

Abstract
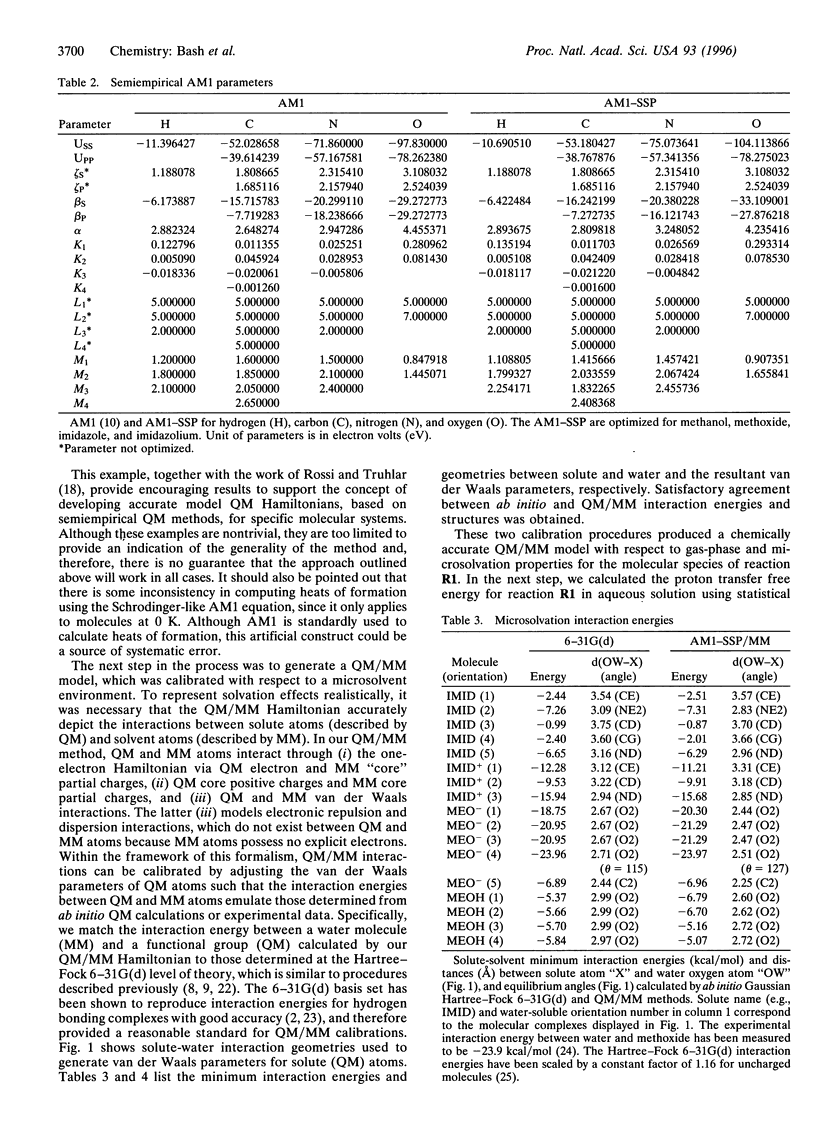
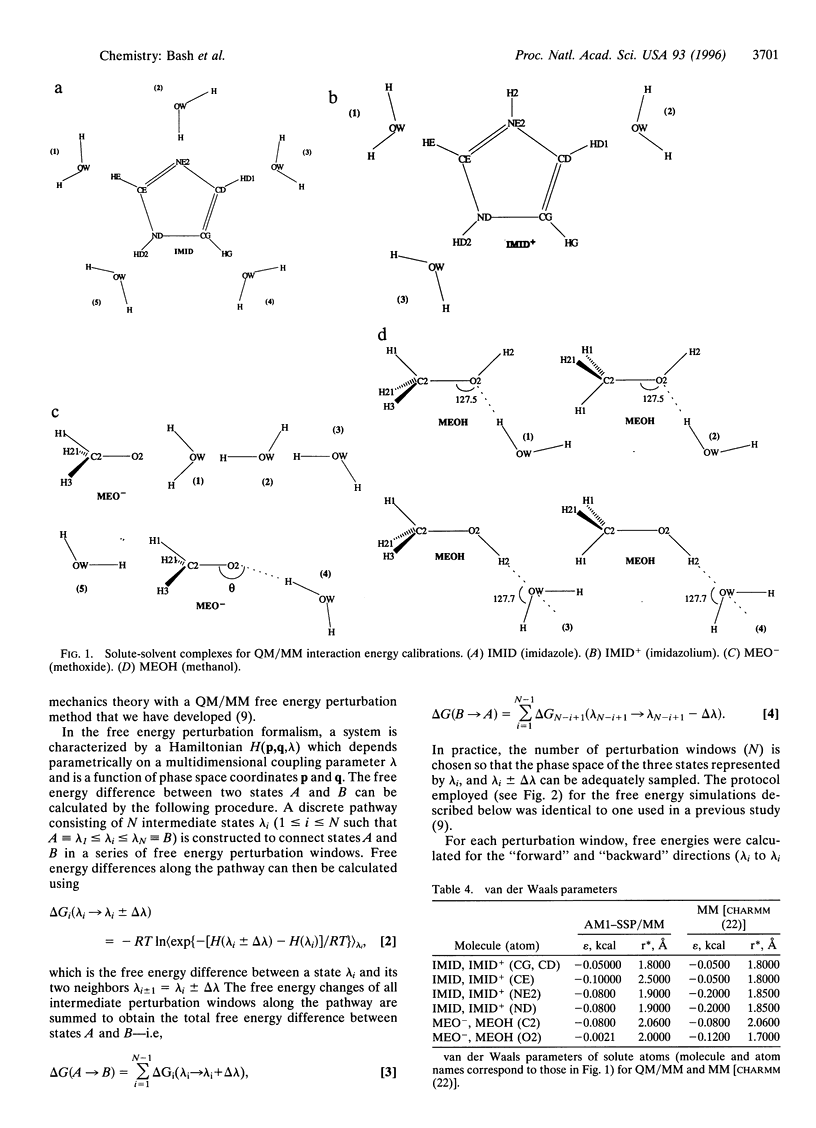
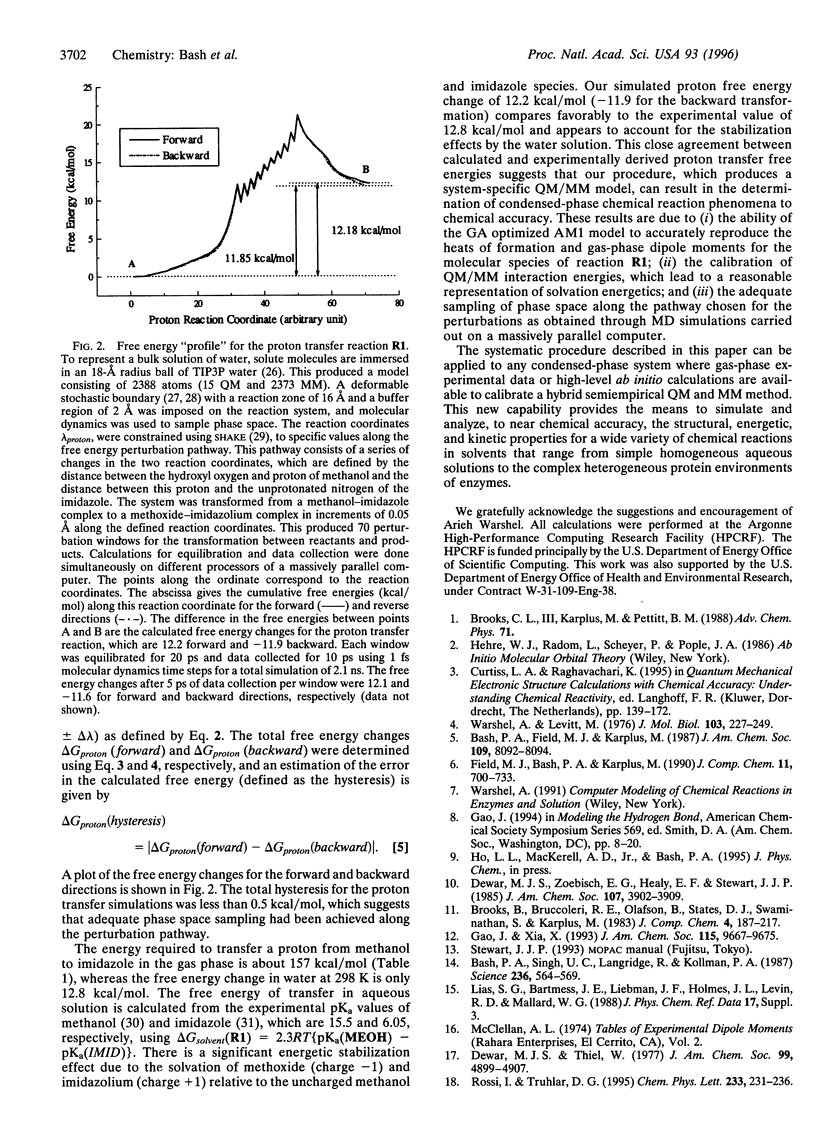
We describe a procedure for the generation of chemically accurate computer-simulation models to study chemical reactions in the condensed phase. The process involves (i) the use of a coupled semiempirical quantum and classical molecular mechanics method to represent solutes and solvent, respectively; (ii) the optimization of semiempirical quantum mechanics (QM) parameters to produce a computationally efficient and chemically accurate QM model; (iii) the calibration of a quantum/classical microsolvation model using ab initio quantum theory; and (iv) the use of statistical mechanical principles and methods to simulate, on massively parallel computers, the thermodynamic properties of chemical reactions in aqueous solution. The utility of this process is demonstrated by the calculation of the enthalpy of reaction in vacuum and free energy change in aqueous solution for a proton transfer involving methanol, methoxide, imidazole, and imidazolium, which are functional groups involved with proton transfers in many biochemical systems. An optimized semiempirical QM model is produced, which results in the calculation of heats of formation of the above chemical species to within 1.0 kcal/mol (1 kcal = 4.18 kJ) of experimental values. The use of the calibrated QM and microsolvation QM/MM (molecular mechanics) models for the simulation of a proton transfer in aqueous solution gives a calculated free energy that is within 1.0 kcal/mol (12.2 calculated vs. 12.8 experimental) of a value estimated from experimental pKa values of the reacting species.
Full text
PDF



Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bash P. A., Singh U. C., Langridge R., Kollman P. A. Free energy calculations by computer simulation. Science. 1987 May 1;236(4801):564–568. doi: 10.1126/science.3576184. [DOI] [PubMed] [Google Scholar]
- Brooks C. L., 3rd, Brünger A., Karplus M. Active site dynamics in protein molecules: a stochastic boundary molecular-dynamics approach. Biopolymers. 1985 May;24(5):843–865. doi: 10.1002/bip.360240509. [DOI] [PubMed] [Google Scholar]
- Brooks C. L., 3rd, Karplus M. Solvent effects on protein motion and protein effects on solvent motion. Dynamics of the active site region of lysozyme. J Mol Biol. 1989 Jul 5;208(1):159–181. doi: 10.1016/0022-2836(89)90093-4. [DOI] [PubMed] [Google Scholar]
- Warshel A., Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol. 1976 May 15;103(2):227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]