

Abstract
Cancer is associated with an increased prevalence of depression. Peripheral tumors induce inflammatory cytokine production in the brain and depressive-like behaviors. Mounting evidence indicates that cytokines are part of a pathway by which peripheral inflammation causes depression. Neuroinflammatory responses to immune challenges can be exacerbated (primed) by prior immunological activation associated with aging, early-life infection, and drug exposure. This experiment tested the hypothesis that peripheral tumors likewise induce neuroinflammatory sensitization, or priming. Female rats with chemically-induced mammary carcinomas were injected with either saline or lipopolysaccharide (LPS, 250 μg/kg; i.p.), and expression of mRNAs involved in the pathway linking inflammation and depression (interleukin-1beta [Il-1β], CD11b, IκBα, indolamine 2,3-deoxygenase [Ido]) was quantified by qPCR in the hippocampus, hypothalamus, and frontal cortex, 4 or 24 h post-treatment. In the absence of LPS, hippocampal Il-1β and CD11b mRNA expression were elevated in tumor-bearing rats, whereas Ido expression was reduced. Moreover, in saline-treated rats basal hypothalamic Il-1β and CD11b expression were positively correlated with tumor weight; heavier tumors, in turn, were characterized by more inflammatory, necrotic, and granulation tissue. Tumors exacerbated CNS proinflammatory gene expression in response to LPS: CD11b was greater in hippocampus and frontal cortex of tumor-bearing relative to tumor-free rats, IκBβ was greater in hippocampus, and Ido was greater in hypothalamus. Greater neuroinflammatory responses in tumor-bearing rats were accompanied by attenuated body weight gain post-LPS. The data indicate that neuroinflammatory pathways are potentiated, or primed, in tumor-bearing rats, which may exacerbate future negative behavioral consequences.
Keywords: microglia; cytokine; indolamine 2,3-deoxygenase; Nf-κB; depression; cancer
1. Introduction
Depression and anxiety disorders are significantly more prevalent in cancer patients than in the general population (Mitchell et al., 2011; Waraich et al., 2004). Cognitive impairments, including learning and memory deficits, are also reported in cancer patients (Joly et al., 2011; Wefel et al., 2008). The causes of these emotional and behavioral comorbidities are multifactorial in nature, likely reflecting interactions among the biological sequelae of the cancer itself, the toxicity due to cancer treatment, and psychological stress associated with cancer diagnosis (Miller et al., 2008). Depression is negatively correlated with cancer survival (Giese-Davis et al., 2011; Pinquart and Duberstein, 2010) and positively correlated with treatment non-compliance (DiMatteo et al., 2003; Kissane et al., 2007); therefore, understanding the mechanisms underlying cancer-associated depression is of clinical relevance.
One of the mechanisms by which cancers may cause negative affect is through neuroinflammation (Cleeland et al., 2003). Similar to patients with major depressive disorder, cancer patients exhibit elevated pro-inflammatory cytokines in the circulation and CSF (Miller et al., 2008). Cytokine-based immunotherapies trigger transient depressive episodes in cancer and hepatitis patients, which remit once treatment is terminated (Bower et al., 2011; Capuron et al., 2004). Furthermore, in rodent models of acute infection, peripheral inflammation upregulates cytokine (IL-1β, IL-6, TNFα) production in brain regions that regulate emotional behavior (e.g., hypothalamus, hippocampus, frontal cortex), and are responsible for the subsequent activation of Nf-κB (Nadjar et al., 2003) and indolamine-2,3-deoxygenase (IDO; Konsman et al., 2002) mRNA expression in the brain. Inflammation-induced IDO expression may play a key role in the genesis of depressive-like syndromes, as IDO depletes synaptic serotonin, an effect that is reversible by pharmacological treatment with anti-depressants (Dantzer et al., 2008).
In rodent models, mammary and ovarian carcinomas induce the production of circulating and CNS proinflammatory cytokines (Lamkin et al., 2011; Pyter et al., 2009); tumors alone are sufficient to trigger depressive-like behaviors and cognitive impairments (Lamkin et al., 2011; Pyter et al., 2010; Pyter et al., 2009). Whether tumors initiate other factors in the CNS inflammatory signaling cascade (e.g., NF-κB, IDO) that have been functionally-linked to negative affective-like behavior remains to be examined.
One mechanism by which brain proinflammatory cytokine expression may be upregulated in animals with cancer is via priming, or sensitization, of CNS neuroinflammatory response pathways. This hypothesis stems from the experience-dependent functional and structural plasticity exhibited by CD11b+ microglia, resident immune cells in the CNS which are the primary source of cytokine production in the brain. For example, early-life infection alters microglia in rats such that, when challenged with lipopolysaccharide (LPS) in adulthood, microglial neuroinflammatory responses are enhanced, and downstream learning and memory impairments are exacerbated (Bilbo et al., 2005; Bilbo and Schwarz, 2009). Advanced aging similarly gives rise to functional changes in microglia (Henry et al., 2009). If tumor-induced increases in constitutive cytokine expression in the CNS reflect the activity of primed microglia, then one would predict: 1) greater expression of CD11b in the CNS of rats with tumors absent any acute inflammatory challenge, and 2) greater LPS-induced increases in the expression of brain proinflammatory cytokines, NF-κB, and IDO.
This experiment tested the hypotheses that neuroinflammatory signaling pathways associated with cytokine-induced depressive-like behavior are activated in rats with mammary cancer, and that a peripheral immune challenge (LPS) elicits greater CNS neuroinflammatory signaling responses in rats with tumors compared to tumor-free controls.
2. Results
2.1 Tumor induction
All NMU-treated rats developed mammary tumors, whereas no tumors were evident in saline-injected rats. The average timing of tumor detection was 6.25 weeks after NMU injection (range 4–10 weeks); mean number of tumors/rat was 3.3 (range 1–7), and the mean total tumor mass was 6.98 g (range 0.3–20.0 g).
2.2 LPS-induced proinflammatory mRNA expression
In the absence of LPS, hippocampal Il-1β mRNA was significantly higher in tumor-bearing relative to tumor-free rats (U=56, p≤0.05; Fig. 1A; cf, Pyter et al., 2010). LPS treatment increased Il-1β in the hippocampus (F2,72=16.4, p≤0.001; Fig. 1A), hypothalamus (F2,64=7.7, p≤0.01; Fig. 1B), and frontal cortex (F2,70=12.0, p≤0.001; Fig. 1C) of all rats. The magnitude of the increases observed in the hypothalamus and frontal cortex were greater for tumor-bearing rats 4 h post-LPS compared with tumor-free controls (p≤0.05). A significant interaction was evident between injection (LPS/saline) and tumor condition (tumor/tumor-free) in hypothalamic Il-1β mRNA (F2,64=3.9, p≤0.05) such that the increase in Il-1β in tumor-bearing rats was greater 4 h post-LPS and lower 24 h post-LPS.
Figure 1.
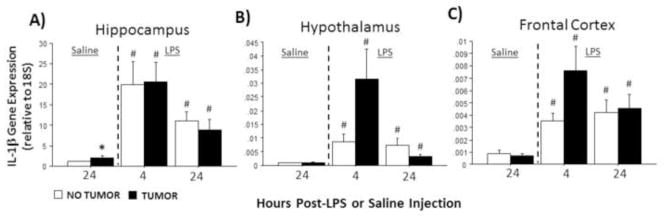
Tumors alter LPS-induced Il-1β mRNA expression in the brain. Mean ± SEM Il-1β mRNA in the (A) hippocampus, (B) hypothalamus, and (C) frontal cortex of rats with or without mammary carcinomas 4 or 24 h after i.p. injection with LPS or 24 h after i.p. injection with saline (n=10–15 rats/treatment). *p ≤ 0.05 between tumor and no tumor values #p ≤ 0.05 between LPS and respective saline values
In the absence of LPS, CD11b mRNA expression was significantly greater in the hippocampus of tumor-bearing relative to tumor-free rats (U=51, p≤0.05; Fig. 2A). LPS treatment affected CD11b expression in the hippocampus (F2,70=4.5, p≤0.05; Fig. 2A), hypothalamus (F2,70=7.9, p≤0.001; Fig. 2B), and frontal cortex (F2,70=7.6, p≤0.01; Fig. 2C) of all rats. Specifically, LPS-induced increases in CD11b mRNA expression were significantly higher in tumor-bearing relative to tumor-free rats in the hippocampus (p≤0.01) and frontal cortex (p≤0.05). Interactions between tumor status and LPS treatment were not observed for CD11b mRNA (p>0.05, all comparisons).
Figure 2.
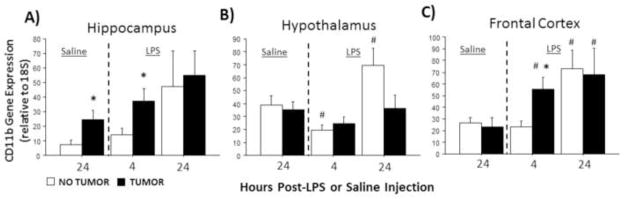
Tumors alter LPS-induced CD11b mRNA expression in the brain. Mean ± SEM CD11b mRNA in the (A) hippocampus, (B) hypothalamus, and (C) frontal cortex of rats with or without mammary carcinomas 4 or 24 h after i.p. injection with LPS or 24 h after i.p. injection with saline (n=11–16 rats/treatment). *p ≤ 0.05 between tumor and no tumor values #p ≤ 0.05 between LPS and respective saline values
Without LPS stimulation, IκBα expression remained comparable in all brain regions examined between tumor-bearing and tumor-free rats (p>0.05 in all cases). IκBα expression was significantly affected by LPS treatment in all brain regions examined (hippocampus: F2,70=16.8, p≤0.0001; hypothalamus: F2,67=6.2, p≤0.01; frontal cortex: F2,73=14.1, p≤0.0001; Fig. 3A–C). Injection treatment and tumor condition interacted to affect patterns of hippocampal IκBα expression (F2,70=6.8; p≤0.01) such that hippocampal IκBα expression was significantly greater in tumor-bearing relative to tumor-free rats 4 h after LPS injection (p≤0.05), but 24 h after LPS, hippocampal IκBα mRNA was significantly reduced relative to controls (p≤0.05; Fig. 3A).
Figure 3.
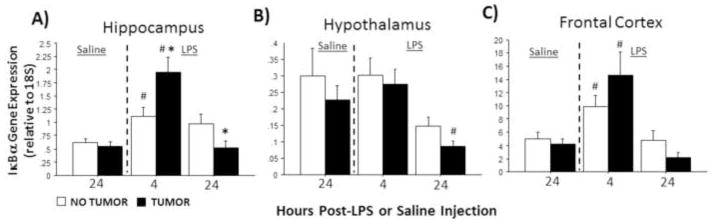
Tumors alter LPS-induced IκBa mRNA expression in the brain. Mean ± SEM IκBa mRNA in the (A) hippocampus, (B) hypothalamus, and (C) frontal cortex of rats with or without mammary carcinomas 4 or 24 h after i.p. injection with LPS or 24 h after i.p. injection with saline (n=11–17 rats/treatment). *p ≤ 0.05 between tumor and no tumor values #p ≤ 0.05 between LPS and respective saline values
Finally, in the absence of LPS, hippocampal Ido expression was significantly lower in tumor-bearing rats relative to tumor-free controls (p≤0.05). LPS significantly influenced Ido expression in all brain regions examined (hippocampus: F2,51=13.1, p≤0.0001; hypothalamus: F2,58=12.4, p≤0.0001; frontal cortex: F2,42=4.8, p ≤0.05; Fig. 4A–C) regardless of tumor treatment. Tumor status (F1,58=4.6; p≤0.05), and the interaction between tumor status and injection treatment (F2,58=4.6; p≤0.05), each affected hippocampal Ido expression (Fig. 3A). Four h after LPS treatment, hypothalamic Ido expression was significantly greater in tumor-bearing relative to tumor-free rats (p≤0.05).
Figure 4.
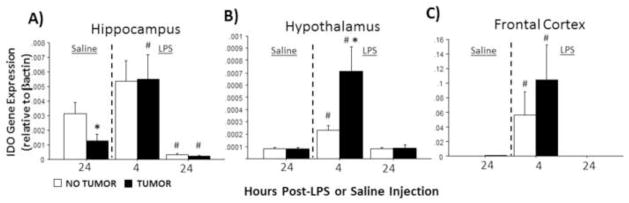
Tumors alter LPS-induced indolamine 2,3-deoxygenase (IDO) mRNA expression in the brain. Mean ± SEM Ido mRNA in the (A) hippocampus, (B) hypothalamus, and (C) frontal cortex of rats with or without mammary carcinomas 4 or 24 h after i.p. injection with LPS or 24 h after i.p. injection with saline (n=7–12 rat/treatment). Ido was undetectable for the majority 72% of samples in the 24 h post-LPS groups (see Methods). *p ≤ 0.05 between tumor and no tumor values #p ≤ 0.05 between LPS and respective saline values
2.3 LPS-induced changes in body mass
Weekly weight gain following LPS treatment was significantly reduced in tumor-bearing relative to tumor-free rats (p≤0.01; Fig. 5), whereas weekly weight gain following saline injection was comparable between tumor-bearing and -free rats (p>0.05; Fig. 5).
Figure 5.
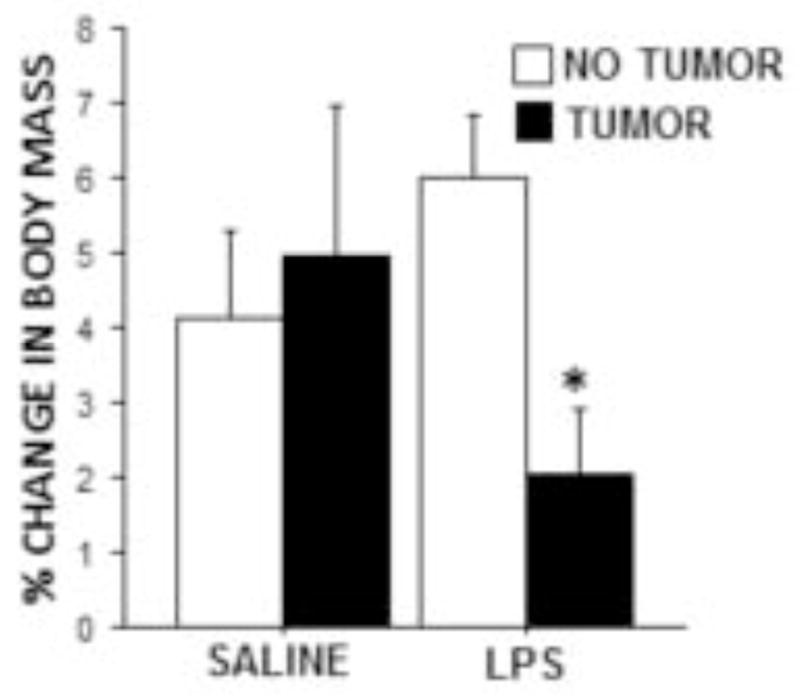
Tumors attenuated LPS-induced weight loss. Mean ± SEM percent change in body mass of tumor-free and tumor-bearing rats 24 h after i.p. sterile saline or LPS injection compared with body mass one week prior (n=9–10 rats/group). *p ≤ 0.05 between tumor and no tumor values
2.4 Relations between total tumor mass and proinflammatory mRNA expression without LPS treatment
Exploratory analyses were performed to evaluate whether mammary tumor burden was positively associated with baseline Il-1β, CD11b, IκBα, and Ido mRNA expression by assessing the linear effects of tumor burden (total tumor mass) on brain mRNA levels in the absence of LPS treatment (Fig. 6). Total tumor mass significantly and positively correlated with hypothalamic Il-1β (p≤0.05, Fig. 6A) and CD11b (p≤0.05, Fig. 6B) mRNA expression in saline-treated rats, but not with IκBα (p=0.1; Fig. 6C) or Ido (p>0.05; Fig. 6D). Significant correlations were not observed in other brain regions.
Figure 6.

Tumor burden correlates positively with hypothalamic proinflammatory transcripts in the absence of LPS. Linear regressions between total mammary tumor mass and hypothalamic (A) Il-1β, (B) CD11b, (C) IκBα, and (D) Ido mRNA expression in non-LPS-injected, tumor-bearing rats (n=9–11). *p ≤ 0.05, **p ≤ 0.005
2.5 Tumor histopathology
Representative mammary tumors are depicted in Figure 7. All tumors were invasive carcinomas with moderate differentiation. Smaller tumors (~0.3 g; Fig. 7A) presented with minimal regions of inflammation, necrosis, and granulation, whereas in larger tumors (~6.0 g; Fig. 7B) these regions were extensive and numerous. Histological evidence of inflammation was localized to the necrotic and granulation tissue regions as opposed to the epithelial component of the carcinomas.
Figure 7.
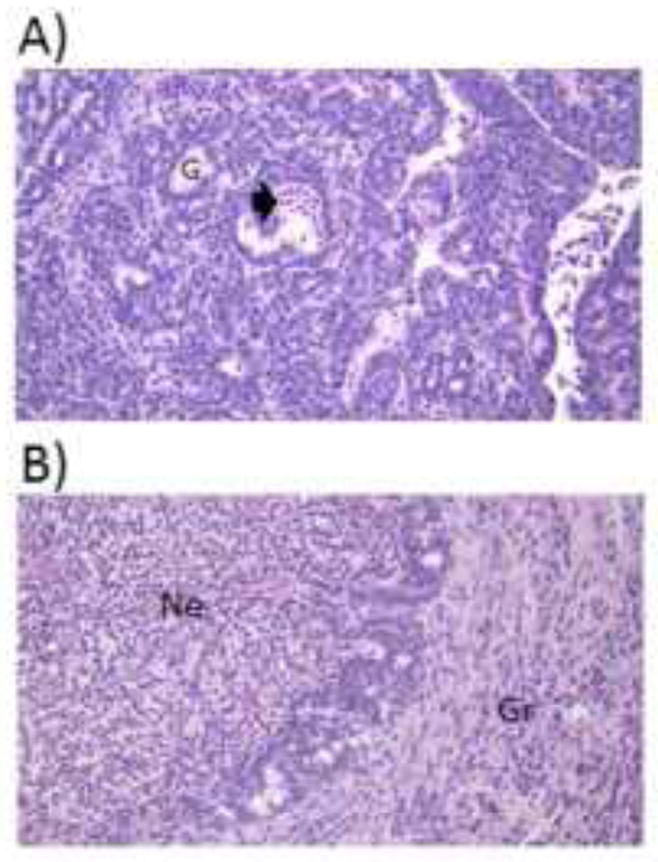
Tumor inflammation and necrosis. Representative 5 μm sections stained with hematoxylin and eosin of (A) a small (0.3 g) and (B) a large (6.0 g) tumor. All tumors were pathologically similar: invasive ductal carcinomas with moderate differentiation. Smaller tumors exhibited minimal necrotic and inflammatory tissue, whereas larger tumors consisted of widespread necrosis with acute inflammation, as well as granulation tissue with chronic inflammation. Magnification = 400X. Arrowhead=acute inflammation and necrosis, G=gland, Ne=necrotic tissue with acute inflammation, Gr=granulation tissue with chronic inflammation
3. Discussion
The present report confirms and extends previous observations (Pyter et al., 2010; Turrin et al., 2004), indicating that peripheral tumors constitutively increase both Il-1β and the microglial cell marker, CD11b, mRNA expression in the hippocampus. Microglia are the main sources of de novo cytokine production in the brain, although astrocytes, neurons, and endothelial cells may also contribute (Hanisch, 2002; Kim and de Vellis, 2005). Based on the assumption that parenchymal microglia are the main sources of the inflammatory factors measured in this study, these data suggest that either microglial cell priming or proliferation is induced by tumors in the periphery or that overall neuroinflammatory signaling is elevated in the hippocampus of tumor-bearing rats. In the former scenario, microglial cell activation would likely be responsible for the modest increases in constitutive Il-1β expression observed in the hippocampus of tumor-bearing rats. Multiple lines of convergent evidence indicate that environmental events, broadly-defined, can prime the brain microglial system. Neonatal infection (Bilbo et al., 2005), adolescent morphine exposure (Schwarz and Bilbo, 2013), adult environmental enrichment (Williamson et al., 2012), and advanced aging (Rosczyk et al., 2008) are each associated with increases in brain neuroinflammatory markers (e.g., Il-1β, CD11b, TLR4). The present data imply that peripheral cancers may constitute yet another event that primes hippocampal microglia. Further characterization of microglial morphology and function is warranted in future studies, however.
Constitutively increased levels of IL-1β and CD11b expression in rats with tumors were modest relative to the changes unmasked by the addition of a neuroinflammatory trigger: peripheral LPS injection. Hippocampal CD11b and IκBa were >2-fold higher in tumor-bearing rats after LPS treatment, and hypothalamic Ido mRNA was >3-fold increased at this early time point. Exacerbated proinflammatory responses in the CNS are the first evidence, to our knowledge, that peripheral cancer can alter the neuroinflammatory response to an acute innate immune challenge. Prior work, which used different models to elicit neuroinflammatory signaling, reported results consistent with the present data. For example, LPS (i.p.) elicits greater increases in cortical Il-1β and Il-10 mRNA in CD11b+/MHC+ cells in aged relative to young mice (Henry et al., 2009). Similarly, exaggerated cytokine and microglial responses have been observed following a peripheral LPS injection in adult rats that experienced a bacterial infection as neonates (Bilbo et al., 2005). Other, inflammatory triggers (e.g., surgical incisions [Beggs et al., 2012], high fat diet [Bolton et al., 2012], morphine exposure [Schwarz & Bilbo, 2013], seizures [Somera-Molina et al., 2007]) also trigger exaggerated microglial responses in the CNS of adult rodents that had previously (typically during the neonatal interval) experienced insults (e.g., surgery, air pollution exposure, kainic acid, morphine treatment). Therefore, it is unlikely that the exaggerated proinflammatory response to LPS evident in tumor-bearing rats is unique to LPS as an eliciting stimulus, although this conjecture awaits direct assessment. The tumor model described here likely requires significant peripheral-to-brain communication during tumor progression in order to increase gene expression of microglial marker, CD11b, however, the mechanisms by which this occurs are presently unknown.
The functional consequences of these elevated neuroinflammatory responses have not yet been fully characterized in this model, but it stands to reason that they may drive more severe behavioral responses to illness (McLinden et al., 2012). Thermoregulatory, metabolic, emotional and cognitive symptoms of infection are driven by proinflammatory cytokine expression in the CNS (Bluthe et al., 1995; Bluthe et al., 1992; Bluthe et al., 2000a; Bluthe et al., 2000b; Frenois et al., 2007; Gibertini et al., 1995). Although a thorough assay of LPS-induced sickness behaviors was beyond the scope of the present study, and incompatible with the cross-sectional nature of the study design, preliminary data in support of the above conjecture is evident in the impaired body mass recovery following LPS treatment in tumor-bearing rats relative to tumor-free controls (Fig. 5).
The three brain regions examined in this report were chosen based on their coordinated and overlapping roles in cytokine-induced depressive-like behavior and because of their rapid and robust inflammatory responsiveness to LPS treatment (Dantzer et al., 1996; Kent et al., 1992; Laye et al., 1994; Quan et al., 1998). The hypothalamus may be more relevant to metabolic and endocrine consequences of neuroinflammation, whereas the frontal cortex and hippocampus have been more directly linked to cognitive and affective-like consequences, respectively (Fu et al., 2010; Ilyin et al., 1998; Jenrow et al., 2013). Therefore, the slight differences observed in neuroinflammatory responses among brain regions may predict selective behavioral outcomes, although better temporal resolution and more detailed analyses of sickness behaviors would be required to fully elaborate these relations. Regional variance in microglial abundance (Lawson et al., 1990) and relatively low sample sizes (n=7–12) may also contribute to the variation in LPS responses observed among brain regions.
Most neuroinflammatory transcripts elicited by LPS subsided within 24 hours; however, these 24-h reductions were of greater magnitude in tumor-bearing rats than controls, given that they followed a higher 4-h post-LPS level. This pattern of response resulted in significant statistical interactions between LPS and tumor treatments in hippocampal and hypothalamic Il-1β, IκBα, or Ido gene expression. Anti-inflammatory IL-10 production is elevated in hippocampi of tumor-bearing rats (Pyter et al., 2009), which may contribute to the rapid resolution of exacerbated neuroinflammation in rats with tumors. Alternatively, the transient nature of the exaggerated neuroinflammatory rise in tumor-bearing rats may be a consequence of disease-burdened metabolic constraints. Inflammation, and in particular, excessive inflammation, requires significant energetic resources (Belloni et al., 2010), which may be unsustainable in rats that are simultaneously supporting the growth of a tumor.
The CD11b expression pattern was noteworthy in that it remained elevated for at least 24 h after LPS treatment. Microglial cells display significant morphological and functional plasticity in adult the brain, and such changes can endure for days to weeks (Town et al., 2005). Therefore, it is possible that the persistently elevated CD11b mRNA expression may reflect a sustained activation of microglia in response to LPS. The magnitude of LPS-induced CD11b mRNA expression did not differ as a function of tumor status, although the present experiment did not examine the time course of the eventual reduction in CD11b.
Lastly, an exploratory correlational analysis indicated that greater tumor burden predicted increased baseline hypothalamic IL-1β and CD11b mRNA expression in rats that were not treated with LPS. These data raise numerous causal hypotheses related to humoral factors generated by tumors and metabolic consequences of larger tumors in their modulation of microglial priming and CNS inflammation. The limited histopathological data in the present study, which suggest that larger tumors were characterized by greater inflammation and necrosis, were consistent with the former hypothesis, but a more detailed and quantitative assessment of this conjecture is required. Few studies have addressed the possibility that tumor size relates to production of tumor inflammatory factors (but see Day et al., 2013), although prognosis and tumor invasiveness are negatively related to circulating proinflammatory cytokine concentrations (Lippitz, 2013).
Taken together, the present data indicate that peripheral tumors alter basal neuroinflammatory tone (hippocampal IL-1β and CD11b expression) and CNS neuroinflammatory responses to subsequent innate immune challenges. The activation of microglia or neuroinflammation, in general, suggested by these data bears superficial similarities to lifespan developmental changes in microglia that are conferred by early-life infection (neonatal priming), but the present work adds to an emerging literature (e.g., Bilbo et al., 2005; Henry et al., 2009; Schwarz & Bilbo, 2013) that identifies events occurring well past the neonatal period (i.e., peripubertal tumor induction) that are nevertheless capable of inducing constitutive and latent changes in hippocampal microglia. A better understanding of the mechanisms by which chronic disease affects brain cytokine signaling may yield deeper insights into the causal mechanisms underlying psychiatric comorbidities common in cancer patients and survivors.
4. Experimental Procedures
4.1 Animals
Female, nulliparous Wistar rats (Harlan, Indianapolis, IN) were maintained in 16 h photocycle (lights on at 20:00 h C.S.T.) and housed 2–3/cage in polypropylene cages (25.9 × 47.6 × 20.9 cm) at temperature and humidity of 22 ± 1 °C and 50 ± 5%, respectively, and with ad libitum access to food (Harlan 2018) and filtered tap water. Rats were pseudo-randomly distributed among treatment groups (see below).
4.2 Tumor induction
Rats (28–37 day old) were injected with a chemical carcinogen (N-nitroso-N-methylurea [NMU]; Sigma, St. Louis, MO; i.p.; 50 mg/kg; “TUMOR”) or 0.2 ml sterile saline (“NO TUMOR”) as described in detail elsewhere (Pyter et al., 2009). All cagemates received the same treatment. NMU induces >90% malignant mammary carcinomas in Wistar rats (Thompson et al., 2000). Mammary gland palpation and measurement were performed beginning 4 weeks after carcinogen/saline treatment by an experienced researcher who was blind to injection treatments.
4.3 Experimental Design
One to two weeks after tumor detection, rats were injected i.p. with bacterial lipopolysaccharide (LPS; E. coli, 127:8B; 250 μg/kg; Sigma) or 0.2 ml sterile saline (n=12–17/group) at 10:00 h. Brain tissues were collected 4 or 24 h after injection treatments. Body masses were determined (to 0.1 g) in all rats that were euthanized at the 24 h post-injection time point. Percentage body mass loss was calculated relative to body mass values taken one week prior to LPS/saline injection.
4.4 Tissue Collection
Four or 24 h after LPS treatment, and 24 h after saline treatment, rats were anesthetized using sodium pentobarbital (50 mg/kg; i.p.) and transcardially perfused with ~50 ml cold sterile 0.9% saline in order to flush circulating leukocytes from the brain microvasculature. Rapid microdissections were performed to remove the hypothalamus, hippocampus and frontal cortex (~20 mg) under RNAse-free conditions; samples were frozen on dry ice and stored for gene expression analyses. At the time of necropsy, tumors were excised, weighed, and preserved in 10% buffered formalin. After paraffin embedding, 5 μm tumor sections were stained with hematoxylin and eosin were histologically analyzed at the University of Chicago Pathology Department.
4.5 qPCR
Total RNA (RNeasy, Qiagen) from dissected brain tissues was used to measure gene expression of interleukin 1-beta (IL-1β, a proinflammatory cytokine), CD11b (a microglial activation marker), IκBα (an indicator of NF-κB activation), and indolamine 2,3-deoxygenase (IDO). RNA quality was verified by A260/A280 ratios >1.8 on a spectrophotometer. Following reverse transcription, commercial qPCR primer and probe sets for rat Il-1β (Rn0058432_m1), CD11b (Rn01506865), IκBα (Rn01473657_g1), Ido (Rn01482210_m1) and 18S rRNA control gene (Hs99999901_s1) were used to quantify mRNA expression on an ABI 7900HT platform (Applied Biosystems). The probes were labeled with 6-FAM (VIC for 18S rRNA) and MGB (non-fluorescent quencher) at the 5′ and 3′ ends, respectively. Amplification was performed using Taqman® Universal PCR Master Mix. The universal two-step RT-PCR cycling conditions used were: 50° C for 2 min, 95° for 10 min, followed by 40 cycles of 95° C for 15 sec and 60° C for 1 min. Relative mRNA expression of individual samples run in duplicate was calculated by comparison to relative standard curves consisting of serial dilutions of pooled rat hippocampal cDNA (1:1, 1:10, 1:100, 1:1000, 1:10,000) followed by normalization to 18S rRNA or β-actin gene expression. Ido mRNA levels were not detectable in the majority of frontal cortex samples at the 24-h post-LPS sampling interval (tumor-free controls: 12 of 13 were undetectable; tumor-bearing rats: 6 of 12 were undetectable), therefore these samples were not included in the subsequent analyses.
4.6 Statistical Analyses
Differences in mRNA expression were assessed using two-way ANOVAs, and a priori planned pairwise comparisons between either tumor/no tumor treatment or saline/LPS treatment were performed using student’s t-tests or nonparametric tests (Mann-Whitney U) as warranted. Linear regressions between gene expression and tumor burden were assessed using Pearson’s correlations. StatView 5.0 software (v. 5.0.1, SAS, Cary, NC, USA) was used to perform all statistical calculations. Differences were considered significant if p ≤ 0.05.
Highlights.
Effects of peripheral tumors on LPS-induced neuroinflammatory signaling were examined in rats.
Without LPS, tumors increased hippocampal IL-1beta and microglial marker, CD11b.
With LPS, neuroinflammatory mRNAs were elevated in the brain beyond that of tumor-free controls.
Cancer-induced behavioral comorbidities may be due to enhanced neuroinflammatory sensitivity.
Acknowledgments
We thank Priyesh Patel, Dr. Staci Bilbo, August Kampf-Lassin, Jill McKay-Fleish, Jaclyn Tamaroff, Ron Barthelemy, Ken Onishi, Dr. Katja Gwin, and Dr. Betty Theriault for technical advice and assistance. This project was supported by an American Cancer Society fellowship (PF-08-086-TBE), NIH grant AI-67406, and a grant from the Brain Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beggs S, Currie G, Salter MW, Fitzgerald M, Walker SM. Priming of adult pain responses by neonatal pain experience: maintenance by central neuroimmune activity. Brain. 2012;135:404–417. doi: 10.1093/brain/awr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloni V, Faivre B, Guerreiro R, Arnoux E, Bellenger J, Sorci G. Suppressing an anti-inflammatory cytokine reveals a strong age-dependent survival cost in mice. PloS one. 2010;5:e12940. doi: 10.1371/journal.pone.0012940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci. 2005;119:293–301. doi: 10.1037/0735-7044.119.1.293. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM. Early-life programming of later-life brain and behavior: a critical role for the immune system. Frontiers in behavioral neuroscience. 2009;3:14. doi: 10.3389/neuro.08.014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluthe RM, Beaudu C, Kelley KW, Dantzer R. Differential effects of IL-1ra on sickness behavior and weight loss induced by IL-1 in rats. Brain Res. 1995;677:171–176. doi: 10.1016/0006-8993(95)00194-u. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Dantzer R, Kelley KW. Effects of interleukin-1 receptor antagonist on the behavioral effects of lipopolysaccharide in rat. Brain Res. 1992;573:318–320. doi: 10.1016/0006-8993(92)90779-9. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Laye S, Michaud B, Combe C, Dantzer R, Parnet P. Role of interleukin-1beta and tumour necrosis factor-alpha in lipopolysaccharide-induced sickness behaviour: a study with interleukin-1 type I receptor-deficient mice. Eur J Neurosci. 2000a;12:4447–4456. [PubMed] [Google Scholar]
- Bluthe RM, Michaud B, Poli V, Dantzer R. Role of IL-6 in cytokine-induced sickness behavior: a study with IL-6 deficient mice. Physiol Behav. 2000b;70:367–373. doi: 10.1016/s0031-9384(00)00269-9. [DOI] [PubMed] [Google Scholar]
- Bolton JL, Smith SH, Huff NC, Gilmour MI, Foster WM, Auten RL, Bilbo SD. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J. 2012;26:4743–4754. doi: 10.1096/fj.12-210989. [DOI] [PubMed] [Google Scholar]
- Bower JE, Ganz PA, Irwin MR, Kwan L, Breen EC, Cole SW. Inflammation and behavioral symptoms after breast cancer treatment: do fatigue, depression, and sleep disturbance share a common underlying mechanism? J Clin Oncol. 2011;29:3517–3522. doi: 10.1200/JCO.2011.36.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Ravaud A, Miller AH, Dantzer R. Baseline mood and psychosocial characteristics of patients developing depressive symptoms during interleukin-2 and/or interferon-alpha cancer therapy. Brain Behav Immun. 2004;18:205–213. doi: 10.1016/j.bbi.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Cleeland CS, Bennett GJ, Dantzer R, Dougherty PM, Dunn AJ, Meyers CA, Miller AH, Payne R, Reuben JM, Wang XS, Lee BN. Are the symptoms of cancer and cancer treatment due to a shared biologic mechanism? A cytokine-immunologic model of cancer symptoms. Cancer. 2003;97:2919–2925. doi: 10.1002/cncr.11382. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Bluthe RM, Aubert G, Goodall J-L, Bret-Dibat JL, Kent P, Goujon E, Laye S, Parnet P, Kelley KW. Cytokines actions on behavior. In: Rothwell NJ, editor. Cytokines in the Nervous System. Springer; Austin: 1996. pp. 117–144. [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day SD, Enos RT, McClellan JL, Steiner JL, Velazquez KT, Murphy EA. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine. 2013 doi: 10.1016/j.cyto.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMatteo MR, Robinson JD, Heritage J, Tabbarah M, Fox SA. Correspondence among patients’ self-reports, chart records, and audio/videotapes of medical visits. Health Commun. 2003;15:393–413. doi: 10.1207/S15327027HC1504_02. [DOI] [PubMed] [Google Scholar]
- Frenois F, Moreau M, O’Connor J, Lawson M, Micon C, Lestage J, Kelley KW, Dantzer R, Castanon N. Lipopolysaccharide induces delayed FosB/DeltaFosB immunostaining within the mouse extended amygdala, hippocampus and hypothalamus, that parallel the expression of depressive-like behavior. Psychoneuroendocrinology. 2007;32:516–531. doi: 10.1016/j.psyneuen.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Zunich SM, O’Connor JC, Kavelaars A, Dantzer R, Kelley KW. Central administration of lipopolysaccharide induces depressive-like behavior in vivo and activates brain indoleamine 2,3 dioxygenase in murine organotypic hippocampal slice cultures. Journal of neuroinflammation. 2010;7:43. doi: 10.1186/1742-2094-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibertini M, Newton C, Friedman H, Klein TW. Spatial learning impairment in mice infected with Legionella pneumophila or administered exogenous interleukin-1-beta. Brain Behav Immun. 1995;9:113–128. doi: 10.1006/brbi.1995.1012. [DOI] [PubMed] [Google Scholar]
- Giese-Davis J, Collie K, Rancourt KM, Neri E, Kraemer HC, Spiegel D. Decrease in depression symptoms is associated with longer survival in patients with metastatic breast cancer: a secondary analysis. J Clin Oncol. 2011;29:413–420. doi: 10.1200/JCO.2010.28.4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyin SE, Gayle D, Flynn MC, Plata-Salaman CR. Interleukin-1beta system (ligand, receptor type I, receptor accessory protein and receptor antagonist), TNF-alpha, TGF-beta1 and neuropeptide Y mRNAs in specific brain regions during bacterial LPS-induced anorexia. Brain Res Bull. 1998;45:507–515. doi: 10.1016/s0361-9230(97)00437-1. [DOI] [PubMed] [Google Scholar]
- Jenrow KA, Brown SL, Lapanowski K, Naei H, Kolozsvary A, Kim JH. Selective inhibition of microglia-mediated neuroinflammation mitigates radiation-induced cognitive impairment. Radiat Res. 2013;179:549–556. doi: 10.1667/RR3026.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly F, Rigal O, Noal S, Giffard B. Cognitive dysfunction and cancer: which consequences in terms of disease management? Psychooncology. 2011;20:1251–1258. doi: 10.1002/pon.1903. [DOI] [PubMed] [Google Scholar]
- Kent S, Bluthe RM, Kelley KW, Dantzer R. Sickness behavior as a new target for drug development. Trends Pharmacol Sci. 1992;13:24–28. doi: 10.1016/0165-6147(92)90012-u. [DOI] [PubMed] [Google Scholar]
- Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- Kissane DW, Grabsch B, Clarke DM, Smith GC, Love AW, Bloch S, Snyder RD, Li Y. Supportive-expressive group therapy for women with metastatic breast cancer: survival and psychosocial outcome from a randomized controlled trial. Psychooncology. 2007;16:277–286. doi: 10.1002/pon.1185. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci. 2002;25:154–159. doi: 10.1016/s0166-2236(00)02088-9. [DOI] [PubMed] [Google Scholar]
- Lamkin DM, Lutgendorf SK, Lubaroff D, Sood AK, Beltz TG, Johnson AK. Cancer induces inflammation and depressive-like behavior in the mouse: modulation by social housing. Brain Behav Immun. 2011;25:555–564. doi: 10.1016/j.bbi.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience. 1990;39:151–170. doi: 10.1016/0306-4522(90)90229-w. [DOI] [PubMed] [Google Scholar]
- Laye S, Parnet P, Goujon E, Dantzer R. Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res Mol Brain Res. 1994;27:157–162. doi: 10.1016/0169-328x(94)90197-x. [DOI] [PubMed] [Google Scholar]
- Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. The lancet oncology. 2013;14:e218–228. doi: 10.1016/S1470-2045(12)70582-X. [DOI] [PubMed] [Google Scholar]
- McLinden KA, Kranjac D, Deodati LE, Kahn M, Chumley MJ, Boehm GW. Age exacerbates sickness behavior following exposure to a viral mimetic. Physiol Behav. 2012;105:1219–1225. doi: 10.1016/j.physbeh.2011.04.024. [DOI] [PubMed] [Google Scholar]
- Miller AH, Ancoli-Israel S, Bower JE, Capuron L, Irwin MR. Neuroendocrine-immune mechanisms of behavioral comorbidities in patients with cancer. J Clin Oncol. 2008;26:971–982. doi: 10.1200/JCO.2007.10.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AJ, Chan M, Bhatti H, Halton M, Grassi L, Johansen C, Meader N. Prevalence of depression, anxiety, and adjustment disorder in oncological, haematological, and palliative-care settings: a meta-analysis of 94 interview-based studies. The lancet oncology. 2011;12:160–174. doi: 10.1016/S1470-2045(11)70002-X. [DOI] [PubMed] [Google Scholar]
- Nadjar A, Combe C, Laye S, Tridon V, Dantzer R, Amedee T, Parnet P. Nuclear factor kappaB nuclear translocation as a crucial marker of brain response to interleukin-1. A study in rat and interleukin-1 type I deficient mouse. J Neurochem. 2003;87:1024–1036. doi: 10.1046/j.1471-4159.2003.02097.x. [DOI] [PubMed] [Google Scholar]
- Pinquart M, Duberstein PR. Depression and cancer mortality: a meta-analysis. Psychol Med. 2010;40:1797–1810. doi: 10.1017/S0033291709992285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyter LM, Cochrane SF, Ouwenga RL, Patel PN, Pineros V, Prendergast BJ. Mammary tumors induce select cognitive impairments. Brain Behav Immun. 2010 doi: 10.1016/j.bbi.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyter LM, Pineros V, Galang JA, McClintock MK, Prendergast BJ. Peripheral tumors induce depressive-like behaviors and cytokine production and alter hypothalamic-pituitary-adrenal axis regulation. Proc Natl Acad Sci U S A. 2009;106:9069–9074. doi: 10.1073/pnas.0811949106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan N, Whiteside M, Herkenham M. Time course and localization patterns of interleukin-1beta messenger RNA expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience. 1998;83:281–293. doi: 10.1016/s0306-4522(97)00350-3. [DOI] [PubMed] [Google Scholar]
- Rosczyk HA, Sparkman NL, Johnson RW. Neuroinflammation and cognitive function in aged mice following minor surgery. Exp Gerontol. 2008;43:840–846. doi: 10.1016/j.exger.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Bilbo SD. Adolescent morphine exposure affects long-term microglial function and later-life relapse liability in a model of addiction. J Neurosci. 2013;33:961–971. doi: 10.1523/JNEUROSCI.2516-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somera-Molina KC, Robin B, Somera CA, Anderson C, Stine C, Koh S, Behanna HA, Van Eldik LJ, Watterson DM, Wainwright MS. Glial activation links early-life seizures and long-term neurologic dysfunction: evidence using a small molecule inhibitor of proinflammatory cytokine upregulation. Epilepsia. 2007;48:1785–1800. doi: 10.1111/j.1528-1167.2007.01135.x. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Singh M, McGinley J. Classification of premalignant and malignant lesions developing in the rat mammary gland after injection of sexually immature rats with 1-methyl-1-nitrosourea. J Mammary Gland Biol Neoplasia. 2000;5:201–210. doi: 10.1023/a:1026495322596. [DOI] [PubMed] [Google Scholar]
- Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. Journal of neuroinflammation. 2005;2:24. doi: 10.1186/1742-2094-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrin NP, Ilyin SE, Gayle DA, Plata-Salaman CR, Ramos EJ, Laviano A, Das UN, Inui A, Meguid MM. Interleukin-1beta system in anorectic catabolic tumor-bearing rats. Current opinion in clinical nutrition and metabolic care. 2004;7:419–426. doi: 10.1097/01.mco.0000134373.16557.92. [DOI] [PubMed] [Google Scholar]
- Waraich P, Goldner EM, Somers JM, Hsu L. Prevalence and incidence studies of mood disorders: a systematic review of the literature. Can J Psychiatry. 2004;49:124–138. doi: 10.1177/070674370404900208. [DOI] [PubMed] [Google Scholar]
- Wefel JS, Witgert ME, Meyers CA. Neuropsychological sequelae of non-central nervous system cancer and cancer therapy. Neuropsychol Rev. 2008;18:121–131. doi: 10.1007/s11065-008-9058-x. [DOI] [PubMed] [Google Scholar]
- Williamson LL, Chao A, Bilbo SD. Environmental enrichment alters glial antigen expression and neuroimmune function in the adult rat hippocampus. Brain Behav Immun. 2012;26:500–510. doi: 10.1016/j.bbi.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]