

Abstract
Ca2+ release from intracellular stores and influx from extracellular reservoir regulate a wide range of physiological functions including muscle contraction and rhythmic heartbeat. One of the most ubiquitous pathways involved in controlled Ca2+ influx into cells is store-operated Ca2+ entry (SOCE), which is activated by the reduction of Ca2+ concentration in the lumen of endoplasmic or sarcoplasmic reticulum (ER/SR). Although SOCE is pronounced in non-excitable cells, accumulating evidences highlight its presence and important roles in skeletal muscle and heart. Recent discovery of STIM proteins as ER/SR Ca2+ sensors and Orai proteins as Ca2+ channel pore forming unit expedited the mechanistic understanding of this pathway. This review focuses on current advances of SOCE components, regulation and physiologic and pathophysiologic roles in muscles. The specific property and the dysfunction of this pathway in muscle diseases, and new directions for future research in this rapidly growing field are discussed. [BMB Reports 2014; 47(2): 69-79]
Keywords: Aging, Junctophilin, Mitsugumin29, Muscular dystrophy, Muscle fatigue, Orai1, Sarcopenia, STIM1
INTRODUCTION
Intracellular Ca2+ is an important second messenger that regulates a wide range of cellular events and physiological functions. It has long been recognized that dysregulation of Ca2+ homeostasis is associated with a plethora of pathological conditions including neurodegenerative diseases, skeletal muscular and cardiovascular disorders. In non-excitable cells, one of the most ubiquitous pathways involved in regulated Ca2+ influx across plasma membrane (PM) is the store-operated Ca2+ entry (SOCE). It was first described by Putney nearly three decades ago in salivary glands cells termed as capacitative Ca2+ entry (1).
In this pathway, intracellular Ca2+ release through IP3 receptor stimulated by a variety of agonists, results in reduction of Ca2+ concentration in the lumen of endoplasmic reticulum (ER); the depleted Ca2+ stores in turn activates the SOCE channel to allow extracellular Ca2+ across PM, which eventually refills the empty ER Ca2+ stores. The molecular players mediating SOCE were discovered rather recently. Large scale unbiased RNA interference (RNAi)-based screen identified stromal interaction molecule 1 (STIM1) as the ER Ca2+ sensor (2-4). Shortly, the same RNAi screen together with a finding of severe combined immune deficiency (SCID) that is caused by a defect in Ca2+ entry of T cells, led to a groundbreaking discovery of a membrane protein as the SOCE channel (5-8). This protein was named Orai, after the Keeper of Heaven’s gate in Greek mythology. STIM1 is an ER membrane protein with a luminal EF-hand allowing it to detect changes in the ER Ca2+ content. Orai1 is an integral membrane protein with four trans-membrane domains and cytoplasmic N- and C-termini. Upon ER Ca2+ store depletion or reduction in some cases, STIM1 molecules cluster at the ER/PM junctional region, where they directly interact with Orai1 and send retrograde signals for the assembled Orai1s and consequent channel opening (9,10). Genetic mutations in Orail1 or STIM1 in patients and knock-out strains of either protein in mice abolish SOCE in lymphocytes and other cells in the body. These findings provide compelling evidence that Orai1 and STIM1 comprise the minimal functional unit of SOCE. In addition to STIM1 and Orai1, mammalian genomes also encode a STIM homologue, STIM2, and two other Orai genes, Orai2 and Orai3. Since the identification of STIMs and Orais, the structural-functional understanding of SOCE components and mechanism in a variety of cell types, especially in immune cells have greatly advanced.
The discovery of SOCE in excitable cells, especially in skeletal muscle is a Johnny-comes-lately. Although intracellular Ca2+ is a central player in excitation-contraction coupling (E-C coupling) to fulfill muscle’s mission, extracellular Ca2+ is not required for normal contraction in skeletal muscle or is required for contraction but permeated through L-type voltage-gated Ca2+ channel in cardiac muscle cells (11). Thus, for a long time, it had been believed that there was no room for SOCE in muscles cells, especially in adult skeletal muscle. This belief was cracked by several studies in early of this century. Kurebayashi et al. demonstrated that depletion of the SR Ca2+ stores by repetitive stimulation of Ca2+ release in combination with inhibitors of the SERCA Ca2+ pump resulted in activation of SOCE in skeletal muscle fibers isolated from adult mice (12). We further demonstrated that the activation of SOCE is coupled to SR Ca2+ release channel in a graded manner and it is important to keep muscle contractility during fatiguing stimulation, when the SR Ca2+ are quickly depleted (13). Later, a strong body of evidence indicates that SOCE impacts muscle physiology in many aspects. The importance of SOCE is further confirmed in mice lacking STIM1 which die perinatally and in patients with mutations on STIM1 or Orai1 who exhibit a myopathy or hypotonia (14-16).
Since the molecular structure of STIM1 and Orai1 has received extensive attention and many recent excellent review articles have provided details about the advances in this field (17-21), this review only focuses on five topics related to the role of SOCE in skeletal and cardiac muscles. First, we introduce the detection of SOCE in skeletal muscles and the distinct properties of SOCE in muscle compared with that in non-excitable cells. Second, we outline the SOCE complex in the context of unique architecture of muscle junctional membranes. Third, we review the physiological functions of Orai1 and STIM1 in skeletal muscle, such as its involvement in muscle contractility, fatigue and muscle aging. Fourth, we summarize the confirmed muscle diseases resulted from malfunction of SOCE and the mutations in STIM or Orai. Lastly, we discuss the role of SOCE in heart with emphasis on areas of controversy and uncertainty as well as future challenges in this exciting growing field of research.
DETECTION OF SOCE IN SKELETAL MUSCLE
Skeletal muscle utilizes a highly specialized Ca2+ signaling apparatus to couple electrical excitation across the PM to myofibril contraction. It contains two major protein components: the L-type voltage-gated Ca2+ channel (dihydropyridine receptors, DHPR), which senses the depolarization of the sarcolemma, and the ryanodine receptor 1 (RyR1), which mediates Ca2+ release from SR to trigger rapid and coordinated muscle contraction throughout the muscle fiber (11,22,23). During an action potential (AP, 2-5 ms in a fast-twitch muscle), DHPRs relay a charge signal to the RyR1s through direct interaction but themselves do not mediate extracellular Ca2+ entry since the kinetics of activation of DHPR are at least 1 order of magnitude slower than the AP itself (24,25). In additional to the DHPR/RYR1 interaction, the skeletal muscle fiber also has an efficient system to recycle Ca2+ into SR stores for relaxation and for utilization on the next contraction-relaxation cycle, which is achieved mainly by the SR/ER Ca2+-ATPase 1 (SERCA1) (26-28). There are three SERCA isoforms and SERCA1 is the skeletal muscle isoform with fastest kinetics (29). The reuptake of cytosolic Ca2+ to SR stores is efficient enough that the normal skeletal muscle contractions can be sustained in the absence of external Ca2+, therefore, it is not surprise to see that SOCE had been largely overlooked as a resource for SR Ca2+ store refilling in skeletal muscle. However, the efficiency of this Ca2+ cycling apparatus is really challenged to support all Ca2+ needs for muscle contractions involving repetitive bursts of tetanic stimulations in strenuous exercise or fatigue, or perhaps during hypoxia, or other situations of higher metabolic demand and also under pathological conditions. Indeed, several studies demonstrated that SOCE is called-in under these conditions. Repeated application of high concentration of K+ combined with SERCA inhibitor without external Ca2+ was shown to result in the depletion of SR Ca2+ stores, which in turn activated SOCE in isolated muscle fibers from adult mice (12). In primary cultured myotubes, we observed the activation of SOCE in response to depletion of SR Ca2+ stores via either inhibition of SERCA by thapsigargin or by locking of RyR1 Ca2+ release channel at the open configuration via combination of caffeine and ryanodine (13). These studies clearly demonstrated the existence of SOCE in skeletal muscle. Besides SOCE, excitation coupled Ca2+ entry (ECCE) has been detected as another form of Ca2+ entry (30,31). ECCE is activated by prolonged membrane depolarization or stimulatory trains rather than by the reduction/depletion of SR Ca2+ stores. However, considerable overlap may exist between the two forms of Ca2+ entry, for example, both involve coupling to RyR1 (32). Furthermore, it can be argued that these long stimulatory trains might work as a form of fatiguing stimulation, which in turn might reduce the SR Ca2+ stores. In addition, these long trains might open the L-type Ca2+ channel long enough for Ca2+ entry to occur through these channels also.
After the initial reports on SOCE in skeletal muscles, increasing evidence has accumulated to reveal the important roles of this Ca2+ entry in normal skeletal muscle physiology, muscle development and aging (17,33-35). These studies were largely facilitated by the convenient methods for detection and measurement of SOCE in skeletal muscles. The kinetics and functional properties of SOCE in non-excitable cells are routinely revealed by electrophysiology patch-clamp studies. However, the whole-cell recording in adult muscle fiber or in myotube has been extremely challenging since the small conductance of SOCE channels can be easily missed in activate and complex Ca2+ currents in these excitable cells. In addition, it is much easier to patch cells that do not respond with contractions upon physical manipulation. Only recently, the electrophysiology measurements of ICRAC-like currents in skeletal myotubes were successfully conducted by a few research groups (14,36).
A large body of knowledge about SOCE in muscle cells has been gained by fluorescence-imaging based intracellular Ca2+ measurements because of its convenience and feasibility in muscle cells (37). The most common one of these fluorescence methods is to directly monitor the dynamics of intracellular Ca2+ using the ratio of F340 nm and F380 nm (510 nm for emission wavelength) of the ratiometric Ca2+ indicator Fura-2. To isolate the activity of unidirectional SOCE from intracellular Ca2+ release and Ca2+ extrusion, a Mn2+ quenching assay is also frequently used. Although it is not for sure, Mn2+ is generally believed to cross PM through SOCE while it is impervio us to the surface membrane extrusion processes or to ER uptake by Ca2+ pumps due to its very high Fura-2 affinity. As a result, the quenching of Fura-2 fluorescence induced by the entry of extracellular Mn2+ into the cells represents a measurement of activity of SOCE (38). To gain the spatial and temporal information of activity of SOCE in the structurally specialized skeletal muscle, a high-resolution laser scanning confocal microscopy imaging method has been developed (32,38,39). This method takes advantage of two low-affinity Ca2+ indicators sequentially trapped into specific compartments of a mechanically skinned muscle fiber, i.e. Rhod-5N in the transverse-tubules (T-tubules) and Fluo-5N in the SR. This dual-dye method allows simultaneous measurement of SR Ca2+ content and Ca2+ concentration in T-tubules; the threshold in SR Ca2+ storage for SOCE activation and the kinetics of SOCE thus can be conveniently studied in adult skeletal muscles.
These approaches have informed that SOCE in skeletal muscle was sensitive to blockade by Ni2+, resistant to nifedipine and can be suppressed by SOCE blockers, such as skf-96365, 2-aminoethoxydiphenyl borate (2-APB) and YM-58483/BTP-2 (12,13,40). These characters are the same as those in non-excitable cells; however, many differences are obvious. In T-cells, the IP3 receptor is required for the activation of SOCE through direct conformational coupling. Using primary cultured myotubes derived from neonates of ryr1(−/−) ryr3(−/−) mice, we demonstrated that SOCE was significantly impaired in myotubes lacking RyR1 and RyR3, which provided direct evidence to support a model of RyR-coupled SOCE in muscle cells. This RyR-dependent SOCE can be activated by combination of caffeine and ryanodine in both myotubes and adult skeletal muscle fibers and is sensitive to azumolene (32), a water-soluble equipotent analog of dantrolene (the only available treatment for malignant hyperthermia [MH]). While thapsigargin-induced depletion of SR Ca2+ stores leads to maximal activation of SOCE, this TG-induced SOCE is insensitive to azumolene in muscle cells. These findings highlight that there are two distinct portions of SOCE in skeletal muscles, i.e. RyR1-dependent and RyR1-independent, which can be distinguished by azumolene. Another different property of SOCE in skeletal muscle is the fast kinetics. In non-excitable cells, the development of SOCE requires tens of seconds for full activation; whereas in skeletal muscle, SOCE is activated within a second (14,41). The possible mechanisms underlying the fast kinetics of skeletal muscle SOCE are discussed in next section.
SOCE SIGNALING COMPLEX IN TRIAD JUNCTION
The discoveries of ER Ca2+ sensor STIM1 and PM channel pore unit Orai1 have provided molecular tools to understand the mechanism of SOCE regulation in both non-excitable cells and skeletal muscles. We now know that the interaction between STIM1 and Orai1 is essential for transduction of the retrograde signal from ER/SR lumen to activation of SOCE at the PM in lymphocytes and other non-excitable cells. Recent crystal structure of Orai determined at 3.35A resolution further revealed that the channel gating requires Orai1-STIM1 coupling (42). In muscle cells, a large body of evidence convincingly demonstrated that Orai1 and STIM1 are the major players in SOCE signaling complex even though other PM Ca2+ permeable channels, e.g. TRPC may participate (43,44). Both Orai1 and STIM1 are abundantly expressed in neonatal myotubes and adult skeletal muscle fibers as consistently shown by RT-PCR, Western blot and immunostaining (14,35,45,46). Orai1 apparently is required for the activation of SOCE in myotubes since expression of human dominant-negative Orai1 (dnOrai1, E106Q) resulted in abolished SOCE (47). In the same study, the authors also used transgenic mice with muscle-specific expression of dnOrai1 and characterized Orai1-dependent SOCE in adult skeletal muscle. STIM1 is expressed at such a high level in skeletal muscle that only T cells and the cerebellum have comparable expression levels (14). During myogenesis, STIM1 not only increases in expression but also aggregates and redistributes to the cellular periphery. Myotubes lacking STIM1 fail to exhibit SOCE and mice lacking STIM1 die perinatally from a skeletal myopathy. These findings and other reports highlight both Orai1 and STIM1 as essential components of the SOCE machinery in skeletal muscle.
E-C coupling occurs in the junction between the T-tubule, an invagination of sarcolemma, and the terminal cisternae of SR in a structure known as the “triad” (48,49) (see Fig. 1). The triad junction provides a unique architecture for the direct interaction between DHPRs and RyRs, which mediates signal transduction of voltage-induced Ca2+ release to initiate consequent myofilament contraction (22). The triad junction also seems to provide a specialized frame structure for rapid activation of SOCE, which is supported by several lines of evidence. Endogenous Orai1 co-localizes with STIM1 at triad junction to form STIM1–Orai1 complexes upon store depletion in muscle fibers isolated from adult mice (47). Rosenberg and colleagues showed that STIM1 locates in both the terminal cisternae and the para-junctional SRs in electron micrograph, which is consistent with immunostaining study on partial co-localization of STIM1 and RYR1 (14). Based on these findings, they proposed that there are two pools of STIM1 proteins: a fast pool, in which STIM1s are ready to couple with Orai1s for a quick response; a reserve pool, in which STIM1 are away from triad junction but are able to translocate to terminal cisternae membranes, perhaps these pools are used under different pathophysiological conditions, for example exacerbated SOCE occurs in muscles from dystrophic animals and humans. It is possible that STIM1 already interacts with Orai1 in the absence of SR store depletion in the fast pool, even though a recent study interprets otherwise (47). Using a bimolecular fluorescence complementation (BiFC) approach, Wei-LaPierre et al. delivered complementary DNAs encoding cherry-STIM1-N (1-158)YFP and cherry-Orai1-C (159-283)YFP into FDB muscles by electroporation (47). Their results demonstrated that the transfected muscle fibers failed to show basal YFP fluorescence, whereas a strong YFP signal could be observed following thapsigargin-induced SR store depletion. While this provides evidence to support the notion that there are dramatic conformational changes in STIM1 and Orai1 after SR store depletion, more rigorous and direct examinations will warrant the conclusion of whether or not STIM1 and Orai1 form preassembled complexes in muscle cells.
Fig. 1. Model of SOCE machinery in triad junction of skeletal muscle. SOCE in skeletal muscle displays rapid kinetics compared to non-excitable cells, which is likely attributed to a unique architecture between the plasma membrane (PM) and the sarcoplasmic reticulum (SR). In skeletal muscle, T-tubule invagination of the PM touches SR membranes. The close PM-SR junctional structure, known as “triad”, enables direct control of the RyR1/Ca2+ release channel by the DHPR via voltage-induced Ca2+ release. The released Ca2+ can be rapidly reuptake into SR by SERCA pump to complete the rapid contraction and relaxation cycle in skeletal muscle. STIM1s, as SR luminal Ca2+ binding proteins, can detect the reduction of Ca2+ concentration inside the SR lumen, undergo conformational changes, which in turn serves as a retrograde signal to activate Orai1 channel at T-tubule via direct contact interaction. Junctophilins (JP) and mitsugumin29 (MG29) are essential protein components of the junctional membrane structure between PM and SR. Disruption of JP or MG29 expression results in uncoupling of PM with SR, leading to dysfunction of SOCE. Alternatively, JP or MG29 may directly interact with RyR or other molecules involving in SOCE. Calsequestrin (CSQ) is a SR luminal Ca2+ binding protein, which influences the activation of SOCE by either altering the function of RyR or altering the Ca2+ homeostasis inside the SR lumen.
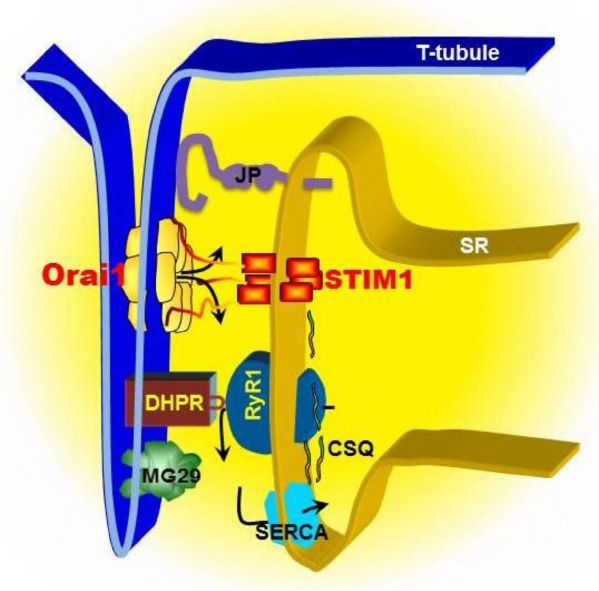
The fast kinetics of SOCE was also proposed to be attributed to a new splice variant of STIM1, STIM1L (L for long since it contains extra 106 a.a than the already well-described 90-kD STIM1) (50). STIM1L is abundant in skeletal muscle, heart and central nervous system, but much less in non-excitable cells. It co-localizes with Orai1 even when the SR store is full and interacts with actin to form permanent clusters at the triad junction. Silencing of endogenous STIM1L results in delayed SOCE activation and rapid depletion of SR Ca2+ stores in respond to repetitive depolarization-induced Ca2+ releases. Perhaps, the two mechanisms, i.e. STIM1L and fast pool of STIM1 at terminal cisternae membranes, are not excluding each other in supporting the fast kinetics of SOCE in skeletal muscle. Nevertheless, both mechanisms likely require the triad junction to provide the essential structure basis. A tantalizing possibility is that the specific SR Ca2+ level at the time of contraction could dictate which pool to be used or that metabolic status could directly influence on the accessibility of specific pools. Furthermore, the new expanding field of tissue endocrine crosstalk might imply that secreted factors from other tissue (for example bone secreted factors that influence muscle function) could influence the utilization of these pools (51,52).
If the specialized frame structure in triad junction is essential for the interaction between STIM1 and Orai1 in activation of SOCE in skeletal muscles, any disruption of this triad complex is expected to result in dysfunction of SOCE. To test this hypothesis, we have used two approaches to alter the tight coupling between triad junction membranes and examined the activity of SOCE in muscle cells. The formation and maintenance of triad junction require two major supportive proteins: mitsugumin29 (MG29) and junctophilin (JP). MG29 is a synaptophysin-family member protein with specific location in triads of skeletal muscle (53). In adult muscle fiber, MG29 is present predominantly on the T-tubule membranes in the form of homohexamer (Fig. 1) (48,54). Abnormalities of membrane ultrastructure around the triad junction were detected in skeletal muscle from the mg29(-/-) mice: the T-tubules were swollen and sometimes missing and the SR networks were poorly formed with vacuolated and fragmented structures, leading to mis-alignment of triad junctions (55,56). These ultra-structural defects occurred during early myogenesis and remained in adult skeletal muscles. JP is a major protein component at junctional membrane complexes in excitable cells with a unique secondary structure: a large cytoplasmic region and a carboxyl-terminal transmembrane segment spanning the SR/ER (Fig. 1) (54,57,58). The cytoplasmic region of JP contains repeated motifs of 14 amino acid residues termed “membrane occupation and recognition nexus” or MORN motifs, and exhibit selective binding affinity to the plasma membrane. So far, four subtypes of JPs are identified: JP1, predominantly expressed in skeletal muscle; JP2, distributed in both skeletal and heart muscles; JP3 and JP4, abundantly expressed in the brain and testis (59,60). In skeletal muscle, JP1 and JP2 link the T-tubules and SR membranes together to form triad junctions via their specific anchorage to the SR/ER membrane and MORN motifs selective interaction with the PM (Fig. 1). Knockout of JP1 reduces number of triad junctions and abnormal SR structure in skeletal muscle (61); overexpression of JP1 induces rolled up T-tubules and SR membranes and abnormal junctional membranes in cardiac muscle (62). When the triad junctional structures are disrupted by either removal of MG29 or knockdown of JP1 and JP2, the SOCE machinery indeed is impaired. We used individual myotubes derived from mg29(-/-) or control mg29 (+/+) mice and identified a defective SOCE in skeletal muscle (13). The skf-96365 sensitive-SOCE in mg29(-/-) cells were only about 30% of that in mg29(+/+) cells regardless of the mode of depletion of SR stores, either through sustained opening of the RyR channel by caffeine and ryanodine or through complete inhibition of SERCA by thapsigargin. In cultured C2C12 myotubes transfected with plasmid and freshly isolated FDB muscle fibers infected with adenovirus containing shRNA against JP1 and JP2 expression, we observed disruption of T-tubules/SR structure as a result of silencing of jp1 and jp2 genes and a loss of graded-activation of SOCE as well as impaired SR Ca2+ stores (38). In an ex vivo model using the skinned EDL muscle fibers with adenoviral expression of shRNA against both JP1 and JP2, we further showed that acute suppression of JP1 and JP2 led to a remarkable uncoupling of SOCE activated by exposure to thapsigargin and caffeine, which component is sensitive to skf-96365 and 2-APB in control fibers (38).
As illustrated in Fig. 1, it is currently believed that the SOCE machinery as a signaling complex locates at triad of skeletal muscle and contains several components, e.g. STIM1, Orai1 and RyR1. In addition to their functions as structural protein to maintain the triad junction membranes, MG29 and JP1 may also actively regulate other components in SOCE complex directly. For example, we found that the purified MG29 protein enhanced activity of the RyR channel incorporated into the lipid bilayer membrane (63). Co-expression of MG29 and RyR in CHO cells caused cell apoptosis due to depletion of ER Ca2+ stores and activation of SOCE, suggesting a functional interaction between MG29 and RyR. Others also reported that MG29 might direct interact with TRPC3, another PM Ca2+ channel possibly involved in SOCE in muscle cells (64). Recently, JP2 was reported to interact with TRPC3 or Orai1 and thus to modulate SOCE directly (65,66).
The retrograde signaling from STIM1 to Orai1 likely resembles well the coupling between RyR and DHPR. Thus, the SR lumen Ca2+ binding proteins, which are known to regulate E-C coupling may play similar roles in regulation of SOCE. Calsequestrin (CSQ) might be the best example of such proteins. CSQ, a SR-resident protein in muscle cells, has previous known function as Ca2+ buffer in the lumen of SR and was recently recognized to participate in the active Ca2+ release process from SR by actively modulating RyR/Ca2+ release channel (22). Several studies including ours demonstrated that CSQ also involves in the regulation of SOCE in muscle cells. Overexpression of the full-length CSQ in cultured skeletal myotubes led to inhibition of SOCE, which was absent with overexpression of CSQ lacking the Ca2+-binding motif located on the carboxyl-terminal region (67). Using small-hairpin RNA, we further demonstrated that knockdown of CSQ caused elevation of extracellular Ca2+ entry into the myoplasm via SOCE (68). And more importantly, this was accompanied by alteration of Ca2+ release from SR and sensitivity of SOCE to elevated temperature, which may explain MH-like symptoms observed in the CSQ knockout mice (69).
PHYSIOLOGICAL FUNCTIONS OF SOCE IN SKELETAL MUSCLES
The primary function of skeletal muscle is to perform rapid Ca2+-mediated contraction to generate force. What is the role of SOCE in this process? Although SOCE has relatively faster kinetics than non-excitable cells, it is still too slow in a single contraction-relaxation cycle to account for any significant contribution of the elevation of cytosolic Ca2+ for twitch contraction during a brief AP or SR Ca2+ refilling during repolarization. As mentioned previously, SOCE likely participates the muscle contractions during tetanic stimulations in strenuous exercise or fatigue, and also under common conditions where a contraction might need to be sustained for long period. Fatigue is defined as a reversible decrease in the isometric contractile force in response to an increase in the frequency or duration of stimulation (intermittent fatigue) or due to the prolongation of stimulation (continuous fatigue) (70). Evidences from isolated intact muscle fiber show that the size of the free Ca2+ store declines during fatigue and recovers upon rest, whereas extracellular Ca2+ influx is required to replenish the depleted SR Ca2+ stores. Our earlier work showed that the fatigue pattern of isolated muscle fibers clearly depended upon extracellular Ca2+ since removal of Ca2+ from bath solution significantly reduced the sustained force output at the end of the tetanic stimulation (13). This extracellular Ca2+ influx is mainly through SOCE since application of skf-96365, 2-APB or BTP-2 can increase muscle fatigue even in an extracellular solution containing 2 mM Ca2+. Another strong evidence to support the role of SOCE is obtained from studies using soleus muscles isolated from MG29 null mice. Compared with wild type muscle, soleus muscle without MG29 demonstrated susceptibility to muscle fatigue and the difference between the two muscles was diminished in bath solution containing either 0 mM Ca2+ or SOCE channel blockers, such as skf-96365, which indicates that the dysfunction of SOCE in MG29 null muscles results in the susceptibility to fatigue.
Since our original publication on SOCE in muscle cells lacking mg29(-/-), more studies from multiple groups including us have been reported to further support the function of SOCE in muscle contractility. Using mice lacking sarcalumenin, a Ca2+-binding protein located in the SR of skeletal and cardiac muscle cells, we showed that sarcalumenin ablation significantly elevated expression of MG29 and resulted in altered SOCE and enhanced muscle fatigue resistance (71). EDL muscles lacking functional STIM1 fail to show SOCE and fatigue rapidly (47). Moreover, transgenic mice with muscle-specific expression of dnOrai1 demonstrate defect in SOCE and rapid fatigue in adult skeletal muscle (47). It is worth noting that the contribution of SOCE to muscle contractility may not limit to only through SR Ca2+ stores refilling, it is also possible through modulatory role on E-C coupling, such as the channel function of RyR1.
In addition to controlling E-C coupling, changes in intracellular Ca2+ also serve as signals to regulate muscle development, differentiation and remodeling through downstream Ca2+-dependent pathways, such as calcineurin, NFAT, mitogen-activated protein (MAP) kinases, extracellular signal-regulated kinase 1 and 2 (ERK1/2), canonical Wnt and AKT (33,72). Interestingly enough, the signaling involved cytosolic Ca2+ ions largely come from extracellular Ca2+ reservoir in skeletal muscle, presumably through STIM1 and Orai1 mediated-SOCE. After birth, skeletal muscle undergoes an enormous period of growth and differentiation whereas myoblasts fuse together and differentiate into myotubes, and then muscle fibers. In both cultured mouse muscle cell line C2C12 and cultured human myoblast derived from muscle biopsies, the expression levels of STIM1 and Orai1 are dramatically increased accompanied by enhanced SOCE activity during differentiation of myoblasts into myotubes (73). Silencing of STIM1 or Orai1 greatly reduces SOCE and impairs myoblast differentiation, whereas overexpression of STIM1 and Orai1 accelerates differentiation in the myoblasts by increasing fusion events (73). Using conditional skeletal muscle STIM1-/- knockout mice, Li et al. demonstrated that STIM1-mediated SOCE is required for cellular processes that are necessary for neonatal muscle growth and differentiation (74). Together these observations implicate that STIM1 and Orai1-mediated SOCE are likely to regulate processes necessary for myoblast fusion and differentiation.
Skeletal muscle has various muscle types with distinct physiological and metabolic parameters to provide a variety of functional properties. In response to environmental demands, skeletal muscle remodels by activating signaling pathways to reprogram gene expression to sustain muscle performance. Although lack of direct evidence, it is reasonable to believe that STIM1 and Orai1-medated SOCE may also involve in the various Ca2+-dependent signaling pathways and multiple transcription factors in adult skeletal muscle remodeling. An inducible skeletal muscle specific transgenic mouse model, in which the expression of STIM1 or Orai1 in skeletal muscle can be controlled at will, will greatly facilitate the research in this area. The details about the SOCE-dependent signal pathways in muscle growth and differentiation can be found in a recent comprehensive review (33).
Whether STIM1 and Orai1-medated SOCE also involve in the significant decline in contractile force during muscle aging is still debating. Besides muscle atrophy, compromised E-C coupling, lack of sufficient available Ca2+ to allow for repetitive muscle contractility, likely all contribute to muscles weakness. We showed that SOCE is compromised in aged skeletal muscle but not in young ones and the reduction of SOCE activity correlates with loss of contractile function during aging (45,75). Moreover, we found that the expression of STIM1 and Orai1 do not change during muscle aging, but the expression of MG29 is significantly reduced in aged muscle, indicating that the compromised SOCE may be associated with reduction of MG29. From a conceptual point of view one would expect SOCE to be reduced during aging in muscles and other tissues since a natural response of aging is the downregulation of channels, transporters, co-transporters mechanisms (76). Perhaps it is not just a mere coincidence that aged neuronal have been reported to display reduced SOCE (77). Despite all these lines of evidence that favor the view of reduction in SOCE during aging in skeletal muscles, further studies, especially the availability of aged mice with skeletal muscle conditional knockdown or knockout of STIM1 and/or Orai1 will provide valuable tools to elucidate the functions of STIM1 and Orai1 in skeletal muscle aging.
SOCE AND SKELETAL MUSCLE DISEASES
The involvement of SOCE in skeletal muscle disease was clear when Orai1 was identified as the pore-forming subunit of SOCE channel. A missense mutation (R91W) in the first transmembrane domain of Orai1 was found in patients with SCID and these patients also suffer from myopathy with symptoms of global hypotonia and respiratory muscle weakness (8,19). The results of a muscle biopsy from one of the patients revealed atrophy of type II fibers. Then later, a mutation in STIM1 was also identified in patients with a syndrome of immunodeficiency and nonprogressive muscular hypotonia (16). More recently, other dominant STIM1 mutations have also been found as a genetic cause of tubular-aggregate myopathy (TAM) (78). Tubular aggregates are regular arrays of tubules membrane accumulating in muscle during aging and TAM is characterized by progressive muscle weakness. Compared with wild-type STIM1 that clusters and interacts with Orai1 for SOCE activation only after SR Ca2+ stores depletion, the TAM-related STIM1 mutations appear to have constitutive aggregation even with a filled Ca2+ store. A significant higher basal Ca2+ level and a dysregulation of SOCE were observed in the muscles cells derived from TAM patients. These phenotypes from patients bearing mutations in Orai1 or STIM1 are consistent with the observations from Orai1 and STIM1 knockout mice although muscle dysfunction is more severe in such mice. STIM1-deficient mice often die perinatally and the muscle weakness is obvious during tetanic stimulations, whereas twitch contractions are normal (14).
Increased Ca2+ influx and activation of cytosolic proteases play a key role in the pathogenesis of Duchenne muscular dystrophy (DMD). Although the exact mechanism underlying this abnormal Ca2+ influx remains elusive, SOCE is proposed to be involved. Both STIM1 and Orai1 are found to be significantly elevated in the adult dystrophic muscles in mdx mice, a well-known DMD animal model (40). Parallel increases in SOCE activity and SR Ca2+ storage are also detected in these dystrophic muscle fibers. When high-efficient shRNA probes against Orai1 were delivered into the flexor digitorum brevis muscle in live mice, knockdown of Orai1 eliminated the differences in SOCE activity and SR Ca2+ storage between the mdx and wild type muscle fibers. Furthermore, intraperitoneal injection of BTP-2 could suppress SOCE activity and cytosolic calpain1 activity. Together, these data indicate that upregulation of Orai1-mediated SOCE contributes to the disrupted Ca2+ homeostasis and elevated proteolytic activity in mdx muscles.
MH is a potentially fatal pharmacogenetic syndrome in which exposure to volatile anesthetics triggers uncontrolled elevation of cytosolic Ca2+, skeletal muscle hypercontracture, and hypermetabolism, resulting in a dramatic rise in body temperature (79). The hyperactivity or leakiness of the RyR1 channel has been described as the primary physiological defect in MH-susceptible muscle. Similar as MH, central core disease (CCD) is another muscle diseases associated with mutations in RyR1. Patients with CCD suffer from progressive muscle weakness from a young age and are also at increased risk for the development of MH. To date, the only effective treatment for MH is dantrolene sodium, a skeletal muscle relaxant, which suppresses the uncontrolled rise in intracellular Ca2+. It was previously assumed that the uncontrolled elevation of cytosolic Ca2+ seen in MH was through Ca2+ release from the SR. However, several recent studies suggest that SOCE may contribute to the sustained increase in cytosolic Ca2+ in MH skeletal muscle. We showed that dantrolene analog azumolene inhibits SOCE both in skeletal muscle fibers and in cultured cells expressing RyR1 (32). Duke et al. demonstrated that halothane treatment resulted in activation of SOCE in myofibers from patients with MH but not in control (80). SOCE could be blocked in MH muscle fibers by application of a STIM1 antibody to the t-tubular system. Accelerated activation of SOCE current is also observed in myotubes derived from MH mouse models. These findings provide strong evidences to support the functional role of STIM1 and Orai1-mediated SOCE in the pathophysiology of MH.
Sarcopenia is defined as muscle wasting associated with functional impairment during aging. Patients possess significant reduction in muscle strength, caused by muscle protein wasting, and loss of functionality. Various conditions leading to muscle wasting involve different pathways of intracellular signaling that trigger apoptosis, Ca2+-dependent proteases (calpains and caspases) and decreased satellite cell activation/responsible for muscle regeneration, etc. While sarcopenia is considered a normal part of healthy aging, studies suggest that the progression of sarcopenia can be slowed if the specific molecular process responsible for its pathophysiological effects can be determined and specifically targeted (81). In fact, the complexity of aging muscle wasting comes with an intriguing signature where muscle mass does not necessarily match functional status (i.e. muscle contractile force and muscle power) (82-86). Multiple factors contribute to this phenomenon, including increased fatty infiltration and an increased prevalence of myosin type I expression in aging skeletal muscle. We have proposed that another key reason is the reduced SOCE with aging, since a reduced SOCE will naturally lead into a reduced SR Ca2+ stores and lesser amount of Ca2+ available for contractions (45).
SOCE IN CARDIAC MUSCLE
Comparing with the advances in skeletal muscle, the physiological function of SOCE in heart is much less clear and even the existence of SOCE in adult cardiomyocytes is still debating (87). Cardiac DHPR is able to mediate extracellular Ca2+ entry and triggers Ca2+ induced Ca2+ release during contractility in cardiomyocytes. In addition, STIM1 has very low expression in cardiomyocytes and is even undetectable in isolated adult mouse cardiomyocytes using RT-PCR and Western blot assay (our unpublished data). Moreover, neither patients nor mice lacking functional STIM1 or Orai1 show any obvious cardiac muscle-related phenotypes, which is consistent with the idea that the SOCE pathway is less important in heart at least in normal physiology. On the other hand, SOCE appears to be developmentally regulated in heart, with activity prominent in embryonic and neonatal cardiomyocytes and undetected in adult cardiomyocytes. In a mouse atrial cardiomycyte cell line, HL-1, the expression of STIM1 and Orai1 and the existence of SOCE have been reported (88). Interestingly, HL-1 cells present more neonatal cardiomyocyte phenotype including spontaneous contractions. As with skeletal muscles, these authors found a potential correlation of a role of SOCE under pathological conditions in the heart.
A recent study using zebrafish model demonstrated that isolated Orai1-deficient cardiomyocytes exhibit loss of the calcineurin-associated protein calsarcin from the z-discs and defected mechanosignal transduction (89). Furthermore, the authors also discovered that inactivation of Orai1 resulted in the development of heart failure in zebrafish. Lou, et. al provided evidence to show that the expression of STIM1 reappear in cardiomyocytes during extreme physical activity and STIM1/Orai1-mediated SOC is required for enhanced cardiac output (90). This study revealed that neonatal cardiomyocytes express significant amounts of STIM1 and STIM1L, which decrease with cardiomyocyte maturation. Consistent with the idea of a prominent role of STIM1 in cardiomyocytes under stressful situations, STIM1L expression in mature cardiomyocytes reappears with agonist- or after load-induced cardiac stress. A heart phenotype in STIM1- and Orai1-deficient mice or patients bearing mutations in stim1 and orai1 genes might become apparent after long-term processes such as aging, stressful stimuli, or enduring exercise, although patients with mutations in stim1 and orai1 may succumb to immunodeficiency in their childhood and the impact of defect SOCE on the heart may never be revealed.
Several independent groups reported the important role of STIM1-mediated SOCE in regulation of cardiac hypertrophy (87,90-92). Ohba et al. demonstrated that endothelin-1 induced activation of SOCE in neonatal cardiomyocytes, which in turn stimulates NFAT nuclear translocation and cardiomyocyte hypertrophy (91). More importantly, the NFAT translocation and cardiomyocytes hypertrophy could be diminished by silencing STIM1. Later, similar results were observed in cardiomyocytes following knockdown of Orai1 (92). Transfection of cardiomyocytes with constitutively active-STIM1 alone could increase hypertrophy and silencing of STIM1 gene in vivo protected the heart from pressure overload-induced hypertrophy. Clearly, the roles of STIM1 and Orai1-mediated SOCE in cardiac physiology and diseases are emerging.
CONCLUDING REMARKS
Over the past few years we have gained significantly the basic molecular insight of STIM/Orai-mediated SOCE. However, many questions regarding the contribution of STIM and Orai proteins to skeletal muscle and cardiac physiology and pathophysiology remain. Understanding of the mechanisms by which STIM and Orai proteins exert their physiological roles in the muscle specific environs will be a major challenge over the coming years. While STIMs and Orais have already been proposed as potential therapeutic targets for treatment of certain immune diseases associated with dysregulation of SOCE, the related exciting developments in skeletal muscle and heart area are expected as well.
Acknowledgments
This work was supported by the American Diabetes Association Grant (1-13-IN-40-BD to Z.P.), American Heart Association Grant (N5505355), NIH-NCI TREC Award U54-116867, NIH-NIA P01 AG039355 and the Thompson Endowment Fund (to M.B.), NIH grants (RO1-AG-028614, RO1-AG-028856, RO1-AR-061385, and RO1-HL-069000 (to J.M.).
References
- 1.Putney J. W. Jr. A model for receptor-regulated calcium entry. Cell Calcium. (1986);7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 2.Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., Cahalan M. D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. (2005);437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E. Jr., Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. (2005);15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roos J., DiGregorio P. J., Yeromin A. V., Ohlsen K., Lioudyno M., Zhang S., Safrina O., Kozak J. A., Wagner S. L., Cahalan M. D., Velicelebi G., Stauderman K. A. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. (2005);169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeromin A. V., Zhang S. L., Jiang W., Yu Y., Safrina O., Cahalan M. D. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. (2006);443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P. G. Orai1 is an essential pore subunit of the CRAC channel. Nature. (2006);443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 7.Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., Kinet J. P. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. (2006);312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S. H., Tanasa B., Hogan P. G., Lewis R. S., Daly M., Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. (2006);441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 9.Luik R. M., Wu M. M., Buchanan J., Lewis R. S. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J. Cell Biol. (2006);174:815–825. doi: 10.1083/jcb.200604015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang G. N., Zeng W., Kim J. Y., Yuan J. P., Han L., Muallem S., Worley P. F. STIM1 carboxyl-terminus activates native SOC, I (crac) and TRPC1 channels. Nat. Cell Biol. (2006);8:1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- 11.Rios E., Pizarro G., Stefani E. Charge movement and the nature of signal transduction in skeletal muscle excitation-contraction coupling. Annu. Rev. Physiol. (1992);54:109–133. doi: 10.1146/annurev.ph.54.030192.000545. [DOI] [PubMed] [Google Scholar]
- 12.Kurebayashi N., Ogawa Y. Depletion of Ca2+ in the sarcoplasmic reticulum stimulates Ca2+ entry into mouse skeletal muscle fibres. J. Physiol. (2001);533:185–199. doi: 10.1111/j.1469-7793.2001.0185b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pan Z., Yang D., Nagaraj R. Y., Nosek T. A., Nishi M., Takeshima H., Cheng H., Ma J. Dysfunction of store-operated calcium channel in muscle cells lacking mg29. Nat. Cell Biol. (2002);4:379–383. doi: 10.1038/ncb788. [DOI] [PubMed] [Google Scholar]
- 14.Stiber J., Hawkins A., Zhang Z. S., Wang S., Burch J., Graham V., Ward C. C., Seth M., Finch E., Malouf N., Williams R. S., Eu J. P., Rosenberg P. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. (2008);10:688–697. doi: 10.1038/ncb1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCarl C. A., Picard C., Khalil S., Kawasaki T., Rother J., Papolos A., Kutok J., Hivroz C., Ledeist F., Plogmann K., Ehl S., Notheis G., Albert M. H., Belohradsky B. H., Kirschner J., Rao A., Fischer A., Feske S. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. (2009);124:1311–1318 e1317. doi: 10.1016/j.jaci.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Picard C., McCarl C. A., Papolos A., Khalil S., Luthy K., Hivroz C., LeDeist F., Rieux-Laucat F., Rechavi G., Rao A., Fischer A., Feske S. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N. Engl. J. Med. (2009);360:1971–1980. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis R. S. Store-operated calcium channels: new perspectives on mechanism and function. Cold Spring Harb. Perspect. Biol. (2011);3:a003970. doi: 10.1101/cshperspect.a003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Putney J. W. The physiological function of storeoperated calcium entry. Neurochem. Res. (2011);36:1157–1165. doi: 10.1007/s11064-010-0383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol. Rev. (2009);231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feske S. CRAC channelopathies. Pflugers. Arch. (2010);460:417–435. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feske S. Immunodeficiency due to defects in store-operated calcium entry. Ann. N. Y. Acad. Sci. (2011);1238:74–90. doi: 10.1111/j.1749-6632.2011.06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma J., Pan Z. Retrograde activation of storeoperated calcium channel. Cell Calcium. (2003);33:375–384. doi: 10.1016/S0143-4160(03)00050-2. [DOI] [PubMed] [Google Scholar]
- 23.Rios E., Ma J. J., Gonzalez A. The mechanical hypothesis of excitation-contraction (EC) coupling in skeletal muscle. J. Muscle. Res. Cell Motil. (1991);12:127–135. doi: 10.1007/BF01774031. [DOI] [PubMed] [Google Scholar]
- 24.Cota G., Stefani E. Voltage-dependent inactivation of slow calcium channels in intact twitch muscle fibers of the frog. J. Gen. Physiol. (1989);94:937–951. doi: 10.1085/jgp.94.5.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakai J., Dirksen R. T., Nguyen H. T., Pessah I. N., Beam K. G., Allen P. D. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. (1996);380:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- 26.MacLennan D. H. Ca2+ signalling and muscle disease. Eur. J. Biochem. (2000);267:5291–5297. doi: 10.1046/j.1432-1327.2000.01566.x. [DOI] [PubMed] [Google Scholar]
- 27.Hovnanian A. SERCA pumps and human diseases. Subcell. Biochem. (2007);45:337–363. doi: 10.1007/978-1-4020-6191-2_12. [DOI] [PubMed] [Google Scholar]
- 28.MacLennan D. H., Rice W. J., Odermatt A., Green N. M. Structure-function relationships in the Ca (2+)-binding and translocation domain of SERCA1: physiological correlates in Brody disease. Acta. Physiol. Scand. (1998);643(Suppl):55–67. [PubMed] [Google Scholar]
- 29.Periasamy M., Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle. Nerve. (2007);35:430–442. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 30.Bannister R. A., Pessah I. N., Beam K. G. The skeletal L-type Ca (2+) current is a major contributor to excitation-coupled Ca (2+) entry. J. Gen. Physiol. (2009);133:79–91. doi: 10.1085/jgp.200810105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cherednichenko G., Hurne A. M., Fessenden J. D., Lee E. H., Allen P. D., Beam K. G., Pessah I. N. Conformational activation of Ca2+ entry by depolarization of skeletal myotubes. Proc. Natl. Acad. Sci. U. S. A. (2004);101:15793–15798. doi: 10.1073/pnas.0403485101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao X., Weisleder N., Han X., Pan Z., Parness J., Brotto M., Ma J. Azumolene inhibits a component of store-operated calcium entry coupled to the skeletal muscle ryanodine receptor. J. Biol. Chem. (2006);281:33477–33486. doi: 10.1074/jbc.M602306200. [DOI] [PubMed] [Google Scholar]
- 33.Stiber J. A., Rosenberg P. B. The role of store-operated calcium influx in skeletal muscle signaling. Cell Calcium. (2011);49:341–349. doi: 10.1016/j.ceca.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brotto M. Aging, sarcopenia and store-operated calcium entry: a common link? Cell Cycle. (2011);10:4201–4202. doi: 10.4161/cc.10.24.18645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dirksen R. T. Checking your SOCCs and feet: the molecular mechanisms of Ca2+ entry in skeletal muscle. J. Physiol. (2009);587:3139–3147. doi: 10.1113/jphysiol.2009.172148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yarotskyy V., Dirksen R. T. Temperature and RyR1 regulate the activation rate of store-operated Ca (2)+ entry current in myotubes. Biophys J. (2012);103:202–211. doi: 10.1016/j.bpj.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan Z., Zhao X., Brotto M. Fluorescencebased measurement of store-operated calcium entry in live cells: from cultured cancer cell to skeletal muscle fiber. J. Vis. Exp. (2012) doi: 10.3791/3415. pii: 3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirata Y., Brotto M., Weisleder N., Chu Y., Lin P., Zhao X., Thornton A., Komazaki S., Takeshima H., Ma J., Pan Z. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys. J. (2006);90:4418–4427. doi: 10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Launikonis B. S., Barnes M., Stephenson D. G. Identification of the coupling between skeletal muscle store-operated Ca2+ entry and the inositol trisphosphate receptor. Proc. Natl. Acad. Sci. U S. A. (2003);100:2941–2944. doi: 10.1073/pnas.0536227100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao X., Moloughney J. G., Zhang S., Komazaki S., Weisleder N. Orai1 mediates exacerbated Ca (2+) entry in dystrophic skeletal muscle. PLoS One. (2012);7:e49862. doi: 10.1371/journal.pone.0049862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Launikonis B. S., Rios E. Store-operated Ca2+ entry during intracellular Ca2+ release in mammalian skeletal muscle. J. Physiol. (2007);583:81–97. doi: 10.1113/jphysiol.2007.135046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hou X., Pedi L., Diver M. M., Long S. B. Crystal structure of the calcium release-activated calcium channel Orai. Science. (2012);338:1308–1313. doi: 10.1126/science.1228757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Louis M., Zanou N., Van Schoor M., Gailly P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. (2008);121:3951–3959. doi: 10.1242/jcs.037218. [DOI] [PubMed] [Google Scholar]
- 44.Brinkmeier H. TRP channels in skeletal muscle: gene expression, function and implications for disease. Adv. Exp. Med. Biol. (2011);704:749–758. doi: 10.1007/978-94-007-0265-3_39. [DOI] [PubMed] [Google Scholar]
- 45.Zhao X., Weisleder N., Thornton A., Oppong Y., Campbell R., Ma J., Brotto M. Compromised store-operated Ca2+ entry in aged skeletal muscle. Aging Cell. (2008);7:561–568. doi: 10.1111/j.1474-9726.2008.00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lyfenko A. D., Dirksen R. T. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J. Physiol. (2008);586:4815–4824. doi: 10.1113/jphysiol.2008.160481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wei-Lapierre L., Carrell E. M., Boncompagni S., Protasi F., Dirksen R. T. Orai1-dependent calcium entry promotes skeletal muscle growth and limits fatigue. Nat. Commun. (2013);4:2805. doi: 10.1038/ncomms3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma J., Pan Z. Junctional membrane structure and store operated calcium entry in muscle cells. Front. Biosci. (2003);8:d242–255. doi: 10.2741/977. [DOI] [PubMed] [Google Scholar]
- 49.Flucher B. E., Takekura H., Franzini-Armstrong C. Development of the excitation-contraction coupling apparatus in skeletal muscle: association of sarcoplasmic reticulum and transverse tubules with myofibrils. Dev. Biol. (1993);160:135–147. doi: 10.1006/dbio.1993.1292. [DOI] [PubMed] [Google Scholar]
- 50.Darbellay B., Arnaudeau S., Bader C. R., Konig S., Bernheim L. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. (2011);194:335–346. doi: 10.1083/jcb.201012157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mo C., Romero-Suarez S., Bonewald L., Johnson M., Brotto M. Prostaglandin E2: from clinical applications to its potential role in bone- muscle crosstalk and myogenic differentiation. Recent. Pat. Biotechnol. (2012);6:223–229. doi: 10.2174/1872208311206030223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jahn K., Lara-Castillo N., Brotto L., Mo C. L., Johnson M. L., Brotto M., Bonewald L. F. Skeletal muscle secreted factors prevent glucocorticoid-induced osteocyte apoptosis through activation of beta-catenin. Eur. Cell Mater. (2012);24:197–209. doi: 10.22203/ecm.v024a14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takeshima H., Shimuta M., Komazaki S., Ohmi K., Nishi M., Iino M., Miyata A., Kangawa K. Mitsugumin29, a novel synaptophysin family member from the triad junction in skeletal muscle. Biochem. J. (1998);331(Pt 1):317–322. doi: 10.1042/bj3310317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao X., Yamazaki D., Kakizawa S., Pan Z., Takeshima H., Ma J. Molecular architecture of Ca2+ signaling control in muscle and heart cells. Channels (Austin) (2011);5:391–396. doi: 10.4161/chan.5.5.16467. [DOI] [PubMed] [Google Scholar]
- 55.Komazaki S., Nishi M., Takeshima H., Nakamura H. Abnormal formation of sarcoplasmic reticulum networks and triads during early development of skeletal muscle cells in mitsugumin29-deficient mice. Dev. Growth Differ. (2001);43:717–723. doi: 10.1046/j.1440-169X.2001.00609.x. [DOI] [PubMed] [Google Scholar]
- 56.Nishi M., Komazaki S., Kurebayashi N., Ogawa Y., Noda T., Iino M., Takeshima H. Abnormal features in skeletal muscle from mice lacking mitsugumin29. J. Cell Biol. (1999);147:1473–1480. doi: 10.1083/jcb.147.7.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takeshima H., Komazaki S., Nishi M., Iino M., Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol. Cell. (2000);6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 58.Weisleder N., Takeshima H., Ma J. Immunoproteomic approach to excitation--contraction coupling in skeletal and cardiac muscle: molecular insights revealed by the mitsugumins. Cell Calcium. (2008);43:1–8. doi: 10.1016/j.ceca.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishi M., Mizushima A., Nakagawara K., Takeshima H. Characterization of human junctophilin subtype genes. Biochem. Biophys. Res. Commun. (2000);273:920–927. doi: 10.1006/bbrc.2000.3011. [DOI] [PubMed] [Google Scholar]
- 60.Nishi M., Sakagami H., Komazaki S., Kondo H., Takeshima H. Coexpression of junctophilin type 3 and type 4 in brain. Brain Res. Mol. Brain Res. (2003);118:102–110. doi: 10.1016/S0169-328X(03)00341-3. [DOI] [PubMed] [Google Scholar]
- 61.Ito K., Komazaki S., Sasamoto K., Yoshida M., Nishi M., Kitamura K., Takeshima H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J. Cell Biol. (2001);154:1059–1067. doi: 10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Komazaki S., Nishi M., Takeshima H. Abnormal junctional membrane structures in cardiac myocytes expressing ectopic junctophilin type 1. FEBS Lett. (2003);542:69–73. doi: 10.1016/S0014-5793(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 63.Pan Z., Hirata Y., Nagaraj R. Y., Zhao J., Nishi M., Hayek S. M., Bhat M. B., Takeshima H., Ma J. Co-expression of MG29 and ryanodine receptor leads to apoptotic cell death: effect mediated by intracellular Ca2+ release. J. Biol. Chem. (2004);279:19387–19390. doi: 10.1074/jbc.C400030200. [DOI] [PubMed] [Google Scholar]
- 64.Woo J. S., Cho C. H., Kim do H., Lee E. H. TRPC3 cation channel plays an important role in proliferation and differentiation of skeletal muscle myoblasts. Exp. Mol. Med. (2010);42:614–627. doi: 10.3858/emm.2010.42.9.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woo J. S., Hwang J. H., Ko J. K., Weisleder N., Kim do H., Ma J., Lee E. H. S165F mutation of junctophilin 2 affects Ca2+ signalling in skeletal muscle. Biochem. J. (2010);427:125–134. doi: 10.1042/BJ20091225. [DOI] [PubMed] [Google Scholar]
- 66.Woo J. S., Hwang J. H., Ko J. K., Kim do H., Ma J., Lee E. H. Glutamate at position 227 of junctophilin- 2 is involved in binding to TRPC3. Mol. Cell Biochem. (2009);328:25–32. doi: 10.1007/s11010-009-0070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shin D. W., Pan Z., Kim E. K., Lee J. M., Bhat M. B., Parness J., Kim D. H., Ma J. A retrograde signal from calsequestrin for the regulation of store-operated Ca2+ entry in skeletal muscle. J. Biol. Chem. (2003);278:3286–3292. doi: 10.1074/jbc.M209045200. [DOI] [PubMed] [Google Scholar]
- 68.Zhao X., Min C. K., Ko J. K., Parness J., Kim do H., Weisleder N., Ma J. Increased store-operated Ca2+ entry in skeletal muscle with reduced calsequestrin-1 expression. Biophys. J. (2010);99:1556–1564. doi: 10.1016/j.bpj.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yarotskyy V., Protasi F., Dirksen R. T. Accelerated activation of SOCE current in myotubes from two mouse models of anesthetic- and heat-induced sudden death. PLoS One. (2013);8:e77633. doi: 10.1371/journal.pone.0077633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Allen D. G., Lamb G. D., Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol. Rev. (2008);88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
- 71.Zhao X., Yoshida M., Brotto L., Takeshima H., Weisleder N., Hirata Y., Nosek T. M., Ma J., Brotto M. Enhanced resistance to fatigue and altered calcium handling properties of sarcalumenin knockout mice. Physiol. Genomics. (2005);23:72–78. doi: 10.1152/physiolgenomics.00020.2005. [DOI] [PubMed] [Google Scholar]
- 72.Kiviluoto S., Decuypere J. P., De Smedt H., Missiaen L., Parys J. B., Bultynck G. STIM1 as a key regulator for Ca2+ homeostasis in skeletal-muscle development and function. Skelet Muscle. (2011);1:16. doi: 10.1186/2044-5040-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Darbellay B., Arnaudeau S., Konig S., Jousset H., Bader C., Demaurex N., Bernheim L. STIM1- and Orai1-dependent store-operated calcium entry regulates human myoblast differentiation. J. Biol. Chem. (2009);284:5370–5380. doi: 10.1074/jbc.M806726200. [DOI] [PubMed] [Google Scholar]
- 74.Li T., Finch E. A., Graham V., Zhang Z. S., Ding J. D., Burch J., Oh-hora M., Rosenberg P. STIM1-Ca (2+) signaling is required for the hypertrophic growth of skeletal muscle in mice. Mol. Cell Biol. (2012);32:3009–3017. doi: 10.1128/MCB.06599-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thornton A. M., Zhao X., Weisleder N., Brotto L. S., Bougoin S., Nosek T. M., Reid M., Hardin B., Pan Z., Ma J., Parness J., Brotto M. Store-operated Ca(2+) entry (SOCE) contributes to normal skeletal muscle contractility in young but not in aged skeletal muscle. Aging (Albany NY) (2011);3:621–634. doi: 10.18632/aging.100335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zahn J. M., Sonu R., Vogel H., Crane E., Mazan-Mamczarz K., Rabkin R., Davis R. W., Becker K. G., Owen A. B., Kim S. K. Transcriptional profiling of aging in human muscle reveals a common aging signature. PLoS Genet. (2006);2:e115. doi: 10.1371/journal.pgen.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vanterpool C. K., Pearce W. J., Buchholz J. N. Advancing age alters rapid and spontaneous refilling of caffeine-sensitive calcium stores in sympathetic superior cervical ganglion cells. J. Appl. Physiol. (2005);99:963–971. doi: 10.1152/japplphysiol.00343.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bohm J., Chevessier F., Maues De Paula A., Koch C., Attarian S., Feger C., Hantai D., Laforet P., Ghorab K., Vallat J. M., Fardeau M., Figarella-Branger D., Pouget J., Romero N. B., Koch M., Ebel C., Levy N., Krahn M., Eymard B., Bartoli M., Laporte J. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. (2013);92:271–278. doi: 10.1016/j.ajhg.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parness J., Bandschapp O., Girard T. The myotonias and susceptibility to malignant hyperthermia. Anesth. Analg. (2009);109:1054–1064. doi: 10.1213/ane.0b013e3181a7c8e5. [DOI] [PubMed] [Google Scholar]
- 80.Duke A. M., Hopkins P. M., Calaghan S. C., Halsall J. P., Steele D. S. Store-operated Ca2+ entry in malignant hyperthermia-susceptible human skeletal muscle. J. Biol. Chem. (2010);285:25645–25653. doi: 10.1074/jbc.M110.104976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Romanick M., Thompson L. V., Brown-Borg H. M. Murine models of atrophy, cachexia and sarcopenia in skeletal muscle. Biochim. Biophys. Acta. (2013);1832:1410–1420. doi: 10.1016/j.bbadis.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lowe D. A., Husom A. D., Ferrington D. A., Thompson L. V. Myofibrillar myosin ATPase activity in hindlimb muscles from young and aged rats. Mech. Ageing. Dev. (2004);125:619–627. doi: 10.1016/j.mad.2004.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lowe D. A., Thomas D. D., Thompson L. V. Force generation, but not myosin ATPase activity, declines with age in rat muscle fibers. Am. J. Physiol. Cell Physiol. (2002);283:C187–192. doi: 10.1152/ajpcell.00008.2002. [DOI] [PubMed] [Google Scholar]
- 84.Romero-Suarez S., Shen J., Brotto L., Hall T., Mo C., Valdivia H. H., Andresen J., Wacker M., Nosek T. M., Qu C. K., Brotto M. Muscle-specific inositide phosphatase (MIP/MTMR14) is reduced with age and its loss accelerates skeletal muscle aging process by altering calcium homeostasis. Aging (Albany NY) (2010);2:504–513. doi: 10.18632/aging.100190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Russ D. W., Gregg-Cornell K., Conaway M. J., Clark B. C. Evolving concepts on the age-related changes in "muscle quality". J. Cachexia. Sarcopenia. Muscle. (2012);3:95–109. doi: 10.1007/s13539-011-0054-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Manini T. M., Clark B. C. Dynapenia and aging: an update. J. Gerontol. A Biol. Sci. Med. Sci. (2012);67:28–40. doi: 10.1093/gerona/glr010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Collins H. E., Zhu-Mauldin X., Marchase R. B., Chatham J. C. STIM1/Orai1-mediated SOCE: current perspectives and potential roles in cardiac function and pathology. Am. J. Physiol. Heart. Circ. Physiol. (2013);305:H446–458. doi: 10.1152/ajpheart.00104.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Touchberry C. D., Elmore C. J., Nguyen T. M., Andresen J. J., Zhao X., Orange M., Weisleder N., Brotto M., Claycomb W. C., Wacker M. J. Store-operated calcium entry is present in HL-1 cardiomyocytes and contributes to resting calcium. Biochem. Biophys. Res. Commun. (2011);416:45–50. doi: 10.1016/j.bbrc.2011.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Volkers M., Dolatabadi N., Gude N., Most P., Sussman M. A., Hassel D. Orai1 deficiency leads to heart failure and skeletal myopathy in zebrafish. J. Cell Sci. (2012);125:287–294. doi: 10.1242/jcs.090464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Luo X., Hojayev B., Jiang N., Wang Z. V., Tandan S., Rakalin A., Rothermel B. A., Gillette T. G., Hill J. A. STIM1-dependent store-operated Ca (2) (+) entry is required for pathological cardiac hypertrophy. J. Mol. Cell Cardiol. (2012);52:136–147. doi: 10.1016/j.yjmcc.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ohba T., Watanabe H., Murakami M., Sato T., Ono K., Ito H. Essential role of STIM1 in the development of cardiomyocyte hypertrophy. Biochem. Biophys. Res. Commun. (2009);389:172–176. doi: 10.1016/j.bbrc.2009.08.117. [DOI] [PubMed] [Google Scholar]
- 92.Voelkers M., Salz M., Herzog N., Frank D., Dolatabadi N., Frey N., Gude N., Friedrich O., Koch W. J., Katus H. A., Sussman M. A., Most P. Orai1 and Stim1 regulate normal and hypertrophic growth in cardiomyocytes. J. Mol. Cell Cardiol. (2010);48:1329–1334. doi: 10.1016/j.yjmcc.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]