

Abstract
Motivation: Generating accurate transcription factor (TF) binding site motifs from data generated using the next-generation sequencing, especially ChIP-seq, is challenging. The challenge arises because a typical experiment reports a large number of sequences bound by a TF, and the length of each sequence is relatively long. Most traditional motif finders are slow in handling such enormous amount of data. To overcome this limitation, tools have been developed that compromise accuracy with speed by using heuristic discrete search strategies or limited optimization of identified seed motifs. However, such strategies may not fully use the information in input sequences to generate motifs. Such motifs often form good seeds and can be further improved with appropriate scoring functions and rapid optimization.
Results: We report a tool named discriminative motif optimizer (DiMO). DiMO takes a seed motif along with a positive and a negative database and improves the motif based on a discriminative strategy. We use area under receiver-operating characteristic curve (AUC) as a measure of discriminating power of motifs and a strategy based on perceptron training that maximizes AUC rapidly in a discriminative manner. Using DiMO, on a large test set of 87 TFs from human, drosophila and yeast, we show that it is possible to significantly improve motifs identified by nine motif finders. The motifs are generated/optimized using training sets and evaluated on test sets. The AUC is improved for almost 90% of the TFs on test sets and the magnitude of increase is up to 39%.
Availability and implementation: DiMO is available at http://stormo.wustl.edu/DiMO
Contact: rpatel@genetics.wustl.edu, ronakypatel@gmail.com
1 INTRODUCTION
Transcription factors (TFs) can modulate gene expression patterns and hence are key components of cellular regulatory networks. TFs bind to DNA in a sequence-specific manner. The relative preference of TFs to various nucleotide sequences is conveniently expressed in the form of position weight or frequency matrices (PWM or PFM, respectively), often referred as TF binding site motifs. A typical experiment to determine TF specificity, especially using next-generation sequencing, generates information about binding sites at a relatively low resolution. The binding site is identified within a region of the genome that is significantly larger than TF binding site. Identified peaks can be as long as few hundreds to thousands of base pairs, and the total number of peaks can also be in a similar range. Most traditional motif finders are slow to identify motifs from such an enormous amount of unaligned sequences.
As an attempt to overcome this problem, a few hundreds of top-scored peaks are used for identifying TF binding site motifs leaving a large number of sequences unused to generate accurate motifs. Alternatively, in recent years motif finders have been developed that can handle relatively large amount of sequences and identify motifs faster with the use of some approximations. For example, a recently developed method, Discriminative Regular Expression Motif Elicitation (DREME), rapidly identifies multiple short eukaryotic motifs for a large number of sequences. To achieve higher speed, it searches for motifs over degenerate sequence (IUPAC) space and estimates Fisher’s P-value rather than rescanning input sequences (Bailey, 2011). Another method, Discriminative Motif Enumerator (DME) is fast in identifying motifs by use of precomputed matrices. However, the number of precomputed matrices increases significantly and speed decreases for identification of motifs at fine resolutions (Smith et al., 2005). MDScan uses few sequences from large input set to identify motifs and calculates significance on a number of sequences that are larger than those used for finding motifs (Liu et al., 2002). Automated Motif Discovery (AMD) uses multistep processing and filtering to identify significant core motifs in IUPAC space. The core motifs are then extended and refined based on selected sites with one mutation from core motifs (Shi et al., 2011). DECOnvolved Discriminative motif discovery (DECOD) uses a heuristic hill climbing approach to identify enriched motifs from a limited set of k-mers (specified by motif cardinality) (Huggins et al., 2011). All these methods can identify significant seeds motifs from a large database leaving room for further optimization. Like a few other methods (Leung and Chin, 2006; Redhead and Bailey, 2007), Contrast Motif Finder (CMF) uses two step strategies: seed identification and iterative optimization. Both steps use Z-score to identify significant motifs (Mason et al., 2010). Discriminative PWM Search (DIPS), one of the earliest discriminative motif finder, uses an iterative optimization process to find a set of sites that maximizes discrimination score (Sinha, 2006). XXmotif is a recently developed method that uses three step procedures—seed finding, merging and refinement—to find motifs enriched in positive sequences (Hartmann et al., 2013). Both seed searching and refinement are performed based on enrichment P-value. Peak motifs of regulatory sequence analysis tool (RSAT) are fast and capable of handling several thousands of sequences. It also generates user friendly and rich output (Thomas-Chollier et al., 2011, 2012). It rapidly searches overrepresented hexa/heptamer followed by merging significant patterns. DIPS, CMF and XXmotif compared with DME and MDScan are slower for an input of several hundred sequences. However, they can handle small number of sequences efficiently and produce motif models. Such motifs can again be used as seeds and further re-optimized on a larger set of sequences. In addition to these, several other methods are developed using discriminative principle-based on different scoring functions and search procedures (da Piedade et al., 2009; Davis et al., 2012; Elemento et al., 2007; Fauteux et al., 2008; Fu et al., 2009; Linhart et al., 2008; Sharan and Myers, 2005; Siddharthan, 2008; Sinha, 2003; Wang et al., 2005) and vary on a scale of computational efficiency.
In this article, we present a discriminative motif optimizer, DiMO, that takes seed motifs described earlier in the text and upgrades them with a systematic, rapid and local search. DiMO can be used for the same number of sequences that is used for identifying motifs or on larger databases. DiMO uses intuitive AUC under ROC curve as a measure of discrimination by a motif and improves it using a strategy based on the logic of perceptron learning. With the use of DiMO statistically significant improvements in AUCs were achieved on a large test set of 87 TFs motifs. The motifs generated using multiple software, on sequences from ChIP-seq and ChIP-chip experiments on human K562 cell lines, drosophila and yeast, were improved. In addition, applicability of DiMO in generating more complete motifs by optimizing width of motifs is also suggested. In addition to this, DiMO is fast and converges in median of nine epochs when seed motifs were optimized with same motif lengths.
2 METHODS
2.1 AUC as a discrimination score and algorithm to improve
A machine learning method can correctly identify true positives by compromising with false positives. If the input data are heterogeneous with a fuzzy boundary of separation, then depending on the boundaries or cutoffs, different compromises between true positives and false positives can be achieved. If a boundary is drawn or a cutoff is used to classify all positives correctly, then the chance of classifying negatives incorrectly increases. Simple measures or scores describing classification accuracy like sensitivity (true-positive rate; TPR), specificity [1—false-positive rate (FPR); or true-negative rate], likelihood ratios and other related variants are measured for a selected boundary or cutoff. Hence, these measures are not a general measure to gauge classification accuracy because they vary depending on the cutoff used. Instead, ROC curve plots the trade-off between two important measures of classification performance, TPR and FPR at all possible cutoff values. Visual inspection of ROC provides an intuitive way to see trade-off between TPR and FPR at various cutoffs, and the area under the ROC curve with few limitations provides an accurate measure of classification performance (Bewick et al., 2004; Grzybowski and Younger, 1997). The AUC under ROC has been used to gauge and compare the performance of different motifs given a motif and set of sequences bound by TFs and not bound by TFs in previous studies (Weirauch et al., 2013). The AUC under ROC has also been used as a scoring function to optimize motifs using genetic algorithm, given a foreground and background set of sequences (Li et al., 2007).
Given a PFM/PWM, a positive (P) and a negative set (N) of sequences, ROC can be computed by varying cutoffs systematically to find the fraction of sequences that exceed it in P and N. The area under ROC curve is AUC. The best possible PFM/PWM gives AUC of 1. AUC of 1 for a PFM/PWM is obtained when a cutoff exists such that all the sequences in P have that motif with no occurrence in N. However, owing to experimental noise and limitations of PWM/PFM models, finding a motif with AUC of 1 is nearly impossible except when both P and N are a small set of short sequences.
Various reported methods, described in the introduction, use different scoring functions and search procedures and produce motifs by compromising between speed and accuracy. The motifs reported might be a precise solution that describes the input data or speed compromised solution with discrete numbers. Such PFM/PWMs are referred to as a seed matrix. This seed matrix serves as an input to our method and is refined using perceptron learning. Perceptron learning originally reported by Frank Rosenblatt (1962) has been used to generate a weight matrix that discriminated between ribosome binding sites (RBS) from the non-RBS (Stormo et al., 1982). Weight matrix to distinguish RBS from random sites is updated by randomly picking a site followed by adding to weight matrix if it is an RBS and subtracting if it is not an RBS. The approach reported in the present study is different from original approach in the respect that the weight matrix is updated based on multiple sites simultaneously.
If the seed matrix is a PFM, it is converted to a PWM by taking a natural logarithm and denoted by W. W is composed of matrix of 4 × L elements, where L is the width of a motif. Given a W, P and N, for each sequence in P and N the best scoring site is identified and denoted by S+ and S−, respectively. A new PWM for next iteration is generated in the following manner adapted from perceptron learning:
where, α is the learning rate and δ is computed in the following way:
δ in perceptron learning represents the difference between current and target value of variable to be optimized. In the current work, where the problem is of discrimination in nature, δ was defined by the difference in two weight matrices denoted by ω+ and ω−.
To construct ω+ and ω−, first sites in S+ and S− were combined and sorted based on the score. The sites were scored by adding corresponding elements from W (4 × L). If a set of positive and negative sites has an identical score then positive sites were put first. Such sorting results into a list of sites with the best scoring positive sites at the top and the worst scoring negative sites at the bottom. In that sorted list, the sites in region marked by the first occurrence of a negative site and the last occurrence of a positive site are the ones in error state (a perfect classifier will have no S− scoring higher than any S+), and if these are classified correctly then the AUC can be improved. The positive sites in this region were collected and used for computing ω+. Similarly, negative sites in this region were collected and used for computing ω−.
The new PWM, 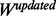 , is generated for user-defined maximum number of epochs (default is 150). During each epoch, different α are tried. For the results presented here, three equidistantly placed α, between 1 to 0.1, were used. If the AUC increases, in a given epoch with one of the learning rate, the new PWM is accepted and the process is repeated. At the start of optimization when the motif is far from solution, new PWM generated with learning rate 1 is accepted. When close to solution, PWMs generated using smaller learning rates are successful in updating PWM. If no improvement in AUC is achieved then further optimization is stopped and the results are reported.
, is generated for user-defined maximum number of epochs (default is 150). During each epoch, different α are tried. For the results presented here, three equidistantly placed α, between 1 to 0.1, were used. If the AUC increases, in a given epoch with one of the learning rate, the new PWM is accepted and the process is repeated. At the start of optimization when the motif is far from solution, new PWM generated with learning rate 1 is accepted. When close to solution, PWMs generated using smaller learning rates are successful in updating PWM. If no improvement in AUC is achieved then further optimization is stopped and the results are reported.
2.2 Computation of ROC and AUC
Points on the ROC curve represent the fraction of negative sequences (x-axis) versus fraction of positive sequences (y-axis) containing a site that exceeds a threshold and are denoted as FPR and TPR, respectively. The number and spacing of cutoffs to compute points on ROC are not known a priori. The AUC computed under a line joined by such points with approximations might change depending on different number and spacing of cutoffs used. To overcome this problem, first all the sites in S+ and S− were combined and sorted. This is followed by going down the sorted list and computing the fraction of total positive and total negative sequence at or above it. If several sites in S+ and S− have identical scores, then area under diagonal line is plotted. AUC represents area under this curve.
2.3 Databases for motif finding
To evaluate the extent of improvements that can be achieved by our method, sequence data were collected from ChIP-seq and ChIP-chip experiments in the public domain. The sequences are from the drosophila (Drosophila melanogaster), human and yeast (Saccharomyces cerevisiae) genomes. The number of sequences in positive and negative sets range from 102–1000 (Supplementary Table S1). Optimizing motifs on small set of sequences (typically <100) in the training set using DiMO result in significant improvement in AUC, however, perform poorly on test set as a result of over fitting. We generally recommend at least 100 sequences of ∼500 nt each in positive and negative sets. Detailed information about data collection is given in Supplementary Information. Overall, sequences for 87 TFs (34 drosophila, 43 human and 10 yeast) were collected. To generate a negative set, equal number of non-overlapping sequences bound by TFs other than TF under consideration were collected. This is a good choice of negative database, as it is composed of sequences that are accessible to the TF under consideration but are bound by other TFs under the same environmental conditions and/or same cell line. Difference in percent GC content of positive and negative set of sequences is given in Supplementary Figure S1. It is evident that for the most TFs, percent GC difference for sequences in positive and negative dataset is <8%.
2.4 Software to identify preliminary motifs
The collected set of positive and negative sequences was randomly split into three sets of similar sizes, P1 to P3 and N1 to N3. Two of the three are combined to form a training set and one forms the test set for 3-fold cross-validation. This results in three training (TR1, TR2 and TR3) and three test sets (TE1, TE2 and TE3) in the ratio of ∼66:33. On each of the training set, DREME, DECOD, MDScan, DME, ChIPMunk and RSAT/peak motifs were run (motif finder set I, MF1). ChIPMunk, (Kulakovskiy et al., 2010) although not a discriminative motif finder, was included in the present study as it is fast and capable of handling large number of sequences. Considering the speed of CMF, DIPS and XXmotif, only 10% of the aforementioned training sets sequences from drosophila and human was used for motif finding (motif finder set 2, MF2).
The motif finders were run with a positive and negative set of sequences generated as mentioned in Section 2.3 (except ChIPMunk). Default parameters were used for motif finders unless mentioned. For DREME, DECOD, MDScan, DME, ChIPMunk and DIPS, TF binding site models of width eight were used. For CMF TF motifs of width 7–8 were used considering technical restraints. For similar reason, maximum motif width of 15 was used with XXmotif. Software was run with default parameters except specifying motif width. For DECOD, one motif was identified, for DIPS, one iteration was used to find motifs and for MDScan, 30 and 1000 sequences were used to find and confirm motifs, respectively. DREME, CMF and XXmotif produced multiple motifs. E-score, t-score and P-values were used to identify the best motif from the output of DREME, CMF and XXmotif, respectively. For XXmotif, only five-mer seeds were generated to start motif finding, leaving out palindromic and repeat seeds, to be consistent with other motif finders. RSAT/peak motifs were run using the server at http://rsat.ulb.ac.be/, and the top motif was selected for optimization. For each TF, using sequences in TR1, TR2 and TR3, three motifs were identified using different motif finders and evaluated on TE1, TE2 and TE3. The 3-fold cross-validation AUC for training set is the average performance of motif on TR1, TR2 and TR3. Similarly, for test set, the 3-fold cross-validation AUC is the average on TE1, TE2 and TE3.
2.5 Validation of identified motifs
Motifs identified using nine software were used as inputs to DiMO along with the positive and negative training sets (TR1 to TR3). The motifs were re-optimized using DiMO by keeping the same width as well as by optimizing motif width on both 5′ and 3′ ends. When the motifs were optimized using same motif length, the improvements were gauged by comparing AUC from 3-fold cross-validation with and without optimizing motifs using DiMO. The student’s t-test was used to evaluate the significance of improvements in AUC.
2.6 Motif width optimization
From the three motifs per TFs generated by the aforementioned procedure, one motif that gives the best AUC on the corresponding test set was selected for width optimization. The corresponding training sets of sequences (TR) were split into training and validation sets in the ratio of ∼80:20. The DiMO-optimized motif with the same motif length was extended to around width 20 (19–21) by adding equal number of non-specific position on 5′ and 3′ sides (with equal frequency of bases A, C, G and T). The extended motif was then optimized using DiMO with 80% of sequences. This results in optimized-extended motif of width 19–21. If the motif finder reports motif that is of width >20, no extension is performed.
This extended motif was then shortened to a motif of width six. This six-long motif is central part of extended-optimized motif. With this short motif, AUC is calculated on left out 20% of sequences. The six-long motif was then extended by a width one on 5′ side. This is done by restoring corresponding position from the large motif generated using procedure described in previous paragraph. AUC on validation set is then computed with this motif of width seven. If the seven-long motif improves AUC on validation set by at least 0.3% then the extension is accepted. Motif is restored on 5′ side till AUC on validation set increase at least by 0.3% per position addition. After that the motif is extended on 3′ side in a similar manner. The motif extension penalty of 0.3% is necessary to prevent over-fitting on smaller database. No penalty or penalty <0.3% will produce motifs that are of large width without much gain in enrichment in terms of AUC. A brief summary of workflow of motif finding and optimization is summarized in Supplementary Figure S2.
2.7 Graphical rendering of weight matrix
During optimization DiMO improves the discriminative weights and does not have physical meaning. We assume that the weights are energy (in the unit of kT) and use Boltzmann distribution to convert PWM to PFM. PFMs generated in this way were used to generate logos. Although DiMO produces PFMs without scaling the PWM, a scaling factor can be used and result in PFM with different information content.
2.8 Selection of motifs for human, drosophila and yeast TFs
Nine original (as reported by the MF1 and MF2) and nine-optimized motifs (DiMO-optimized motifs) per TF were used for evaluating AUC on combined training and test sets. The motif that gave the best AUC was considered as the motif that causes the best discrimination in the positive/negative sequences for that TF.
3 RESULTS
3.1 Summary of improvements with a constant motif length
Motifs produced by software in MF1 (ChIPMunk, DREME, DECOD, MDScan, RSAT and DME) were optimized on exact same set of sequences using DiMO. DiMO in most cases improves AUC on training set when same motif length was used (Fig. 1). The extent of increase in 3-fold cross-validation AUC is up to 32% on training set. This clearly indicates that it is possible to improve the AUC under ROC curve with the perceptron training strategy used in the present study. The quality of motifs is improved by adjusting information content as well as by change of preferred bases. When the original and optimized motifs are evaluated on the test set, the 3-fold cross-validation AUC improves with a significant student's paired t-test P-value (Table 1).
Fig. 1.

Plots of 3-fold cross-validation AUC before (original AUC) and after optimization (improved AUC) with DiMO. The left and right panels show 3-fold cross-validation AUCs on training and test sets, respectively. The top panels show AUCs when motifs were identified using software from set MF1, whereas the bottom panels from set MF2
Table 1.
Statistical summary of improvements in AUC on test sets using 3-fold cross-validation
| Optimization with same motif width |
||
|---|---|---|
| MF1 | MF2 | |
| Students t-test P-valuea | <2.2e-16 | <2.2e-16 |
| AUC increased (% of runs) | 91.05 | 95.02 |
| AUC increased by 0.03 (% of runs) | 34.63 | 43.3 |
| AUC increased by 0.07 (% of runs) | 11.09 | 13.03 |
| Maximum increase in AUC | 0.32 | 0.23 |
| AUC decreased (% of runs) | 8.95 | 4.98 |
| AUC decreased by 0.03 (% of runs) | 0.58 | 0.77 |
| AUC decreased by 0.07 (% of runs) | 0 | 0 |
| Maximum decrease in AUC | 0.05 | 0.05 |
aPaired t-test with alternative hypothesis of differences in means of optimized and original motif is <0.
Overall on the test set, the 3-fold cross-validation AUC was increased for 90% of TF motifs. The 3-fold cross-validation AUC increased at least by 7% for 11% of TFs. Various statistical measures used to gauge the extent of improvements achieved with optimization and the trade-off with cases when the 3-fold cross-validation AUC was decreased on optimization on test sets are summarized in Table 1. It is evident that the extent and cases of increase in AUC are much greater than those where the AUC decreases on test sets. Time required for optimization of motifs reported from DREME is given in Supplementary Figure S3. Although time required by DiMO to optimize motifs depends on starting seed, for most cases the runs required <2 min for typical input size used in present study. Median number of epochs required for DiMO to converge is nine and distribution is given in Supplementary Figure S4.
3.2 Improving motifs with increased size of database
CMF, XXmotifs and DIPS are relatively slow compared with MDScan and DME for typical input used in current study; however, they can generate motifs on a small set of sequences. Using CMF, XXmotifs and DIPS, the motifs were identified on only 10% of sequences in training set for drosophila and human TFs, but optimized on entire corresponding training set (TR1, TR2 or TR3) to show DiMO’s applicability in optimizing motifs on dataset that is different or larger than the one on which motifs were discovered. The performance of improvement in AUC is summarized in Table 1 and Figure 1. Again using DiMO, on test set a significant increase in AUC is achieved (Table 1) for 95% of TFs. The improvement is by adjusting information content and base specificity. Here, we show applicability of DiMO in using greater number of sequences to optimize motifs than it was originally identified on. With DiMO, it is also possible to re-optimize motifs with different positive and/or negative sets.
3.3 Summary of improvements by optimizing width of motifs
Seed motifs identified by software used in current study are mostly of width eight. This motif length is the upper limit suggested for the speed efficiency of DREME (Bailey, 2011). The computation time required for the identification of motif increases exponentially for DECOD (Huggins et al., 2011). When the motif width of eight is used to identify motifs, it might happen that the software reports a part of a motif for the TFs with binding site width >8. The motif width is not known a priori. From MF1 and MF2, DREME, CMF and ChIPMunk allow for using variable width during motif finding and rank them based on score. RSAT and XXmotifs produce motifs that are of different widths.
We used DiMO for finding optimum width of motifs based on AUC on validation set (Section 2). The comparison of AUCs computed from motifs reported by various motif finders and selected length-optimized motifs on test set is given in Table 2 and Figure 2. The total number of cases where the AUC was improved on test set is almost 85–90% with 50 and 25% of cases with AUC improved on test cases by 3 and 7%, respectively. The improvement in AUC achieved by DiMO on test set is a general trend and not limited to the seeds generated by particular software (Figs 1, 2 and Table 3). Irrespective of difference in seed motifs, the AUC optimization using perceptron training leads to reasonably similar AUCs and motifs if the starting seed is relatively close to final motifs (Supplementary File S2). This shows that one can use a fast motif finder (like DREME, DME or MDScan), as long as the quality of motif is reasonably good, and then optimize it to get a motif that is as good as those obtained by slower motif discovery algorithms.
Table 2.
Statistical summary of improvements in AUC on test sets when length of motif may be different from original motifs
| Optimization when motif width may be different from the original motifs |
||
|---|---|---|
| MF1 | MF2 | |
| Students t-test P-value | <2.2e-16 | <2.2e-16 |
| AUC increased (% of runs) | 84.82 | 89.66 |
| AUC increased by 0.03 (% of runs) | 51.95 | 59 |
| AUC increased by 0.07 (% of runs) | 22.76 | 31.42 |
| Maximum increase in AUC | 0.39 | 0.31 |
| AUC decreased (% of runs) | 15.18 | 10.34 |
| AUC decreased by 0.03 (% of runs) | 2.72 | 1.15 |
| AUC decreased by 0.07 (% of runs) | 0.78 | 0.38 |
| Maximum decrease in AUC | 0.14 | 0.24 |
Fig. 2.

Plots of AUC reported on test sets with motifs before (original AUC) and after DiMO optimization (improved AUC). The optimized motifs may have length different than the original motifs. The top and bottom compares AUCs reported by original and improved motifs generated using software in MF1 and MF2, respectively
Table 3.
Mean, median and standard deviations of improvement in AUC on optimization using seeds from different software
| Methods | Mean | Median | Standard deviation |
|---|---|---|---|
| ChIPMunk | 0.039 | 0.035 | 0.052 |
| CMF | 0.045 | 0.030 | 0.055 |
| DECOD | 0.034 | 0.022 | 0.054 |
| DIPS | 0.067 | 0.050 | 0.061 |
| DME | 0.058 | 0.044 | 0.067 |
| DREME | 0.039 | 0.026 | 0.040 |
| MDScan | 0.060 | 0.028 | 0.077 |
| RSAT/peak motifs | 0.046 | 0.040 | 0.058 |
| XXmotif | 0.055 | 0.039 | 0.071 |
In a time benchmark study of finding motifs using motif length 8–20, we selected 34 drosophila TFs and run DREME, CMF, XXmotifs and DiMO. Time required by different software is given in supplementary information (Supplementary Fig. S5). Overall performance of DiMO is comparable with other software, in some cases slower and in some cases faster than DREME, CMF and XXmotifs. The performance of DiMO depends on the nature of seed motifs, but the time benchmarking gives an overall idea. DREME and CMF although take different width as input, they produce motifs of width 7–8.
In Figure 3, a histogram of difference in lengths of original and selected motifs is given. It is evident that in several cases original length of motif was good enough for optimization; however, for most cases, the selected motif is of size that is different from original. There are few cases where the motifs extended or trimmed by width of >5.
Fig. 3.

Histogram of distribution of selected lengths that gives the best AUC
3.4 Extent of improvements
As described previously, the improvement in AUC by DiMO is achieved by changing the information content and preferred base. In Figure 4, two quantities: symmetric Kullback–Leibler (sKL) divergence averaged over motifs and numbers of preferred bases different that describe difference in original and optimized motifs are shown. The per position average sKL divergence is mostly <0.5; however, there are >25% cases, when the new motifs are different from original motifs with distance greater than this. Similarly, at least for 65% of cases, the preferred base of optimized and original motifs differs from each other by at least one position. Overall, the two quantities plotted in Figure 4 show that it is possible to upgrade motifs to better utilize information in training data. To show the visual difference in motifs, binding site logos for few selected TFs are shown in Figure 5.
Fig. 4.

Distribution of sKL distances (top panel) and number of preferred based different (bottom panel) between original and optimized motifs obtained when optimization was performed with identical motif widths
Fig. 5.

Binding site logos generated using original (left panel), optimized motifs (middle panel) and length-optimized motifs (right panel). AUC of original motifs on a test set is 0.70, 0.87, 0.58, 0.55, 0.69 and 0.79, and on optimization increased to 0.71, 0.97, 0.76, 0.66, 0.78 and 0.84, respectively, for Ctcf, Mafk, hkb, run, prd and RAP1, respectively. The AUC on length optimization on test set is 0.92, 0.98, 0.78, 0.68, 0.83 and 0.92, for Ctcf, Mafk, hkb, run, prd and RAP1, respectively. The sKL divergence between original and optimized motifs with same motif length per base pair is 0.07, 1.44, 2.74, 1.33, 1.29 and 0.65, and number of preferred base different are 0, 2, 2, 3, 1 and 0, for Ctcf, Mafk, hkb, run, prd and RAP1, respectively
Recently, it has been observed that the positional distribution of sites bound by TFs in Chip-seq peaks is uni-modal and centrally enriched (Bailey and Machanick, 2012). We computed centrality enrichment using CentriMo for original and length-optimized motifs generated using Chip-seq peaks from human TFs. The distribution of sites reported by CentriMo is given in supplementary information. In the cases where the motif seed showed some central enrichment, DiMO improves the centrality to a minor extent; however, there are examples where centrality of DiMO-reported motifs is much better than original motifs (Atf1/CMF, Ccnt2/DIPS, MDScan/c-myc, JunD/XXmotifs, Max/RSAT-peak motifs, Znf384/RSAT-peak motifs; Supplementary File S3).
3.5 Selected motifs for drosophila, human and yeast TFs based on the best AUC on combined dataset
All original (as reported by the MF1 and MF2) and optimized motifs (DiMO-optimized motifs) that gave best performance on corresponding test set were used for computing AUC on entire set of sequences. The motif that gives the best AUC was selected as a motif for that TF. In the supplementary information (Supplementary File S4), binding site logos of selected PFMs for 87 TFs are given. For all TFs but two, the optimized motifs by DiMO were selected as the best PFM, indicating the usefulness of optimization of seed motifs.
4 DISCUSSIONS
4.1 Concept of motif optimization
Generating TF binding site models is challenging, but recent technological advances offer the opportunity for rapid determinations of specificity from in vitro studies (Stormo and Zhao, 2010). In vivo location analysis can also be used effectively to determine TF binding site motifs. ChIP followed by microarray or next-generation sequencing reports few thousand sequences at low resolution that are bound by a TF under consideration. The resolution of identified TF binding site and number of sequences reported are expected to increase in near future with the advent of improved technology (Furey, 2012). With this consideration, it is imperative to develop motif finding tools that can handle large number of sequences. Identifying accurate TF binding site motifs from few thousand sequences is computationally expensive. On other hand, software that rapidly identifies motifs uses certain approximations in scoring and/or search space. Different methods use varying levels of compromise between speed and accuracy. Methods that search for accurate binding site models using global optimization can only be applied on a small number of sequences. On other hand, methods using approximation for searching motifs result in motifs that might be a suboptimal use of the depth of data generated. In either case, the generated motifs can be evaluated and/or improved on the same or larger positive and/or negative datasets. In this article, we developed a tool, DiMO, for this purpose.
Inputs for DiMO are a seed motif and a database of sequences expected or not expected to contain binding sites. DiMO uses perceptron learning algorithm to improve the AUC of the motif. The new motif can be of similar or different width. The capability of DiMO to improve AUC and hence discrimination with same and width-optimized motifs are summarized in Figures 1 and 2, respectively. Key statistics to describe improvement of quality of motifs in terms of AUC on test dataset are summarized in Tables 1 and 2. It should be noted that Tables 1 and 2 summarize data of improvements of quality of motifs reported by nine software on a test datasets for 87 experiments from three different organisms. DiMO improves motifs by not only optimizing information content of motif but also by changing preferred base, if necessary (Figs 4 and 5). Finally, using AUC under ROCs as a criterion, one motif per TF enriched in ChIP-chip/seq data is presented for TFs of human, drosophila and yeast.
4.2 ROC-AUC as scoring function and perceptron learning for motif optimization
DiMO uses intuitive AUC under ROC as a score to gauge discriminating power of a motif. AUC under ROC (partial) has been used as a scoring in combination with genetic algorithm for optimization for optimizing PWMs in GAPWM (Li et al., 2007). The major advantage of AUC compared with other discrimination score mostly based on log-likelihood ratio is that there is no need of user or developer imposed cutoffs. AUC under ROC has been used to compare performance of various motifs given a set of sequences bound and not bound by TFs (Weirauch et al., 2013). The AUC can be improved analytically using well-known neural-network/perceptron-based learning. The perceptron-based optimization under current implementation is a local optimization procedure. If the combinatorial parameter space is steep with single minimum, then it can report the globally optimized motifs. Other techniques like support vector machine or variants can also be used to improve AUC; however, the use of kernels might not be suitable for motif optimization problem.
4.3 Applications of DiMO
In this article, we present two specific applications of DiMO along with motif optimization. (i) To optimize motifs on a positive/negative dataset that is larger than the set used for identifying motifs. (ii) Optimization of motifs with suitable motif widths compared with identified motifs. However, DiMO can also be used to understand how the binding site preferences change when different background/negative set or foreground/positive sets are used given a starting/seed motif. This is often true when a TF interacts with different cofactors to generate different specificity. Applicability of DiMO in identifying motifs based on dynamically changing negative set based on availability/unavailability of other TFs is under evaluation. Although in the present study, perceptron training and discriminative learning are used for improving motifs, it can be generally applicable for other discriminative learning problems frequently encountered in science and technology.
5 CONCLUSIONS
In the present article, a novel concept of motif optimization as an independent part of motif identification is suggested. The motif optimization as a distinct part of motif finding problem is inevitable in the light of using depth and width of data generated from next-generation sequencing experiments. DiMO is presented as a tool to cater this need. Using a large test of 87 TFs from human, drosophila and yeast, we show the applicability of DiMO in improving motifs identified using nine methods.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are thankful to Saurabh Sinha for the help with sequences for drosophila TFs. They are also thankful to anonymous reviewers for helpful comments.
Funding: NIH (HG000249 to G.D.S.).
Conflict of Interest: none declared.
REFERENCES
- Bailey TL. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics. 2011;27:1653–1659. doi: 10.1093/bioinformatics/btr261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Machanick P. Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res. 2012;40:e128. doi: 10.1093/nar/gks433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick V, et al. Statistics review 13: receiver operating characteristic curves. Crit. Care. 2004;8:508–512. doi: 10.1186/cc3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Piedade I, et al. DISPARE: DIScriminative PAttern REfinement for position weight matrices. BMC Bioinformatics. 2009;10:388. doi: 10.1186/1471-2105-10-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, et al. POWRS: position-sensitive motif discovery. PLoS One. 2012;7:e40373. doi: 10.1371/journal.pone.0040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elemento O, et al. A universal framework for regulatory element discovery across all genomes and data types. Mol. Cell. 2007;28:337–350. doi: 10.1016/j.molcel.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauteux FO, et al. Seeder:discriminative seeding DNA motif discovery. Bioinformatics. 2008;24:2303–2307. doi: 10.1093/bioinformatics/btn444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, et al. DISCOVER: a feature-based discriminative method for motif search in complex genomes. Bioinformatics. 2009;25:i321–i329. doi: 10.1093/bioinformatics/btp230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 2012;13:840–852. doi: 10.1038/nrg3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzybowski M, Younger JG. Statistical methodology: III. Receiver operating characteristic (ROC) curves. Acad. Emerg. Med. 1997;4:818–826. doi: 10.1111/j.1553-2712.1997.tb03793.x. [DOI] [PubMed] [Google Scholar]
- Hartmann H, et al. P-value-based regulatory motif discovery using positional weight matrices. Genome Res. 2013;23:181–194. doi: 10.1101/gr.139881.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins P, et al. DECOD: fast and accurate discriminative DNA motif finding. Bioinformatics. 2011;27:2361–2367. doi: 10.1093/bioinformatics/btr412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulakovskiy IV, et al. Deep and wide digging for binding motifs in ChIP-Seq data. Bioinformatics. 2010;26:2622–2623. doi: 10.1093/bioinformatics/btq488. [DOI] [PubMed] [Google Scholar]
- Leung HCM, Chin FYL. Finding motifs from all sequences with and without binding sites. Bioinformatics. 2006;22:2217–2223. doi: 10.1093/bioinformatics/btl371. [DOI] [PubMed] [Google Scholar]
- Li L, et al. GAPWM: a genetic algorithm method for optimizing a position weight matrix. Bioinformatics. 2007;23:1188–1194. doi: 10.1093/bioinformatics/btm080. [DOI] [PubMed] [Google Scholar]
- Linhart C, et al. Transcription factor and microRNA motif discovery: the Amadeus platform and a compendium of metazoan target sets. Genome Res. 2008;18:1180–1189. doi: 10.1101/gr.076117.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, et al. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 2002;20:835–839. doi: 10.1038/nbt717. [DOI] [PubMed] [Google Scholar]
- Mason MJ, et al. Identification of context-dependent motifs by contrasting ChIP binding data. Bioinformatics. 2010;26:2826–2832. doi: 10.1093/bioinformatics/btq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redhead E, Bailey TL. Discriminative motif discovery in DNA and protein sequences using the DEME algorithm. BMC Bioinformatics. 2007;8:385. doi: 10.1186/1471-2105-8-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt F. Principles of Neurodynamics: Perceptrons and the Theory of Brain Mechanisms. Washington, DC: Spartan Books; 1962. [Google Scholar]
- Sharan R, Myers EW. A motif-based framework for recognizing sequence families. Bioinformatics. 2005;21(Suppl. 1):i387–i393. doi: 10.1093/bioinformatics/bti1002. [DOI] [PubMed] [Google Scholar]
- Shi J, et al. AMD, an automated motif discovery tool using stepwise refinement of gapped consensuses. PLoS One. 2011;6:e24576. doi: 10.1371/journal.pone.0024576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddharthan R. PhyloGibbs-MP: module prediction and discriminative motif-finding by Gibbs sampling. PLoS Comput. Biol. 2008;4:e1000156. doi: 10.1371/journal.pcbi.1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. Discriminative motifs. J. Comput. Biol. 2003;10:599–615. doi: 10.1089/10665270360688219. [DOI] [PubMed] [Google Scholar]
- Sinha S. On counting position weight matrix matches in a sequence, with application to discriminative motif finding. Bioinformatics. 2006;22:e454–e463. doi: 10.1093/bioinformatics/btl227. [DOI] [PubMed] [Google Scholar]
- Smith AD, et al. Identifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc. Natl Acad. Sci. USA. 2005;102:1560–1565. doi: 10.1073/pnas.0406123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo GD, et al. Use of the ‘Perceptron' algorithm to distinguish translational initiation sites in E. coli. Nucleic Acids Res. 1982;10:2997–3011. doi: 10.1093/nar/10.9.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo GD, Zhao Y. Determining the specificity of protein-DNA interactions. Nat. Rev. Genet. 2010;11:751–760. doi: 10.1038/nrg2845. [DOI] [PubMed] [Google Scholar]
- Thomas-Chollier M, et al. RSAT peak-motifs: motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res. 2011;40:e31. doi: 10.1093/nar/gkr1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M, et al. A complete workflow for the analysis of full-size ChIP-seq (and similar) data sets using peak-motifs. Nat. Protoc. 2012;7:1551–1568. doi: 10.1038/nprot.2012.088. [DOI] [PubMed] [Google Scholar]
- Wang G, et al. WordSpy: identifying transcription factor binding motifs by building a dictionary and learning a grammar. Nucleic Acids Res. 2005;33:W412–W416. doi: 10.1093/nar/gki492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013;31:126–134. doi: 10.1038/nbt.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.