

Abstract
Recently, autophagy has drawn more attention in cardiovascular disease as it has important roles in lipid metabolism. Mammalian target of rapamycin (mTOR) is a key regulator of autophagy; however, its effect on atherosclerosis and the underlying mechanism remains undefined. In this study, an obvious upregulation of mTOR and p-mTOR protein was observed in macrophage-derived foam cells. Blocking mTOR expression with specific small interference RNA (siRNA) dramatically suppressed foam cell formation, accompanied by a decrease of lipid deposition. Further mechanistic analysis indicated that suppressing mTOR expression significantly upregulated autophagic marker LC3 expression and downregulated autophagy substrate p62 levels, indicating that mTOR silencing triggered autophagosome formation. Moreover, blocking mTOR expression obviously accelerated neutral lipid delivery to lysosome and cholesterol efflux from foam cells, implying that mTOR could induce macrophage foam cell formation by suppressing autophagic pathway. Further, mTOR silencing significantly upregulated ULK1 expression, which was accounted for mTOR-induced foam cell formation via autophagic pathway as treatment with ULK1 siRNA dampened LC3-II levels and increased p62 expression, concomitant with lipid accumulation and decreased cholesterol efflux from foam cells. Together, our data provide an insight into how mTOR accelerates the pathological process of atherosclerosis. Accordingly, blocking mTOR levels may be a promising therapeutic agent against atherosclerotic complications.
Introduction
Cardiovascular disease (CVD) is known as the biggest killer around the world, representing nearly 29% of mortalities globally (McLaren et al., 2011; Verhagen and Visseren, 2011). The principal cause of cardiovascular complications, such as myocardial infarction, stokes, and coronary heart disease, is atherosclerosis, which is characterized by lipid accumulation within the arteries. It is generally believed that lipid-laden foam cell accumulation is the hallmark of the early stage of atherosclerosis, and then triggers a series of atherosclerotic complications (Lu et al., 2010; Mendis et al., 2011).
Lipid droplet (LD) formation confers one of the most cardiovascular risks, and initiates atherosclerotic progression. Macrophages are ranked as a critical regulator to induce the lipid-laden foam cell formation by the uptake of oxidized low-density lipoprotein (oxLDL), following the appearance of fatty streaks (Moore and Tabas, 2011). Blocking the macrophage foam cell formation significantly attenuates atherosclerotic development, indicating a promising therapeutic strategy against atherosclerotic complications via the mobilization of lipophagy (Li et al., 2004; Qiu et al., 2010). Lipophagy results in lipid breakdown, and its defect contributes to metabolic disorders, such as obesity and atherosclerosis (Singh and Cuervo, 2012). During the process of lipophagy, autophagy pathway is recognized as being pivotal in regulating cholesterol flux from macrophages (Ouimet and Marcel, 2012). It is generally believed that autophagy can deliver LDs to lysosome, followed by the hydrolyzation and release of cholesterol from LDs (Mizushima, 2007). Cholesterol efflux from macrophages is the potentially most prominent step in reverse cholesterol transport, a process especially involved in atherosclerosis and the regression of atherosclerotic plaques (Rosenson et al., 2012).
Recently, a role for autophagy and lysosomal acid lipase (LAL) in macrophage lipolysis has been demonstrated, indicating that manipulating the autophagic pathway may have therapeutic benefits in preventing the development of atherosclerosis through the regulation of cholesterol efflux (Ouimet et al., 2011). It is known that autophagic process involves the formation of autophagosomes, and subsequently fuse with lysosome for the degradation of LDs. Increasing evidences have corroborated a key role of mammalian target of rapamycin (mTOR) in the regulation of autophagy (Jung et al., 2010; Nyfeler et al., 2012). mTOR is a conserved serine/threonine kinase that mediates various cellular processes, including cell growth, proliferation, and autophagy. Delivery of rapamycin derivative everolimus, an mTOR inhibitor, obviously activates the autophagy process and reduces the macrophage content in cholesterol-fed rabbits, suggesting an important role in plaque stabilization and the prevention of acute coronary syndrome (Martinet et al., 2007). Abundant researches have confirmed that mTOR can negatively regulate ULK1 protein kinase activation, a crucial initiator of autophagic process, and ultimately inhibit the formation of autophagosomes (Kim et al., 2011; Roach, 2011). However, the pivotal role of mTOR in atherosclerosis and the underlying molecular mechanism remains undefined.
Based on the important roles of autophagic pathway in lipolysis, we well linked the mTOR to pro-atherosclerosis by regulating the lipid-laden foam cell formation via autophagic pathway. The aim of this study was therefore to investigate the correlation between mTOR and macrophage foam cell formation, as well as the underlying mechanism of this process.
Materials and Methods
Reagents and antibodies
Primary antibodies including antibodies against mTOR, phosphorylated mTOR (Ser2448), S6K1, phosphorylated S6K1 (Thr389), 4E-BP1, and phosphorylated 4E-BP1 (Thr37), were obtained from Cell Signaling Technology (Beverly, MA). The antibody against mouse LC3 was from Bio-Rad Laboratories (Richmond, CA). Anti-p62 antibody was provided by Progen Biotechnik (Heidelberg, Germany).
Preparation of oxLDL
The preparation of oxLDL was performed as previously described (Wang et al., 2012). To obtain oxLDL, about 200 μg/mL of LDL was exposed to 20 μM CuSO4 in phosphate-buffered saline (PBS) for oxidation. Then, 40 μM butylhydroxytoluene in ethanol was introduced to block the oxidative reactions. Following dialysis against culture medium, the obtained oxidized LDL (CuoxLDL) was then filtered sterilely.
Cell culture
The mouse RAW 264.7 monocyte/macrophage-like cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum, 5 mM sodium pyruvate, and 100 U/mL streptomycin–penicillin at 37°C with 5% CO2. Cells from the third to fifth passage were used in this experiment.
Silencing mTOR and ULK1 expression with small interference RNA
For targeted knockdown of mTOR and ULK1 expression, the specific mTOR and ULK1 small interference RNA (siRNA) sequences were designed previously (Verheye et al., 2007; Hosokawa et al., 2009). All the sense and antisense sequences were synthesized by Genetimes Technology (Shanghai, China). Further, the scrambled siRNA was designed by Genetimes Technology, which was used as control in our study. Cells were transfected with a mixture of 2 μg/mL mTOR siRNA or ULK1 siRNA using the GeneSilencer® siRNA transfection reagent (GeneTherapy System, San Diego, CA). Twenty-four hours later, cells were treated with CuoxLDL. The transfection efficiency was assessed by Western blotting.
Western blotting
To obtain the total protein extracts of RAW 246.7 cells, RIPA lysis buffer (Beyotime, Nantong, China) was used, and the collected protein concentration was assessed by the BCA kit (Pierce, Rockford, IL). After electrophoresis on a 12% polyacrylamide gel, about 100 μg of proteins was transferred onto a polyvinylidene difluoride membrane. After blocking with buffer containing 5% nonfat dry milk in Tris-buffered saline with Tween at 4°C overnight, the target proteins were probed with the primary antibodies for 1 h. Then, HRP-conjugated second antibodies were added for 1 h followed by the visualization with LumiGLo reagent (Pierce). Quantification of these protein levels was done with densitometer and Imagequant software (Shenteng, Shanghai, China).
Foam cell formation assay with oil red O staining
RAW 246.7 cells were cultured with CuoxLDL for 10 h, and then washed three times with PBS. After fixation with 10% formalin, cells were stained with freshly diluted 0.5% oil red O solution for 10 min at 37°C to evaluate the characteristic lipid accumulation in macrophage-derived foam cells. Cells were then rinsed with water, and hematoxylin was introduced to label the cell nuclei. Foam cell formation was observed under a microscope, and the oil red O staining was assessed using color density assay using iVision software.
Lipid assay by high-performance liquid chromatography
To analyze lipid accumulation in macrophage-derived foam cells, total cholesterol (TC) and cholesterol ester (CE) contents were assessed by high-performance liquid chromatography (HPLC). After exposure to CuoxLDL, cells were lysed with 0.9% NaOH solution followed by sonication in an ice bath for 10 s. The total protein concentration was detected by BCA kit. About 150 μL stigmasterol was introduced; the residues were resuspended in 100 μL of isopropanol-acetonitrile (v/v, 20:80) for further analysis. All the samples were performed by Agilent 1100 series HPLC (Wilmington, DE). The ratio of CE/TC was shown to analyze the lipid-deposition levels.
Assessment of cholesterol efflux
Cells were incubated for 35 h with 3H-cholesterol (5 μCi/mL) and 50 mg/mL oxLDL in basic medium. Following equilibration of 3H-cholesterol with intracellular cholesterol, cells were cultured for 24 h in serum-free media containing bovine serum albumin (2 g/L). Then, 10 mg/mL of apoA-I was introduced to initiate cholesterol efflux for 12 h. After these treatments, all the samples were subjected to FJ-2107P-type liquid scintillator to ascertain the radioactivity. Efflux is shown as a percentage of 3H-cholesterol in medium/(3H-cholesterol in medium+3H-cholesterol in cells)×100%.
Fluorescence microscopy
After washing three times with Hanks buffered salt solution (HBSS), cells were cultured with LysoTracker® Red (100 nM) for 30 min to label lysosome. Then, Bodipy 493/503 (10 μg/mL) was added to visualize the neutral lipids in CuoxLDL-laden RAW 246.7 cells. The living cells were ultimately mounted in HBSS on microscope slides, following observation on a DM2000 Leica microscope (Leica Microsystems, Groot Bijgaarden, Belgium). All images were captured by a Leica DFC 290 camera.
Data analysis
All numerical results are shown as mean±SD. SPSS 11.0 was used to analyze all the data. A typical image from at least three similar experiments is provided. Student's t-test was used to assess the statistical significance of differences. p<0.05 was considered statistically significant.
Results
The mTOR pathway was dramatically activated during foam cell formation
Following treatment with CuoxLDL to induce macrophage foam cell formation, abundant protein levels of mTOR were demonstrated in RAW 246.7 cells, as well as the phosphorylation of mTOR (Fig. 1). It is widely accepted that S6K1 and 4E-BP1 are the critical downstream effectors of the mTOR pathway to regulate various cellular signals (Sarbassov et al., 2005). To further clarify the activation of the mTOR pathway during foam cell formation, we analyzed the phosphorylation levels of S6K1 and 4E-BP1. As expected, incubation of macrophages with CuoxLDL notably upregulated S6K1 and 4E-BP1 phosphorylation, suggesting that the mTOR pathway was strikingly activated during foam cell formation.
FIG. 1.
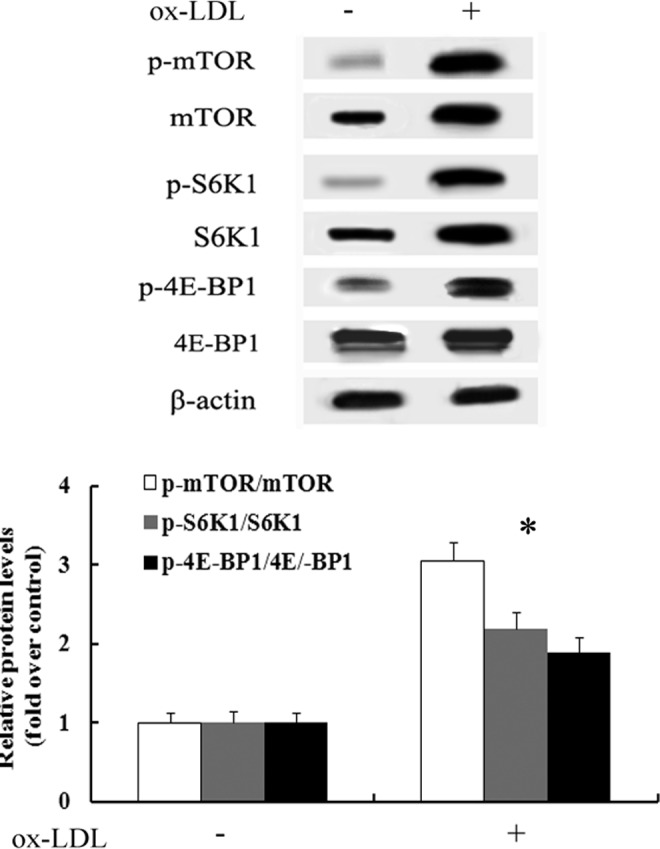
The mammalian target of rapamycin (mTOR) pathway was dramatically activated during macrophage-derived foam cell formation. Following incubation with CuoxLDL for 10 h, the expression levels of mTOR and p-mTOR were analyzed by western blotting. The corresponding expression levels of downstream effectors S6K1 and 4E-BP1 were also detected. Data were normalized based on the β-actin levels and were represented as relative expression levels. *p<0.05 versus oxidized low-density lipoprotein (oxLDL)–untreated group.
Blocking mTOR levels significantly dampened foam cell formation
To clarify the relationship between mTOR activation and foam cell formation, we silenced mTOR expression by mTOR siRNA, and found that most of mTOR expression was silenced (Fig. 2A). Further analysis confirmed that the absence of mTOR significantly dampened foam cell formation (Fig. 2B). Additionally, the TC levels were obviously attenuated after pretreatment with mTOR siRNA, concomitant with the gradual decrease of CE/TC ratio from 68.12% to 27.86% (Fig. 2C), suggesting that suppressing mTOR levels obviously inhibited lipid accumulation. Taken together, these results indicated that mTOR could mediate macrophage foam cell formation.
FIG. 2.
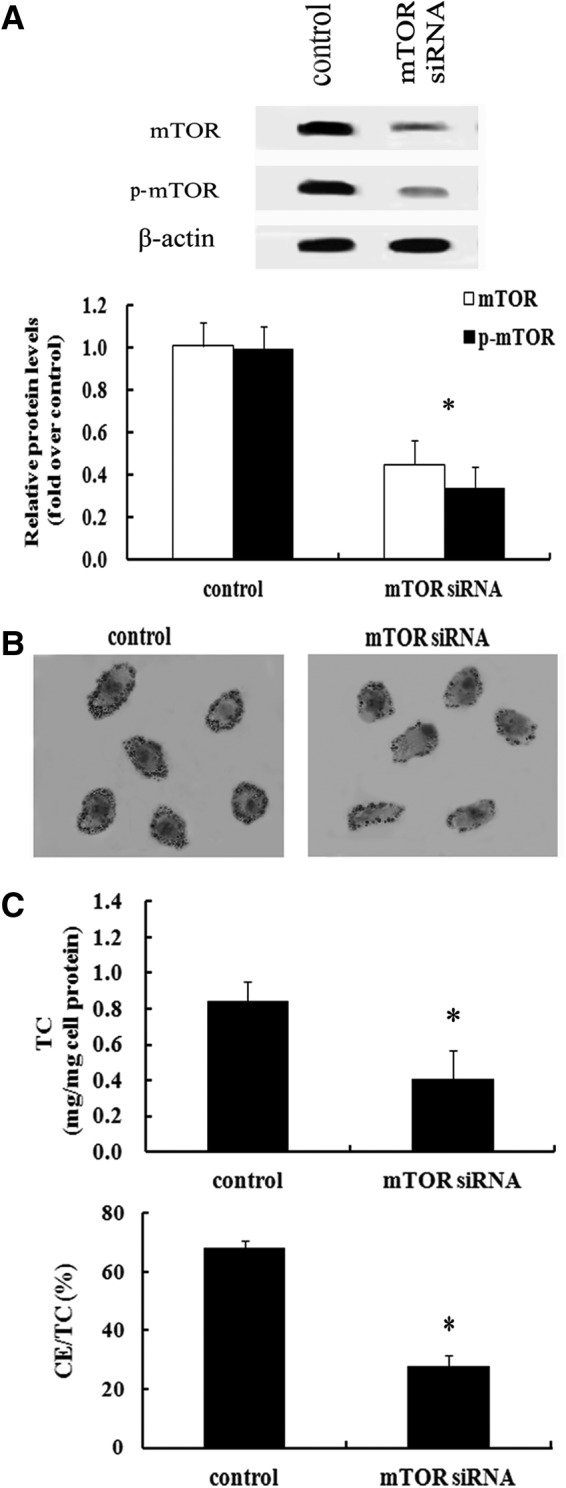
mTOR enhanced foam cell formation. After preconditioning with 2 μg/mL of mTOR small interference RNA (siRNA) or scrambled siRNA (used as control group), RAW 264.7 cells were exposed to CuoxLDL for 10 h. The silencing effect of mTOR was examined by western blotting (A), and oil red O staining was used to label foam cells (B). Additionally, the total cholesterol (TC) level and ratio of cholesterol ester (CE)/TC was introduced to assess lipid accumulation (C). *p<0.05 versus control group.
mTOR negatively regulated the activation of the autophagic machinery responding to lipid loading in macrophage-derived foam cells
It is widely accepted that mTOR is a key regulator for autophagic processes that is related to cholesterol efflux from foam cells (Singh et al., 2009; Ouimet et al., 2011). We were compelled to investigate whether mTOR-induced foam cell formation is associated with the activation of autophagy pathway. To address this hypothesis, we silenced mTOR expression levels by mTOR siRNA. As shown in Figure 3A, absence of mTOR expression significantly upregulated LC3-I and LC3-II expression, which is essential for autophagy (Tanida et al., 2004). As a common autophagic substrate, p62 serves as a useful marker for measuring autophagic flux. After blocking the expression of mTOR, the high expression levels of p62 were obviously attenuated, compared with the control group. Chloroquine is known as a common inhibitor for autophagy pathway and its treatment attenuated mTOR-silence-induced LC3 levels and increased p62 levels. Therefore, these results indicated that inhibition of mTOR significantly activated autophagosomes.
FIG. 3.
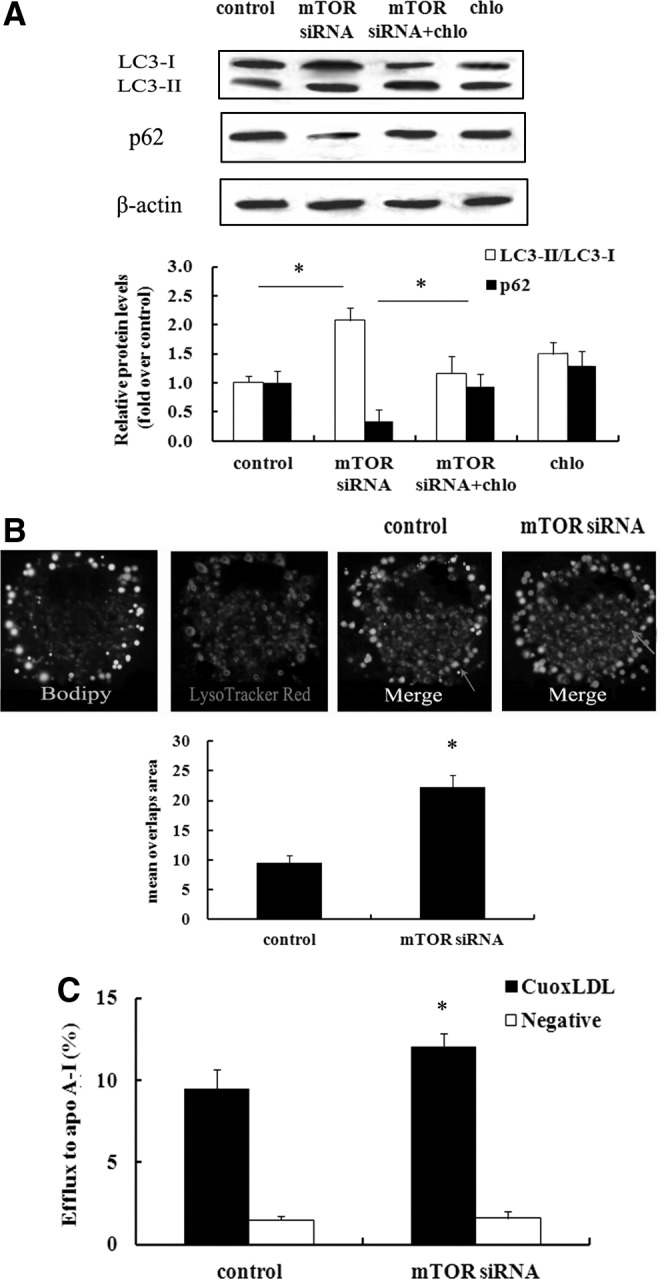
Autophagic machinery was responsible for mTOR-induced lipid loading in macrophage-derived foam cells. RAW 264.7 cells were transiently transfected with mTOR siRNA, and then incubated with CuoxLDL. Autophagosomes were visualized by assessing the expression levels of LC3 and p62. Chloroquine treatment reversely upregulated mTOR-silence-induced expression levels of p62 and downregulated LC3 levels (A). After washing with Hanks buffered salt solution (HBSS), cells were cultured with LysoTracker® Red (100 nM) to label lysosome, and Bodipy 493/503 (10 μg/mL) to visualize the neutral lipids by fluorescence microscopy (B). To further analyze cholesterol efflux, cells were labeled with 3H-cholesterol (5 μCi/mL) in the absence of mTOR expression. Further, the effect of mTOR inhibition on cholesterol efflux in unloaded (negative group) versus CuoxLDL-loaded macrophages was also discussed (C). Arrows indicated the overlapping signals. *p<0.05.
Autophagic process involves the autophagosome formation, followed by fusion with lysosomes for lipid degradation via LAL (Ouimet et al., 2011). As shown in Figure 3B, abundant neutral lipids were observed by Bodipy staining in CuoxLDL-incubated macrophage. After staining with LysoTracker Red to label lysosomes, numerous overlapping signals were demonstrated, suggesting that neutral lipid was delivered to lysosomes via autophagy. Importantly, more overlapping fluorescence signals were examined when suppressing mTOR expression. Agreement with the hydrolysis role of lysosome in regulating cytoplasmic CE, increased autophagy activation through mTOR silencing dramatically increased cholesterol efflux (Fig. 3C). Moreover, there is no effect on cholesterol efflux of the mTOR siRNA in oxLDL-unloaded cells, indicating that mTOR knockdown did not alter the basic cell efflux machinery. Taken together, our data confirmed that mTOR could negatively regulate the activation of the autophagic machinery, as response to lipid metabolism in macrophage-driven foam cells.
ULK1 was responsible for mTOR-regulated foam cell formation by autophagic pathway
ULK1 is a key initiator of the autophagic pathway (Kim et al., 2011). To further clarify the underlying mechanisms involved in mTOR-induced foam cell formation, the expression levels of ULK1 were assessed. Compared with the control group, mTOR silencing remarkably upregulated the expression of ULK1 and resulted in 2.58-fold increase in ULK1 expression levels (Fig. 4A), which led us to investigate whether ULK1 is indispensable for mTOR-regulated autophagic pathway during foam cell formation. After blocking ULK1 expression, a significant decrease in LC3-II expression was detected, as well as upregulation of p62 levels (Fig. 4B), implying that ULK1 was responsible for mTOR-triggered activation of autophagic pathway. Further analysis confirmed that blocking ULK1 expression strikingly suppressed cholesterol efflux (Fig. 4C), accompanied with the increase in lipid deposition (Fig. 4D). All of these results suggested that ULK1 was accounted for negatively regulated mTOR autophagic machinery during the formation of foam cells.
FIG. 4.

ULK1 was responsible for mTOR-regulated foam cell formation by autophagic machinery. Following treatment with mTOR siRNA, the expression levels of ULK1 were detected by western blotting (A). To further clarify the correlation between ULK1 and mTOR-regulated autophagic machinery during foam cell formation, the expression levels of LC3 and p62 were examined to evaluate the autophagic pathway in the absence of ULK1 (B). The corresponding cholesterol efflux from lipid-loading macrophage foam cells was evaluated (C). Lipid accumulation was also measured by TC levels and CE/TC ratio (D). *p<0.05.
Discussion
Atherosclerosis and its complications continue to cause considerable morbidity and mortality worldwide (Mendis et al., 2011). It is known that atherosclerosis is characterized by the accumulation of lipids and fibrous elements in the large arteries, constituting the major contributor to the growing burden of CVDs (Libby et al., 2010). The concept that lipid-laden foam cell formation is a key step to trigger atherosclerotic plaque formation has gained widespread acceptance. Aggressive lowering of lipid deposition obviously reduces atherosclerotic coronary lesion, suggesting a new approach to prevent atherosclerotic complications (Glass, 2002; Arai et al., 2010). Autophagic pathway is related with cholesterol efflux from macrophages, a process relevant to atherosclerosis (Ouimet and Marcel, 2012; Sene et al., 2013). Increasing evidence suggests that mTOR is a critical negative regulator for autophagy (Ravikumar et al., 2004; Jung et al., 2010). In this study, we discussed the potential roles of mTOR on atherosclerosis, and the correlation with autophagic pathway.
Lipid deposition triggers macrophage-derived foam cell formation, leading to the initiation of atherosclerosis and its complications (McLaren et al., 2011; Kushiyama et al., 2012). In this study, high expression levels of mTOR and p-mTOR were observed during the formation of macrophage-derived foam cells. Moreover, the corresponding downstream effectors S6K1 and 4E-BP1 were also upregulated in CuoxLDL-treated macrophages, suggesting that the mTOR pathway was activated during macrophage foam cell formation. Whether the high activation of mTOR signaling is associated with foam cell formation? To address this question, we silenced the expression of mTOR, and found that less foam cells were examined in the absence of mTOR, concomitant with a decreased CE/TC ratio. Accordingly, these results told that mTOR could regulate foam cell formation, indicating a potential role of mTOR in proatherosclerosis.
Autophagy is a lysosomal pathway that can degrade intracellular organelles and proteins to maintain cellular homeostasis (Mizushima, 2007). Recently, studies have provided supporting evidence for a connection between autophagy and lipid metabolism (Dong and Czaja, 2011; Uchiyama and Kominami, 2013). Inhibiting the autophagic pathway significantly diminishes the lipolysis of LDs in hepatocytes, implying a notable role of autophagy in LD clearance (Ding, 2010). Thus, we well linked the autophagy to mTOR-induced foam cell formation. As demonstrated in our study, pretreatment with mTOR siRNA obviously increased LC3 expression, a representative marker for autophagosomes. Simultaneously, the expression levels of p62 were dramatically decreased, indicating an increased formation of autophagosomes. After culture with CuoxLDL, large neutral lipids were sequestered in autophagosomes, followed by the fusion with lysosomes for the breakdown of LD components by acid proteases and hydrolases in the autolysosome. When blocking mTOR expression, more lipids were corroborated in lysosomes, accompanied with more cholesterol efflux from lipid-loaded macrophage foam cells. All of these results support a notion that mTOR possesses pro-foam cell formation potential by impeding autophagic pathway. Chloroquine is known as a common inhibitor for autophagy pathway and is expected to enhance LC3-II accumulation and p62 levels, compared with control group. However, its treatment attenuated mTOR-silence-induced LC3 levels and increased p62 levels. Therefore, the underlying mechanism involved in how mTOR silencing attenuate chloroquine-triggered LC-II level upregulation needs to be further explored. ULK1 is ranked as a prominent initiator for autophagic pathway (Kim et al., 2011). To further clarify the precise mechanism of mTOR-regulated autophagy pathway during foam cell formation, we analyzed the expression levels of ULK1. As expected, suppressing mTOR expression remarkably increased ULK1 expression. Further analysis indicated that ULK1 silencing blocked autophagy pathway as shown by decreased levels of LC3, as well as increased p62 expression. Followed by the suppression of autophagy pathway, more lipid deposition was observed compared with control group, accompanied with the reduced cholesterol efflux, indicating that ULK1 was accounted for mTOR-regulated foam cell formation by the autophagy pathway. However, the accurate mechanism about how mTOR regulates ULK1-triggered autophagic pathway needs to be discussed in the further.
In conclusion, our study has elucidated that mTOR can enhance lipid-laden macrophage foam cell formation by inhibiting ULK1-mediated autophagic pathway, which will facilitate the pathological process of atherosclerosis. Accordingly, our research has suggested a promising therapeutic agent against atherosclerotic complications.
Acknowledgment
This work was supported by National Natural Science Foundation of China (NSFC; No. 81070045).
Disclosure Statement
The authors have no financial conflicts of interest.
References
- Arai H., Hiro T., Kimura T., Morimoto T., Miyauchi K., Nakagawa Y., et al. (2010). More intensive lipid lowering is associated with regression of coronary atherosclerosis in diabetic patients with acute coronary syndrome—sub-analysis of JAPAN-ACS study. J Atheroscler Thromb 17, 1096–1107 [DOI] [PubMed] [Google Scholar]
- Ding W.-X. (2010). Role of autophagy in liver physiology and pathophysiology. World J Biol Chem 1, 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H., and Czaja M.J. (2011). Regulation of lipid droplets by autophagy. Trends Endocrinol Metab 22, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass C. (2002). The macrophage foam cell as a target for therapeutic intervention. Nat Med 8, 1235–1242 [DOI] [PubMed] [Google Scholar]
- Hosokawa N., Hara T., Kaizuka T., Kishi C., Takamura A., Miura Y., et al. (2009). Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol Biol Cell 20, 1981–1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C.H., Ro S.-H., Cao J., Otto N.M., and Kim D.-H. (2010). mTOR regulation of autophagy. FEBS Lett 584, 1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Kundu M., Viollet B., and Guan K.-L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushiyama A., Okubo H., Sakoda H., Kikuchi T., Fujishiro M., Sato H., et al. (2012). Xanthine oxidoreductase is involved in macrophage foam cell formation and atherosclerosis development. Arterioscler Thromb Vasc Biol 32, 291–298 [DOI] [PubMed] [Google Scholar]
- Li A.C., Binder C.J., Gutierrez A., Brown K.K., Plotkin C.R., Pattison J.W., et al. (2004). Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ. J Clin Invest 114, 1564–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P., Okamoto Y., Rocha V.Z., and Folco E. (2010). Inflammation in atherosclerosis. Circ J 74, 213–220 [DOI] [PubMed] [Google Scholar]
- Lu K.-Y., Ching L.-C., Su K.-H., Yu Y.-B., Kou Y.R., Hsiao S.-H., et al. (2010). Erythropoietin suppresses the formation of macrophage foam cells role of liver X receptor α. Circulation 121, 1828–1837 [DOI] [PubMed] [Google Scholar]
- Martinet W., Verheye S., and De Meyer G.R. (2007). Everolimus-induced mTOR inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy 3, 241–244 [DOI] [PubMed] [Google Scholar]
- McLaren J.E., Michael D.R., Ashlin T.G., and Ramji D.P. (2011). Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog Lipid Res 50, 331–347 [DOI] [PubMed] [Google Scholar]
- Mendis S., Puska P., and Norrving B. (2011). Global atlas on cardiovascular disease prevention and control. World Health Organization [Google Scholar]
- Mizushima N. (2007). Autophagy: process and function. Genes Dev 21, 2861–2873 [DOI] [PubMed] [Google Scholar]
- Moore K.J., and Tabas I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyfeler B., Bergman P., Wilson C.J., and Murphy L.O. (2012). Quantitative visualization of autophagy induction by mTOR inhibitors. Methods Mol Biol 821, 239–250 [DOI] [PubMed] [Google Scholar]
- Ouimet M., and Marcel Y.L. (2012). Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol 32, 575–581 [DOI] [PubMed] [Google Scholar]
- Ouimet M., Franklin V., Mak E., Liao X., Tabas I., and Marcel Y.L. (2011). Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 13, 655–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y., Yanase T., Hu H., Tanaka T., Nishi Y., Liu M., et al. (2010). Dihydrotestosterone suppresses foam cell formation and attenuates atherosclerosis development. Endocrinology 151, 3307–3316 [DOI] [PubMed] [Google Scholar]
- Ravikumar B., Vacher C., Berger Z., Davies J.E., Luo S., Oroz L.G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36, 585–595 [DOI] [PubMed] [Google Scholar]
- Roach P.J. (2011). AMPK→ULK1→Autophagy. Mol Cell Biol 31, 3082–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenson R.S., Brewer H.B., Jr., Davidson W.S., Fayad Z.A., Fuster V., Goldstein J., et al. (2012). Cholesterol efflux and atheroprotection advancing the concept of reverse cholesterol transport. Circulation 125, 1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov dD, Ali S.M., and Sabatini D.M. (2005). Growing roles for the mTOR pathway. Curr Opin Cell Biol 17, 596–603 [DOI] [PubMed] [Google Scholar]
- Sene A., Khan A.A., Cox D., Nakamura R.E., Santeford A., Kim B.M., et al. (2013). Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab 17, 549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., and Cuervo A.M. (2012). Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol 2012, 282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., et al. (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Ueno T., and Kominami E. (2004). LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama Y., and Kominami E. (2013). Autophagy regulates lipid droplet formation and adipogenesis [Google Scholar]
- Verhagen S.N., and Visseren F.L. (2011). Perivascular adipose tissue as a cause of atherosclerosis. Atherosclerosis 214, 3–10 [DOI] [PubMed] [Google Scholar]
- Verheye S., Martinet W., Kockx M.M., Knaapen M.W., Salu K., Timmermans J.-P., et al. (2007). Selective clearance of macrophages in atherosclerotic plaques by autophagy. J Am Coll Cardiol 49, 706–715 [DOI] [PubMed] [Google Scholar]
- Wang J., Si Y., Wu C., Sun L., Ma Y., Ge A., et al. (2012). Lipopolysaccharide promotes lipid accumulation in human adventitial fibroblasts via TLR4-NF-κB pathway. Lipids Health Dis 11, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]