplex-9a interaction. Compound 9a demonstrated slower dissociation kinetics, a stronger interaction and ca 3-fold selectivity for the c-myc quadruplex (Table 2). While the affinity constants for the hTel telomeric quadruplex are reduced relative to BRACO-19,91 the G-quadruplex:duplex DNA selectivity is significantly enhanced, with no duplex DNA interaction detected (KA = < 1 × 103) under the experimental conditions used. This further confirms the G-quadruplex selectivity of ligands 9a-c, which is > 400 – 14000 fold (ratio KQuadruplex/KDuplex).
Molecular Modeling
Ligand structures were build using the Insight II package92 with the CVFF forcefield,93 prior to assessment of the lowest energy conformations of the core structure of ligands 9a-i and 15a-c by manual bond rotation and minimization to convergence (root-mean-square differences (RMSD) < 0.01 kcal mol-1Å-1). Throughout this process the urea and amide bonds were maintained in their more stable trans-conformations.77, 78 This resulted in core ligand structures with all four phenyl rings coplanar. Each low-energy structure differed by 1-5 kcal mol-1 suggesting that several rotamer forms may co-exist (Figure 5a). The lowest-energy conformation of four in-plane phenyl rings has a square arrangement of similar dimensions to that of a G-quartet, and was subsequently used for ligand building.
Figure 5.
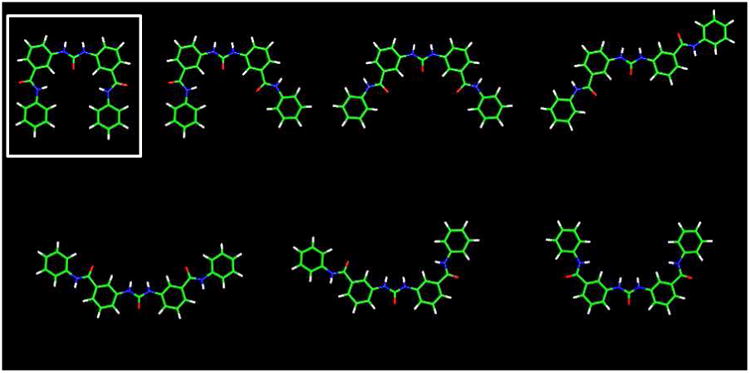

Molecular models of ligands and quadruplex-ligand complexes.
a) The energy minimized all-trans conformation (RMSD < 0.01 kcal mol-1Å-1) cores of ligands 9a-i and 15a-c. Each structure falls within a 1 – 5 kcal mol-1 energy range, and the lowest energy structure is indicate (hydrogen atoms are shown in white, carbon green, nitrogen blue and oxygen red).
b) The low-energy ligand – telomeric G-quadruplex complexes found for ligands 9a-i and 15a-c.
Following the identification of a potent single binding-site mode of G-quadruplex DNA interaction by SPR, a multistage Monte Carlo minimization simulated annealing protocol94, 95 was used to dock the synthesized ligands to the 3′ G-quartet of the parallel crystal form of the human telomeric intramolecular G-quadruplex,68 using the CVFF forcefield93 in the Docking module of Insight II.96 This structure has been used for several previous ligand modeling studies and is supported by the consistent observation of the parallel quadruplex topology in all X-ray crystal structures of human telomeric quadruplex-ligand complexes reported to date.75, 76 A number of low-energy complex structures were predicted by this docking methodology; the urea oxygen atom was consistently predicted to locate over the ion channel of the G-quadruplex. This resulted in the optimal positioning of the diphenyl urea moiety for π-stacking on the exposed G-quartet and allowed the charged side chains to penetrate the G-quadruplex grooves forming electrostatic and hydrogen-bonding interactions. Two low-energy binding modes were predicted for these ligands: the first was demonstrated by ligands 9a-c, 9h, 9i and 15a-c where the ligand core adopted a square conformation allowing three or four phenyl rings to simultaneously π-stack onto the G-quartet (Figures 1b, 5b). The alternative binding mode was predicted for ligands 9d-g where a more extended conformation was observed so that the diphenyl urea was itself optimally positioned for π-stacking, and allowed the side chains to deeply penetrate the grooves (Figure 5b). It was also observed that the n = 2/3 side-chain lengths were optimal since the n = 1 side chain appeared to disrupt π-stacking of the ligand core, and that the sterically compact dimethylamino and pyrrolidino basic groups were better accommodated within the grooves relative to the more bulky piperidino group. The ligand with the uncharged morpholino end-group (9i) did not have electrostatic interactions with the G-quadruplex (Table 3).
Table 3.
Calculated ligand – telomeric quadruplex DNA docking interaction energies (kcal mol-1).
| G-Quadruplex Interaction Energy | |||
|---|---|---|---|
| Total Energy | van der Waals | Electrostatic | |
| 9a | - 1103.23 ± 13.61 | - 101.06 ± 3.21 | - 1002.17 ± 12.83 |
| 9b | - 1157.49 ± 7.33 | - 102.39 ± 3.02 | - 1055.07 ± 7.42 |
| 9c | - 1177.01 ± 16.24 | - 105.31 ± 3.79 | - 1071.66 ± 18.56 |
| 9d | - 1102.44 ± 10.98 | - 94.20 ± 5.04 | - 1008.24 ± 14.99 |
| 9e | -1188.99 ± 14.71 | - 96.98 ± 3.10 | - 1092.01 ± 12.96 |
| 9f | - 1171.68 ± 28.58 | - 103.69 ± 3.56 | - 1069.80 ± 27.58 |
| 9g | - 1140.28 ± 34.06 | - 89.54 ± 4.37 | - 1050.74 ± 31.76 |
| 9h | - 1132.65 ± 25.47 | - 102.92 ± 2.94 | - 1029.72 ± 25.30 |
| 9i | - 134.71 ± 5.52 | - 92.02 ± 2.39 | - 42.68 ± 6.48 |
| 15a | - 1174.23 ± 17.09 | - 100.89 ± 3.44 | - 1073.36 ± 16.11 |
| 15b | - 1182.22 ± 19.06 | - 102.35 ± 2.98 | - 1079.88 ± 20.41 |
| 15c | - 1167.67 ± 16.91 | - 103.31 ± 2.66 | - 1064.37 ± 16.35 |
The calculated interaction energies of the ligands with the G-quadruplex suggest a dependence upon the electrostatic contribution to the total energy. This also showed that all ligands have comparable overall interaction energies, with the exception of ligand 9i with uncharged morpholino end-groups, and ligands 9a and 9d with n = 1 side chains (Table 3).
The G-quadruplex vs duplex DNA selectivity of ligands 9a-c was also examined using a multistage Monte Carlo minimization simulated-annealing procedure, docking the ligands to both the duplex DNA major and minor grooves, as well as to a pseudo-intercalation site built at the GC-step of a 10-mer B-form duplex DNA of sequence analogous to the FRET-based duplex DNA oligonucleotide.95 It was found that the planar aromatic core of ligands 9a-c was sterically too large (∼10.6 – 13.3 Å) relative to the pseudo-intercalation site in the DNA duplex (∼9.2 Å) to allow significant DNA intercalation. The ligands were well accommodated in the duplex DNA grooves; however docking into the minor groove resulted in disruption of Watson-Crick DNA base pairs97 leading to duplex DNA destabilization (Supplementary Information). Hence interaction with the DNA minor groove was discounted on structural stability grounds. Assessment of the interaction energies of these docked models demonstrated that binding to the G-quadruplex was significantly favored relative to DNA duplex intercalation and major groove binding (Table 4). This is consistent with the proposed G-quadruplex: duplex DNA selectivity demonstrated by these ligands in the biophysical assays.
Table 4.
Calculated ligand–duplex DNA docking interaction energies (kcal mol-1), for three binding modes.
| Duplex Interaction Energy | |||
|---|---|---|---|
|
| |||
| Intercalation | Major Groove | Minor Groove | |
| 9a | - 966.99 ± 15.02 | - 1011.11 ± 18.65 | nra |
| 9b | - 956.87 ± 10.39 | - 971.71 ± 28.95 | nra |
| 9c | nrb | - 1027.70 ± 10.63 | nra |
nr represents no result due to:
duplex DNA destabilization, or
no suitably pseudo-intercalated structure observed.
TRAP Assay of Telomerase Activity
The ability of ligands 9a-c, 9f and 9g to inhibit telomerase activity in vitro was assessed by a modified TRAP assay, the TRAP-LIG assay.98 This enables the quantification of telomerase inhibition by removal of the ligand which can inhibit the PCR amplification step of the TRAP assay. Ligands 9a-c, 9f and 9g were all found to be inactive telomerase inhibitors up to the limit of solubility, with EC50 values of >50 µM. This indicates that neither changing the linkage from para to meta nor altering the length of the terminal side-chain results in telomerase inhibition.
Cell Biology
Effects of ligands on cell growth were assessed using a panel of cancer cell lines by means of the SRB assay,99, 100 to give IC50 values. The cancer cell lines screened include the non-small cell lung carcinoma cell line A549 and the breast adenocarcinoma cell line MCF7. The foetal lung somatic cell line WI38 was used as a model for normal human cells to allow assessment of cancer cell selectivity.
The SRB assay results show that the MCF7 cell line was generally the most sensitive to the ligands, with 9b, 9h and 10e having low micromolar IC50 values, comparable to BRACO-1922 (Table 5). A more limited growth inhibition response was observed with the A549 cell line, with ligands 9b and 10e again demonstrating behavior comparable to BRACO-19.22 Selective toxicity for cancer cell lines was assessed by the ratio of the IC50 values for the MCF7 and A549 cell lines, relative to that of the somatic control line WI38. This showed that compounds 9c, 9h and 10e are the most cancer cell selective agents; however a potential “therapeutic window” does exist for several other ligands. Compounds 9d-f and 15a-c show little discrimination between cancer and normal cell lines.
Table 5.
SRB IC50 data (μM). nd represents values not determined due to insolubility.
| SRB IC50 | Selectivity Ratios | |||
|---|---|---|---|---|
| MCF7 | A549 | WI38 | MCF7:A549:WI38 | |
| BRACO-1922 | 2.5 | 2.4 | 10.7 | 4.3 : 4.5 : 1.0 |
| 9a | 14.4 ± 2.8 | > 50.0 | 16.4 ± 1.3 | 1.1 : 0.3 : 1.0 |
| 9b | 2.6 ± 0.3 | 5.3 ± 1.0 | 7.5 ± 2.9 | 2.9 : 1.4 : 1.0 |
| 9c | 11.1 ± 0.8 | > 50.0 | > 50.0 | 4.5 : 1.0 : 1.0 |
| 9d | 34.1 ± 2.0 | > 50.0 | 22.8 ± 2.3 | 0.7 : 0.5 : 1.0 |
| 9e | 14.4 ± 1.5 | > 50.0 | 19.5 ± 1.9 | 1.4 : 0.4 : 1.0 |
| 9f | 49.6 ± 6.0 | > 50.0 | 26.4 ± 5.4 | 0.5 : 0.5 : 1.0 |
| 9g | 12.2 ± 0.6 | > 50.0 | > 50.0 | 4.1 : 1.0 : 1.0 |
| 9h | 5.1 ± 1.4 | > 50.0 | > 50.0 | 9.8 : 1.0 : 1.0 |
| 9i | 40.6 ± 8.4 | > 50.0 | > 50.0 | 1.2 : 1.0 : 1.0 |
| 10a | > 25.0 | > 25.0 | > 25.0 | 1.0 : 1.0 : 1.0 |
| 10b | n.d. | n.d. | n.d. | n.d. |
| 10c | n.d. | n.d. | n.d. | n.d. |
| 10d | > 50.0 | > 50.0 | > 50.0 | 1.0 : 1.0 : 1.0 |
| 10e | 3.2 ± 1.1 | 4.3 ± 1.1 | > 50.0 | 15.6 :11.6: 1.0 |
| 10f | 19.5 ± 2.0 | 19.4 ± 1.7 | > 25.0 | 1.3 : 1.3 : 1.0 |
| 15a | 21.9 ± 2.8 | > 50.0 | 13.8 ± 1.6 | 0.6 : 0.3 : 1.0 |
| 15b | 32.4 ± 2.2 | 41.5 ± 3.0 | 16.2 ± 1.1 | 0.5 : 0.4 : 1.0 |
| 15c | 24.2 ± 1.9 | 30.6 ± 5.6 | 11.0 ± 1.2 | 0.5 : 0.4 : 1.0 |
Longer-term (1 week) exposure of MCF7 cells to sub-toxic concentrations of ligands 9a-c, 9h and 10e resulted in a 0 – 60% reduction in cellular population doublings relative to an untreated control, which was associated with a 1 – 4% incidence of cellular senescence as assessed by the β-galactosidase assay101 (Table 6). Assessment of chromosomal end-end fusions64 was undertaken for ligand 9b and did not show significant changes relative to an untreated control (data not shown).
Table 6.
Long-term (1 week) effects on MCF7 cell growth. vc represents experiments performed solely with a vehicle control.
| Concentration (μM) | PDa | % of Control (vc)b | % Senescencec | |
|---|---|---|---|---|
| vc | - | 4.11 ± 0.34 | 100.0 ± 8.3 | 0.7 ± 0.2 |
| 9a | 5.00 | 3.79 ± 0.12 | 92.2 ± 3.0 | 1.0 ± 0.3 |
| 10.00 | 3.00 ± 0.38 | 73.1 ± 9.2 | 1.7 ± 1.6 | |
| 9b | 1.00 | 4.53 ± 0.07 | 110.4 ± 1.8 | 1.1 ± 1.1 |
| 1.75 | 4.50 ± 0.03 | 109.6 ± 0.73 | 3.1 ± 0.2 | |
| 2.25 | 2.73 ± 0.20 | 66.5 ± 5.0 | 2.4 ± 0.7 | |
| 9c | 3.50 | 3.94 ± 0.24 | 96.0 ± 5.9 | 1.6 ± 0.9 |
| 7.00 | 3.35 ± 0.03 | 81.5 ± 0.8 | 3.1 ± 0.4 | |
| 9h | 2.00 | 3.66 ± 0.00 | 89.2 ± 0.0 | 1.29 ± 0.0 |
| 3.50 | 3.46 ± 0.34 | 84.3 ± 8.2 | 1.33 ± 0.8 | |
| 11e | 1.00 | 3.40 ± 0.26 | 82.9 ± 6.3 | 3.0 ± 0.4 |
| 2.00 | 1.81 ± 0.10 | 44.0 ± 2.4 | 4.1 ± 1.2 |
PD, population doublings ± sd for 1 week MCF7;
percentage of 1 week population doublings normalized to the control (vc = 100%) ± sd;
the percentage of senescent cells assessed by the β-galactosidase assay ± sd.101
Discussion
This study has validated the design concept of non-polycyclic ligands based on the diarylurea skeleton as effective quadruplex-binding ligands coupled with a high level of selectivity over duplex DNA, as shown by molecular modeling and biophysical data. The exceptionally low level of duplex DNA binding is due to poor shape complementarity and consequent disruption of duplex DNA structure. Although ΔTm and affinities are not the highest reported for quadruplex-binding ligands,19, 20 the data presented here suggests that future analogue development to improve affinity is a desirable goal, especially since diarylureas have an inherently drug-like skeleton.
Chemistry
The synthesis of the target ligands 9a-i, 10a-f and 15a-c was achieved in reasonable (unoptimized yields) and high purity, suitable for biological analysis. The synthesis of the 2-substituted target ligands was however not achieved by this methodology, unexpectedly leading instead to the quinazoline dione compounds 8a-f (see the Supplementary Information - structure confirmed by IR spectroscopy 102-105). Alternative synthetic strategies were considered for the synthesis of the 2-subtituted targets. However the cyclization reaction was thought to be favored in all cases due to the proximity of the ortho-substituted phenylcarbamoyl amide nitrogen of the alkylaminoanilino side chain to the imidazole- 1-carboxamide intermediate in the urea forming step.
Biophysical Assays and Molecular Modeling of DNA Interactions
The ligands were screened by a FRET assay, to give an indirect measure of G-quadruplex binding ability through assessment of thermal stabilizing potential at a specified concentration.34, 88, 106 The trend in ATm1μM values for each ligand (the change in melting temperature induced by a ligand concentration of 1μM) is in broad accord with the requirement for ligand planarity; thus the non-planar quinazoline dione ligands 8a-f give only low ΔTm values with all G-quadruplex DNA sequences (Supplementary Information). The global conformation of the ligand core was also shown to be an important factor for efficient G-quadruplex DNA interaction, with the linear analogues 10a-f having only a very weak stabilizing capability with all G-quadruplex DNA sequences (some compounds were insoluble under the experimental conditions used). These particular ligands also have comparable duplex and G-quadruplex DNA affinities, which may result from the structural similarity which these ligands have with duplex DNA groove binding ligands.107
The CD studies show that interactions of the human telomeric intramolecular quadruplex with a representative set of compounds results in a retention of the solution topologies, which indicates that both parallel and antiparallel forms may be present110-112. Molecular modeling has been performed on the parallel form, in accord with several ligand-quadruplex crystal structures75, 76, 114 and the finding that the parallel form is preferred in concentrated solution akin to a cellular environment115. Modeling predicts that the core of ligands 9a-i and 15a-c can form a “square planar” conformation of similar dimensions to a G-quartet, however several other core conformations of comparable energy were also predicted (Figure 5a). The low-energy square planar core would however result in optimal G-quadruplex DNA affinity due to enhanced π-stacking interactions between the four phenyl rings and the guanine bases of the G-quartet, in accord with the experimental findings in the FRET assay of enhanced G-quadruplex DNA stabilizing ability relative to ligands 8a-f and 10a-f. This is generally in accord with SPR binding data for ligands 9a-c, which fits a single strong binding-site mode of G-quadruplex interaction. Further assessment of the potential molecular interactions was obtained by a molecular docking protocol, which found several low-energy binding conformations for these ligands; however the square planar conformation in which the urea oxygen atom was consistently orientated over the ion channel of the G-quadruplex was often observed. The positioning of the urea bond is possibly the result of a weak electrostatic attraction to the cationic ion channel, or a secondary result of the optimal orientation of the π-stacked diphenyl urea moiety. It should be noted that a more extended conformation for bound ligands was also observed, which may represent the conformational diversity predicted for the ligand core. Qualitative assessment of the two predicted core substituted urea conformations indicated that the more extended core is not optimal for π-stacking interactions on the G-quartet, however this is not reflected in the calculated interaction energies of the complexes, which is possibly the result of the force field over-emphasizing electrostatic contributions to the binding. In general calculated energies in this study can only provide semi-quantitative indications of properties since implicit solvent is used in the simulations and the effects of metal ions in quadruplexes are challenging to model precisely.
The FRET assay results for ligands 9a-i and 15a-c show that the G-quadruplex stabilization ability of these ligands depends to some extent upon the length of the alkylamino side chains, with the side-chain length of 9a and 9d (n = 1) always being inferior to those of 9b/c or 9e/f (n = 2/3), which are approximately comparable. This pattern is not however shown by ligands 15a-c, which have reduced side-chain length dependence. The results of the docking study are consistent with these results with a qualitative assessment of the generated low-energy complexes showing that the side chains are generally able to be deeply buried in the G-quadruplex DNA grooves making several electrostatic and hydrogen-bonding contacts, however the n = 1 side chain was inferior to the n = 2/3 since forming favorable interactions within the grooves affected the orientation of the aromatic core on the G-tetrad surface altering the π-stacking of the ligand. This effect is shown by the order of calculated interaction energies for these complexes. The high correlation between calculated interaction energies and the FRET-based ΔTm1µM values (Supplementary Information) (r = - 0.833) indicates the predictive value of this docking method for the design of further ligands in this series.
The side-chain length dependence of G-quadruplex interaction was also assessed by SPR measurements. The results did not correlate with the FRET observations, although this is probably too small a sample from which to draw firm conclusions. SPR results indicate that the n = 1 side chain of 9a is optimal for general G-quadruplex DNA interaction over 9b (n = 2) and 9c (n = 3). It may be that in this instance affinity and thermal stability cannot be directly compared. For the hTel, c-kit1/2 and duplex DNAs where FRET and SPR results can be compared, there is however good agreement (Tables 1 and 2).
Variations in the basic end-groups were also assessed by the FRET assay. This showed that for the telomeric and c-kit2 G-quadruplexes, the pyrrolidino (9b) and the dimethylamino basic groups (9g) resulted in superior quadruplex stabilization compared to the piperidino group (9h). Qualitative examination of the predicted molecular interactions of the ligand – telomeric G-quadruplex complexes indicated that this may result from the decreased steric bulk of the pyrrolidino and dimethylamino basic groups, which the modeling suggests are well accommodated in the DNA grooves relative to the piperidino group. The FRET assay results also indicate that the introduction of the non-basic morpholino group (compound 9i) is deleterious for all G-quadruplex sequences examined here, with docking to the telomeric quadruplex suggesting that this may result from poor groove penetration, a direct result of the absence of cationic charge at physiological pH, thus minimizing electrostatic contribution to the predicted interaction energy. The dimethylamino ligand 9g also has a low ΔTm1µM value with the c-kit1 G-quadruplex, which has no explanation in the absence of a detailed structural model for this complex.
Examination of potential selectivity between different G-quadruplexes as assessed in the FRET assay (comparison of the ΔTm1µM values) shows for any one compound a broad correlation with G-quadruplex DNA affinity, which may be expected since the ligands were initially designed to interact generally with G-quadruplexes (focusing on G-quartet recognition) and not for a specific G-quadruplex. However selectivity, generally in the order c-kit2 > F21T > c-kit1 G-quadruplexes is apparent. The c-kit1 G-quadruplex was observed to melt in a non-sigmoidal manner (Supplementary Information), which may result from the structural difference between the c-kit1 and other G-quadruplex folds. The SPR results indicate a distinct order of selectivity with c-myc > c-kit2 > c-kit1 > F21T, with the interaction of ligand 9a being particularly enhanced for the c-myc quadruplex, which also has slower dissociation kinetics. It should be noted that the FRET-based assessment of the interactions of these ligands with the c-myc G-quadruplex is not possible under the experimental conditions used, due to the exceptional stability of this quadruplex fold.
The best agreement between the FRET and SPR assays is for G-quadruplex vs duplex DNA selectivity, which shows that ligands 9a-i and 15a-c, and especially the para-substituted ligands 9a-h, are significantly more G-quadruplex selective than BRACO-19. The lack of stabilization of a duplex DNA oligonucleotide complex (ΔTm1µM < 2.8°C) indicates a high level of selectivity for G-quadruplex forming sequences, which was confirmed by a competition assay using calf-thymus DNA at a 200-fold excess of duplex DNA. A sub-set of these ligands (9a-c) were assessed for duplex DNA interactions by SPR, with no significant duplex DNA interaction found (KA <1.0 × 103 M-1). This represents at least 400 – 14000 fold selectivity for G-quadruplex vs duplex DNA. These selectivity results have been rationalized by the molecular modeling studies with ligands 9a-c and duplex DNA, which show that the core of the ligands is sterically too large for intercalation. Although the ligands were observed to dock tightly into the duplex DNA grooves, these modes have lower calculated interaction energies relative to that for G-quadruplex interaction.
Examination of the predicted interaction energies for these ligands indicates a dependence upon the electrostatic contribution to the binding, and independence from the predicted binding mode, with all interaction energies comparable across this series.
Cell Biology
There are few clear patterns in the short-term cell growth inhibtion data for the two cancer cell lines used here (IC50 values). However it is apparent that altering the basic group from pyrrolidino (9b) as is the case for ligands 9g-i results in improved MCF7 selectivity albeit with reduced potency. It is also evident that the linear ligands 10e and 10f have selectivity for cancer cells over a normal cell line, and that the n = 2 side chain of 10e confers enhanced potency. These effects do not correlate with the predicted G-quadruplex or duplex DNA ΔTm1µM results from the FRET assay. We have not measured cellular uptake of these compounds. However the potency shown by some compounds in the short-term SRB cell viability assay (Table 5) indicates that some at least are readily able to cross cell membranes, although at this stage we cannot discount the possibility that the less potent are not able to do so.
Investigation into the longer-term effects (1 week) of the more toxic ligands (for example, 9b) in the MCF7 cell line used sub-cytotoxic concentrations of ligands. Over this period, the ligands had a significant effect on population growth, which was associated with a small induction of cellular senescence. No significant levels of telomere end – end fusions were observed over this time period. These results, taken with the lack of telomerase inhibitory activity, suggest that these ligands are at best only modest inducers of telomere uncapping/dysfunction. This further suggests that a minimum threshold level of telomeric quadruplex affinity is required for effective interference with telomere function in cancer cells, which is not reached by these particular diarylurea ligands, but is achieved by compounds such as BRACO-19,22, 24, 32, 61, 76 telomestatin20, 56, 62, 63, 67 and the more recently-developed naphthalene diimides19.
There is a strong suggestion from the IC50 data that the compounds reported here fall into several distinct biological classes, that may reflect distinct modes of action (though differences in cellular uptake cannot be discounted): (i) the para-substituted set 9a-c/g-i, which are moderately selective for the cancer cell lines, especially MCF7; (ii) the smaller set 10a-f, with compound 10e showing exceptional selectivity; and (iii) the non-selective meta-substituted sets 9d-f and 15a-c. The diarylurea skeleton is represented in many kinase inhibitors, for example, multi-targeted tyrosine kinase inhibitors108 and inhibitors of insulin-like growth factor I receptor signaling.109 Even though the substitution patterns in the present compounds have not been previously reported for kinase inhibitors, we cannot discount at present the possibility that the biologically active compounds reported here may be acting directly on kinase binding sites.
Conclusions
The purpose of this study was to design and evaluate a novel series of non-polycyclic compounds that would show effective binding to quadruplex DNAs. This goal has been achieved through the diphenyl urea-based scaffold. Even though initial studies of their cellular effects indicate that the derivatives discussed here do not show potent telomerase inhibitory activity, further analogue development is warranted. A number of the present compounds did show significant effects on cancer cell growth which are not associated with significant telomere uncapping/dysfunction and are unlikely to be effects on duplex DNA. We introduce the concept that a threshold level of quadruplex affinity is required for a compound to show such effects. It is also possible that interactions with non-telomeric quadruplexes may be involved, and screening is planned on larger libraries of derivatives with a more extensive set of quadruplexes from oncogenic promoter sequences37, as well as use of a larger panel of cancer cell lines.
Experimental
Fluorescence Resonance Energy Transfer (FRET) Assays
FRET oligonucleotides (Eurogentec Ltd., U.K.) have the following sequences: F21T, 5′FAM-d[G3(T2AG3)3]-TAMRA3′; c-kit1, 5′FAM-d[AGAG3AG2GCGCTG3AG2AG3GCT]-TAMRA3′; c-kit2, 5′FAM-d[C3G3CG3CGCGAG3AG4AG2]-TAMRA3′; duplex, 5′FAM-d[(TA)2GC(TA)2T6(TA)2GC(TA)2]-TAMRA3′ where FAM is 6-carboxyfluoresein and TAMRA 6-carboxytetramethylrhodamine. FRET Assay: The required oligonucleotide was suspended in FRET buffer (60mM KCl, Kcacodylate, pH 7.4; 400nM DNA) and heated to 85ºC for 10 minuets prior to cooling to room temperature. DNA was distributed (50µl) across a 96 well RT-PCR plate (Bio-Rad) to which ligand was added (50µl; stored as a 20mM DMSO stock, -20°C; diluted to 1mM in HPLC grade DMSO) to afford the required concentration. FRET buffer was used as a negative control. DNA melting was assessed upon a MJ Research Opticon DNA Engine Continuous Fluorescence Detector exciting at 450 – 495nm. Fluorescence values were recorded at 515 – 545nm at 0.5ºC intervals as the plate was heated from 30 – 100ºC. The data was analysis in the Origin 7.0 software package (Origin Lab Corp., Northampton, MA). In the case of the c-kit1 oligonucleotide, melting curves were fitted to sigmoid curves prior to analysis. The change in melting temperature at 1µM ligand concentration (ΔTm1µM) was calculated from four experiments by subtraction of the averaged negative control from the averaged 1µM ligand melting temperature ± the maximum standard deviation (sd). Competition Assay: To F21T DNA (50µl) was added calf thymus DNA (CT-DNA; 25µl; 533.3µM CT-DNA bp stock in 0.5mM EDTA/30mM K+ cacodylate buffer) to afford the required CT-DNA bp concentration. To this was added ligand (25µl, 4µM) to afford the required ligand concentration (1µM). FRET buffer represented no competitor. The percentage retained stabilization was calculated from three experiments, and normalized to the ΔTm1µM for that ligand with no CT-DNA competitor (100%) ± normalized sd. BRACO-19 was prepared in-house30, 32 to a HPLC purity of >98% (data not shown).
Circular dichroism (CD) studies
CD spectra of the 24-mer telomeric DNA sequence d(TTAGGG TTAGGGTTAGGGTTAGGG) both in the absence of ligands and with ligands 9b, 9d and 15b together with two triazole-containing diarylurea derivatives synthesized in a separate project110 were acquired on an Applied Photophysics Ltd Chirascan spectrometer at King's College London. All samples were prepared at 100µM in 100mM potassium chloride/phosphate pH 7.4 and heated to 95ºC and slowly annealed overnight to room temperature. The samples were further diluted, with buffer to 0.8 optical density unit prior to data collection and where appropriate ligand was added to give a 1:1 molar ratio. UV absorbance & CD spectra were measured between 320-220 nm in a 10 mm path length cell. Spectra were recorded with a 0.5 nm step size, a 1.5s time-per-point and a spectral bandwidth of 1 nm. All spectra were acquired at room temperature and buffer baseline corrected. The concentration of the above oligonucleotide was determined by using the absorbance value at 260nm and the Beer-Lambert law.
Surface Plasmon Resonance (SPR)
Biosensor experiments were conducted in filtered, degassed HEPES buffer (10mM HEPES, 100mM KCl, 3mM EDTA, 0.00005v/v of 10% P20 BIACORE surfactant, pH 7.3) at 25°C. The 5′-biotin labeled DNA sequences (Integrated DNA Technologies) were HPLC purified and comprise sequences: hTel, 5′biotin – d[AG3(T2AG3)3]3′; c-kit1, 5′biotin – d[(AG3)2CGCTG3AG2AG3]3′; c-kit2, 5′biotin – d[(CG3)2CGCGAG3AG4]3′; c-myc, 5′biotin – d[(AG3TG4)2A]3′; duplex: 5′biotin – d[CGA2T2CGTCTC2GA2T2CG]3′. The experiments were conducted upon a BIAcore 2000 optical biosensor instrument (BIAcore Inc.). Flow cell 1 was left blank as a reference while flow cells 2 – 4 were immobilized with DNA on a streptavidin-derivatized gold chip (SA chip from BIAcore) by manual injection of DNA stock solutions (flow rate of 1µl/min) until the desired value of DNA response was obtained (350-400 RU). Typically, a series of different ligand concentrations (1nM to 10µM from 20mM DMSO stock) were injected onto the chip (flow rate of 50µl/min, 5 – 10 min) until a constant steady-state response was obtained followed by a dissociation period (buffer, 10 min). After every cycle, the chip surface was regenerated (20 sec injection of 10mM glycine solution, pH 2.0) followed by running buffer flow. The data was processed as previously described,30, 91 using BIAevaluation (BIAcore Inc.) and Kaleidagraph (Synergy Software) software for nonlinear least squares optimization of the binding parameters.
Molecular Modeling
Molecular Modeling was performed on an SGI Octane workstation (2 × 225MHz MIPS R12000 CPUs; IRIX64 6.5 OS; Silicon Graphics Ltd.) with the Insight II software package (Accelerys Software Inc., 2000) utilizing the Builder, Discover and Docking modules. G-quadruplex DNA was prepared from the 22-mer crystal structure of the G-quadruplex formed from the human telomere of sequence d[AG3(T2AG3)3] (PDB 1KF1),68 and a self-complementary duplex DNA was build in Insight II of the sequence d[(TA)2GC(TA)2]. An intercalation site with 6.76Å base-pair separation was introduced at the GC step of the DNA duplex. Ligands were built with atomic potentials and partial charges assigned in the CVFF force field prior to minimization (500 steps steepest descents; 5000 steps conjugate gradients; RMSD of 0.001 kcal mole-1 Å-1) in the Discover module. Basic side chains consisting of the pyrrolidino, dimethylamino and piperidino groups were built as protonated species by addition of a formal atomic charge. Morpholino groups were kept uncharged. Core conformational analysis was achieved by manual rotation prior to minimization (500 steps steepest descent; successive 2000 step conjugate gradients until converged to an RMSD of 0.01 kcal mole-1 Å-1) in the Discover module of Insight II.
A multistage Monte Carlo minimization simulated-annealing docking protocol analogous to those previously reported94, 95 was utilized in the Docking module of Insight II to determine the potential low energy ligand – DNA complexes and their energy, using default program settings unless specified. For G-quadruplex interaction, the ligand was positioned ∼5 Å above the centre of the 3′-G-quartet, defining their hydrogen atoms as the binding site about which a Monte Carlo minimization methodology was employed to generate 200 random orientations of the starting structure, with van der Waals radii scaled to 10% of their full values and charges not considered. The maximum allowable energy change of successive random structures was 10000 kcal mol-1 with an energy range of 40 kcal mol-1. Each conformation was minimized (300 steps of conjugate gradients) and the 75 lowest total system energy conformations filtered by Insight II for further analysis. For duplex DNA interaction, an analogous procedure was used defining the binding site as all of the hydrogen atoms which project into the required DNA groove, or by inserting the ligand into the DNA pseudo-intercalation site in the best perceived conformation, defining the hydrogen atoms of the GC base pairs as the binding site. These 75 structures were subjected to 800 steps of conjugate gradients minimization in which charges were included, van der Waals radii were set to 100% and with a distant dependant dielectric constant of 1.r, prior to simulated annealing between 500 – 300K over 10 ps to the predefined binding site. The resulting structures were energy-minimized (800 steps of conjugate gradients) and the 25 lowest total system energy conformations filtered by Insight II for further manual evaluation. These docked structures were analyzed based upon the chemical correctness of the ligand ensuring trans urea and amide integrity, the lowest total interaction energy of the complex assessed for all atoms with no cut-off, the number of hydrogen bonding interactions observed, the binding modes which best represent the populations and the perceived correctness of the proposed ligand binding mode. This allowed the selection of two lead low energy binding conformations for further analysis. Each low energy binding conformation was used to define a flexible binding site containing all DNA atoms residing 7 – 8 Å from the ligand, fixing all other atoms and tethering the 3′-G-quartet or the terminal duplex DNA TA base pairs. A further 30 rounds of [300 steps of conjugate gradients minimization prior to simulated annealing between 800 – 200K over 10 ps] was performed using a distance-dependent dielectric constant of 4r. Final minimization (1000 steps conjugate gradients) allowed the filtering of the 15 lowest total system energy conformations by Insight II for final analysis. These bound ligand – DNA complexes were manually assessed by the predefined criteria, averaging the total interaction energy observed for the 10 lowest energy binding conformations ± sd, assessed between all atoms with no cut-off. Figures were constructed from a docked binding conformation selected to best represent the optimal low energy binding conformations of the population.
Cell Culture
Cell lines were supplied by ATCC-LGC Promochem and viability maintained in a Heraeus Hera Cell 240 incubator (37ºC, 5% CO2; 75 cm2 plates: TPP). Cells were removed for experimentation as required. Sterile work was conducted in a Heraeus Hera Safe hood. Dulbecco's Modified Eagles Media (DMEM; Invitrogen) supplemented with foetal bovine serum (10% v/v; Invitrogen), hydrocortisone (0.5µg/ml; Acros Organics), L-glutamine (2mM; Invitrogen) and non-essential amino acids (1×; Invitrogen) was used for the MCF7 and A549 cell lines, and Minimal Essential Medium (MEM; Sigma-Aldrich) supplemented with foetal bovine serum (10% v/v; Invitrogen), L-glutamine (2mM; Invitrogen) and non-essential amino acids (1×; Invitrogen) was used for the WI38 cell line.
SRB Cytotoxicity Assay
Short term growth inhibition was measured using the SRB assay as described previously.22, 30, 100 Briefly, cells were seeded (4000 cells/wells) into the wells of 96 well-plates in appropriate medium and incubated overnight to allow the cells to attach. Subsequently cells were exposed to freshly-made solutions of drug at increasing concentrations between 0.25 – 50 µM in quadruplicate and incubated for a further 96h. Following this the cells were fixed with ice cold trichloaceticacid (10% w/v) for 30 min and stained with 0.4% SRB dissolved in 1% acetic acid for 15min. All incubations were carried out at room temperature. The IC50 value, concentration required to inhibit cell growth by 50%, was determined from the mean absorbance at 540nm for each drug concentration expressed as a percentage of the well absorbance in untreated control cells.
Sub-Cytotoxic Induction of Cellular Senescence (β-Galactosidase Assay).30, 101
MCF7 Cells (1×105, 10ml media, ATCC-LGC Promochem) were exposed to two independent sub-cytotoxic concentrations of the required ligand over a 1 week period, with a biweekly treatment. A media negative control was also screened. Cells were counted upon a Neybauer haemocytometer (Assistant, Germany) and the number of cellular population doublings assessed by the equation n = (log Pn – log P0)/log2 where Pn is the number of cells collected and P0 the initial seeding density. Cells were stained for senescence using the β-galactosidase staining kit (Cell Signaling Technology) according to the manufacturer's instructions. In short, cells were seeded (1×105, 2ml) in a 6 well plate (Fisher-Scientific) with the required ligand concentration and incubated overnight. The medium was removed, the well washed with PBS (2ml) prior to fixing (1× fixative solution, 10 min). The fixative was removed and the well washed with PBS (2 × 2ml) prior to the addition of the staining solution (1ml) and the plates incubated overnight. Three independent fields of cells were visualized (200× magnification) from both repeats, with the mean percentage of blue senescent cells reported ± sd.
TRAP-LIG assay
This was performed as recently described.98
Chemistry
All reagents were reagent grade and were used as supplied without further purification (Sigma-Aldrich, Alfa Aesar, Avocado Organics and Lancaster Synthesis). Palladium catalyst was 10% wt loading on activated carbon. Ammonia in methanol (NH3/MeOH, ca 7N; Sigma-Aldrich) was diluted in MeOH as required. Solvents (BDH and Fisher Scientific), anhydrous solvents (Sigma-Aldrich) and HPLC grade solvents (Fisher Scientific) were used as supplied. Microwave reactions were conducted in a Biotage Initiator Microwave, software version 1.1, in Biotage vials (0.5ml - 2.0ml or 2.0ml - 5.0ml) sealed with Biotage caps with septa. All chemistry was conducted in clean, oven dried glassware. 1H NMR and 3C NMR spectra were recorded at 295K upon on a Bruker Avance 400 spectrometer using the specified deuterated solvent (GOSS Scientific and Sigma-Aldrich). NMR spectra were analyzed by the MestReC 4.5.6.0 program with chemical shifts calibrated to the residual proton and carbon resonance of the solvent. NMR multiplicity and coupling constants (J) are reported as observed. Melting points (mp) were measured on a Bibby Stuart Scientific SMP3 melting point apparatus, where ‘dec’ indicates decomposition. Mass spectra were recorded upon a ThermoQuest Navigator Mass Spectrometer using electrospray ionization in positive ([M+H]+) or negative ([M-H]-) modes. High resolution accurate mass spectra were recorded upon a Micromass Q-TTOF Ultima Global Tandem Mass Spectrometer using electrospray ionization mode and 50% acetonitrile in water and 0.1% FA as solvent, and processed using the MassLab 3.2 software. Infrared spectra were recorded from neat samples upon a Nicolet Smart Golden Gate spectrometer (Avatar 360 FT-IR E.S.P.) and processed using the software package OMNIC E.S.P. 5.1. Flash chromatography was conducted on silica gel (partial size 33 - 70µm; BDH). Analytical HPLC was performed on a Gilson Chromatograph with a YMC C18 5µ (100 × 4.6mm) column and an Agilent 1100 series Photo Diode array detector. Spectra were processed in the Unipoint 5.11 software and compound purity and retention times (RT) were assessed at 254nm unless stated. Several HPLC solvent systems and gradients were employed as specified: Method A, 0.1% TFA in MeOH and 0.1% aqueous TFA, 25% - 75% organic over 28 min (1ml/min); Method B, 0.1% TFA in MeOH and 0.1% aqueous TFA, 25% - 50% organic over 18 min (1ml/min); Method C, 0.1% FA in acetonitrile and 0.1% aqueous FA, 5% - 50% organic over 28 min (1ml/min); Method D, 0.1% FA in acetonitrile and 0.1% aqueous FA, 5% - 50% organic over 18 min (1ml/min); Method E, 0.1% FA in MeOH and 0.1% aqueous FA, 25% - 75% organic over 18 min (1ml/min). Semi-preparative reversed-phase HPLC (semi-prep. HPLC) was performed on a Gilson Chromatograph with a Gilson 215 Liquid Handler, a Gilson 845Z injection module coupled to a Gilson UV/VIS 155 detector and a YMC C18 5µ (100 × 20mm) column. Several HPLC solvent systems, gradients and methods of sample isolation were employed as specified: Method F, 0.1% TFA in MeOH and 0.1% aqueous TFA, 25% -75% organic over 25 min (10ml/min), injected from 25% MeOH in 0.1% aqueous TFA (3ml); Method G, 0.1% TFA in MeOH and 0.1% aqueous TFA, 25% - 50% organic over 60 min, 50% - 75% organic over 5 min (10ml/min), injected from 25% MeOH in 0.1% aqueous TFA (3ml). Purified compound were isolated by reduction of fraction volumes (5 ml), precipitation with 5% NH3 (aq.) and filtration.
Method 1 (1a-f and 12a-c)
a) The required nitroaniline was dissolved in THF (150ml) and cooled to 0ºC prior to the addition of TEA (2 equiv.) and the required acid chloride (1.5 – 2 equiv.), and the mixture stirred overnight at room temperature. The precipitate was filtered and the solvent evaporated in vacuo. The product was either precipitated with sat. NaHCO3 (aq.) (200ml), or dissolved in EtOAc (50ml), washed with 5% NH3 (aq.) (50ml), brine (50ml) and dried over MgSO4; b) The required nitroaniline (15.0g, 0.109 mol) was added to the appropriate acid chloride (45ml, 0.402 – 0.471 mol) in portions at room temperature, followed by stirring at 50ºC overnight. The mixture was cooled to 0ºC and the product isolated by filtration, washing with ether (50ml).
Method 2 (2a-i and 13 a-c)
1a-b/d-e/g-i or 12a-c was dissolved in MeOH/THF (5 – 100ml) and the required amine base (2 – 3.6 equiv.) was added. The mixture was either: a) stirred overnight at room temperature prior to the solvent being removed, 5% NH3 (aq.) (50ml) was added and the product extracted with EtOAc (50ml), washed with brine (25ml) and dried over MgSO4; b) heated under reflux for 4h prior to workup as above; c) stirred at 30ºC overnight prior to workup as above. 1c/f was reacted with neat pyrrolidine (20ml, 0.240 mol) at room temperature overnight, followed by evaporation of the excess pyrrolidine and workup as described above.
Method 3 (3a-i and 14a-c)
a) 2a-i was dissolved in MeOH (100ml) then ammonium formate (10 equiv.) and Pd/C (0.1 equiv. w/w) was added. The mixture was stirred overnight at room temperature prior to filtration through celite and evaporation of the solvent. The residue was dissolved in CHCl3 (100ml), washed with 5% NH3 (aq.) (2 × 50ml), brine (50ml) and dried over MgSO4. b) 13a-c was suspended in absolute EtOH (4ml) prior to the addition of ammonium formate (4 equiv.) and Pd/C (0.1 equiv. w/w). The mixture was heated by microwave irradiation (120ºC, 10 min, 7 bar) followed by workup as described above.
Method 4 (4j-k)
The required nitrobenzoic acid (5.0g, 0.030 mol) was dissolved in anhydrous DCM (150ml) and DMF was added as required. DMAP (500mg, 0.1 equiv. w/w) and tBuOH (4.29ml, 0.045 mol, 1.5 equiv.) was added prior to cooling the mixture to 0ºC and adding DCC (6.79g, 0.033 mol, 1.1 equiv.) in two portions. The mixture was warmed to room temperature and stirred for 4h. The precipitate was filtered and the DCM washed with 1M HCl (aq.) (2 × 50ml), brine (50ml) and dried over MgSO4. The resulting oil was purified by flash chromatography.
Method 5 (5j-k)
4j-k was dissolved in MeOH (100ml) and Pd/C (0.1 equiv. w/w) was added prior to purging with H2 (g), and the mixture stirred at room temperature until complete when it was filtered through celite and the solvent evaporated in vacuo.
Method 6 (6j-k and 15a-c)
a) 5j-k was dissolved in anhydrous THF to a concentration of 0.4 M and CDI (0.6 – 1.2 equiv.) was added and the mixture heated at reflux for 24h under N2 (g). The THF was evaporated and the residue dissolved in EtOAc (200ml), washed with 1M HCl (aq.) (2 × 100ml), brine (50ml) and dried over MgSO4; b) as above 14a-c was reacted with CDI (1.0 equiv.) by microwave irradiation (75ºC, 20 min, N2 (g)). The mixture was cooled to 0°C and the product precipitated with EtOAc (10ml).
Method 7 (7j-k)
6j-k was suspended in TFA (30ml) and stirred vigorously at room temperature for 1h. The precipitate was isolated by filtration, washed with DCM (50ml) and oven dried. If no precipitate formed, precipitation was induced by cooling the mixture to 0ºC and treatment with ether (50ml).
Method 8 (8a-f, 9a-i and 10a-f)
7j-k was dissolved in anhydrous DMF (8ml) and to this was added the required side chain 3a-i (4 equiv.) and PyBOP (3 equiv.) and the mixture stirred at room temperature under N2 (g) for 22h. The DMF was evaporated and the resulting oil suspended in CHCl3 (20ml) with sonication. The resulting precipitate was isolated by filtration, however when an oily residue formed the organic solvent was decanted. In either instance, this crude material was dissolved in NH3/MeOH (1M, 3ml), the product precipitated with 5% NH3 (aq.) (30ml) and isolated by filtration.
Detailed experimental procedures and analytical data for the side-chain building-blocks 1a-f, 2a-i, 3a-i, 4j-l, 5j-l and the quinazoline dione ligands 8a-f are given in the Supplementary Information.
2,2′-ureylene-di-(tbutyl benzoate) (6j)
Following method 6a, 5j (3.38g, 17.488 mmol) was reacted with CDI (3.40g, 20.986 mmol) to yield a yellow solid (3.59g, 8.703 mmol, quant.) which was used without further purification. 1H NMR (400MHz, CDCl3) 10.69 (2H, s), 8.46 (2H, dd, J=8.5, 0.9 Hz), 7.96 (2H, dd, J=8.0, 1.6 Hz), 7.49 (2H, ddd, J=8.7, 7.3, 1.7 Hz), 6.99 (2H, ddd, J=8.2, 7.3, 1.1 Hz), 1.64 (18H, s) ppm. 3C NMR (100MHz, CDCl3) 167.8, 152.4, 142.4, 133.8, 131.0, 121.1, 120.0, 116.4, 82.3, 28.3 ppm. HRMS m/z calc. C23H28N2O5 [M+H]+ 413.2071, found 413.2065.
3,3′-ureylene-di-(tbutyl benzoate) (6k)
Following method 6a, 5k (4.01g, 20.744 mmol) was reacted with CDI (2.02g, 12.458 mmol) to yield a white solid (3.97g, 9.625 mmol, 93%). mp 296ºC dec. 1H NMR (400MHz, d6-DMSO) δ 8.97 (2H, s), 8.09 (2H, t, J=1.8 Hz), 7.75 (2H, ddd, J=8.0, 2.1, 1.0 Hz), 7.58 (2H, d, J=7.8 Hz), 7.46 (2H, t, J=7.9 Hz), 1.61 (18H, s) ppm. 13C NMR (100MHz, d6-DMSO) δ 164.8, 152.5, 139.8, 131.9, 129.0, 122.5, 122.5, 118.7, 80.7, 27.8 ppm. HRMS m/z calc. C23H28N2O5 [M+H]+ 413.2071, found 413.2075.
4,4′-ureylene-di-(tbutyl benzoate) (6l)
Following method 6a, 5l (3.71g, 19.202 mmol) was reacted with CDI (3.74g, 23.042 mmol) to yield a white solid (3.94g, 9.552 mmol, 99%) which was used without further purification. 1H NMR (400MHz, CDCl3) δ 9.23 (2H, s), 7.89 (4H, d, J=8.8 Hz), 7.63 (4H, d, J=8.8 Hz), 1.59 (18H, s) ppm. HRMS m/z calc. C23H28N2O5 [M+H]+ 413.2071, found 413.2092.
2,2′-ureylene-di-benzoic acid (7j)
Following method 7, 6j (3.11g, 7.546 mmol) was deprotected to yield a white soild (1.05g, 3.483 mmol, 46%). mp 192 – 194ºC. 1H NMR (400MHz, d6-DMSO) 13.48 (2H, s), 10.73 (2H, s), 8.29 (2H, dd, J=8.4, 0.8 Hz), 7.95 (2H, dd, J=7.9, 1.6 Hz), 7.56 (2H, ddd, J=8.7, 7.3, 1.7 Hz), 7.09 (2H, m) ppm. 13C NMR (100MHz, d6-DMSO) 169.7, 151.7, 141.6, 133.8, 131.0, 121.5, 119.7, 116.4 ppm. HRMS m/z calc. C15H12N2O5 [M+H]+ 301.0819, found 301.0814.
3,3′-ureylene-di-benzoic acid (7k)
Following method 7, 6k (3.01g, 7.266 mmol) was deprotected to yield a white solid (2.18g, 7.260 mmol, quant.). mp 321 – 323ºC. 1H NMR (400MHz, d6-DMSO) δ 12.84 (2H, s), 8.97 (2H, s), 8.15 (2H, t, J=1.7 Hz), 7.66 (2H, ddd, J=8.1, 2.0, 0.9 Hz), 7.57 (2H, d, J=7.8 Hz), 7.41 (2H, t, J=7.9 Hz) ppm. 13C NMR (100MHz, d6-DMSO) δ 167.2, 152.5, 139.8, 131.3, 129.0, 122.8, 122.5, 119.0 ppm. HRMS m/z calc. C15H12N2O5 [M+H]+ 301.0819, found 301.0807.
4,4′-ureylene-di-benzoic acid (7l)
Following method 7, 6l (3.12g, 7.558 mmol) was deprotected to yield a fine white powder (2.15g, 7.172 mmol, 95%). mp >350ºC [Lit. >400ºC]. 1H NMR (400MHz, DMSO) δ 12.62 (2H, s), 9.21 (2H, s), 7.88 (4H, d, J=8.7 Hz), 7.58 (4H, d, J=8.7 Hz) ppm. 13C NMR (100MHz, DMSO) δ 166.9, 151.9, 143.6, 130.5, 123.9, 117.3 ppm. HRMS m/z calc. C15H12N2O5 [M+H]+ 301.0819, found 301.0817.
1,3-bis(3-(4-(2-(pyrrolidin-1-yl)acetamido)phenylcarbamoyl)phenyl)urea (9a)
Following method 8, 7k (150.0mg, 0.500 mmol) was reacted with 3a (438.1mg, 1.998 mmol) to yield a pale brown solid (333.0mg, 0.474 mmol, 95%), which was purified by semi-prep. HPLC (method F). HPLC (method A) 97%, RT 17.69 min. mp 286 – 288ºC. 1H NMR (400MHz, d6-DMSO) δ 10.20 (2H, s), 9.73 (2H, s), 8.98 (2H, s), 7.98 (2H, s), 7.70 (6H, d, J=8.9 Hz), 7.61 (4H, d, J=8.9 Hz), 7.56 (2H, d, J=7.8 Hz), 7.44 (2H, t, J=7.9 Hz), 3.32 (4H, s), 2.67 (8H, m), 1.77 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 167.9, 165.2, 152.4, 139.7, 135.7, 134.6, 134.3, 128.6, 121.1, 120.8, 120.6, 119.6, 117.6, 59.0, 53.7, 23.3 ppm. HRMS m/z calc. C39H42N8O5 [M+H]+ 703.3351, found 703.3386.
1,3-bis(3-(4-(3-(pyrrolidin-1-yl)propanamido)phenylcarbamoyl)phenyl)urea (9b)
Following method 8, 7k (150.0mg, 0.500 mmol) was reacted with 3b (466.2mg, 1.998 mmol) to yield a white solid (346.0mg, 0.473 mmol, 95%). HPLC (method A) 98%, RT 19.00 min. mp 330ºC dec. 1H NMR (400MHz, d6-DMSO) δ 10.18 (2H, s), 10.05 (2H, s), 8.95 (2H, s), 7.97 (2H, s), 7.70 (6H, m), 7.55 (6H, m), 7.44 (2H, t, J=7.9 Hz), 2.75 (4H, t, J=7.1 Hz), 2.50 (12H, m), 1.70 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 169.6, 165.2, 152.4, 139.6, 135.7, 135.0, 134.3, 128.7, 121.1, 120.8, 120.7, 119.1, 117.6, 53.3, 51.4, 35.7, 23.0 ppm. HRMS m/z calc. C41H46N8O5 [M+H]+ 731.3664, found 731.3690.
1,3-bis(3-(4-(4-(pyrrolidin-1-yl)butanamido)phenylcarbamoyl)phenyl)urea (9c)
Following method 8, 7k (150.0mg, 0.500 mmol) was reacted with 3c (494.2mg, 1.998 mmol) to yield a white solid (344.8mg, 0.454 mmol, 91%). HPLC (method A) 96%, RT 19.86 min. mp 239 – 241ºC. 1H NMR (400MHz, d6-DMSO) δ 10.20 (2H, s), 9.89 (2H, s), 9.00 (2H, s), 7.99 (2H, s), 7.70 (6H, m), 7.57 (6H, m), 7.44 (2H, t, J=7.9 Hz), 2.50 (12H, m), 2.35 (4H, t, J=7.4 Hz), 1.77 (4H, m), 1.70 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 170.7, 165.2, 152.4, 139.7, 135.7, 135.1, 134.2, 128.6, 121.1, 120.8, 120.6, 119.1, 117.6, 54.9, 53.4, 45.7, 34.1, 23.0 ppm. HRMS m/z calc. C43H50N8O5 [M+H]+ 759.3977, found 759.3940.
1,3-bis(3-(3-(2-(pyrrolidin-1-yl)acetamido)phenylcarbamoyl)phenyl)urea (9d)
Following method 8, 7k (150.0mg, 0.500 mmol) was reacted with 3c (438.1mg, 1.998 mmol) to yield a pale yellow solid (310.1mg, 0.441 mmol, 88%). HPLC (method A) 91%, RT 18.69 min. mp 154 – 157ºC. 1H NMR (400MHz, d6-DMSO) δ 10.27 (2H, s), 9.78 (2H, s), 8.96 (2H, s), 8.12 (2H, s), 7.79 (2H, s), 7.71 (2H, d, J=8.0 Hz), 7.57 (2H, d, J=7.7 Hz), 7.44 (4H, m), 7.38 (2H, d, J=8.3 Hz), 7.26 (2H, t, J=8.0 Hz), 3.31 (4H, s), 2.65 (8H, m), 1.76 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 168.3, 165.6, 152.6, 139.7, 139.4, 138.8, 135.8, 128.8, 128.7, 121.3, 121.0, 117.8, 115.7, 115.0, 111.8, 59.2, 53.7, 23.4 ppm. HRMS m/z calc. C39H42N8O5 [M+H]+ 703.3351, found 703.3344.
1,3-bis(3-(3-(3-(pyrrolidin-1-yl)propanamido)phenylcarbamoyl)phenyl)urea (9e)
Following method 8, 7k (150.0mg, 0.500 mmol) was reacted with 3e (466.2mg, 1.998 mmol) to yield a pale yellow solid (292.4mg, 0.400 mmol, 80%). HPLC (method A) 93%, RT 19.39 min. mp 160 – 163ºC. 1H NMR (400MHz, d6-DMSO) δ 10.26 (2H, s), 10.13 (2H, s), 9.00 (2H, s), 8.10 (2H, s), 7.98 (2H, s), 7.72 (2H, d, J=7.9 Hz), 7.57 (2H, d, J=7.7 Hz), 7.44 (4H, m), 7.38 (2H, d, J=7.9 Hz), 7.25 (2H, t, J=7.7 Hz), 2.72 (4H, t, J=6.9 Hz), 2.49 (12H, m), 1.70 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 170.0, 165.5, 152.6, 139.6, 139.3, 139.3, 135.7, 128.6, 128.5, 121.2, 120.9, 117.7, 115.2, 114.5, 111.2, 53.3, 51.4, 35.9, 23.1 ppm. HRMS m/z calc. C41H46N8O5 [M+H]+ 731.3664, found 731.3651.
1,3-bis(3-(3-(4-(pyrrolidin-1-yl)butanamido)phenylcarbamoyl)phenyl)urea (9f)
Following method 8, 7k (150.0mg, 0.500 mmol) was reacted with 3f (494.2mg, 1.998 mmol) to yield a white solid (189.6mg, 0.249 mmol, 50%). HPLC (method A) 96%, RT 19.56 min. mp 234 – 236ºC. 1H NMR (400MHz, d6-DMSO) δ 10.26 (2H, s), 9.94 (2H, s), 9.01 (2H, s), 8.11 (2H, s), 7.99 (2H, s), 7.72 (2H, d, J=8.0 Hz), 7.57 (2H, d, J=7.6 Hz), 7.43 (4H, m), 7.37 (2H, d, J=7.9 Hz), 7.25 (2H, t, J=8.1 Hz), 2.48 (12H, m), 2.37 (4H, t, J=7.3 Hz), 1.77 (4H, m), 1.69 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 171.0, 165.5, 152.5, 139.6, 139.4, 139.3, 135.7, 128.6, 128.5, 121.2, 120.9, 117.7, 115.1, 114.5, 111.3, 54.9, 53.4, 34.2, 24.1, 23.0 ppm. HRMS m/z calc. C43H50N8O5 [M+H]+ 759.3977, found 759.3942.
1,3-bis(3-(4-(3-(dimethylamino)propanamido)phenylcarbamoyl)phenyl)urea (9g)
Following method 8, 7k (100.0mg, 0.333 mmol) was reacted with 3g (276.1mg, 1.333 mmol) to yield a white solid (222.8mg, 0.328 mmol, 99%), which was purified by filtration from boiling MeOH (2ml). HPLC (method C) 95%, RT 17.28 min. mp 322 – 324ºC. 1H NMR (400MHz, d6-DMSO) δ 10.18 (2H, s), 10.00 (2H, s), 8.94 (2H, s), 7.98 (2H, s), 7.70 (6H, m), 7.56 (6H, m), 7.44 (2H, t, J=7.9 Hz), 2.57 (4H, t, J=7.0 Hz), 2.44 (4H, t, J=6.9 Hz), 2.19 (12H, s) ppm. 13C NMR (100MHz, d6-DMSO) δ 169.9, 165.3, 152.5, 139.7, 135.8, 135.1, 134.4, 128.8, 121.2, 120.9, 120.7, 119.2, 117.7, 55.1, 44.9, 34.7 ppm. HRMS m/z calc. C37H42N8O5 [M+H]+ 679.3351, found 679.3334.
1,3-bis(3-(4-(3-(piperidin-1-yl)propanamido)phenylcarbamoyl)phenyl)urea (9h)
Following method 8, 7k (100.0mg, 0.333 mmol) was reacted with 3h (329.8mg, 1.333 mmol) to yield a white solid (221.1mg, 0.291 mmol, 87%), which was purified by filtration from boiling MeOH (2ml). HPLC (method C) 100%, RT 18.71 min. mp 322 – 324ºC. 1H NMR (400MHz, d6-DMSO) δ 10.18 (2H, s), 10.14 (2H, s), 8.95 (2H, s), 7.97 (2H, s), 7.70 (6H, m), 7.55 (6H, m), 7.44 (2H, t, J=7.9 Hz), 2.60 (4H, t, J=7.0 Hz), 2.45 (4H, t, J=7.0 Hz), 2.39 (8H, m), 1.51 (8H, m), 1.40 (4H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 167.0, 165.3, 152.5, 139.7, 135.8, 135.1, 134.4, 128.7, 121.2, 120.9, 120.8, 119.2, 117.7, 54.4, 53.6, 34.0, 25.6, 24.0 ppm. HRMS m/z calc. C43H50N8O5 [M+2H]++ 380.2025, found 380.2018.
1,3-bis(3-(4-(3-(morpholino)propanamido)phenylcarbamoyl)phenyl)urea (9i)
Following method 8, 7k (100.0mg, 0.333 mmol) was reacted with 3i (332.4mg, 1.333 mmol) to yield a pale purple solid (239.2mg, 0.314 mmol, 94%), which was purified by filtration from boiling MeOH (2ml). HPLC (method C) 97%, RT 17.20 min. mp 313 – 315ºC. 1H NMR (400MHz, d6-DMSO) δ 10.19 (2H, s), 10.00 (2H, s), 8.92 (2H, s), 7.98 (2H, s), 7.70 (6H, m), 7.56 (6H, m), 7.44 (2H, t, J=7.9 Hz), 3.59 (8H, t, J=4.5 Hz), 2.64 (4H, t, J=7.0 Hz), 2.48 (4H, t, J=7.1 Hz), 2.42 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 169.8, 165.3, 152.5, 139.7, 135.8, 135.1, 134.4, 128.8, 121.2, 120.9, 120.8, 119.2, 117.7, 66.2, 54.2, 53.0, 33.8 ppm. HRMS m/z calc. C41H46N8O7 [M+H]+ 763.3562, found 763.3539.
1,3-bis(4-(4-(2-(pyrrolidin-1-yl)acetamido)phenylcarbamoyl)phenyl)urea (10a)
Following method 8, 7l (150.0mg, 0.500 mmol) was reacted with 3a (438.1mg, 1.998 mmol) to yield a dark brown solid (325.3mg, 0.463 mmol, 93%), which was purified by semi-prep. HPLC (method F). HPLC (method A; 280nm) 98%, RT 19.23 min. mp 296 – 298ºC. 1H NMR (400MHz, d6-DMSO) δ 10.04 (2H, s), 9.65 (2H, s), 9.14 (2H, s), 7.94 (4H, d, J=8.8 Hz), 7.69 (4H, d, J=9.0 Hz), 7.61 (4H, d, J=8.8 Hz), 7.60 (4H, d, J=9.0 Hz), 3.25 (4H, s), 2.61 (8H, m), 1.76 (8H, m) ppm. HRMS m/z calc. C39H42N8O5 [M+H]+ 703.3351, found 703.3353.
1,3-bis(4-(4-(3-(pyrrolidin-1-yl)propanamido)phenylcarbamoyl)phenyl)urea (10b)
Following method 8, 7l (150.0mg, 0.500 mmol) was reacted with 3b (466.2mg, 1.998 mmol) to yield an off white solid (298.6mg, 0.409 mmol, 82%). HPLC (method A; 280nm) 99%, RT 18.65 min. mp 299 – 301ºC. 1H NMR (400MHz, d6-DMSO) δ 10.04 (4H, s), 9.15 (2H, s), 7.94 (4H, d, J=8.6 Hz), 7.69 (4H, d, J=8.8 Hz), 7.62 (4H, d, J=8.6 Hz), 7.55 (4H, d, J=8.8 Hz), 2.77 (4H, t, J=6.4 Hz), 2.50 (12H, m), 1.72 (8H, m) ppm. HRMS m/z calc. C41H46N8O5 [M+H]+ 731.3664, found 731.3653.
1,3-bis(4-(4-(4-(pyrrolidin-1-yl)butanamido)phenylcarbamoyl)phenyl)urea (10c)
Following method 8, 7l (150.0mg, 0.500 mmol) was reacted with 3c (494.2mg, 1.998 mmol) to yield a brown crystalline solid (332.3mg, 0.438 mmol, 88%). HPLC (method A; 280nm) 91%, RT 20.01 min. mp 289 – 291ºC. 1H NMR (400MHz, d6-DMSO) δ 10.02 (2H, s), 9.85 (2H, s), 9.16 (2H, s), 7.93 (4H, d, J=8.7 Hz), 7.67 (4H, d, J=8.9 Hz), 7.61 (4H, d, J=8.7 Hz), 7.55 (4H, d, J=8.9 Hz), 2.5 (12H, m), 2.35 (4H, t, J=7.3 Hz), 1.77 (4H, m), 1.69 (8H, m) ppm. HRMS m/z calc. C43H50N8O5 [M+H]+ 759.3977, found 759.3992.
1,3-bis(4-(3-(2-(pyrrolidin-1-yl)acetamido)phenylcarbamoyl)phenyl)urea (10d)
Following method 8, 7l (150.0mg, 0.500 mmol) was reacted with 3d (438.1mg, 1.998 mmol) to yield a pale yellow solid (265.0mg, 0.377 mmol, 75%), which was purified by semi-prep. HPLC (method F). HPLC (method A) 99%, RT 18.35 min. mp 232 – 234ºC. 1H NMR (400MHz, d6-DMSO) δ 10.10 (2H, s), 9.71 (2H, s), 9.15 (2H, s), 8.11 (2H, s), 7.95 (4H, d, J=8.8 Hz), 7.62 (4H, d, J=8.8 Hz), 7.46 (2H, d, J=8.2 Hz), 7.36 (2H, d, J=8.5 Hz), 7.25 (2H, t, J=8.1 Hz), 3.28 (4H, s), 2.63 (8H, m), 1.76 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 168.5, 164.8, 152.1, 142.6, 139.5, 138.7, 128.8, 128.6, 127.9, 117.2, 115.6, 114.7, 111.7, 59.3, 53.6, 23.4 ppm. HRMS m/z calc. C39H42N8O5 [M+H]+ 703.3351, found 703.3375.
1,3-bis(4-(3-(3-(pyrrolidin-1-yl)propanamido)phenylcarbamoyl)phenyl)urea (10e)
Following method 8, 7l (150.0mg, 0.500 mmol) was reacted with 3e (466.2mg, 1.998 mmol) to yield a pale yellow solid (233.1mg, 0.319 mmol, 64%), which was purified by semi-prep. HPLC (method F). HPLC (method A) 99%, RT 19.01 min. mp 231 – 233ºC. 1H NMR (400MHz, d6-DMSO) δ 10.11 (2H, s), 10.10 (2H, s), 9.17 (2H, s), 8.11 (2H, s), 7.96 (4H, d, J=8.8 Hz), 7.62 (4H, d, J=8.8 Hz), 7.42 (2H, d, J= 8.3 Hz), 7.35 (2H, d, J=8.2 Hz), 7.25 (2H, t, J=8.1 Hz), 2.76 (4H, t, J=7.0 Hz), 2.50 (12H, m), 1.71 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 169.9, 164.8, 152.0, 142.5, 139.5, 139.3, 128.7, 128.5, 127.9, 117.2, 115.2, 114.3, 111.3, 53.3, 51.4, 35.8, 23.0 ppm. HRMS m/z calc. C41H46N8O5 [M+H]+ 731.3664, found 731.3668.
1,3-bis(4-(3-(4-(pyrrolidin-1-yl)butanamido)phenylcarbamoyl)phenyl)urea (10f)
Following method 8, 7l (150.0mg, 0.500 mmol) was reacted with 3f (494.2mg, 1.998 mmol) to yield a white solid (173.4mg, 0.228 mmol, 46%), which was purified by semi-prep. HPLC (method G). HPLC (method A) 97%, RT 20.40 min. mp 249 – 252ºC. 1H NMR (400MHz, d6-DMSO) δ 10.09 (2H, s), 9.91 (2H, s), 9.31 (2H, s), 8.11 (2H, s), 7.95 (4H, d, J=8.5 Hz), 7.63 (4H, d, J=8.5 Hz), 7.41 (2H, d, J=8.1 Hz), 7.34 (2H, d, J=7.9 Hz), 7.23 (2H, t, J=8.0 Hz), 2.52 (12H, m), 2.37 (4H, t, J=7.2 Hz), 1.78 (4H, m), 1.70 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 170.9, 164.7, 152.1, 142.6, 139.4, 139.4, 128.7, 128.4, 127.9, 117.2, 115.2, 114.3, 111.4, 54.9, 53.4, 34.1, 23.9, 22.9 ppm. HRMS m/z calc. C43H50N8O5 [M+H]+ 759.3977, found 759.3975.
3-amino-5-nitro-N-phenylbenzamide (11)
3-amino-5-nitrobenzoic acid (1.00g, 5.490 mmol) was dissolved in DMF (25ml) and to this was added aniline (0.55ml, 6.039 mmol, 1.1 equiv.), HOBt (816.1mg, 6.039 mmol, 1.1 equiv.) and DCC (1.25g, 6.039 mmol, 1.1 equiv.) and the mixture stirred under N2 (g) at room temperature for 24h. The precipitate was filtered from the reaction, the solvent evaporated and to the residue was added 5% NH3 (aq.) (100ml). The product was extracted into EtOAc (3 × 100ml), washed with brine (50ml) and dried over MgSO4. The desired product was purified by flash chromatography to yield an orange powder (1.29g, 5.019 mmol, 91%). mp 202 – 204ºC. 13H NMR (400MHz, d6-DMSO) δ10.40 (1H, s), 7.90 (1H, t, J=1.8 Hz), 7.77 (2H, d, J=7.6 Hz), 7.57 (1H, t, J=2.2 Hz), 7.51 (1H, t, J=1.8 Hz), 7.37 (2H, t, J=7.9 Hz), 7.12 (1H, t, J= 7.4 Hz), 6.09 (2H, s) ppm. 3C NMR (100MHz, d6-DMSO) δ 164.2, 150.2, 148.7, 138.8, 136.9, 128.6, 123.9, 120.5, 118.8, 109.5, 108.6 ppm. HRMS m/z calc. C13H11N3O3 [M+H]+ 258.0873, found 258.0867.
3-(2-chloroacetamido)-5-nitro-N-phenylbenzamide (12a)
Following method 1a, 11 (150.0mg, 0.583 mmol) was reacted with 2-chloroacetyl chloride (0.10ml, 1.255 mmol) to yield a pink solid (127.8mg, 0.383 mmol, 66%). mp 187ºC dec. 1H NMR (400MHz, d6-DMSO) δ 11.03 (1H, s), 10.65 (1H, s), 8.82 (1H, t, J=2.0 Hz), 8.59 (1H, t, J=2.0 Hz), 8.46 (1H, t, J=2.0 Hz), 7.78 (2H, dd, J=8.6, 1.3 Hz), 7.40 (2H, m), 7.15 (1H, t, J=7.3 Hz), 4.37 (2H, s) ppm. 13C NMR (100MHz, d6-DMSO) δ 165.6, 163.1, 148.0, 139.7, 138.6, 136.8, 128.7, 124.6, 124.2, 120.6, 117.1, 116.0, 43.4 ppm. HRMS m/z calc. C15H12ClN3O4 [M+H]+ 334.0589, found 334.0582.
3-(acrylamido)-5-nitro-N-phenylbenzamide (12b)
Following method 1a, 11 (1.50g, 5.831 mmol) was reacted with 3-chloropropanoyl chloride (1.12ml, 11.732 mmol) to yield a cream solid (1.22g, 3.928 mmol, 67%). mp 197 – 199ºC. 1H NMR (400MHz, d6-DMSO) δ 10.67 (1H, s), 10.61 (1H, s), 8.93 (1H, t, J=2.1 Hz), 8.55 (1H, t, J=1.8 Hz), 8.51 (1H, t, J=1.8 Hz), 7.76 (2H, dd, J=8.6, 1.0 Hz), 7.39 (2H, m), 7.15 (1H, t, J=7.4 Hz), 6.48 (1H, dd, J=17.0, 9.9 Hz), 6.36 (1H, dd, J=17.0, 2.0 Hz), 5.88 (1H, dd, J=9.9, 2.0 Hz) ppm. 13C NMR (100MHz, d6-DMSO) δ 163.8, 163.2, 147.9, 140.2, 138.5, 136.7, 131.0, 128.6, 128.4, 124.5, 124.1, 120.5, 116.6, 115.9 ppm. HRMS m/z calc. C16H13N3O4 [M+H]+ 312.0976, found 312.0965.
3-(4-chlorobutanamido)-5-nitro-N-phenylbenzamide (12c)
Following method 1a, 11 (1.50g, 5.831 mmol) was reacted with 4-chlorobutanoyl chloride (1.30 ml, 11.617 mmol) to yield a cream solid (1.94g, 5.360 mmol, 92%). mp 213 – 215ºC. 1H NMR (400MHz, d6-DMSO) 10.69 (1H, s), 10.60 (1H, s), 8.85 (1H, t, J=2.1 Hz), 8.52 (1H, t, J=1.8 Hz), 8.44 (1H, t, J=1.8 Hz), 7.78 (2H, dd, J=8.6, 1.0 Hz), 7.39 (2H, m), 7.15 (1H, t, J=7.4 Hz), 3.74 (2H, t, J=6.5 Hz), 2.58 (2H, t, J=7.3 Hz), 2.09 (2H, m) ppm. 13C NMR (100MHz, d6-DMSO) 171.2, 163.3, 147.9, 140.4, 138.6, 136.7, 128.7, 124.3, 124.1, 120.6, 116.3, 115.7, 44.9, 33.4, 27.6 ppm. HRMS m/z calc. C17H16ClN3O4 [M+H]+ 362.0908, found 362.0900.
3-(2-(pyrrolidin-1-yl)acetamido)-5-nitro-N-phenylbenzamide (13a)
Following method 2a, 12a (102.2mg, 0.306 mmol) was reacted with pyrrolidine (0.05ml, 5.990 mmol) to yield a beige solid (102.9mg, 0.279 mmol, 91%). mp 181 – 184ºC. 1H NMR (400MHz, d6-DMSO) δ 10.57 (1H, s), 10.41 (1H, s), 8.92 (1H, t, J=2.0 Hz), 8.58 (1H, t, J=1.7 Hz), 8.51 (1H, t, J=1.8 Hz), 7.77 (2H, m), 7.39 (2H, m), 7.15 (1H, t, J=7.3 Hz), 3.33 (2H, s), 2.57 (4H, m), 1.72 (4H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 170.0, 163.4, 147.9, 140.1, 138.7, 136.7, 128.7, 124.9, 124.1, 120.6, 116.5, 116.2, 59.5, 53.7, 23.5 ppm. HRMS m/z calc. C19H20N4O4 [M+H]+ 369.1557, found 369.1539.
3-(3-(pyrrolidin-1-yl)propanamido)-5-nitro-N-phenylbenzamide (13b)
Following method 2a, 12b (1.00g, 3.212 mmol) was reacted with pyrrolidine (0.54ml, 6.469 mmol) to yield a cream solid (947.8mg, 2.478 mmol, 77%). mp 151- 153ºC. 1H NMR (400MHz, d6-DMSO) δ 10.69 (1H, s), 10.59 (1H, s), 8.85 (1H, t, J=1.8 Hz), 8.51 (1H, s), 8.41 (1H, s), 7.77 (2H, d, J=8.0 Hz), 7.38 (2H, t, J=7.8 Hz), 7.14 (1H, t, J=7.4 Hz), 2.75 (2H, t, J=6.9 Hz), 2.55 (2H, t, J=6.9 Hz), 2.48 (4H, m), 1.68 (4H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 171.0, 163.2, 147.8, 140.4, 138.6, 136.6, 128.6, 124.1, 124.0, 120.5, 116.1, 115.5, 53.3, 51.2, 36.0, 23.0 ppm. HRMS m/z calc. C20H22N4O4 [M+H]+ 383.1714, found 383.1727.
3-(4-(pyrrolidin-1-yl)butanamido)-5-nitro-N-phenylbenzamide (13c)
Following method 2b, 12c (1.00g, 2.764 mmol) was reacted with pyrrolidine (0.69ml, 8.266 mmol) to yield a pale yellow solid (861.1mg, 2.172 mmol, 79%). mp 157 – 161°C. 1H NMR (400MHz, d6-DMSO) δ 10.58 (2H, s), 8.84 (1H, t, J=2.0 Hz), 8.50 (1H, t, J=1.8 Hz), 8.42 (1H, t, J=1.7 Hz), 7.77 (2H, d, J=8.6 Hz), 7.38 (2H, m), 7.14 (1H, t, J=7.7 Hz), 2.43 (8H, m), 1.79 (2H, m), 1.66 (4H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 172.1, 163.2, 147.8, 140.5, 138.5, 136.6, 128.6, 124.1, 124.0, 120.5, 116.0, 115.5, 54.9, 53.4, 34.4, 24.0, 23.0 ppm. HRMS m/z calc. C21H24N4O4 [M+H]+ 397.1876, found 397.1860.
3-(2-(pyrrolidin-1-yl)acetamido)-5-amino-N-phenylbenzamide (14a)
Following method 3b, 13a (300.0mg, 0.814 mmol) was reduced to yield a brown solid (254.1mg, 0.751 mmol, 92%). mp 88 – 91ºC. 1H NMR (400MHz, d6-DMSO) δ 10.16 (1H, s), 9.61 (1H, s), 7.80 (2H, d, J=7.7 Hz), 7.39 (2H, t, J=7.8 Hz), 7.29 (1H, s), 7.20 (1H, s), 7.13 (1H, t, J=7.4 Hz), 6.82 (1H, s), 5.43 (2H, s), 3.28 (2H, s) 2.65 (4H, m), 1.81 (4H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 168.6, 166.4, 149.0, 139.4, 139.1, 136.5, 128.5, 123.3, 120.1, 108.4, 107.5, 106.7, 59.4, 53.7, 23.5 ppm. HRMS m/z calc. C19H22N4O2 [M+H]+ 339.1815, found 339.1797.
3-(3-(pyrrolidin-1-yl)propanamido)-5-amino-N-phenylbenzamide (14b)
Following method 3b, 13b (300.0mg, 0.784 mmol) was reduced to yield a white solid (225.7mg, 0.640 mmol, 82%). mp 196 – 198ºC. 1H NMR (400MHz, d6-DMSO) δ 10.12 (1H, s), 9.97 (1H, s), 7.74 (2H, d, J=7.7 Hz), 7.32 (2H, t, J=7.9 Hz), 7.16 (1H, s), 7.10 (1H, s), 7.06 (1H, t, J=7.4 Hz), 6.72 (1H, s), 5.35 (2H, s), 2.64 (2H, t, J=7.0 Hz), 2.45 (6H, m), 1.68 (4H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 170.0, 166.5, 148.9, 139.7, 139.3, 136.5, 128.4, 123.2, 119.9, 107.9, 107.1, 106.4, 53.3, 51.5, 36.0, 23.1 ppm. HRMS m/z calc. C20H24N4O2 [M+H]+ 353.1972, found 353.1955.
3-(4-(pyrrolidin-1-yl)butanamido)-5-amino-N-phenylbenzamide (14c)
Following method 3b, 13c (801.0mg, 2.021 mmol) was reduced to yield a white solid (646.9mg, 1.765 mmol, 87%). mp 159 – 161ºC. 1H NMR (400MHz, d6-DMSO) δ 10.07 (1H, s), 9.75 (1H, s), 7.74 (2H, d, J=8.4 Hz), 7.32 (2H, t, J=8.0 Hz), 7.16 (1H, t, J=1.8 Hz), 7.12 (1H, t, J=1.6 Hz), 7.06 (1H, t, J=7.4 Hz), 6.72 (1H, t, J=1.7 Hz), 5.30 (2H, s), 2.42 (4H, m), 2.40 (2H, t, J=7.2 Hz), 2.30 (2H, t, J=7.4 Hz), 1.74 (2H, m), 1.67 (4H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 171.0, 166.6, 148.8, 139.8, 139.3, 136.5, 128.4, 123.2, 119.9, 107.9, 107.2, 106.4, 55.0, 53.4, 34.4, 24.3, 23.0 ppm. HRMS m/z calc. C21H26N4O2 [M+H]+ 367.2134, found 367.2088.
1,3-bis(3-(phenylcarbamoyl)-5-(2-(pyrrolidin-1-yl)acetamido)-phenyl)urea (15a)
Following method 6b, 14a (500.0mg, 1.478 mmol) was reacted with CDI (239.7mg, 1.478 mmol) to yield a white solid (231.0mg, 0.329 mmol, 44%), which was purified by semi-prep. HPLC (method F). HPLC (method A) 96%, RT 21.02 min. mp 179 – 181ºC. 1H NMR (400MHz, d6-DMSO) δ 10.28 (2H, s), 9.92 (2H, s), 8.94 (2H, s), 8.14 (2H, t, J=1.7 Hz), 7.77 (4H, d, J=7.8 Hz), 7.72 (2H, t, J=1.7 Hz), 7.70 (2H, t, J=1.7 Hz), 7.36 (4H, t, J=7.9 Hz), 7.11 (2H, t, J=7.4 Hz), 3.33 (4H, s), 2.66 (8H, m), 1.78 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 168.7, 165.7, 152.2, 139.8, 139.1, 139.0, 136.4, 128.5, 123.5, 120.1, 112.5, 112.5, 111.9, 59.2, 53.6, 23.4 ppm. HRMS m/z calc. C39H42N8O5 [M+H]+ 703.3351, found 703.3369.
1,3-bis(3-(phenylcarbamoyl)-5-(3-(pyrrolidin-1-yl)propanamido)phenyl)urea (15b)
Following method 6b, 14b (500.0mg, 1.419 mmol) was reacted with CDI (230.1mg, 1.419 mmol) to yield a white solid (245.8mg, 0.336 mmol, 47%), which was purified by semi-prep. HPLC (method F). HPLC (method A) 99%, RT 21.01 min. mp 167 – 169ºC. 1H NMR (400MHz, d6-DMSO) δ 10.27 (2H, s), 10.22 (2H, s), 8.90 (2H, s), 8.08 (2H, s), 7.74 (4H, d, J=8.6 Hz), 7.65 (2H, s), 7.62 (2H, s), 7.33 (4H, t, J=7.8 Hz), 7.07 (2H, t, J=7.4 Hz), 2.75 (4H, t, J=6.9 Hz), 2.50 (12H, m), 1.68 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 170.2, 165.8, 152.2, 139.8, 139.6, 139.1, 136.5, 128.5, 123.4, 120.1, 112.1, 112.1, 111.4, 53.3, 51.3, 35.8, 23.0 ppm. HRMS m/z calc. C41H46N8O5 [M+H]+ 731.3664, found 731.3674.
1,3-bis(3-(phenylcarbamoyl)-5-(4-(pyrrolidin-1-yl)butanamido)phenyl)urea (15c)
Following method 6b, 14c (500.0mg, 1.364 mmol) was reacted with CDI (221.2mg, 1.364 mmol) to yield a white solid (288.9mg, 0.381 mmol, 56%), which was purified by semi-prep. HPLC (method F). HPLC (method A) 97%, RT 21.89 min. mp 176 – 178ºC. 1H NMR (400MHz, d6-DMSO) δ 10.28 (2H, s), 10.09 (2H, s), 8.91 (2H, s), 8.10 (2H, s), 7.76 (4H, d, J=7.7 Hz), 7.69 (2H, s), 7.65 (2H, s), 7.36 (4H, t, J=7.4 Hz), 7.10 (2H, t, J=7.4 Hz), 2.46 (12H, m), 2.39 (4H, t, J=7.4 Hz), 1.78 (4H, m), 1.69 (8H, m) ppm. 13C NMR (100MHz, d6-DMSO) δ 171.3, 165.9, 152.2, 139.7, 139.7, 139.1, 136.4, 128.5, 123.4, 120.1, 112.1, 112.0, 111.5, 55.0, 53.4, 34.3, 24.1, 23.0 ppm. HRMS m/z calc. C43H50N8O5 [M+H]+ 759.3977, found 759.4016.
Supplementary Material
Acknowledgments
We are grateful to CRUK (program Grant No. C129/A4489) and the EU (FP6 Molecular Cancer Medicine) for support (S.N.), and to NIH for grant GM61587 (W.D.W.). We are also grateful to various colleagues for useful advice and discussions.
Glossary
- FRET
Fluorescent Resonance Energy Transfer
- CD
circular dichroism
- SPR
surface Plasmon resonance
- DCC
N,N′-dicyclohexyl-carbodiimide
- DMAP
4-dimethylaminopyridine
- CDI
1,1′-carbonyldiimidazole
- TFA
trifluoroacetic acid
- PyBOP
benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
- TRAP
Telomerase Repeat Amplification Protocol
- SRB
Sulforhodamine B
Footnotes
Supporting Information Available: Experimental procedures and characterization of side-chain building-blocks, quinazoline dione ligands, HPLC purity analysis, FRET-based correlations, TRAP-LIG data for 9a-c, MCF7 population growth, senescence and telomere end – end fusion data, plus additional modeled ligand – DNA complexes. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Burge S, Parkinson GN, Hazel P, Todd AK, Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gellert M, Lipsett MN, Davies DR. Helix formation by guanylic acid. Proc Natl Acad Sci USA. 1962;48:2013–2018. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel DJ, Phan AT, Kuryavyi V. Human telomere, oncogenic promoter and 5′-UTR G-quadruplexes: diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimmerman SB, Cohen GH, Davies DR. X-ray fiber diffraction and model-building study of polyguanylic acid and polyinosinic acid. J Mol Biol. 1975;92:181–192. doi: 10.1016/0022-2836(75)90222-3. [DOI] [PubMed] [Google Scholar]
- 5.Meyne J, Ratliff RL, Moyzis RK. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci USA. 1989;86:7049–7053. doi: 10.1073/pnas.86.18.7049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, Meyne J, Ratliff RL, Wu JR. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makarov VL, Hirose Y, Langmore JP. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 1997;88:657–666. doi: 10.1016/s0092-8674(00)81908-x. [DOI] [PubMed] [Google Scholar]
- 8.Sfeir AJ, Chai W, Shay JW, Wright WE. Telomere-end processing the terminal nucleotides of human chromosomes. Mol Cell. 2005;18:131–138. doi: 10.1016/j.molcel.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 9.Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol. 2004;11:1223–1229. doi: 10.1038/nsmb867. [DOI] [PubMed] [Google Scholar]
- 10.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 11.Loayza D, de Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003;423:1013–1018. doi: 10.1038/nature01688. [DOI] [PubMed] [Google Scholar]
- 12.Loayza D, Parsons H, Donigian J, Hoke K, de Lange T. DNA binding features of human POT1: a nonamer 5′-TAGGGTTAG-3′ minimal binding site, sequence specificity, and internal binding to multimeric sites. J Biol Chem. 2004;279:13241–13248. doi: 10.1074/jbc.M312309200. [DOI] [PubMed] [Google Scholar]
- 13.Harley CB, Futcher AB, Greider CW. Telemeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 14.Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–529. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- 15.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 16.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 17.Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 18.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi: 10.1016/S0959-8049(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 19.Cuenca F, Greciano O, Gunaratnam M, Haider S, Munnur D, Nanjunda R, Wilson WD, Neidle S. Tri- and tetra-substituted naphthalene diimides as potent G-quadruplex ligands. Bioorg Med Chem Lett. 2008;18:1668–1673. doi: 10.1016/j.bmcl.2008.01.050. [DOI] [PubMed] [Google Scholar]
- 20.Kim MY, Vankayalapati H, Shin-Ya K, Wierzba K, Hurley LH. Telomestatin, a potent telomerase inhibitor that interacts quite specifically with the human telomeric intramolecular G-quadruplex. J Am Chem Soc. 2002;124:2098–2099. doi: 10.1021/ja017308q. [DOI] [PubMed] [Google Scholar]
- 21.Gowan SM, Harrison JR, Patterson L, Valenti M, Read MA, Neidle S, Kelland LR. A G-quadruplex-interactive potent small-molecule inhibitor of telomerase exhibiting in vitro and in vivo antitumor activity. Mol Pharmacol. 2002;61:1154–1162. doi: 10.1124/mol.61.5.1154. [DOI] [PubMed] [Google Scholar]
- 22.Gunaratnam M, Greciano O, Martins C, Reszka AP, Schultes CM, Morjani H, Riou JF, Neidle S. Mechanism of acridine-based telomerase inhibition and telomere shortening. Biochem Pharmacol. 2007;74:679–689. doi: 10.1016/j.bcp.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Harrison RJ, Gowan SM, Kelland LR, Neidle S. Human telomerase inhibition by substituted acridine derivatives. Bioorg Med Chem Lett. 1999;9:2463–2468. doi: 10.1016/s0960-894x(99)00394-7. [DOI] [PubMed] [Google Scholar]
- 24.Harrison RJ, Cuesta J, Chessari G, Read MA, Basra SK, Reszka AP, Morrell J, Gowan SM, Incles CM, Tanious FA, Wilson WD, Kelland LR, Neidle S. Trisubstituted acridine derivatives as potent and selective telomerase inhibitors. J Med Chem. 2003;46:4463–4476. doi: 10.1021/jm0308693. [DOI] [PubMed] [Google Scholar]
- 25.Harrison RJ, Reszka AP, Haider SM, Romagnoli B, Morrell J, Read MA, Gowan SM, Incles CM, Kelland LR, Neidle S. Evaluation of by disubstituted acridone derivatives as telomerase inhibitors: the importance of G-quadruplex binding. Bioorg Med Chem Lett. 2004;14:5845–5849. doi: 10.1016/j.bmcl.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 26.Heald RA, Modi C, Cookson JC, Hutchinson I, Laughton CA, Gowan SM, Kelland LR, Stevens MF. Antitumor polycyclic acridines. 8.(1) Synthesis and telomerase-inhibitory activity of methylated pentacyclic acridinium salts. J Med Chem. 2002;45:590–597. doi: 10.1021/jm011015q. [DOI] [PubMed] [Google Scholar]
- 27.Leonetti C, Amodei S, D'Angelo C, Rizzo A, Benassi B, Antonelli A, Elli R, Stevens MF, D'Incalci M, Zupi G, Biroccio A. Biological activity of the G-quadruplex ligand RHPS4 (3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate) is associated with telomere capping alteration. Mol Pharmacol. 2004;66:1138–1146. doi: 10.1124/mol.104.001537. [DOI] [PubMed] [Google Scholar]
- 28.Martins C, Gunaratnam M, Stuart J, Makwana V, Greciano O, Reszka AP, Kelland LR, Neidle S. Structure-based design of benzylamino-acridine compounds as G-quadruplex DNA telomere targeting agents. Bioorg Med Chem Lett. 2007;17:2293–2298. doi: 10.1016/j.bmcl.2007.01.056. [DOI] [PubMed] [Google Scholar]
- 29.Mergny JL, Lacroix L, Teulade-Fichou MP, Hounsou C, Guittat L, Hoarau M, Arimondo PB, Vigneron JP, Lehn JM, Riou JF, Garestier T, Helene C. Telomerase inhibitors based on quadruplex ligands selected by a fluorescence assay. Proc Natl Acad Sci USA. 2001;98:3062–3067. doi: 10.1073/pnas.051620698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore MJ, Schultes CM, Cuesta J, Cuenca F, Gunaratnam M, Tanious FA, Wilson WD, Neidle S. Trisubstituted acridines as G-quadruplex telomere targeting agents. Effects of extensions of the 3,6- and 9-side chains on quadruplex binding, telomerase activity, and cell proliferation. J Med Chem. 2006;49:582–599. doi: 10.1021/jm050555a. [DOI] [PubMed] [Google Scholar]
- 31.Moorhouse AD, Santos AM, Gunaratnam M, Moore M, Neidle S, Moses JE. Stabilization of G-quadruplex DNA by highly selective ligands via click chemistry. J Am Chem Soc. 2006;128:15972–15973. doi: 10.1021/ja0661919. [DOI] [PubMed] [Google Scholar]
- 32.Read M, Harrison RJ, Romagnoli B, Tanious FA, Gowan SH, Reszka AP, Wilson WD, Kelland LR, Neidle S. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors. Proc Natl Acad Sci USA. 2001;98:4844–4849. doi: 10.1073/pnas.081560598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Read MA, Wood AA, Harrison JR, Gowan SM, Kelland LR, Dosanjh HS, Neidle S. Molecular modeling studies on G-quadruplex complexes of telomerase inhibitors: structure-activity relationships. J Med Chem. 1999;42:4538–4546. doi: 10.1021/jm990287e. [DOI] [PubMed] [Google Scholar]
- 34.Schultes CM, Guyen B, Cuesta J, Neidle S. Synthesis, biophysical and biological evaluation of 3,6-bis-amidoacridines with extended 9-anilino substituents as potent G-quadruplex-binding telomerase inhibitors. Bioorg Med Chem Lett. 2004;14:4347–4351. doi: 10.1016/j.bmcl.2004.05.090. [DOI] [PubMed] [Google Scholar]
- 35.Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH. Inhibition of human telomerase by a G-quadruplex-interactive compound. J Med Chem. 1997;40:2113–2116. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 36.Wheelhouse RT, Sun DK, Han HY, Han FXG, Hurley LH. Cationic porphyrins as telomerase inhibitors: the interaction of tetra-(N-methyl-4-pyridyl)porphine with quadruplex DNA. J Am Chem Soc. 1998;120:3261–3262. [Google Scholar]
- 37.Huppert JL, Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phan AT, Modi YS, Patel DJ. Propeller-type parallel-stranded G-quadruplexes in the human c-myc promoter. J Am Chem Soc. 2004;126:8710–8716. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phan AT, Kuryavyi V, Gaw HY, Patel DJ. Small-molecule interaction with a five-guanine-tract G-quadruplex structure from the human MYC promoter. Nature Chem Biol. 2005;1:167–173. doi: 10.1038/nchembio723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simonsson T, Pecinka P, Kubista M. DNA tetraplex formation in the control region of c-myc. Nucleic Acids Res. 1998;26:1167–1172. doi: 10.1093/nar/26.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernando H, Reszka AP, Huppert J, Ladame S, Rankin S, Venkitaraman AR, Neidle S, Balasubramanian S. A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry. 2006;45:7854–7860. doi: 10.1021/bi0601510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phan AT, Kuryavyi V, Burge S, Neidle S, Patel DJ. Structure of an unprecedented G-quadruplex scaffold in the human c-kit promoter. J Am Chem Soc. 2007;129:4386–4392. doi: 10.1021/ja068739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rankin S, Reszka AP, Huppert J, Zloh M, Parkinson GN, Todd AK, Ladame S, Balasubramanian S, Neidle S. Putative DNA quadruplex formation within the human c-kit oncogene. J Am Chem Soc. 2005;127:10584–10589. doi: 10.1021/ja050823u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dexheimer TS, Sun D, Hurley LH. Deconvoluting the structural and drug-recognition complexity of the G-quadruplex-forming region upstream of the bcl-2 P1 promoter. J Am Chem Soc. 2006;128:5404–5415. doi: 10.1021/ja0563861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cogoi S, Xodo LE. G-quadruplex formation within the promoter of the KRAS proto-oncogene and its effect on transcription. Nucleic Acids Res. 2006;34:2536–2549. doi: 10.1093/nar/gkl286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumari S, Bugaut A, Huppert JL, Balasubramanian S. An RNA G-quadruplex in the 5′ UTR of the NRAS proto-oncogene modulates translation. Nature Chem Biol. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soldatenkov VA, Vetcher AA, Duka T, Ladame S. First evidence of a functional interaction between DNA quadruplexes and poly(ADP-ribose) polymerase-1. ACS Chem Biol. 2008;3:214–219. doi: 10.1021/cb700234f. [DOI] [PubMed] [Google Scholar]
- 49.Blackburn EH. Telomere states and cell fates. Nature. 2000;408:53–56. doi: 10.1038/35040500. [DOI] [PubMed] [Google Scholar]
- 50.Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–673. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 51.Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi: 10.1146/annurev.biochem.73.071403.160049. [DOI] [PubMed] [Google Scholar]
- 52.Li GZ, Eller MS, Hanna K, Gilchrest BA. Signaling pathway requirements for induction of senescence by telomere homolog oligonucleotides. Exp Cell Res. 2004;301:189–200. doi: 10.1016/j.yexcr.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 53.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 54.Murnane JP. Telomeres and chromosome instability. DNA Repair. 2006;5:1082–1092. doi: 10.1016/j.dnarep.2006.05.030. [DOI] [PubMed] [Google Scholar]
- 55.Gilley D, Tanaka H, Herbert BS. Telomere dysfunction in aging and cancer. Int J Biochem Cell Biol. 2005;37:1000–1013. doi: 10.1016/j.biocel.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 56.Shin-Ya K, Wierzba K, Matsuo K, Ohtani T, Yamada Y, Furihata K, Hayakawa Y, Seto H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J Am Chem Soc. 2001;123:1262–1263. doi: 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- 57.Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA. Inhibition of telomerase limits the growth of human cancer cells. Nature Med. 1999;5:1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- 58.Harley CB. Telomerase and cancer therapeutic. Nature Rev Cancer. 2008;8:167–179. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 59.Herbert BS, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB, Shay JW, Gryaznov SM. Lipid modification of GRN163, an N3′-->P5′ thio-phosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene. 2005;24:5262–5268. doi: 10.1038/sj.onc.1208760. [DOI] [PubMed] [Google Scholar]
- 60.Kelland L. Targeting the limitless replicative potential of cancer: the telomerase/telomere pathway. Clin Cancer Res. 2007;13:4960–4963. doi: 10.1158/1078-0432.CCR-07-0422. [DOI] [PubMed] [Google Scholar]
- 61.Burger AM, Dai F, Schultes CM, Reszka AP, Moore MJ, Double JA, Neidle S. The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res. 2005;65:1489–1496. doi: 10.1158/0008-5472.CAN-04-2910. [DOI] [PubMed] [Google Scholar]
- 62.Gomez D, O'Donohue MF, Wenner T, Douarre C, Macadre J, Koebel P, Giraud-Panis MJ, Kaplan H, Kolkes A, Shin-Ya K, Riou JF. The G-quadruplex ligand telomestatin inhibits POT1 binding to telomeric sequences in vitro and induces GFP-POT1 dissociation from telomeres in human cells. Cancer Res. 2006;66:6908–6912. doi: 10.1158/0008-5472.CAN-06-1581. [DOI] [PubMed] [Google Scholar]
- 63.Gomez D, Wenner T, Brassart B, Douarre C, O'Donohue MF, El, K V, Shin-Ya K, Morjani H, Trentesaux C, Riou JF. Telomestatin-induced telomere uncapping is modulated by POT1 through G-overhang extension in HT1080 human tumor cells. J Biol Chem. 2006;281:38721–38729. doi: 10.1074/jbc.M605828200. [DOI] [PubMed] [Google Scholar]
- 64.Incles CM, Schultes CM, Kempski H, Koehler H, Kelland LR, Neidle S. A G-quadruplex telomere targeting agent produces p16-associated senescence and chromosomal fusions in human prostate cancer cells. Mol Cancer Ther. 2004;3:1201–1206. [PubMed] [Google Scholar]
- 65.Phatak P, Cookson JC, Dai F, Smith V, Gartenhaus RB, Stevens MF, Burger AM. Telomere uncapping by the G-quadruplex ligand RHPS4 inhibits clonogenic tumour cell growth in vitro and in vivo consistent with a cancer stem cell targeting mechanism. Br J Cancer. 2007;96:1223–1233. doi: 10.1038/sj.bjc.6603691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Salvati E, Leonetti C, Rizzo A, Scarsella M, Mottolese M, Galati R, Sperduti I, Stevens MF, D'Incalci M, Blasco M, Chiorino G, Bauwens S, Horard B, Gilson E, Stoppacciaro A, Zupi G, Biroccio A. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J Clin Invest. 2007;117:3236–3247. doi: 10.1172/JCI32461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tahara H, Shin-Ya K, Seimiya H, Yamada H, Tsuruo T, Ide T. G-quadruplex stabilization by telomestatin induces TRF2 protein dissociation from telomeres and anaphase bridge formation accompanied by loss of the 3′ telomeric overhang in cancer cells. Oncogene. 2006;25:1955–1966. doi: 10.1038/sj.onc.1209217. [DOI] [PubMed] [Google Scholar]
- 68.Parkinson GN, Lee MP, Neidle S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature. 2002;417:876–880. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 69.Phan AT, Patel DJ. Two-repeat human telomeric d(TAGGGTTAGGGT) sequence forms interconverting parallel and antiparallel G-quadruplexes in solution: distinct topologies, thermodynamic properties, and folding/unfolding kinetics. J Am Chem Soc. 2003;125:15021–15027. doi: 10.1021/ja037616j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Phan AT, Luu KN, Patel DJ. Different loop arrangements of intramolecular human telomeric (3+1) G-quadruplexes in K+ solution. Nucleic Acids Res. 2006;34:5715–5719. doi: 10.1093/nar/gkl726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Phan AT, Kuryavyi V, Luu KN, Patel DJ. Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K+ solution. Nucleic Acids Res. 2007;35:6517–6525. doi: 10.1093/nar/gkm706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y, Patel DJ. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure. 1993;1:263–282. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 73.Gavathiotis E, Heald RA, Stevens MF, Searle MS. Drug recognition and stabilisation of the parallel-stranded DNA quadruplex d(TTAGGGT)4 containing the human telomeric repeat. J Mol Biol. 2003;334:25–36. doi: 10.1016/j.jmb.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 74.Haider SM, Parkinson GN, Neidle S. Structure of a G-quadruplex-ligand complex. J Mol Biol. 2003;326:117–125. doi: 10.1016/s0022-2836(02)01354-2. [DOI] [PubMed] [Google Scholar]
- 75.Parkinson GN, Ghosh R, Neidle S. Structural basis for binding of porphyrin to human telomeres. Biochemistry. 2007;46:2390–2397. doi: 10.1021/bi062244n. [DOI] [PubMed] [Google Scholar]
- 76.Campbell NH, Parkinson GN, Reszka AP, Neidle S. Structural basis of DNA quadruplex recognition by an acridine drug. J Am Chem Soc. 2008;130:6722–6724. doi: 10.1021/ja8016973. [DOI] [PubMed] [Google Scholar]
- 77.Dugave C. Cis-trans Isomerization in Biochemistry. Wiley-VCH; Weinheim: 2006. [Google Scholar]
- 78.Rajnikant, Dinesh, Deshmukh MB, Kamni Synthesis, X-ray strucutre and N-H…O interactions in 1,3-diphenyl urea. Bull Mater Sci. 2006;29:239–242. [Google Scholar]
- 79.Monchaud D, Teulade-Fichou MP. A hitchhiker's guide to G-quadruplex ligands. Org Biomol Chem. 2008;6:627–636. doi: 10.1039/b714772b. [DOI] [PubMed] [Google Scholar]
- 80.Paul R, Anderson GW. N,N′-carbonyldiimidazole, a new peptide forming reagent. J Am Chem Soc. 1960;82:4596–4600. [Google Scholar]
- 81.Smirnova J, Wöll D, Pflediderer W, Steiner UE. Synthesis of caged nucleosides with photoremovable protecting groups linked to intramolecular antennae. Helv Chim Acta. 2005;88:891–904. [Google Scholar]
- 82.Batey RA, Shen M, Santhakumar V, Yoshina-Ishii C. Parallel synthesis of tri- and tetrasubstituted ureas from carbamoyl imidazolium salts. Comb Chem High Throughput Screen. 2002;5:219–232. doi: 10.2174/1386207024607211. [DOI] [PubMed] [Google Scholar]
- 83.Coyne WC, Cusic JW. 3,4-dihydro-2(1H)-quinazolinones. J Med Chem. 1968;11:1208–1213. doi: 10.1021/jm00312a022. [DOI] [PubMed] [Google Scholar]
- 84.Curran DP, Kuo LH. Altering the stereochemistry of allylation reactions of cyclic α-sulfinyl radicals with diarylureas. J Org Chem. 1994;59:3259–3261. [Google Scholar]
- 85.Wuts PGM, Greene TW. Greene's Protective groups in Organic Synthesis. Wiley & Sons Inc.; New Jersey: 2007. [Google Scholar]
- 86.You SL, Kelly JW. Total stnthesis of didomolamides A and B. Tet Lett. 2005;46:2567–2570. [Google Scholar]
- 87.Mas T, Pardo C, Salort F, Elguero J, Torres MR. A new entry to bis-Tröger's bases. Eur J Org Chem. 2004:1104. [Google Scholar]
- 88.Mergny JL, Maurizot JC. Fluorescence resonance energy transfer as a probe for G-quartet formation by a telomeric repeat. Chembiochem. 2001;2:124–132. doi: 10.1002/1439-7633(20010202)2:2<124::AID-CBIC124>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 89.De Cian A, Guittat L, Shin-Ya K, Riou JF, Mergny JL. Affinity and selectivity of G4 ligands measured by FRET. Nucleic Acids Symp Ser. 2005:235–236. doi: 10.1093/nass/49.1.235. [DOI] [PubMed] [Google Scholar]
- 90.Nguyen B, Tanious FA, Wilson WD. Biosensor-surface plasmon resonance: quantitative analysis of small molecule-nucleic acid interactions. Methods. 2007;42:150–161. doi: 10.1016/j.ymeth.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 91.White EW, Tanious F, Ismail MA, Reszka AP, Neidle S, Boykin DW, Wilson WD. Structure-specific recognition of quadruplex DNA by organic cations: influence of shape, substituents and charge. Biophys Chem. 2007;126:140–153. doi: 10.1016/j.bpc.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 92.Insight II: Modeling Environment. Molecular Simulations Inc.; San Diego: 2000. [Google Scholar]
- 93.Duber-Osguthorpe P, Roberts VA, Osguthorpe DJ, Wolff J, Genest M, Hagler AT. Structure and energetics of ligand binding to proteins: Escherichia coli dihydrofolate reductase-trimethoprim, a drug-receptor system. Proteins. 1988;4:31–47. doi: 10.1002/prot.340040106. [DOI] [PubMed] [Google Scholar]
- 94.Franceschin M, Alvino A, Casagrande V, Mauriello C, Pascucci E, Savino M, Ortaggi G, Bianco A. Specific interactions with intra- and intermolecular G-quadruplex DNA structures by hydrosoluble coronene derivatives: a new class of telomerase inhibitors. Bioorg Med Chem. 2007;15:1848–1858. doi: 10.1016/j.bmc.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 95.Ladame S, Schouten JA, Stuart J, Roldan J, Neidle S, Balasubramanian S. Tetrapeptides induce selective recognition for G-quadruplexes when conjugated to a DNA-binding platform. Org Biomol Chem. 2004;2:2925–2931. doi: 10.1039/B409698C. [DOI] [PubMed] [Google Scholar]
- 96.Affinity. Molecular Simulations Inc.; San Diego: 2000. [Google Scholar]
- 97.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 98.Reed JE, Gunaratnam M, Beltran M, Reszka AP, Vilar R, Neidle S. TRAP-LIG, a modified TRAP assay to quantitate telomerase inhibition by small molecules. Anal Biochem. 2008 doi: 10.1016/j.ab.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 99.Papazisis KT, Geromichalos GD, Dimitriadis KA, Kortsaris AH. Optimization of the sulforhodamine B colorimetric assay. J Immunol Methods. 1997;208:151–158. doi: 10.1016/s0022-1759(97)00137-3. [DOI] [PubMed] [Google Scholar]
- 100.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 101.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Choo HY, Kim M, Lee SK, Kim SW, Chung IK. Solid-phase combinatorial synthesis and cytotoxicity of 3-aryl-2,4-quinazolindiones. Bioorg Med Chem. 2002;10:517–523. doi: 10.1016/s0968-0896(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 103.Jo MN, Seo HJ, Kim Y, Seo SH, Rhim H, Cho YS, Cha JH, Koh HY, Choo H, Pae AN. Novel T-type calcium channel blockers: dioxoquinazoline carboxamide derivatives. Bioorg Med Chem. 2007;15:365–373. doi: 10.1016/j.bmc.2006.09.051. [DOI] [PubMed] [Google Scholar]
- 104.Kakuta H, Tanatani A, Nagasawa K, Hashimoto Y. Specific nonpeptide inhibitors of puromycin-sensitive aminopeptidase with a 2,4(1H,3H)-quinazolinedione skeleton. Chem Pharm Bull. 2003;51:1273–1282. doi: 10.1248/cpb.51.1273. [DOI] [PubMed] [Google Scholar]
- 105.Peet NP, Sunder S. Phosgenation of methyl anthranilate. J Org Chem. 1974;39:1931–1935. [Google Scholar]
- 106.Rachwal PA, Fox KR. Quadruplex melting. Methods. 2007;43:291–301. doi: 10.1016/j.ymeth.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 107.Neidle S. DNA minor-groove recognition by small molecules. Nat Prod Rep. 2001;18:291–309. doi: 10.1039/a705982e. [DOI] [PubMed] [Google Scholar]
- 108.Dai Y, Hartandi K, Ji Z, Ahmed AA, Albert DH, Bauch JL, Bouska JJ, Bousquet PF, Cunha GA, Glaser KB, Harris CM, Hickman D, Guo J, Li J, Marcotte PA, Marsh KC, Moskey MD, Martin RL, Olson AM, Osterling DJ, Pease LJ, Soni NB, Stewart KD, Stoll VS, Tapang P, Reuter DR, Davidsen SK, Michaelides MR. Discovery of N-(4-(3-amino-1H-indazol-4-yl)phenyl)-N′-(2-fluoro-5-methylphenyl)urea (ABT-869), a 3-aminoindazole-based orally active multitargeted receptor tyrosine kinase inhibitor. J Med Chem. 2007;50:1584–1597. doi: 10.1021/jm061280h. [DOI] [PubMed] [Google Scholar]
- 109.Gable KL, Maddux BA, Penaranda C, Zavodovskaya M, Campbell MJ, Lobo M, Robinson L, Schow S, Kerner JA, Goldfine ID, Youngren JF. Diarylureas are small-molecule inhibitors of insulin-like growth factor I receptor signaling and breast cancer cell growth. Mol Cancer Ther. 2006;5:1079–1086. doi: 10.1158/1535-7163.MCT-05-0397. [DOI] [PubMed] [Google Scholar]
- 110.Ambrus A, Chen D, Dai J, Bialis T, Jones RA, Yang D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006;34:2723–2735. doi: 10.1093/nar/gkl348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Phan AT, Kuryavyi V, Luu KN, Patel DJ. Structure of two intramolecular G-quadruplexes formed by natural human telomere sequences in K+ solution. Nucleic Acids Res. 2006;35:6517–6525. doi: 10.1093/nar/gkm706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dai J, Carver M, Punchihewa C, Jones RA, Yang D. Structure of the hybrid-2 type intramolecular human telomeric G-quadruplex in K+ solution: insights into structure polymorphism of the human telomeric sequence. Nucleic Acids Res. 2007;35:4927–4740. doi: 10.1093/nar/gkm522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Drewe WC, Neidle S. Click chemistry assembly of G-quadruplex ligands incorporating a diarylurea scaffold and triazole linkers. Chem Commun. 2008 doi: 10.1039/b814576h. in press. [DOI] [PubMed] [Google Scholar]
- 114.Xue Y, Kan ZY, Wang O, Yao Y, Liu J, Hao YH, Tan Z. Human telomeric DNA forms parallel-stranded intramolecular G-quadruplex in K+ solution under molecular crowding condition. J Amer Chem Soc. 2007;129:11185–11191. doi: 10.1021/ja0730462. [DOI] [PubMed] [Google Scholar]
- 115.Parkinson GN, Cuenca F, Neidle S. Topology conservation and loop flexibility in quadruplex-drug recognition: crystal structures of inter- and intramolecular telomeric DNA quadruplex-drug complexes. J Mol Biol. 2008;381:1145–1156. doi: 10.1016/j.jmb.2008.06.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.