

Abstract
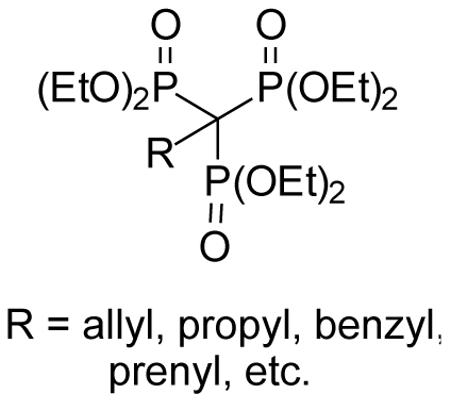
The α–trisphosphonic acid esters provide a unique spatial arrangement of three phosphonate groups, and may represent an attractive motif for inhibitors of enzymes that utilize di- or triphosphate substrates. To advance studies of this unique functionality, a general route to alkyl derivatives of the parent system (R = H) has been developed. A set of new α-alkyl-1,1,1-trisphosphonate esters has been prepared through phosphinylation and subsequent oxidation of tetraethyl alkylbisphosphonates, and the reactivity of these new compounds has been studied in representative reactions that afford additional examples of this functionality.
Introduction
The geminal bisphosphonate moiety is found in a number of drugs that are in widespread clinical use, including alendronate (1, Fosamax®), risedronate (2, Actonel®), and zoledronate (3, Zometa®).1 The clinical applications of these compounds in treatment of various diseases of the bone,1 together with the prevalence of di- and triphosphate intermediates in metabolism, have encouraged studies of many other bisphosphonates, and there are numerous reports on their chemical synthesis2 and biological activity.3 In sharp contrast to the extensive work with geminal bisphosphonates, there are very few reported studies of aryl- or alkyl-1,1,1-trisphosphonate esters (4). Phosphonate esters often are prepared through reaction of a trialkyl phosphite with an alkyl halide, but simple alkyl halides are not very reactive in this classical Michaelis-Arbuzov synthesis and chloroform does not react with triethyl phosphite even under forcing conditions.4 However, trichloromethylamine is known to react with triethyl phosphite to afford the aminotrisphosphonate 55 through a reaction sequence now assumed to be based on elimination-addition reactions.6 A similar strategy with a quinone methide was used to prepare the aryl trisphosphonate 6,7 and ultimately the parent compound 7 was prepared through addition to tetraethyl diazo-bisphosphonate.6 The parent trisphosphonate 7 also has been prepared via C-P bond formation. In this case, the anionic bisphosphonate proved unreactive with diethyl chlorophosphate but phosphinylation of the bisphosphonate anion followed by oxidation to the phosphonate was successful,6 a strategy which already had been applied to preparation of β-keto phosphonates from ketone and ester enolates.8 However, apart from some intriguing studies by Blackburn et al., who prepared adenosine esters derived from compound 7 and its halogenated analogues,9 little has been done with trisphosphonates for some time. The limited information available on trisphosphonates esters and our longstanding interest in C-P bond formation10 led us to investigate the synthesis and reactivity of this functionality.
Results and Discussion
One might reasonably assume that the shortest route from tetraethyl methylenebisphosphonate (8) to a family of alkyl trisphosphonates would involve preparation of the parent trisphosphonate 7 followed by alkylation. To explore this possibility, compound 7 was prepared starting with the literature approach that employed phosphinylation of tetraethyl methylenebisphosphonate, but followed by oxidation of the presumed phosphinate intermediate with hydrogen peroxide8b,10b rather than air (Scheme 1).6 This modified procedure gave an improved yield (48% vs. the 32% in the original report6) when the methylenebisphosphonate is thoroughly dried (vide infra), and the 1H, 13C and 31P spectra of the material prepared this way matched literature data.6 Attempted reaction of the methylenebisphosphonate anion with diethyl chlorophosphate was not successful under the same reaction conditions.
Scheme 1.
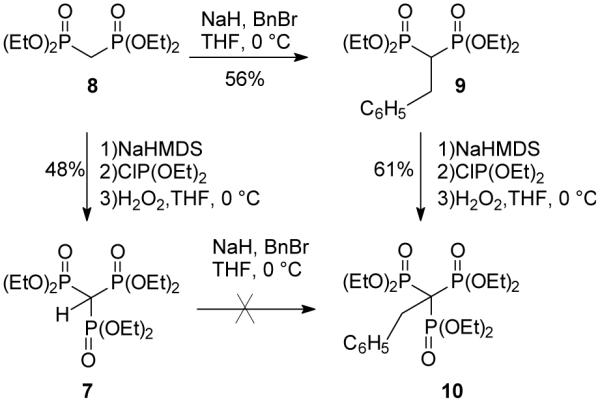
Strategies for trisphosphonate synthesis.
Upon treatment of compound 7 with NaH, formation of the anion was strongly suggested by a downfield shift in the 31P NMR spectrum (from 14 to 32 ppm). However, addition of benzyl bromide did not induce any further change in the 31P NMR resonance, nor did addition of the less sterically encumbered alkylating agent allyl bromide. Presumably the limited reactivity of the anion under these conditions is a consequence of the fact that the carbanionic center is both well stabilized and considerably hindered. Several experiments were conducted to determine the approximate acidity of compound 7. For example, the 31P NMR resonance observed for the anion at 32 ppm persisted even after addition of water or saturated NH4Cl to the NMR sample. Only after addition of acetic acid was a resonance representing the neutral compound 7 again observed at 14 ppm. Titration of the trisphosphonate ester 7 with NaOH gave a pKa of ~6.5, suggesting that the negative charge is highly delocalized and that this ester should be viewed as a strong carbon acid.
To explore alternate approaches to these compounds, tetraethyl benzylbisphosphonate 9 was prepared by alkylation of bisphosphonate 8.2c, 2d No evidence for C-P bond formation could be detected by 31P NMR upon treatment of compound 9 with NaH and diethyl chlorophosphate, but reactions that employed diethyl chlorophosphite as the electrophile6,8 were more encouraging. After treatment of bisphosphonate 9 with NaH and diethyl chlorophosphite, exposure to air under standard conditions afforded just trace amounts of the desired trisphosphonate. However, when reaction of the bisphosphonate 9 with NaHMDS and diethyl chlorophosphite at 0 °C was followed by treatment with H2O2, an exothermic reaction ensued. A new product was detected by TLC analysis, and analysis of the reaction mixture by 31P NMR revealed a new resonance at 18 ppm. After isolation of this product via column chromatography, the 1H NMR spectrum displayed a notable phosphorus coupling to the benzylic hydrogens (q, J = 15.8 Hz). The 13C NMR spectrum was even more striking, with observable couplings to 31P throughout the spectrum and a resonance for the quaternary carbon that appeared as a clear quartet (JCP = 118 Hz). Based on these data, as well as a consistent elemental analysis, the product was assigned the structure of trisphosphonate 10.
The three-step protocol of alkylation, phosphinylation, and oxidation proved to be successful with several other alkyl halides but the isolated yields initially were modest (<20%). Addition of excess base did not result in increased yields. Elemental analyses of alkylbisphosphonates have consistently revealed the presence of water, suggesting that hydroxide generated in situ might complicate formation of the desired intermediate in this case. Thorough drying of the alkylated bisphosphonates via azeotopic distillation with either benzene or toluene prior to deprotonation and phosphinylation resulted in a dramatic increase in reaction yields. Ultimately this strategy resulted in conversions ranging from 64 to 86% by 31P NMR, with isolated yields typically just slightly lower. This methodology was then applied to the synthesis of a variety of alkyl trisphosphonates with good isolated yields (Table 1). Furthermore, there is at least the potential to recover the alkylbisphosphonate in the cases of lower conversion, and that decision can be based on inspection of the 31P NMR spectrum of the reaction mixture (~18 ppm for trisphosphonate 10 versus ~23 ppm for the bisphosphonate 9).
Table 1.
Synthesis of α-alkyl-1,1,1- trisphosphonates
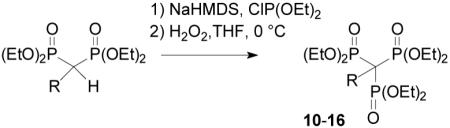
| ||
|---|---|---|
| R = | Conversion (by 31P NMR) (%) |
Isolated Yield (%) |
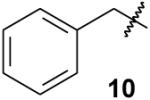
|
67% | 61% |
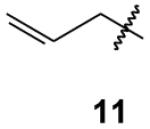
|
77% | 71% |
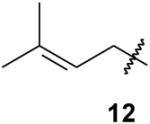
|
74% | 68% |
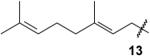
|
69% | 63% |
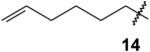
|
84% | 78% |
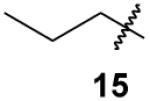
|
86% | 79% |
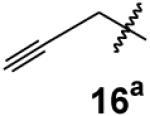
|
64% | 59% |
In this case, the bisphosphonate was prepared via conjugate addition of sodium acetylide to vinyl bisphosphonate.2a
Because so few alkyl-1,1,1-trisphosphonates are known, it was unclear whether it would be possible to carry out various functional group transformations in the presence of this group. Some reactions proved to be routine while others were not. For example, treatment of allyltrisphosphonate 11 with hydrogen over Pd/C resulted in selective reduction of the olefin to afford the saturated compound 15 in good yield (Scheme 2). However, some oxidative transformations proved to be more problematic. After treatment of the allyl trisphosphonate 11 with ozone under typical conditions11 some evidence for formation of the desired aldehyde 17 was obtained, including a resonance appropriate for the aldehyde hydrogen in the 1H NMR spectrum. However, this material was obtained in low yield and there was detectable dephosphorylation to a bisphosphonate. Attempts to avoid decomposition by treatment with a limited amount of ozone under Rubin conditions12 also resulted in decomposition of the allyltrisphosphonate. Similar observations were found upon attempted epoxidation. Treatment of allyl trisphosphonate 11 with m-CPBA resulted in the disappearance of the resonances for the olefinic hydrogens, but again only low amounts of material that appeared to be the epoxide 18 were obtained and decomposition to a bisphosphonate may be competitive.
Scheme 2.
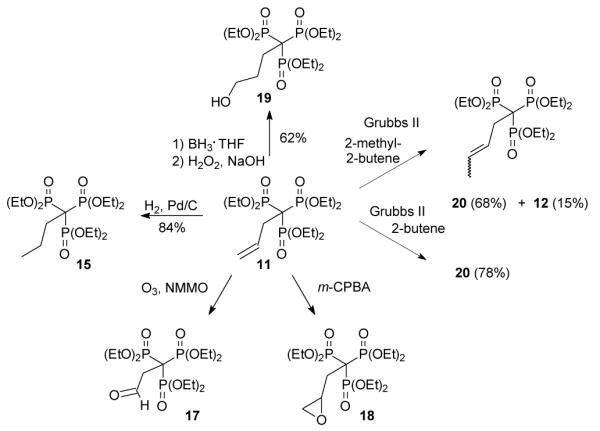
Reactions of trisphosphonate 11.
As one might expect, the trisphosphonate functionality is of considerable size, and several attempted reactions appeared to be difficult due to steric hindrance. For example, the allyl trisphosphonate 11 did not undergo hydroboration readily upon treatment with 9-BBN, but treatment with borane in THF resulted in conversion to the primary alcohol 19 in reasonable yield. The trisphosphonate group was not significantly impacted by this oxidative work-up with H2O2, as might be expected given that hydrogen peroxide was used during the trisphosphonate synthesis. Cross metathesis reactions of trisphosphonate 11 also may be affected by the size and proximity of the trisphosphonate moiety. Attempted cross metathesis with 2-methyl-2-butene and the Grubbs second generation catalyst gave the unexpected cis and trans 1,2-disubstituted olefins 20 as the major product, and only a small amount of the expected trisubstituted alkene 12.13 The identity of the olefins 20 was established unequivocally when the cross metathesis reaction of trisphosphonate 11 with 2-butene also gave a mixture of the same cis and trans olefins 20.
The importance of steric factors in the reactivity of compound 11 may be clarified by consideration of the reactivity of trisphosphonate 14, which can be viewed as a less sterically congested analogue where the number of methylene carbons between the double bond and the trisphosphonate group has been increased. In this case, attempted cross metathesis of compound 14 with 2-methyl-2-butene proceeded smoothly and gave the expected trisubstituted olefin 21 in high yield (87%, Scheme 3). In a similar sense, treatment of olefin 14 with 9-BBN followed by standard oxidative work-up gave the primary alcohol 22 in reasonable yield (57%).
Scheme 3.
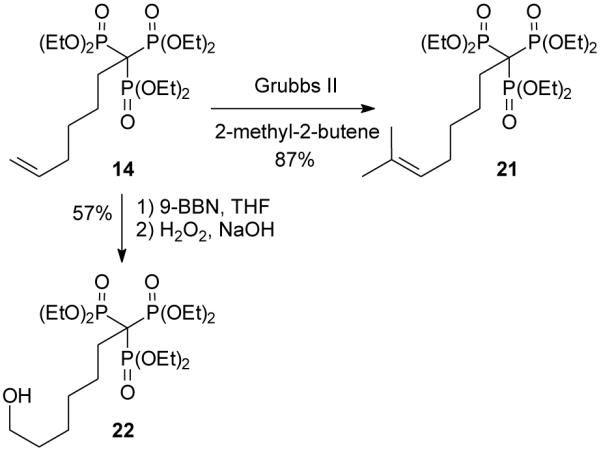
Reactions of trisphosphonate 14.
While metathesis reactions of the trisphosphonate 11 may be limited in their ability to afford a diverse array of new trisphosphonates, this is clearly a promising approach with more distal olefins such as compound 14. Another approach to facile preparation of compound libraries is based on the 1,3-dipolar cycloaddition of azides with acetylenes (or click chemistry).14 Somewhat to our surprise given the results with the metathesis reaction, the copper-catalyzed reaction of the acetylene trisphosphonate 16 with benzyl azide proceeded smoothly to give the triazole 23 in 85% yield (Scheme 4). This reaction clearly demonstrates that the trisphosphonate group will tolerate standard conditions for this cycloaddition, and strongly suggests that more distal acetylenes would react at least as well.
Scheme 4.
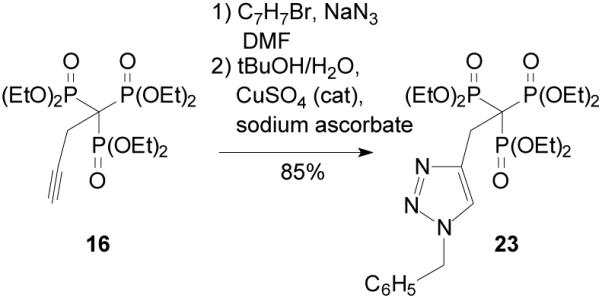
A click reaction with trisphosphonate 16.
Hydrolysis of these trisphosphonate esters could provide a variety of salts depending upon the extent of ester hydrolysis. Initial attempts to bring about complete hydrolysis of benzyl trisphosphonate 10 by treatment with HCl under reflux resulted in decomposition.15 Even though the corresponding benzylbisphosphonate 9 undergoes complete hydrolysis under parallel conditions,2d the more relevant comparison may be with the parent trisphosphonate 7 which also was reported to undergo decomposition when subjected to acid hydrolysis.6 Treatment of trisphosphonate 11 with TMSBr and collidine (Scheme 5)16 resulted in the formation of the TMS esters, as monitored by 31P NMR spectroscopy. Addition of 1N NaOH to a solution of the TMS ester led to complete hydrolysis and formation of the mixed sodium and collidinium salt17 (24) which could be isolated by standard work-up.
Scheme 5.
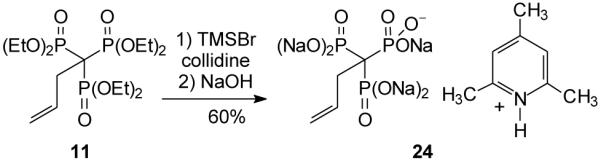
Hydrolysis of trisphosphonate 11.
In conclusion, these studies have led to a synthesis of hexaethyl methanetrisphosphonate more efficient than the original report,6 and determined that this compound should be viewed as a strong carbon acid. They also have established a general strategy for preparation of alkyl-1,1,1-trisphosphonates from the corresponding alkyl-1,1-bisphosphonates through phosphinylation and oxidation with hydrogen peroxide. As long as steric factors from the bulky trisphosphonate group are considered, alkyl-1,1,1-trisphosphonates can undergo a variety of functional group transformations although they are sensitive to some oxidative conditions. In particular, steric factors already have led to an interesting variation on the Grubbs metathesis where a disubstituted olefin was observed as the major product from metathesis with 2-methyl-2-butene rather than the expected trisubstituted alkene. Furthermore, the ability of an acetylene trisphosphonate to undergo click chemistry suggests that libraries of trisphosphonates should be readily available. Thus it appears likely that further studies of alkyl-1,1,1-trisphosphonates will unveil other new chemistry, and that screening of trisphosphonate libraries could be used to identify biologically active compounds of this general structure. Investigations along these lines are continuing, and will be reported in due course.
Experimental
General Procedures
Both THF and Et2O were distilled from sodium and benzophenone immediately prior to use. All non-aqueous reactions were performed with either oven-dried or flame-dried glassware under an argon atmosphere. Flash chromatography was performed on silica gel with an average particle size of 40-63 μm. The 1H NMR spectra were recorded at 300 MHz (75 MHz for 13C) with CDCl3 as solvent and (CH3)4Si as internal standard unless otherwise noted. The 1H NMR spectra recorded in D2O used residual H2O (4.80 ppm) as a reference, while 1,4-dioxane (67.0 ppm) was used as a reference for these 13C NMR spectra. Chemical shifts of 31P NMR spectra are reported in ppm relative to H3PO4 as an external standard. Elemental analyses were performed at a commercial facility. High resolution mass spectral analysis was performed with a quadrupole time of flight hybrid mass spectrometer with the capacity for positive and negative ionization modes. Electrospray ionization was employed with acetonitrile or aqueous (24) solutions.
Methylidynetrisphosphonic acid, hexaethyl ester (7)
Tetraethyl methylenebisphosphonate (251 mg, 0.87 mmol) was dissolved in benzene (5 mL) and then concentrated in vacuo to remove traces of water. After three such cycles, the residue was dissolved in THF (10 mL) and cooled to 0 °C in an ice bath. A solution of NaHMDS in THF (1.0 M, 1.3 mL, 1.3 mmol) was added, and the mixture was allowed to stir at 0 °C for 30 min after which ClP(OEt)2 (340 mg, 2.17 mmol) was added. After an additional 30 min, H2O2 (2.0 mL, 17.6 mmol) was very slowly added dropwise to the vessel. The reaction mixture was allowed to stir for one h, then diluted with brine, and extracted with CH2Cl2. The organic portions were combined, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification via flash chromatography (silica gel, 0 to 50% EtOH in EtOAc) gave the desired trisphosphonate 7 as a light yellow oil (176 mg, 48%). Both 31P and 1H NMR data were consistent with previously reported values.5b,6
2-Phenylethylidynetrisphosphonic acid, hexaethyl ester (10)
General procedure for the synthesis of alkyl trisphosphonates. A sample of tetraethyl benzylbisphosphonate (9)18, 19 (518 mg, 1.37 mmol) was dissolved in benzene (5 mL) and then concentrated in vacuo. After three such cycles, the residue was dissolved in THF (6.4 mL) and cooled to 0 °C in an ice bath. A solution of NaHMDS in THF (1.0 M, 2.1 mL, 2.1 mmol) was added, and the mixture was allowed to stir at 0 °C for 30 min, after which ClP(OEt)2 (437 mg, 2.74 mmol) was added. After an additional 30 min, excess H2O2 (2.0 mL, ~30% by titration) was slowly added dropwise (5 – 10 min) to the vessel. The reaction mixture was allowed to stir for one h, then diluted with brine, and extracted with CH2Cl2. The organic portions were combined, dried (MgSO4), and filtered, and the filtrate was concentrated in vacuo. Final purification via flash chromatography (silica gel, 0 to 30% EtOH in EtOAc) gave the desired trisphosphonate 10 as a clear oil (432 mg, 61%): 1H NMR δ 7.60–7.57 (m, 2H), 7.23–7.20 (m, 3H), 4.24–4.13 (m, 12H), 3.59 (q, JPH = 15.8 Hz, 2H), 1.27 (t, J = 8.9 Hz, 18H); 13C NMR (100 MHz) δ 136.3 (q, JPC = 5.9 Hz), 132.5 (2C), 126.9 (2C), 126.6, 63.4 (m, 6C), 52.8 (q, JPC = 117.8 Hz), 36.1 (q, JPC = 5.1 Hz), 16.3 (m, 6C); 31P NMR (121 MHz, CDCl3) +17.7 ppm; HRMS calcd for C20H37O9NaP3 (M+Na)+ 537.1548, found 537.1553. Anal. Calcd for C20H37O9P3·H2O: C, 45.12; H, 7.38. Found: C, 45.26; H, 7.57.
3-Butenylidynetrisphosphonic acid, hexaethyl ester (11)
According to the general procedure for synthesis of alkylated trisphosphonates, allylbisphosphonate20 (985 mg, 3.0 mmol) was treated with NaHMDS (4.5 mL, 4.5 mmol) and ClP(OEt)2 (1.29 g, 8.2 mmol), and then after 30 min H2O2 (3.50 mL, 31 mmol) was added to the reaction mixture. After standard workup the product was purified via column chromatography on silica gel (0 to 40% EtOH in EtOAc) and compound 11 was isolated as a clear oil (987 mg, 71%): 1H NMR δ 6.32–6.18 (m, 1H), 5.15–5.05 (m, 2H), 4.34–4.14 (m, 12H), 2.92 (qd, JPH = 9.0 Hz, J = 6.6 Hz, 2H), 1.34 (t, J = 7.2 Hz, 18H); 13C NMR δ 134.4 (q, JPC = 6.3 Hz), 117.1, 63.4 (m, 6C), 50.5 (q, JPC = 119.8 Hz), 35.0 (q, JPC = 5.5 Hz), 16.5 (m, 6C); 31P NMR +18.0 ppm; HRMS calcd for C16H35O9NaP3 (M+Na)+, 487.1392, found 487.1407. Anal. Calcd for C16H35O9P3·H2O: C, 39.84; H, 7.73. Found: C, 40.19; H, 7.83.
4-Methyl-3-pentenylidynetrisphosphonic acid, hexaethyl ester (12)
According to the general procedure for synthesis of alkylated trisphosphonates, prenylbisphosphonate2e (494 mg, 1.39 mmol) was treated with NaHMDS (2.10 mL, 2.1 mmol) and ClP(OEt)2 (472 mg, 2.77 mmol). After 30 min H2O2 (2.00 mL, 17.6 mmol) was added to the reaction mixture. The product 12 was purified by column chromatography on silica gel (0 to 30% EtOH in EtOAc) and was isolated as a faint yellow oil (461 mg, 68%): 1H NMR δ 5.67 (t, J = 6.6 Hz, 1H), 4.29–4.17 (m, 12H), 2.86 (qd, JPH = 15.9 Hz, J = 6.6 Hz, 2H), 1.72 (s, 3H), 1.64 (s, 3H), 1.36–1.30 (m, 18H); 13C NMR δ 132.5, 120.2 (q, JPC = 6.1 Hz), 63.4–63.3 (m, 6C), 50.4 (q, JPC = 117.6 Hz), 29.5, (q, JPC = 5.4 Hz), 26.0, 17.9, 16.5–16.3 (m, 6C); 31P NMR (121 MHz, CDCl3) +18.6 ppm; HRMS calcd for C18H40O9P3 (M+H)+, 493.1885, found 493.1883.
(3E)-4,9-Dimethyl-3-nonadienylidynetrisphosphonic acid, hexaethyl ester (13)
According to the general procedure for synthesis of alkylated trisphosphonates, geranylbisphosphonate3e (249 mg, 0.6 mmol) was treated with NaHMDS (1.00 mL, 1.0 mmol) and ClP(OEt)2 (125 mg, 0.8mmol) and then after 30 min H2O2 (1.00 mL, 8.8 mmol) was added to the reaction mixture. After standard workup, the product 13 was purified by column chromatography on silica gel (0 to 30% EtOH in EtOAc) and was isolated as a faintly yellow oil (208 mg, 63%): 1H NMR δ 5.72 (t, J = 6.3 Hz, 1H), 5.13 (t, J = 6.2 Hz, 1H), 4.25 (q, J = 7.1 Hz, 12H), 2.87 (qd, JPH = 16.2, J = 7.1 Hz, 2H), 2.09–2.03 (m, 4H), 1.68 (s, 3H), 1.63 (s, 3H), 1.60 (s, 3H), 1.33 (t, J = 6.6 Hz, 18H);13C NMR δ 135.8, 131.1, 124.1, 119.8 (q, JPC = 6.2 Hz), 63.1 (m, 6C), 50.2 (q, JPC = 119.6 Hz), 39.9, 29.2 (q, JPC = 5.6 Hz), 26.5, 25.5, 17.4, 16.3–16.2 (m, 6C), 16.1; 31P NMR +18.6 ppm; HRMS calcd for C23H48O9P3 (M+H)+, 561.2511, found 561.2529. Anal. Calcd for C23H47O9P3·H2O: C, 47.75; H, 8.54. Found: C, 47.98; H, 8.44.
Tetraethyl 6-hepten-1,1-bisphosphonate
Tetraethyl methylenebisphosphonate (5.31 g, 18.4 mmol) was added dropwise to a stirring suspension of NaH (810 mg, 20.2 mmol) in THF (10 mL). After 30 min, 6-bromo-1-hexene (3.00 g, 18.4 mmol) was added and the mixture was heated at reflux overnight. After the reaction mixture had cooled to room temperature, saturated NH4Cl was added and the organic and aqueous portions were separated. The aqueous portion was extracted with Et2O and the organic layers were combined, dried (MgSO4), and concentrated in vacuo. The resulting oil was purified via flash chromatography (silica gel, 10% EtOH in hexanes) and the desired bisphosphonate was isolated in 46% yield (3.10 g): 1H NMR δ 5.84–5.73 (m, 1H), 5.03–4.92 (m, 2H), 4.23–4.12 (m, 8H), 2.27 (tt, JPH = 24.3 Hz, J = 6.3 Hz, 1H), 2.01–1.83 (m, 4H) 1.64–1.54 (m, 2H), 1.45–1.32 (m, 14H); 13C NMR δ 138.6, 114.3, 62.4–62.1 (m, 4C) 36.6 (t, JPC = 132.5 Hz), 28.5 (2C), 25.3, 16.3–16.2 (m, 4C); 31P NMR +23.9 ppm; HRMS calcd for C15H33O6P2 (M+H)+, 371.1752, found 371.1745.
6-Heptenylidynetrisphosphonic acid, hexaethyl ester (14)
According to the general procedure for synthesis of alkylated trisphosphonates, tetraethyl 6-hepten-1,1-bisphosphonate (508 mg, 1.37 mmol) was treated with NaHMDS (2.06 mL, 2.06 mmol) and ClP(OEt)2 (438 mg, 2.75 mmol), and then H2O2 (2.00 mL, 17.6 mmol) was added. After standard workup the product 14 was purified by column chromatography on silica gel (0 to 30% EtOH in EtOAc) and was isolated as a faintly yellow oil (435 mg, 78%): 1H NMR (CDCl3) δ 5.89–5.75 (m, 1H), 5.04–4.92 (m, 2H), 4.29–4.19 (m, 12H), 2.11–2.00 (m, 4H; 2 exchange with D2O) 1.91–1.80, (m, 2H) 1.43–1.13 (m, 22H); 13C NMR (CDCl3) δ 138.8, 114.2, 63.4–63.1 (m, 6C), 50.6 (q, JPC = 119.3 Hz), 33.4, 30.8 (q, JPC = 5.3 Hz), 29.3, 25.2 (q, JPC = 5.0 Hz), 16.5–16.2 (m, 6C); 31P NMR (121 MHz, CDCl3) +18.7 ppm; HRMS calcd for C19H41O9NaP3 (M+Na)+, 529.1861, found 529.1867.
Butylidynetrisphosphonic acid, hexaethyl ester (15)
According to the general procedure for synthesis of alkylated trisphosphonates, propylbisphosphonate19, 21 (292 mg, 0.9 mmol) was treated with NaHMDS (1.30 mL, 1.3 mmol) and ClP(OEt)2 (346 mg, 2.2 mmol). After 30 min H2O2 (2.00 mL, 17.6 mmol) was added to the reaction mixture. Standard workup and purification via column chromatography on silica gel (0 to 35% EtOH in EtOAc) gave compound 15 as a clear oil (324 mg, 79%). Both the 31P and 1H NMR spectra are consistent with material prepared via hydrogenation of compound 11 (vide infra).
3-Butyn-1-ylidynetrisphosphonic acid, hexaethyl ester (16)
According to the general procedure for synthesis of alkylated trisphosphonates, propargylbisphosphonate22 (291 mg, 0.9 mmol) was treated with NaHMDS (0.9 mL, 0.9 mmol) and ClP(OEt)2 (280 mg, 1.8 mmol) and then after 30 min H2O2 (2.00 mL, 17.6 mmol) was added to the reaction mixture. After standard workup the product was purified via column chromatography on silica gel (0 to 30% EtOH in EtOAc) and compound 16 was isolated as a clear oil (243 mg, 59%): 1H NMR δ 4.33–4.21 (m, 12H), 3.03 (qd, JPH = 14.7 Hz, J = 3.3 Hz 2H), 2.09–2.07 (m, 1H), 1.34–1.32 (m, 18H); 13C NMR δ 79.9 (q, JPC = 9.1 Hz), 70.5, 63.9–63.6 (m, 6C), 49.7 (q, JPC = 120.0 Hz), 21.0 (q, JPC = 5.6 Hz), 16.4–16.3 (m, 6C); 31P NMR +16.7 ppm; HRMS calcd for C16H34O9P3 (M+H)+, 463.1416, found 463.1420.
Trisphosphonate 15 via catalytic hydrogenation of compound 11
Trisphosphonate 11 (96 mg, 0.2 mmol) in EtOH (5 mL) was treated with Pd/C (23 mg, 0.2 mmol) under an H2 atmosphere. After 12 h the reaction mixture was filtered through celite, and the filtrate was collected and concentrated in vacuo. The resulting oil was purified using flash chromatography (silica gel, 0 to 25% EtOH in EtOAc) to obtain compound 15 as a clear oil (81 mg, 84%): 1H NMR δ 4.30–4.18 (m, 12H), 2.18–1.77 (m, 4H), 1.34 (t, J = 6.6 Hz, 18H), 0.91 (t, J = 6.9 Hz, 3H); 13C NMR δ 63.4 (m, 6C), 51.6 (q, JPC = 119.8 Hz), 33.0 (q, JPC = 5.5 Hz), 19.2 (q, JPC = 5.2 Hz) 16.4 (m, 6C), 15.0; 31P NMR +18.8 ppm; HRMS calcd for C16H37O9NaP3 (M+Na)+, 489.1548, found 489.1564. Anal. Calcd for C16H37O9P3·H2O: C, 39.67; H, 8.11. Found: C, 39.66; H, 8.02.
4-Hydroxybutylidynetrisphosphonic acid, hexaethyl ester (19)
Trisphosphonate 11 (102 mg, 0.22 mmol) was dried under vacuum in the presence of P2O5 overnight. The remaining oil was dissolved in THF (5 mL) and placed into an ice bath. To the reaction flask, BH3·THF (1M in THF, 0.45 mL, 0.45 mmol) was added and the mixture was allowed to stir. After 1.5 h, MeOH (2 mL) was added to the flask, followed by NaOH (3M, 0.5 mL, 1.5 mmol) and then H2O2 (0.3 mL, 2.7 mmol), and the resulting mixture was heated at 50 °C for 1 h. The reaction mixture was washed with saturated NaCl and the aqueous portions were retained and extracted with CH2Cl2. The organic portions were combined, dried (MgSO4), and concentrated in vacuo. The resulting oil was purified via flash chromatography (silica gel, 0 to 45% EtOH in EtOAc) to obtain compound 19 as a clear oil (66 mg, 62%): 1H NMR δ 4.33–4.20 (m, 12H), 3.64 (t, J = 5.7 Hz, 2H), 2.30–2.05 (m, 7H; 2 exchange with D2O) 1.35 (t, J = 6.6 Hz, 18H); 13C NMR δ 63.5–63.4 (m, 6C), 63.0, 51.6 (q, JPC = 119.8 Hz), 33.0 (q, JPC = 5.5 Hz), 19.2 (q, JPC = 5.2 Hz) 16.4 (m, 6C); 31P NMR +18.8 ppm; HRMS calcd for C16H37O10NaP3 (M+Na)+, 505.1497, found 505.1503.
3-Pentenylidynetrisphosphonic acid, hexaethyl ester (20) and compound 12
Grubbs second generation catalyst (4.9 mg, 3 mol %) was dissolved in 2-methyl-2-butene (1 mL) and placed in a 1-dram vile. The trisphosphonate 11 (88.3 mg, 0.2 mmol) was added to this mixture, along with an additional 1 mL of 2-methyl 2-butene. The vile was sealed and the reaction was allowed to stir at 40 °C overnight. After the solvent was removed in vacuo, the resulting oil was purified via flash chromatography (silica gel, 0 to 30% EtOH in EtOAc). The reaction products (76 mg, 83% total) were isolated as an inseparable mixture of cis and trans isomers of compound 20 (68%, 1.2:4.3 isomer ratio) and prenyl trisphosphonate 12 (15%, 1:5.5 ratio with respect to olefins 20). The 31P, 1H and 13C NMR spectra were consistent with a mixture of compounds 20 and 12, both of which had been prepared independently.
Compound 20 via metathesis with 2-butene
Grubbs second generation catalyst (2.3 mg, 6 mol %) was dissolved in CH2Cl2 (0.5 mL) and placed in a 1-dram vile, and trisphosphonate 11 (24 mg, 0.1 mmol) was added to this mixture. After 2-butene was added to the vessel via balloon, the vessel was sealed and the reaction was allowed to stir at 40 °C overnight. The volatile materials were removed in vacuo and the resulting oil was purified via flash chromatography (silica gel, 0 to 30% EtOH in EtOAc). The olefins 20 were isolated as a mixture of trans and cis isomers (19 mg, 78%) in a 2.8:1 ratio. For the trans isomer: 1H NMR (500 MHz, CDCl3) δ 5.85 (dt, J = 14.0, 7.0 Hz, 1H), 5.57–5.50 (m, 1H), 4.29–4.18 (m, 12H), 2.89–2.84 (m, 2H), 1.67 (dd, J = 7.0, 1.5 Hz, 3H), 1.35–1.32 (m, 18H); 13C NMR (125 MHz, CDCl3) δ 128.0, 126.7 (q, JPC = 6.3 Hz), 63.4–63.3 (6C), 50.7 (q, JPC = 119.5 Hz), 33.9 (q, JPC = 5.3 Hz), 17.9, 16.5–16.3 (6C); 31P NMR (121 MHz, CDCl3), +18.4 ppm. For the cis isomer: 1H NMR (500 MHz, CDCl3), δ 5.94–5.92 (m, 1H), 5.57–5.50 (m, 1H), 4.29–4.18 (m, 12H), 2.89–2.84 (m, 2H), 1.64 (dd, J = 7.0, 1.0 Hz, 3H), 1.35–1.32 (m, 18H); 13C NMR (125 MHz, CDCl3) 126.1 (q, JPC = 6.0 Hz), 125.0, 63.5–63.4 (6C), 50.1 (q, JPC = 119.6 Hz), 28.3 (q, JPC = 8.0 Hz), 16.5–16.3 (6C), 12.9; 31P NMR (121 MHz, CDCl3) +18.5 ppm; HRMS calcd for C17H37O9NaP3 (M+Na)+, 501.1548, found 501.1554. Anal. Calcd for C17H37O9P3·H2O: C, 41.13; H, 7.92. Found: C, 41.35; H, 7.91.
7-Methyl-6-octenylidynetrisphosphonic acid, hexaethyl ester (21)
Grubbs second generation catalyst (3.1 mg, 3 mol %) was dissolved in 2-methyl-2-butene, placed in a 1-dram vile, and trisphosphonate 14 (62 mg, 0.12 mmol) was added along with an additional 1 mL of 2-methyl-2-butene. The vile was sealed and the reaction was allowed to stir at 40 °C overnight. After concentration in vacuo, the resulting oil was purified via flash chromatography (silica gel, 0 to 30% EtOH in EtOAc), and the desired product 21 was isolated as an oil (57 mg, 87%): 1H NMR δ 5.12 (t, J = 6.0 Hz, 1H), 4.19–4.31 (m, 12H), 1.96–2.24 (m, 6H), 1.77–1.88 (m, 2H), 1.68 (3H), 1.57 (3H), 1.34 (t, J = 6.6 Hz, 18H); 13C NMR δ 131.2, 124.7, 63.5–63.2 (m, 6C), 50.7 (q, JPC = 119.4 Hz), 31.0–30.9 (m), 30.9, 27.8, 25.7, 25.4 (q, JPC = 5.2 Hz) 17.6, 16.4–16.2 (m, 6C); 31P NMR +18.8 ppm; HRMS calcd for C21H46O9P3 (M+H)+, 535.2355, found 535.2357.
7-Hydroxyheptylidynetrisphosphonic acid, hexaethyl ester (22)
Trisphosphonate 14 (109 mg, 0.2 mmol) was dried overnight under vacuum in the presence of P2O5. The remaining oil was dissolved in THF (5 mL) and placed into an ice bath. To the reaction flask, 9-BBN (0.5 M in THF, 1.0 mL, 0.5 mmol) was added and the mixture was allowed to stir. After 1.5 h, MeOH (2 mL) was added to the flask, followed by NaOH (3 M, 0.5 mL, 1.5 mmol) and then H2O2 (0.5 mL, 4.4 mmol), and the resulting mixture was heated at 50 °C for 1 h. The reaction mixture was washed with saturated NaCl and the aqueous portions were retained and extracted with CH2Cl2. The organic portions were combined, dried (MgSO4), and concentrated in vacuo. The resulting oil was purified via flash chromatography (silica gel, 0 to 40% EtOH in EtOAc) to obtain compound 22 as a clear oil (64 mg, 57%): 1H NMR δ 4.30–4.17 (m, 12H), 3.63 (t, J = 6.3 Hz, 2H), 2.18–2.03 (m, 3H), 1.90–1.82 (m, 2H), 1.60–1.53 (m, 2H), 1.44–1.28 (m, 22); 13C NMR δ 63.5–63.2 (m, 6C), 62.8, 50.6 (q, JPC = 119.5 Hz), 32.7, 30.8 (q, JPC = 5.3 Hz), 30.3, 25.5 (q, JPC= 5.3 Hz), 25.3, 16.5–16.2 (6C); 31P NMR +18.8 ppm; HRMS calcd for C19H43O10NaP3 (M+Na)+, 547.1967, found 547.1991.
1-Benzyl-4-[2,2,2-tris(diethyoxyphosphinyl)ethyl-1H-1,2,3-triazole (23)
Benzyl bromide (182 mg, 1.1 mmol) was added to a suspension of sodium azide (83 mg, 1.3 mmol) in DMF (5 mL) and the resulting mixture was allowed to stir. After 10 min, trisphosphonate 16 (164 mg, 0.4 mmol) was added along with 0.1 mL CuSO4 (5M), sodium ascorbate (43 mg, 0.2 mmol), and a solution of tBuOH in water (1:4 ratio, 5 mL), and the reaction mixture was allowed to stir at room temperature. After 24 h EDTA and 1M NH4OH were added, the resulting solution was placed in a continuous liquid-liquid extractor and extracted for 4 h with EtOAc. The organic portion was retained and concentrated in vacuo. The resulting oil was purified via flash chromatography (silica gel, 0 to 50% EtOH in EtOAc) to provide the desired triazole 23 (179 mg, 85%): 1H NMR δ 7.95 (s, 1H), 7.35–7.31 (m, 5H), 5.47 (s, 2H), 4.22–4.07 (m, 12H), 3.66 (q, JPH = 15.9 Hz, 2H), 1.26–1.21(m, 18H); 13C NMR δ 143.3 (q, JPC = 7.4 Hz), 135.2, 128.9 (2C), 128.4, 128.0 (2C), 124.7, 63.7–63.4 (m, 6C), 53.9, 50.6 (q, JPC = 119.2 Hz), 27.9, (q, JPC = 5.5 Hz), 16.4–16.1 (m, 6C); 31P NMR +17.6 ppm; HRMS calcd for C23H40N3O9NaP3 (M+Na)+, 618.1875, found 618.1893.
3-Butenylidynetrisphosphonic acid, pentasodium, 2,4,6-trimethylpyridinium salt (24)
A solution of 2,4,6-collidine (524 mg, 4.3 mmol) and TMSBr (568 mg, 4.3 mmol) was allowed to stir in an ice bath. After 20 min, trisphosphonate 11 (75mg, 0.2 mmol) was added and the reaction was allowed to stir for 24 h with periodic monitoring by 31P NMR spectroscopy. Once the reaction was complete, it was diluted by addition of toluene, the solvent was removed in vacuo, and aqueous sodium hydroxide (1.5 mmol, 9 eq) was added. The mixture was allowed to stir overnight and again was monitored by 31P NMR. The reaction mixture then was lyophilized, the resulting solid was dissolved in a minimum amount of water, then slowly poured into cold acetone and kept at 40 °C overnight. The resulting precipitate was filtered and washed with cold acetone. The remaining residue was dissolved in water and lyophilized to afford compound 24 as a flocculent white residue (51 mg, 60%): 1H NMR (D2O) δ 7.43 (s, 2H), 6.19–6.10 (m, 1H), 5.23–5.05 (m, 2H), 2.92– 2.85 (m, 2H), 2.93 (s, 6H), 2.50 (s, 3H); 13C NMR (D2O) δ 164.1 (2C), 155.8, 139.1–139.0 (m), 129.2, 121.6 (2C), 52.3 (q, JPC = 103.6 Hz), 38.6. 25.3 (2C), 22.5; 31P NMR (121 MHz, D2O) +17.5 ppm; HRMS calcd for C4H10O9P3 (M–H)−, 294.9538, found 294.9542.
Supplementary Material
Figure 1.
Bis- and Trisphosphonates
Acknowledgements
Financial support from the NIH Predoctoral Training Program in the Pharmacological Sciences (2 T32 GM067795), the U.S. Department of Education (GAANN award P200A070526), and the Roy J. Carver Charitable Trust as a Research Program of Excellence is gratefully acknowledged.
Footnotes
Supporting Information. The 1H and 13C NMR spectra for compounds 10–16, and 19–24, and the titration curve for compound 7. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1).For an excellent review of the chemistry and biological activity of bisphosphonates, see: Ebetino FH, Hogan A-M, Sun S, Tsoumpra M, Duan A, Triffitt JT, Kwaasi AA, Dunford JE, Barnettt BL, Oppermann U, Lundy MW, Boyde A, Kashemirov BA, McKenna CE, Russell RGG. Bone. 2011;49:20–33. doi: 10.1016/j.bone.2011.03.774.
- 2).For representative syntheses of bisphosphonates via addition to alkylidene bisphosphonates, see: Sturtz G, Guervenou J. Synthesis. 1991;661 Hutchinson DW, Thornton DM. J. Organomet. Chem. 1988;346:341. For synthesis via alkylation of methylenebisphosphonates, see: Kosolapoff GM. J. Am. Chem. Soc. 1953;75:1500–1501. Quimby OT, Curry JD, Nicholson DA, Prentice JB, Roy CHJ. Organomet. Chem. 1968;13:199–207. Shull LW, Wiemer AJ, Hohl RJ, Wiemer DF. Bioorg. Med. Chem. 2006;14:4130–4136. doi: 10.1016/j.bmc.2006.02.010. Barney RJ, Wasko BM, Dudakovic A, Hohl RJ, Wiemer DF. Bioorg. Med. Chem. 2010;18:7212–7220. doi: 10.1016/j.bmc.2010.08.036. For a synthesis via two C-P bond formations, and lead references to other strategies, see: Du Y, Jung KY, Wiemer DF. Tetrahedron Lett. 2002;43:8665–8668.
- 3).For representative reports of biologically active bisphosphonates, see: Kashemirov BA, Bala JL, Chen X, Ebetino FH, Xia Z, Russell RGG, Coxon FP, Roelofs AJ, Rogers MJ, McKenna CE. Bioconjugate Chem. 2008;19:2308–10. doi: 10.1021/bc800369c. Singh AP, Zhang Y, No JH, Docampo R, Nussenzweig V, Oldfield E. Antimicrobial Agents and Chemotherapy. 2010;54:2987–2993. doi: 10.1128/AAC.00198-10. Holstein SA, Cermak DM, Wiemer DF, Lewis K, Hohl RJ. Bioorg Med. Chem. 1998;6:687–694. doi: 10.1016/s0968-0896(98)00034-0. Wiemer AJ, Yu JS, Lamb KM, Hohl RJ, Wiemer DF. Bioorg. Med. Chem. 2008;16:390–399. doi: 10.1016/j.bmc.2007.09.029. and references cited therein.
- 4).Battacharya AK, Thyagarajan G. Chem. Rev. 1981;81:415–431. [Google Scholar]
- 5).a) Kukhar VP, Pasternak VI, Kirsanov AV. Zh. Obshch. Khim. 1972;42:1169. [Google Scholar]; b) Gross H, Costisella B. J. Prakt. Chem. 1972;314:87–92. [Google Scholar]; c) Gross H, Costisella B, Brennnick L, et al. J. Prakt. Chem. 1972;314:969–974. [Google Scholar]
- 6).Gross H, Costisella B, Keitel I, Ozegowski S. Phosphorus, Sulfur, and Silicon. 1993;83:203–207. [Google Scholar]
- 7).Gross H, Ozegowski S, Costisella B. Phosphorus, Sulfur, and Silicon. 1990;47:7–13. [Google Scholar]
- 8).a) Lee K, Wiemer DF. J. Org. Chem. 1991;56:5556–5560. [Google Scholar]; b) Boeckman RK, Jr., Kamenecka TM, Nelson SG, Pruit JR, Barta TE. Tetrahedron Lett. 1991;32:2581–2584. [Google Scholar]
- 9).a) Liu X, Zhang X, Blackburn GM. Chem. Commun. 1997:87–88. [Google Scholar]; b) Liu X, Adams H, Blackburn GM. Chem. Commun. 1998:2619–2620. [Google Scholar]
- 10).a) Calogeropoulou T, Hammond GB, Wiemer DF. J. Org. Chem. 1987;52:4185–4190. [Google Scholar]; b) Du Y, Wiemer DF. J. Org. Chem. 2002;67:5701–5708. doi: 10.1021/jo0202233. [DOI] [PubMed] [Google Scholar]; c) Chen X, Wiemer DF. J. Org. Chem. 2003;68:6597–6604. doi: 10.1021/jo0300136. and references cited therein. [DOI] [PubMed] [Google Scholar]; d) Barney RJ, Richardson RM, Wiemer DF. J. Org. Chem. 2011;76:2875–2879. doi: 10.1021/jo200137k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).a) Schwartz C, Raible J, Mott K, Dussault PH. Tetrahedron. 2006;62:10747–10752. [Google Scholar]; b) Schwartz C, Raible J, Mott K, Dussault PH. Org. Lett. 2006;8:3199–3201. doi: 10.1021/ol061001k. [DOI] [PubMed] [Google Scholar]
- 12).Rubin MB. J. Chem. Educ. 1964;41:388. [Google Scholar]
- 13).a) Chatterjee AK, Sanders DP, Grubbs RH. Org. Lett. 2002;4:1939–1942. doi: 10.1021/ol0259793. [DOI] [PubMed] [Google Scholar]; b) Chatterjee AK, Choi T, Grubbs RH. Synlett. 2001:1034–1037. [Google Scholar]; c) Gibson SE, Haycock PR, Miyazaki A. Tetrahedron. 2009;65:7498–7503. [Google Scholar]; d) Rambabu C, Tan MMK, Hanson PR. J. Org. Chem. 2011;76:3909–3916. doi: 10.1021/jo200337v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Kolb HC, Finn MG, Sharpless KB. Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 15).Hawkins MJ, Powell ET, Leo GC, Gauthier DA, Greco MN, Maryanoff B. Org. Lett. 2006;8:3429–3431. doi: 10.1021/ol060519l. [DOI] [PubMed] [Google Scholar]
- 16).McKenna CE, Higa MT, Cheung NH, McKenna MC. Tetrahedron Lett. 1977;18:155–158. [Google Scholar]
- 17).Ramirez F, Marecek JF, Ugi I. J. Am. Chem. Soc. 1975;97:3809–3817. [Google Scholar]
- 18).Nguyen LM, Niesor E, Bentzen CL. J. Med. Chem. 1987;30:1426–1433. doi: 10.1021/jm00391a027. [DOI] [PubMed] [Google Scholar]
- 19).Lolli ML, Lazzarato L, Di Stilo A, Fruttero R, Gasco A. J. Organomet. Chem. 2002;650:77–83. [Google Scholar]
- 20).Dufau C, Sturtz G. Phosphorus, Sulfur, and Silicon. 1992;69:93–102. [Google Scholar]
- 21).Teulade M, Savignac P, Aboujaoude EE, Liétge S, Collignon N. J. Organomet. Chem. 1986;304:283–300. [Google Scholar]
- 22).Skarpos H, Osipov SN, Vorob’eva DV, Odinets IL, Lork E, Roschenthaler G. Org. Biomol. Chem. 2007;5:2361–2367. doi: 10.1039/b705510b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.