

Abstract
Solid phase multiplex-bead arrays for the detection and characterization of HLA antibodies provide increased sensitivity and specificity compared to conventional lymphocyte-based assays. Assay variability due to inconsistencies in commercial kits and differences in standard operating procedures hamper comparison of results between laboratories. The Clinical Trials in Organ Transplantation Antibody Core Laboratories investigated sources of assay variation and determined if reproducibility improved through utilization of standard operating procedures, common reagents and normalization algorithms. Ten commercial kits from two manufacturers were assessed in each of seven laboratories using 20 HLA reference sera. Implementation of a standardized (versus a non-standardized) operating procedure greatly reduced MFI variation from 62% to 25%. Although laboratory agreements exceeded 90% (R2), small systematic differences were observed suggesting center specific factors still contribute to variation. MFI varied according to manufacturer, kit, bead type and lot. ROC analyses showed excellent consistency in antibody assignments between manufacturers (AUC>0.9) and suggested optimal cutoffs from 1000–1500 MFI. Global normalization further reduced MFI variation to levels near 20%. Standardization and normalization of solid phase HLA antibody tests will enable comparison of data across laboratories for clinical trials and diagnostic testing.
Keywords: HLA antibodies, diagnostic markers, arrays, desensitization, immune monitoring, humoral immunity, immunological markers, transplantation immunology, quality assurance, quantitative, multicenter studies, post transplant monitoring, donor-specific antibodies
Introduction
A major task of the Immunogenetics laboratory is to measure patient’s humoral immune responses to HLA class I and class II antigens to guide donor selection and kidney paired exchange programs through virtual crossmatching (1–3), for risk assessment for acute and chronic antibody-mediated rejection (4–13) and for guiding desensitization therapy (14–19). Therefore, the precise and standardized assessment of antibodies (Ab) to HLA is crucial for successful transplant patient management and as a potential clinical indicator to guide immunotherapy.
Solid phase multiplex-bead arrays for the identification of HLA Ab utilize purified HLA class I and class II molecules that are coupled to color-coded polystyrene beads. The beads are labeled with different ratios of two fluorescent dyes yielding discrete bead populations that bear distinct HLA antigens. A human anti-IgG reagent labeled with a third fluorescent dye is used to assess the binding of Ab. The bead arrays provide a semi-quantitative measurement of the HLA Ab bound to each bead, expressed as the mean fluorescence intensity (MFI). Three types of kits have been developed to detect HLA class I (A, B, C) and class II (DR, DQ, DP) Ab: a) mixed antigen screen beads wherein a single bead carries a mixture of purified class I and class II molecules from three or more donors; b) phenotypic or PRA beads consisting of 30 or more beads where each bead carries an HLA class I or class II phenotype purified from a single donor; and c) single antigen beads (SA) where each bead carries a single recombinant HLA class I or class II antigen/allele.
The multiplex-bead arrays offer several advantages over conventional cell-based assays. They are more sensitive, enabling the detection of low levels of HLA Ab (20) and permit the accurate identification of HLA Ab in broadly reactive alloantisera (21, 22). However, there are limitations that impede the widespread use of these solid phase assays in clinical trials, and assay variability and interference from intrinsic factors present in the patient’s serum have been noted (23–25). Notable sources of deviation are run-to-run variability caused by differences in the test conditions and operator performance. Additional assay variance can stem from differences in kit design and manufacturing practices causing variability in both the density and quality of HLA molecules coupled to the beads (22, 26).
For cooperative studies, these technical issues hinder the generation of accurate data within and across clinical trial sites and limit the ability to compare data sets across different studies. The multiplex-bead arrays for HLA Ab identification described above are currently being utilized by core laboratories of the Clinical Trials in Organ Transplantation (CTOT), a collaborative clinical research project headquartered at the National Institute of Allergy and Infectious Diseases. Since assay reproducibility is a prerequisite for correct interpretation of the clinical trial data sets, the CTOT core laboratories sought to determine if variability in the multiplex-bead arrays for measuring HLA Ab could be reduced through implementation of a standardized protocol for assay performance.
Materials and Methods
Luminex Reagents
Multiplex-bead array Luminex kits were purchased from two manufacturers: One Lambda, Inc. and Gen-Probe Inc. (Table 1).
Table 1.
Kits and reagents
| Lot 1 | Lot2 | ||||
|---|---|---|---|---|---|
| Catlog No. | Product Description | Kit type | Lot No. | Lot No. | |
| One Lambda, Inc. | LSM12 | LABScreen Mixed Class I and Class II | Scr | 017 | 016 |
| LS1PRA | LABScreen PRA Class I | PRA1 | 014 | 013 | |
| LS2PRA | LABScreen PRA Class II | PRA2 | 013 | 012 | |
| LS1A04 | LABScreen Single Antigen HLA Class I | SA1 | 006 | 007 | |
| LS2A01 | LABScreen Single Antigen HLA Class II | SA2 | 008 | 009 | |
| LS-NC | LABScreen negative control | 010 | 011 | ||
| LS-AB2 | Goat anti-human IgG | A76 | A76 | ||
| Gen-Probe, Inc | 628215 | Lifecodes LifeScreen Deluxe | Scr | 100510-LMX | 052311-LMX |
| 628200 | Lifecodes Class I ID | PRA1 | 102710-LM1 | 041311-LM1 | |
| 628223 | Lifecodes Class II IDv2 | PRA2 | 090210-LM2Q | 042211-LM2Q | |
| 265100 | Lifecodes LSA Class I | SA1 | 10050M | 04101P | |
| 265200 | Lifecode LSA Class II | SA2 | 10190D | 05311A | |
| Luminex | L100-CAL1 | Calibrator Microspheres I | B21094 | ||
| L100-CAL2 | Calibrator Microspheres II | B20676 | |||
| L100-CON1 | Control Microspheres I | B20770 | |||
| L100-CON2 | Control Microspheres II | B20501 | |||
Serum samples
Twenty sera were selected from an inventory of reference material from UCLA and Emory University. The set covered HLA class I and/or class II specificities of all cross-reactive groups for HLA-A, B, DR, DQ and DP loci (Table 2). One serum had no HLA Ab and two sera contained Abs to a limited number of HLA-C specificities. All 20 sera were tested with mixed antigen screening kits (Cat. LSM12 and 628215), 14 sera were tested with class I phenotype and SA kits (Cat. LS1PRA, LS1A04, 628200 and 265100), and 16 sera were tested with class II phenotype and SA kits (Cat. LS2PRA, LS2A01, 628223 and 265200).
Table 2.
Nature of exchanged sera and kit usage
| Serum | Source1 | HLA antibody specificities | Kit2 | |||||
|---|---|---|---|---|---|---|---|---|
| Class I | Class II | Scr | PRAI | PRAII | SAI | SAII | ||
| 1 | EU | B44,B45,B76,B82 | DR7,DR53,DQ2 | X3 | x | x | x | x |
| 2 | EU | Bw4,A23,A24,A25,A32 | DR1,DR4,DR9,DR10,DR15,DR51,DR53,DR103,DQ5,DP1,DP3,DP5,DP9,DP11,DP14,DP17,DP19 | x | x | x | x | x |
| 3 | EU | A1,A3,A11,A23,A24,A36,A80,B37,B38,B39,B67,B76 | DR7,DR8,DR9,DR11,DR12,DR13,DR14,DR17,DR18,DR52,DQ4,DQ7, DQ8,DQ9 | x | x | x | x | x |
| 4 | EU | A66,B7,B13,B27,B39,B42,B47,B48,B54,B55,B56,B60,B61,B67,B73,B81, B82 | DR15,DR51,DQ5,DQ6 | x | x | x | x | x |
| 5 | EU | A1,A23,A24,A25,A32,A36,A43,A80,Bw4,B45,B62,B72,B76,B82 | DR4,DP1,DP2,DP4,DP5,DP11,DP13, DP19 | x | x | x | x | x |
| 6 | EU | Bw6,A1,A3,A11,A23,A24,A29,A36,A43,A80,B59 | DR1,DR4,DR9,DR15,DR16,DR51,DR103 | x | x | x | x | x |
| 7 | EU | A2,A3,A11,A23,A24,A32,A43,A68,A69,Bw4,B35,B4005,B41,B46,B47, B50,B56,B60,B61,B62,B72,B75,B78 | DR4,DP3,DP9,DP14,DP17 | x | x | x | x | x |
| 8 | EU | A2,A68,A69,A80,B57,B58 | DR103,DR11,DR12,DR13,DR16,DQ4,DQ5,DQ6,DQ8,DQ9,DP2,DP3,DP4,DP9,DP10,DP14,DP17 | x | x | x | x | x |
| 9 | EU | A23,A24,A25,A32,A66,B35,B38,B4005,B46,B48,B49,B51,B52,B53,B54,B56,B57,B58,B59,B62,B63,B71,B72,B75,B77,B78,Cw10 | DR7,DR9,DR12,DR52,DR53,DQ2,DQ4,DQ7,DQ9,DP1,DP3,DP4,DP5,DP10,DP11,DP13,DP14,DP17,DP18, DP19 | x | x | x | x | x |
| 10 | UCLA | B7,B8,B13,B18,B27,B35,B39,B41,B42,B45,B47,B48,B50,B54,B55,B56, B60,B61,B62,B64,B65,B67,B71,B72,B73,B75,B76,B78,B81,B82 | - | x | x | x | ||
| 11 | UCLA | B7,B27,B42,B48,B54,B55,B56,B60, B67,B81,B82 | - | x | x | x | ||
| 12 | UCLA | A23,A24,B13,B35,B38,B44,B45,B46,B49,B50,B51,B52,B53,B56,B57,B58,B62,B63,B71,B72,B75,B76,B77,B78 | - | x | x | x | ||
| 13 | UCLA | A1,A11,A25,A26,A29,A30,A31,A33,A34,A36,A43,A66,A68,A69,A80,B8,B38,B39,B41,B42,B54,B55,B57,B58,B59,B63,B67,B73,Cw6,Cw7,Cw18 | - | x | x | x | ||
| 14 | UCLA | - | DR15,DR16,DR51,DQ4,DQ5,DQ6 | x | x | x | ||
| 15 | UCLA | - | DR7,DR9,DQ2 | x | x | x | ||
| 16 | UCLA | - | DR51,DR7,DR9,DR10,DR15,DR16, DR53,DR103,DR1 | x | x | x | ||
| 17 | UCLA | - | DR11,DR13,DR14,DR17,DR18,DR52,DQ2,DQ7 | x | x | x | ||
| 18 | UCLA | - | DR4,DR53,DQ2,DQ4,DQ7,DQ8,DQ9 | x | x | x | ||
| 19 | UCLA | - | DR4,DR53,DQ8,DQ9 | x | x | x | ||
| 20 | UCLA | - | - | x | x | x | x | x |
Source: EU = Emory University, Atlanta; UCLA = University of California, Los Angeles.
Kit: Scr = screen; PRAI = panel reactive antibody class I; PRAII = panel reactive antibody class II; SAI = single antigen class I; SA2 = single antigen class II.
x: indicates serum was included in kit’s test set.
SOP development
Seven CTOT laboratories (UCLA, Emory, Harvard, Manitoba, Northwestern, Pittsburgh, and Washington University) participated in this study. Although each center followed the manufacturer’s protocol to develop in-house operating procedures (iHOP) for their clinical practice, considerable variation in iHOPs existed. Thus, standard operating procedures (SOP) for use by all seven laboratories were developed and adopted to further harmonize testing methods. The detailed SOP for this project is available at www.ctotstudies.org/SOP_List.htm.
Study Design
Identical lots (Lot1 and Lot2) of kits and their related reagents were sent directly from the manufacturers to the seven centers (Table 1). The set of 20 sera was distributed to each center and tested against the HLA Ab kits following the SOP. Tests were performed on Luminex 100 (5 laboratories) and Luminex 200 (2 laboratories) instruments, which were calibrated before use with identical calibrators (Luminex Inc., Table 1). Results in the form of CSV files containing fluorescent intensity measurements were collected, compiled and analyzed.
We also collected the results of two American Society for Histocompatibility and Immunogenetics proficiency tests (ASHI PT) SA surveys (2011-AC1 and 2011-AC2) from five centers who participated in this CTOT study. Each ASHI PT tested five sera with reagents from a single manufacturer via each centers iHOP. CTOT lots and centers were matched to the ASHI PT tests for analysis.
Statistical analysis
Luminex output files were collected containing information identifying centers, kits, lots, sera and beads. Files also contained median fluorescent intensities (MFI), which represent a central measure of fluorescence per individual test (a serum-by-bead combination). Medians were derived from the underlying micro-bead fluorescent reactions (average of 120 values per test). Mode, mean and trimmed-mean fluorescent intensities were available for analysis; however, they were not reported since their analyses yielded comparable results to MFI values. Data were analyzed using STATA (StataCorp. 2011. Stata Statistical Software: Release 12, College Station, TX), P-values were two-sided, and, when appropriate, a Bonferroni-approach was used to correct significance levels for multiple comparisons.
Center variation was assessed using several statistical methods including random-effects regression modeling, pairwise correlation comparisons and multi-dimensional scaling (MDS) of MFI values. (Details regarding these methods are given in Supplemental Material.) Coefficients of variation [%CV = (SD of the seven center MFIs ÷ mean of the seven center MFIs) X 100%] were computed to directly measure the variability of each test across the seven centers. Using boxplots, nonparametric (Kruskal-Wallis) and quantile (median) regression analyses, CV distributions were compared among operating procedures (SOP vs. iHOP), manufacturers (Vendor A vs. B), kits (Scr vs. PRA vs. SA), beads (HLA-A,B,C,DR,DQ,DP loci) and lots (Lot 1 vs. Lot2). The magnitude of lot-to-lot variation was gauged using Bland-Altman plots of MFI values from pairs of concordant Lot1 and Lot2 SA beads (27).
Validation and normalization studies were performed on SA Kits. Supplementary material provides details regarding the use of receiver operating characteristic (ROC) analysis to validate the Luminex platform between two manufacturers and the strategies used to adjust raw MFI for unwanted, background signals. With regard to the latter, raw MFI values were adjusted either according to the manufacturer’s recommended formulas from their product inserts producing ‘standardized’ values or via LOESS (locally weighted scatterplot smoothing) curve fitting algorithms (28, 29) producing ‘normalized’ values. The manufacturer’s calculations attempt to remove noise due to non-specific binding using fluorescent intensities from negative control beads and negative serum as factors in ratios of the sample-specific raw MFI. LOESS normalization attempts to equate fluorescent intensities globally (over the entire array of data) without the use of specific controls by assuming that the ranks of MFI values for identical bead sets are invariant across centers.
Results
Results are presented in an order consistent with the study’s timeline. Following adoption of SOPs by the seven laboratories, one mixed antigen screen (Scr) kit, two ID or PRA kits (PRA1 for class I and PRA2 for class II) and two SA kits (SA1 and SA2) from both manufacturers (Vendor A and Vendor B) were distributed to seven centers along with 20 reference sera, yielding a total of 9,918 median fluorescent intensity (MFI) values collected from each center. To assess the impact of standardization of the operating procedures we compared MFI variations from the SOP with variations from iHOP. Next, we addressed sources of MFI variation when the same SOP was employed by assessing center, manufacturer, kit, bead and lot effects. ROC analyses were used to measure the agreement in antibody assignments between the two manufacturers. Finally, we investigated the potential of reducing variation using standardized and normalized MFI values.
SOP effect
To gauge the impact of standardizing our testing approach we compared the laboratory variance using this study’s SOP to the results obtained using the laboratory’s iHOP. Fortuitously, 5 CTOT centers participated in two ASHI PT surveys using the same lots of reagents from one of the manufacturers but used their individual iHOPs. For the two SA kits (class I and class II), the ASHI results from 5 centers were collected and analyzed alongside their SOP results.
Boxplots of the %CV for iHOP- versus SOP-test effects for SA1 and SA2 kits are shown in Figure 1. MFI variation between centers was close to 3 times as much in ASHI surveys (~62%) compared with SOP results (~21% and ~20% for Lot1 and Lot2, respectively), P<0.0001. Similar conclusions were drawn when the MFI range was restricted as the follows: 0–500, 501–1000, 1001–3000, 3000–10,000, >10,000 (data not shown). This suggests the difference in %CV between iHOP and SOP is due to procedure difference rather than different choice of reference sera.
Figure 1. Laboratory variance for single antigen assays using standardized operating procedures versus non-standardized procedures.
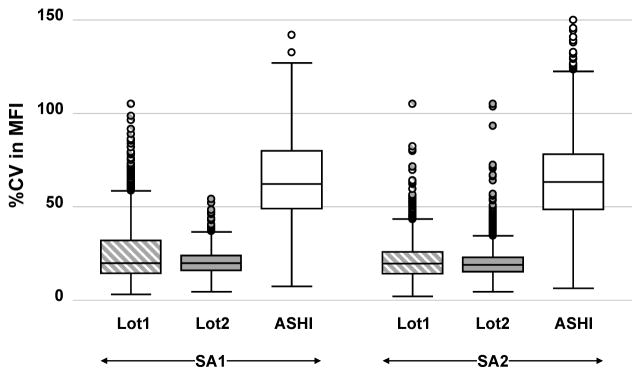
Comparisons of %CV in MFI distributions (depicted as boxplots) from 5 centers testing SA1 (first three plots) and SA2 (second three plots) kits with CTOT materials processed under SOP (Lot1
 and Lot2
and Lot2
 ) or with ASHI Proficiency Test materials processed under iHOP (□).
) or with ASHI Proficiency Test materials processed under iHOP (□).
Sources of MFI variation under SOP
Center effect
When MFI data were plotted on a log-scale, center-specific histograms appeared bimodal and formed near mirror images of one another (Figure 2, Left Panel). The “saddle” values (MFI separating the two modes) ranged from ~1200–3200. As listed in Table 3 and depicted by vertical lines in Figure 2, the average MFI values for the seven centers ranged from 2436 to 3139, producing systematic shifts in the central location of MFI distributions: Center B (3139) had a significantly higher mean (p = 0.001 vs. Center F); Centers D (2436) and G (2569) had significantly lower means (p<0.001 vs. Center F); and Centers A, C and E had similar means as Center F. Table 3 also lists the 21 pairwise correlations among centers. All are high and ranged from 0.953–0.992. After correcting for multiple comparisons, the minimum correlation confidence-bound was 0.950, implying at least 90% (0.952 × 100%) of the MFI variability was in agreement between any two centers.
Figure 2. Center effects on assay variation.

Left Panel: Histograms of MFI data (log scale) for Centers A to G (9,918 values each) and all centers (Total A–G =7×9918=69,426 values). Average values are marked by the vertical bar within each histogram. Right Panel: Plot shows the relative relationships among seven centers following Multidimensional Scaling (MDS) analysis. The original 7-dimensional array of dissimilarities (i.e., 1–correlation values computed between every pair of centers) was reduced into two major components (Dimensions 1 and 2) that preserved original dissimilarities while explaining ~80% of the variance (see Supplementary Material for details).
Table 3.
Pairwise MFI correlations of 9,918 replicate tests among 7 centers testing Lot 1
| Center | MFI (mean±SD)1 | Pearson correlation coefficient [95% CI2] | |||||||
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | |||
| A | 2997±5117 P = 0.33 vs. F |
1 | 0.963 [.961,.965] | 0.986 [.985,.987] | 0.979 [.977,.980] | 0.975 [.974,.977] | 0.986 [.986,.987] | 0.968 [.966,.970] | |
| B | 3139±5429 P = 0.001 vs. F |
1 | 0.972 [.971,.974] | 0.953 [.950,.956] | 0.980 [.979,.981] | 0.981 [.980,.982] | 0.966 [.964,.968] | ||
| C | 2826±5022 P =0.08 vs. F |
1 | 0.989 [.989,.990] | 0.989 [.988,.989] | 0.992 [.992,.993] | 0.978 [.977,.979] | |||
| D | 2436±4438 P < 0.001 vs. F |
1 | 0.972 [.970,.974] | 0.982 [.981,.983] | 0.971 [.969,.973] | ||||
| E | 3007±5354 P = 0.26 vs. F |
1 | 0.988 [.987,.989] | 0.975 [.973,.976] | |||||
| F | 2936±5188 baseline |
1 | 0.981 [.980,.983] | ||||||
| G | 2569±4686 P < 0.001 vs. F |
1 | |||||||
For testing average center differences, Center F was set as baseline based on multi-dimensional scaling analysis (See Figure 2). P-values for center differences were calculated using a random effects regression model, which included 6 dichotomous variables (one for each remaining center A–E and G) and a random component representing replicates of kits and beads.
Overall 95% confidence intervals for correlations calculated using Fisher’s z transform and a Bonferroni-adjustment for 21 comparisons.
Based on the dissimilarity (1-correlation) matrix for the seven centers, multi-dimensional scaling (MDS) analysis confirmed systemic center differences suggesting that Centers B, G and D stray away from a cluster formed by Centers A, C, E and F (Figure 2, Right Panel). Although differences in means existed, overall dissimilarities were quite small (±0.03 units), indicating the agreement among centers was very good. When a different lot of multiplex bead arrays (Lot2) were tested and analyzed, the results were the same as those from Lot1 (Table 4). Pairwise center correlation coefficients ranged from 0.96–0.99, and relative center relationships from MDS analysis were similar (see Supplementary Online Content).
Table 4.
Pairwise MFI correlations of 9,604 replicate tests among 7 centers testing Lot 2
| Center | MFI (mean±SD)1 | Pearson correlation coefficient [95% CI2] | ||||||
|---|---|---|---|---|---|---|---|---|
| A | B | C | D | E | F | G | ||
| A | 2903±5234 P = 0.02 vs. F |
1 | 0.975 [.973,.977] | 0.977 [.975,.978] | 0.953 [.950,.955] | 0.975 [.974,.977] | 0.969 [.967,.971] | 0.966 [.963,.968] |
| B | 2877±5141 P = 0.05 vs. F |
1 | 0.986 [.985,.987] | 0.969 [.967,.971] | 0.986 [.985,.987] | 0.980 [.979,.982] | 0.973 [.971,.975] | |
| C | 2535±4648 P <0.001 vs. F |
1 | 0.970 [.968,.972] | 0.991 [.991,.992] | 0.988 [.987,.988] | 0.979 [.977,.980] | ||
| D | 2555±4638 P = 0.001 vs. F |
1 | 0.981 [.980,.982] | 0.971 [.969,.973] | 0.968 [.966,.970] | |||
| E | 2631±4881 P = 0.05 vs. F |
1 | 0.985 [.984,.986] | 0.974 [.972,.975] | ||||
| F | 2754±4879 baseline |
1 | 0.969 [.967,.971] | |||||
| G | 2519±4626 P < 0.001 vs. F |
1 | ||||||
For testing average center differences, Center F was set as baseline (see Table 2). P-values were calculated using a random effects regression model, which included 6 dichotomous variables (one for each remaining center A–E and G) and a random component representing replicates of kits and beads.
Overall 95% confidence intervals for correlations calculated using Fisher’s z transform and a Bonferroni-adjustment for 21 comparisons.
Manufacturer and kit effects
In order to gauge manufacturer and kit-type variation among centers, coefficients of variation (%CV) were calculated across the 5 kit types (Scr, PRA1, PRA2, SA1 and SA2) and 2 vendors (A and B). Figure 3 shows boxplots of %CV distributions grouped according to five ranges of average MFI (0–500, 501–1000, 1001–3000, 3001–10,000 and 10,001–25,000 units). The %CV distribution for the total cohort of 9,918 tests is also displayed in the lower, right panel of Figure 3. The overall median %CV was 25%. In general, MFI variation was greater for Vendor A (median %CV = 30%) than Vendor B (median %CV = 20%, P<0.001). Vendor A SA kits exhibited the highest MFI variation (%CV= 36% and 29% for class I and class II, respectively, P<0.001 compared to the overall median); whereas Vendor B SA kits exhibited lower MFI variation (%CV = 20%, P<0.001).
Figure 3. Manufacturer and kit effects on assay variation.

Boxplots of %CV in MFI distributions grouped according to manufacturer and kit-type in 5 strata (0–500, 501–1000, 1001–3000, 3001–10,000 and 10,000–25,000) and all strata combined (Total) with Lot1 data. The median %CV for each stratum was shown as a thick horizontal line. Boxes were shaded to indicate whether %CV variations were statistically significant: not different from median (□, P>0.05); less than median (
 , P<0.001); greater than median (
, P<0.001); greater than median (
 , P<0.001).
, P<0.001).
Patterns of kit %CVs across the five MFI strata (0–500, 501–1000, 1001–3000, 3001–10,000 and 10,001–25,000) are presented in the first five panels of Figure 3. As indicated by greater interquartile ranges (longer boxes) and higher medians (black horizontal lines), %CVs increased as the average MFI decreased. However, general patterns exhibited by manufacturers’ kit-types within each MFI stratum were consistent with the pattern for the total cohort. For Vendor A, %CV variability tended to increase in a stepwise fashion for the first four kit types (Scr < PRA1 < PRA2 < SA1) and then drop for the SA2 kit. In contrast, %CV variability tended to be uniformly smaller for Vendor B’s specific kits (PRA1, PRA2, SA1 and SA2) compared to the screening kit (Scr). Within each of Figure 3’s panels, statistically significant deviations in kit-specific %CV distributions from the median were highlighted by shading boxplots gray (less than median) and black (greater than median).
Bead effect
The %CV was further analyzed at the antigen-specificity level in an attempt to identify ‘problematic’ beads in SA kits. Negative reactions (MFI ≤1000) were eliminated and %CV in MFI>1000 was calculated. No significant bead anomalies could be identified for the HLA-B, DR and DP loci. Beads from Vendor A detecting HLA-Cw12 Ab and beads from Vendor B detecting HLA-A2, A68, Cw1 and DQ4 Ab did tend to exhibit more variability than other specificities (Figure 4).
Figure 4. Bead effects on assay variation.

Boxplots of %CV in MFI>1000 values grouped according to HLA locus and manufacturer in SA kits. Beads exhibiting significantly more variability than other specificities are boxed with their corresponding plots highlighted (
). Only the A, C and DQ loci are shown since no significant SA bead anomalies were identified for the B, DR and DP loci.
Lot effect
The study was designed to measure lot-to-lot variation by exchanging two sets of identical kit reagents from the two manufacturers. As described above, the lot-specific center effects were consistent with one another as shown by measures listed in Tables 2 and 3. In Figure 5 (Left Panel), lot-to-lot center variations were further contrasted and summarized by plotting each lot’s %CV distribution, grouped according to manufacturer and kit type. Within each of Vendor A’s kit types, %CV boxplots for Lot1 and Lot2 overlapped and exhibited comparable medians. The overall %CV medians for Vendor A were 29.62% and 30.19% for Lot1 and Lot2 kits, respectively (P = 0.42), with no kit-type differences. The overall %CV medians for Vendor B were 19.78% and 18.99% for Lot1 and Lot2 kits, respectively (P<0.001). Vendor B’s Lot1 Scr and SA1 kits exhibited significantly more center variation compared with Lot2 (P<0.001), but other kits (PRA1, PRA2 and SA2) demonstrated closer agreement in their %CV distributions (P-values of 0.02, 0.02, and 0.06), respectively, which are non-significant for the multiple comparisons).
Figure 5. Lot effects on assay variation.

Left Panel: %CV in MFI in Lot1 and Lot 2 are shown by their boxplots grouped by manufacturer (Vendors A and B) and 5 kit types (Scr, PRA1, PRA2, SA1 and SA2). Right Panel: Bland-Altman plots divided into 3 strata based on average bead MFI magnitude (1001–3000 units, 3001–10,000 units and >10,000 units) showing ΔMFI (Lot1 minus Lot2 differences) among matched beads in SA kits. Lines represent the 95% chance boundaries (i.e., mean±1.96SD): 1001–3000 MFI range = 310±1500; 3001–10,000 MFI range = 620±4000; >10,000 MFI range = 1500±5600.
Since some of the HLA alleles conjugated on the bead changed from lot to lot, the allele specificities in SA kits were matched between Lot1 and Lot2. The pairwise difference in MFI values between lots was analyzed using Bland-Altman plots, which were divided into 3 broad strata where variation was relatively constant over the average (1001–3000 units; 3001–10,000 units; and >10,000 units) (Figure 5, Right Panel). Differences (Lot1 minus Lot2) were positivity biased by 310, 620 and 1500 units for the three MFI ranges, respectively. The limits-of-agreement (i.e., average difference ± 1.96SDdifference) are depicted within each MFI strata and indicate the typical variation in bead MFI values that can be expected in lot-to-lot testing. Values beyond these limits would indicate a possible change in Ab level.
Agreement in HLA Antibody detection
To validate HLA Ab assignments by multiplex-bead array technology, SA data from one manufacturer were used in ROC analysis to assess agreement with predicted positive tests based on data from the other manufacturer. Figure 6 displays ROC curves for both SA kits (class I and II) and their nested loci. At the class level, percent areas under the ROC curves (AUC) equaled 92±1% and 93±1% for class I and II, respectively. Except for the HLA-C-locus, AUCs ranged from 0.87 to 0.98 (with standard errors ≤0.03) across the individual loci. The HLA-C-locus AUC was poor (68%) and was not analyzed further given the dearth of corresponding beads and sera upon which to accurately predict reactions (i.e., standard error = 8%).
Figure 6. Receiver Operating Characteristic (ROC) analysis of manufacturer agreement.

One of the manufacturer’s SA tests and software were used to establish HLA Ab presence (truth). Then, the second manufacturer’s platform was tested for agreement using data from reactions on beads carrying the same antigen specificities. Analyses were performed for each loci nested within class I and II SA kits. Areas under ROC curves (AUC) are listed along with their 95% confidence intervals in parentheses, and the MFI cutoffs (Cut) used to yield maximum correct classification rates (Agreement) are also listed.
The MFI cutoff of 1400 units was found to optimize the correct classification rates for both class I and II kits. Based on this cutoff, accuracy/sensitivity/specificity values were 90%/68%/97% and 90%/75%/94% for SA1 and SA2 kits, respectively. Cutoffs ranging from 1000–1500 units were found to maximize agreement (ranging from 86–93% correct classifications) between the two manufacturers’ SA kits at the HLA-A, B, DR, DQ and DP loci.
Standardization and normalization to account for systematic center variance
In an attempt to further minimize variation, the manufacturer’s recommended formulas for standardizing MFI using values from controls were computed. Normalized MFI values were also obtained from LOESS regression models (see Methods and Supplemental Material). After adjusting raw MFI values, standardized and normalized %CV were recomputed for each SA bead and serum combination over the seven centers and compared in Figure 7. The ‘standardized’ %CV exhibited either larger median or larger interquartile ranges compared to ‘raw’ %CVs. In contrast, the ‘normalized’ %CV demonstrated both smaller medians and interquartile ranges compared to raw ‘%CVs’.
Figure 7. Comparison of manufacturer standardization and global normalization methods for reducing assay variation.

Boxplots show %CV in MFI distributions using raw, manufacturer’s standardized data and LOESS global normalized data from Lot1’s SA1 and SA2 kits. Values inside boxes represent the median %CV in MFI for a particular grouping. From left to right, the boxes’ lengths represent their interquartile ranges equaled 24%, 28%, 22%, 14%, 17% and 14%, respectively.
Discussion
Designing and implementing treatment strategies to individualize care of transplant recipients necessitates identifying and standardizing biomarkers that reliably stratify patient risk. While the accumulating evidence indicates that multiplex-bead array based measurements of anti-HLA antibodies are useful risk assessment tools for transplant recipients, the absence of standardization has limited the clinical utility of the findings. One goal of the CTOT is to assess and minimize the variability of solid-phase HLA Ab testing across laboratories. Our data derived from this analysis provide insights into the key sources of variation in the commercially available HLA Ab testing kits and represent a resource that can be used as a benchmark for further optimization and for normalization algorithm development. As expected, the best approach to lower variability and improve interassay comparison was achieved by employing a standardized protocol. When a single SOP was adopted, test variance among centers was reduced from 62% in a nonstandardized setting to 25% (Figure 1). The lower variability may be attributed in part to elimination of differences in reaction volume, mixing techniques, incubation times, and wash steps.
Although general agreement in test replicates was achieved among the seven laboratories following standardization of the SOP, analysis revealed small, but significant systemic variation in test replicates across laboratories (Figure 2). We found the range of means across the seven labs to be approximately 700 MFI units. This suggests additional laboratory-specific sources of variation independent of the reagents, protocol and instrument calibration that influence test reproducibility. As plate washing is considered a potential source of variability, the use of a robotic platform for automated pipetting and wash steps may minimize operator variability and is currently under investigation.
Analysis of the %CV across the different kit types suggests that MFI variation depends on both the manufacturer and kit type (Figure 3). Overall, we found MFI variation to be greater for Vendor A than Vendor B. Factors that could contribute to the higher variance include the lack of historic experience with one manufacturer, different dilution protocols, wash methods and bead manufacturing procedures. With regard to bead manufacturing processes, it is noteworthy that a limited number of SA beads from each vendor exhibited more variability compared to other specificities (Figure 4).
Recent reports indicate significant lot to lot variation in the quality and quantity of HLA antigens conjugated to beads (23). An analysis of matched SA beads across the two different lots demonstrated that individual beads could have a wide variation and these differences were related to the range in MFI (Figure 5). For example, if a bead tests at 2000 MFI units, then, all else being equal, a lot-retested MFI value could fall between 500 and 3500 MFI units (i.e., value ± 1500, the limit-of-agreement). If the bead tests higher, limits widen so larger differences are needed to determine a significant change in Ab levels to account for lot variation.
The degree of agreement in HLA Ab specificity assignment between manufacturers as assessed by ROC analysis was excellent at the antigen level (Figure 6). The HLA-C locus could not be analyzed due to insufficient HLA-C locus antisera included in the study. MFI positive cutoffs ranging from 1000 to 1500 yielded a high level of agreement (>90%) in antigen specificity assignment. The MFI cutoff of 1400 units was found to optimize the correct classification rates for both class I and II kits. However, examination of %CVs across different MFI strata showed larger differences in lower MFI ranges (0–3000) (Figure 3). Given this finding we can expect to encounter a lower degree of reproducibility in weak reactive sera which jeopardizes assigning Ab specificity with high certainty, especially in dealing with Ab near the positive cutoff value.
Can mathematical adjustment mitigate center variation? We investigated the effects of using manufacturer algorithms for standardization based on controls and found that they increased %CV rather than decreasing it (Figure 7). We show, however, that a normalization process (LOESS curve fitting algorithm – a method applied to remove noise from microarray data without specifying controls) performed better than the manufacturer’s formula. Further study is needed to develop normalization algorithms for individual laboratory data sets or at the patient level of data.
There are limitations to our study that could be improved in future studies. We did not take into account the impact of antigen density on assay variation nor did we assess whether serial dilutions of the HLA Ab result in a uniform linear change in MFI. We also did not characterize different lots of secondary Ab for their effect on test variability. These assays were performed manually by a single laboratory technologist within each laboratory so we have not estimated the full impact of tech to tech variability on assay variance.
Our results provide important implications for a clinical laboratory utilizing multiplex-bead arrays for Histocompatibility testing for transplantation. Typically, the interpretation of HLA Ab testing in clinical laboratories is based on different MFI thresholds established by the individual laboratory. Our data indicates that significant differences in MFI should be expected between laboratories using different protocols, reagents and even different lots of reagents from the same manufacturer. Even with a single protocol adopted by multiple clinical laboratories, our study found that the overall CV equaled 25%, implying a 50% change in MFI (i.e., twice the %CV) is needed to be confident that a definitive change in Ab level has occurred between two tests. In light of these findings, clinical laboratories should follow a standardized protocol and monitor inter-assay and intra-assay variability as part of their quality assurance program.
The multiplex-bead arrays are FDA approved for reporting semi-quantitative measurements of HLA Ab. The FDA guidelines indicate the acceptable levels of CV range from 15–20% to be qualified as a quantitative test (30). Our results suggest that assay variability can be reduced to near quantitative accuracy across different laboratories with assay standardization and use of appropriate normalization strategies (i.e. Figure. 7 below 20% CV).
Supplementary Material
Acknowledgments
The authors wish to acknowledge the expert skills of the following technologists: Brian Smith (BWH); Nancy D Herrera (NWU); Patricia Brannon and Zachary Gebel (Emory); Jon Lomago, Don Foster and Mariann Bigley (UPMC); Nancy Steward (WUSL); Denise Pochinco (Manitoba); Jason Song and George Banzuela (UCLA). The authors thank Dr. David Iklé for his statistical review of the manuscript.
This research was performed as part of an American Recovery and Reinvestment (ARRA) funded project under Award Number U0163594 (to P Heeger), from the National Institute of Allergy and Infectious Diseases. The work was carried out by members of the Clinical Trials in Organ Transplantation (CTOT) and Clinical Trials in Organ Transplantation in Children (CTOT-C) consortia.
Abbreviations
- CTOT
Clinical Trials in Organ Transplantation
- MFI
Median Fluorescence Intensity
- %CV
Coefficient of Variance
- MDS
Multi-Dimensional Scaling
- SOP
Standard Operating Procedure
- iHOP
in-House Operating Procedure
- ROC
Receiver Operating Characteristics
- AUC
area under curve
- PRA
Panel Reactive Antibodies
- SA
Single Antigen
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
References
- 1.Vaidya S. Clinical importance of anti-human leukocyte antigen-specific antibody concentration in performing calculated panel reactive antibody and virtual crossmatches. Transplantation. 2008;85(7):1046–50. doi: 10.1097/TP.0b013e318168fdb5. Epub 2008/04/15. [DOI] [PubMed] [Google Scholar]
- 2.Bray RA, Nolen JD, Larsen C, Pearson T, Newell KA, Kokko K, et al. Transplanting the highly sensitized patient: The emory algorithm. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2006;6(10):2307–15. doi: 10.1111/j.1600-6143.2006.01521.x. Epub 2006/08/31. [DOI] [PubMed] [Google Scholar]
- 3.Cecka JM, Kucheryavaya AY, Reinsmoen NL, Leffell MS. Calculated PRA: initial results show benefits for sensitized patients and a reduction in positive crossmatches. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11(4):719–24. doi: 10.1111/j.1600-6143.2010.03340.x. Epub 2010/12/01. [DOI] [PubMed] [Google Scholar]
- 4.Colvin RB. Antibody-mediated renal allograft rejection: diagnosis and pathogenesis. Journal of the American Society of Nephrology: JASN. 2007;18(4):1046–56. doi: 10.1681/ASN.2007010073. Epub 2007/03/16. [DOI] [PubMed] [Google Scholar]
- 5.Kobashigawa J, Crespo-Leiro MG, Ensminger SM, Reichenspurner H, Angelini A, Berry G, et al. Report from a consensus conference on antibody-mediated rejection in heart transplantation. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2011;30(3):252–69. doi: 10.1016/j.healun.2010.11.003. Epub 2011/02/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stewart S, Fishbein MC, Snell GI, Berry GJ, Boehler A, Burke MM, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2007;26(12):1229–42. doi: 10.1016/j.healun.2007.10.017. Epub 2007/12/22. [DOI] [PubMed] [Google Scholar]
- 7.Loupy A, Hill GS, Jordan SC. The impact of donor-specific anti-HLA antibodies on late kidney allograft failure. Nature reviews Nephrology. 2012;8(6):348–57. doi: 10.1038/nrneph.2012.81. Epub 2012/04/18. [DOI] [PubMed] [Google Scholar]
- 8.Li F, Atz ME, Reed EF. Human leukocyte antigen antibodies in chronic transplant vasculopathy-mechanisms and pathways. Current opinion in immunology. 2009;21(5):557–62. doi: 10.1016/j.coi.2009.08.002. Epub 2009/09/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Leary JG, Kaneku H, Susskind BM, Jennings LW, Neri MA, Davis GL, et al. High mean fluorescence intensity donor-specific anti-HLA antibodies associated with chronic rejection Postliver transplant. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2011;11(9):1868–76. doi: 10.1111/j.1600-6143.2011.03593.x. Epub 2011/06/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farmer DG, Venick RS, Colangelo J, Esmailian Y, Yersiz H, Duffy JP, et al. Pretransplant predictors of survival after intestinal transplantation: analysis of a single-center experience of more than 100 transplants. Transplantation. 2010;90(12):1574–80. doi: 10.1097/TP.0b013e31820000a1. Epub 2010/11/26. [DOI] [PubMed] [Google Scholar]
- 11.Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, et al. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2012;12(5):1157–67. doi: 10.1111/j.1600-6143.2012.04013.x. Epub 2012/03/21. [DOI] [PubMed] [Google Scholar]
- 12.Ho EK, Vlad G, Vasilescu ER, de la Torre L, Colovai AI, Burke E, et al. Pre- and posttransplantation allosensitization in heart allograft recipients: major impact of de novo alloantibody production on allograft survival. Human immunology. 2011;72(1):5–10. doi: 10.1016/j.humimm.2010.10.013. Epub 2010/10/26. [DOI] [PubMed] [Google Scholar]
- 13.Girnita AL, McCurry KR, Iacono AT, Duquesnoy R, Corcoran TE, Awad M, et al. HLA-specific antibodies are associated with high-grade and persistent-recurrent lung allograft acute rejection. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2004;23(10):1135–41. doi: 10.1016/j.healun.2003.08.030. Epub 2004/10/13. [DOI] [PubMed] [Google Scholar]
- 14.Zachary A, Reinsmoen NL. Quantifying HLA-specific antibodies in patients undergoing desensitization. Current opinion in organ transplantation. 2011;16(4):410–5. doi: 10.1097/MOT.0b013e32834899b8. Epub 2011/06/15. [DOI] [PubMed] [Google Scholar]
- 15.Hachem RR, Yusen RD, Meyers BF, Aloush AA, Mohanakumar T, Patterson GA, et al. Anti-human leukocyte antigen antibodies and preemptive antibody-directed therapy after lung transplantation. The Journal of heart and lung transplantation: the official publication of the International Society for Heart Transplantation. 2010;29(9):973–80. doi: 10.1016/j.healun.2010.05.006. Epub 2010/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeevi A, Lunz J. HLA antibody profiling in thoracic transplantation undergoing desensitization therapy. Current opinion in organ transplantation. 2012;17(4):416–22. doi: 10.1097/MOT.0b013e328355f1ab. Epub 2012/07/14. [DOI] [PubMed] [Google Scholar]
- 17.Montgomery RA, Lonze BE, King KE, Kraus ES, Kucirka LM, Locke JE, et al. Desensitization in HLA-incompatible kidney recipients and survival. The New England journal of medicine. 2011;365(4):318–26. doi: 10.1056/NEJMoa1012376. Epub 2011/07/29. [DOI] [PubMed] [Google Scholar]
- 18.Mengel M, Sis B, Haas M, Colvin RB, Halloran PF, Racusen LC, et al. Banff 2011 Meeting report: new concepts in antibody-mediated rejection. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2012;12(3):563–70. doi: 10.1111/j.1600-6143.2011.03926.x. Epub 2012/02/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sis B, Mengel M, Haas M, Colvin RB, Halloran PF, Racusen LC, et al. Banff ‘09 meeting report: antibody mediated graft deterioration and implementation of Banff working groups. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons. 2010;10(3):464–71. doi: 10.1111/j.1600-6143.2009.02987.x. Epub 2010/02/04. [DOI] [PubMed] [Google Scholar]
- 20.Couzi L, Araujo C, Guidicelli G, Bachelet T, Moreau K, Morel D, et al. Interpretation of positive flow cytometric crossmatch in the era of the single-antigen bead assay. Transplantation. 2011;91(5):527–35. doi: 10.1097/TP.0b013e31820794bb. Epub 2010/12/31. [DOI] [PubMed] [Google Scholar]
- 21.Pei R, Lee JH, Shih NJ, Chen M, Terasaki PI. Single human leukocyte antigen flow cytometry beads for accurate identification of human leukocyte antigen antibody specificities. Transplantation. 2003;75(1):43–9. doi: 10.1097/00007890-200301150-00008. Epub 2003/01/25. [DOI] [PubMed] [Google Scholar]
- 22.Taylor CJ, Kosmoliaptsis V, Summers DM, Bradley JA. Back to the future: application of contemporary technology to long-standing questions about the clinical relevance of human leukocyte antigen-specific alloantibodies in renal transplantation. Human immunology. 2009;70(8):563–8. doi: 10.1016/j.humimm.2009.05.001. Epub 2009/05/19. [DOI] [PubMed] [Google Scholar]
- 23.Zachary AA, Lucas DP, Detrick B, Leffell MS. Naturally occurring interference in Luminex assays for HLA-specific antibodies: characteristics and resolution. Human immunology. 2009;70(7):496–501. doi: 10.1016/j.humimm.2009.04.001. Epub 2009/04/15. [DOI] [PubMed] [Google Scholar]
- 24.Schnaidt M, Weinstock C, Jurisic M, Schmid-Horch B, Ender A, Wernet D. HLA antibody specification using single-antigen beads--a technical solution for the prozone effect. Transplantation. 2011;92(5):510–5. doi: 10.1097/TP.0b013e31822872dd. Epub 2011/08/27. [DOI] [PubMed] [Google Scholar]
- 25.Waterboer T, Sehr P, Pawlita M. Suppression of non-specific binding in serological Luminex assays. Journal of immunological methods. 2006;309(1–2):200–4. doi: 10.1016/j.jim.2005.11.008. Epub 2006/01/13. [DOI] [PubMed] [Google Scholar]
- 26.El-Awar N, Terasaki PI, Nguyen A, Sasaki N, Morales-Buenrostro LE, Saji H, et al. Epitopes of human leukocyte antigen class I antibodies found in sera of normal healthy males and cord blood. Human immunology. 2009;70(10):844–53. doi: 10.1016/j.humimm.2009.06.020. Epub 2009/07/08. [DOI] [PubMed] [Google Scholar]
- 27.Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1(8476):307–10. Epub 1986/02/08. [PubMed] [Google Scholar]
- 28.Ballman KV, Grill DE, Oberg AL, Therneau TM. Faster cyclic loess: normalizing RNA arrays via linear models. Bioinformatics. 2004;20(16):2778–86. doi: 10.1093/bioinformatics/bth327. Epub 2004/05/29. [DOI] [PubMed] [Google Scholar]
- 29.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–93. doi: 10.1093/bioinformatics/19.2.185. Epub 2003/01/23. [DOI] [PubMed] [Google Scholar]
- 30.Administration FaD. Guidance for Industry: Bioanalytical Method Validation. 2001. pp. 13–9. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.