

Abstract
The inhibitory Fcγ receptor, FcγRIIB, is widely expressed on B cells, dendritic cells and myeloid effector cells and modulates a variety of antibody-driven in vivo functions. While it has been established that FcγRIIB plays an important role in the maintenance of peripheral tolerance, the responsible cell-specific FcγRIIB expression remains to be determined. In this study, we generated mice with selective deletion of FcγRIIB in B cells, dendritic cells and myeloid effector cells and evaluated these novel strains in models of tolerance and autoimmune diseases. Our results demonstrate that mice with selective deletion of FcγRIIB expression in B cells and dendritic cells have increased antibody and T cell responses, respectively, and display enhanced susceptibility to disease in distinct models, suggesting that FcγRIIB expression in distinct cellular populations contributes to the maintenance of peripheral tolerance through different mechanisms.
Keywords: Inhibitory Fcγ receptor, conditional knockout, tolerance, anti-nuclear antibodies, collagen induced arthritis
INTRODUCTION
The loss of tolerance to self-antigens with the resultant accumulation of autoantibodies and pathogenic immune complexes is a hallmark of autoimmune diseases and contributes to their pathological sequelae with resulting morbidity and mortality. Genetic and biochemical studies have identified multiple tolerance checkpoints that appear to be dysregulated in autoimmune diseases such as lupus and are thus considered to be targets for the development of therapeutic approaches to either prevent or cure this disease. One such candidate, the inhibitory Fcgamma receptor for IgG (FcγRIIB), was initially identified as a member of the wider FcγR family, functioning to balance the ability of IgG immune complexes to activate myeloid effector cells through engagement of the activation members of the FcγR family (1, 2). This inhibitory receptor is widely expressed on cells of the immune system, including myeloid populations, dendritic cells and B cells. It functions to gate signaling from ITAM-containing activation receptors by recruiting phosphatases, such as SHIP, and thus set thresholds for immune complex stimulation of inflammatory responses. On B cells, however, in addition to its ability to gate activation responses triggered by the BCR, crosslinking of FcγRIIB in the absence of an activation partner resulted in apoptosis, and accounted for the role of FcγRIIB in limiting the accumulation of plasma cells (3, 4). Genetic disruption of the gene for FcγRIIB on the mixed C57BL/6 and 129/Sv background, resulted in the spontaneous accumulation of autoantibodies, followed by progression to glomerular disease and premature mortality, identifying the gene as an epistatic modifier of lupus susceptibility (5). In a similar manner, immunization of non-permissive, H-2b strain of mice, such as B6, with bovine collagen type II or IV resulted in loss of tolerance with the development of anti-mouse collagen antibodies and the subsequent development of arthritis and a Goodpasture Disease like phenotype, respectively (6, 7). More recently studies with FcγRIIB-deficient mice derived from B6 embryonic stem (ES) cells confirmed that FcγRIIB-deficiency sensitizes C57BL/6 mice to collagen induced arthritis (8). Previous studies also demonstrated that mouse strains susceptible to the development of autoimmunity, such as NZW, BXSB and NOD, displayed defective regulation of FcγRIIB expression, the result of polymorphisms in the promoter region of the gene, leading to a failure of the normal pattern of upregulation upon B cell activation (9). The autoimmunity-associated polymorphic allele encoding FcγRIIB was recently knocked into C57BL/6 mice and resulted in a number of autoimmune phenotypes including the development of more severe collagen-induced arthritis (10). Tolerance could be restored in these lupus susceptible strains by restoring wild-type levels of FcγRIIB on B cells by gene transfer (11). Combining FcγRIIB dysregulation with other genetic modifiers of autoimmunity, such as yaa, MDA5 or MRL/lpr resulted in exacerbation of autoimmune disease (12–14).
Similar defects in FcγRIIB expression or function were described in human SLE populations where it had been observed that >50% of lupus patients fail to upregulate FcγRIIB upon B cell activation (15). A promoter polymorphism affecting the regulation of FcγRIIB has been identified in some SLE populations in which the common haplotype, -386G/-120T is replaced by -386C/-120A (16). In addition to defects in the appropriate regulation of the FcγRIIB gene, a polymorphism has been identified in the transmembrane region of the gene, I232T (17) which results in a hypomorphic mutation that fails to mediate inhibitory signaling and thus compromises this function of FcγRIIB (18–20). Confirming the importance of this hypomorphic allele in maintaining tolerance was the observation that hematopoeitic stem cells derived from patients homozygous for the I232T polymorphism, when transplanted into immunodeficient recipient mice, resulted in reconstituted immune systems that failed to maintain tolerance and developed anti-DNA antibodies (21).
Therefore, defects in FcγRIIB function and regulation have emerged as a common feature of lupus and other autoimmune diseases, contributing both to disease susceptibility and progression. However, the relative contributions of FcγRIIB expression in different cellular compartments, such as B cells, dendritic cells and myeloid effector cells to these phenotypes have not been firmly established. In the current study we have investigated the contributions of FcγRIIB expression in B cells, dendritic cells and myeloid effector cells to the maintenance of peripheral tolerance through the analysis of mice conditionally deleted for this receptor in these immune cells.
MATERIALS AND METHODS
Generation of mice carrying Fcgr2b− and Fcgr2bfl alleles
In order to generate Fcgr2b germline and conditional knockout mice from B6 ES cells, two homologous arms cloned from the Fcgr2b locus of C57BL/6 genomic DNA were inserted into to an ES cell targeting vector (Supplementary Figure 1). The 5′ homologous arm, a 8.5 kb DNA fragment containing the exons coding for the S2, EC1, EC2, and TM domains of FcγRIIB, was generated by PCR (Expand Long Template PCR, Roche) using primers 5′CCCATCGATATGAACAGTAAAGTTGTCTCTGCAAGGTCACT3′ and 5′ATATTCTTGCGGCCGCCATTTTCCAGACTGGTAAACTGGG3′ and cloned into the Cla1/NotI sites of the pEasyFlox vector. A loxP-Neo-loxP cassette encoding neomycin-resistant gene (neo) was inserted after this 8.5 kb fragment in the NotI/SalI sites of pEasyFlox, and its location in respect to the gene would place it 1300 bp downstream of the TM exon (exon 5) in intron 5. The 3′ homologous arm of the targeting vector, a 4.3 kb DNA fragment containing the exons coding for the 3 intracellular domains, IC1, IC2, and IC3, was generated by PCR (Expand Long Template PCR, Roche) using primers 5′GCCGAGTCGACAACACTATGGGGCCCACCTTACAGGAATA3′ and 5′ATAGCTCTCGAGTCTCCTCTACCTCCTATCTACTGCTACCAG3′ and cloned into the SalI/XhoI sites of pEasyFlox. The third loxP site was inserted in the HindIII site in the 5′ homologous arm, 134 bp upstream to the EC1 exon. Transfection of B6 ES cells with the targeting vector and the subsequent selection and screening were performed in the Rockefeller University Gene Targeting Facility. Clones containing the targeted Fcgr2b allele (Fcgr2bflNeo) were identified by Southern blot analysis of EcoRV digested genomic DNA with a probe that hybridizes outside of the targeting vector. Based on the design of the targeting vector, a hybridized band of 13.6 kb would identify the wild-type Fcgr2b allele and a band of 10.5 kb would identify the targeted Fcgr2bflNeo allele (Supplementary Figures 1A–B). Positive clones that also contain the loxP site inserted into the HindIII site in the 5′ homologous arm (confirmed by PCR and sequencing) were selected for microinjection into C57BL/6 embryos and chimeric male offspring were bred to C57BL/6 females for germline transmissions. The offspring carrying the Fcgr2bflNeo allele identified by Southern blot were crossed to B6 mice expressing Cre under the control of the cytomegalovirus immediate early enhancer-chicken beta-actin hybrid (CAG) promoter(22) for the deletion of the Fcgr2b sequences between the two distal loxP sites to create the Fcgr2b− allele. To create the Fcgr2bfl allele, ES cells carrying the Fcgr2bflNeo allele were transiently transfected with a Cre-expressing plasmid and screened for deletion of the loxP-flanked neo cassette by PCR specific for the resulting Fcgr2bfl allele using primers pR2floxA 5′ AATGGCGGCCGCGGATCCATAACTTCG3′ and pR2delta4.2 5′TGGCTTCCATTGACCTGCCTACAACATTCCTC3′ (Supplementary Figures 1A, 1C). The deletion of the neo cassette was confirmed by two PCR reactions using primers pNeo-cF1 5′GATTCGCAGCGCATCGCCTTCTATCG3′ and pR2delta4.2, or primers pR2floxA and pNeo-R1 5′GCCGATTGTCTGTTGTGCCCAGTCATAG3′. ES clone #39 carrying the Fcgr2bfl allele was selected for microinjection into C57BL/6 embryos and chimeric male offspring bred to C57BL/6 females gave successful germline transmissions, which were confirmed by Southern blots and PCR.
Mice
Wild-type C57BL/6 mice were purchased from Taconic. Mice carrying Fcgr2bfl or Fcgr2b− alleles were generated from B6 ES cells (Supplementary Figure 1). Mb1Cre/Fcgr2bfl/fl, Cg1Cre/Fcgr2bfl/fl, CD11cCre/Fcgr2bfl/fl, LysMCre/Fcgr2bfl/fl mice were generated by crossing mice carrying Fcgr2bfl alleles to Mb1Cre mice (23), Cg1Cre mice (24), CD11cCre mice (25), and LysMCre mice (26), respectively, which have been backcrossed to the B6 background for at least 10 times. In some mice, Mb1Cre and CD11cCre also mediated germline deletion of one Fcgr2bfl allele and gave rise to Mb1Cre/Fcgr2bfl/− and CD11cCre/Fcgr2bfl/− mice. B6.Fcgr2b129−/−(N12) were obtained from Taconic. Fcer1g−/−Fcgr2b−/− mice had been described previously (27). All mice were maintained in The Rockefeller University Comparative Bioscience Center. All experiments were performed in compliance with federal laws and institutional guidelines and had been approved by the Rockefeller University IACUC.
Flow cytometry
In order to analyze the levels of FcγRIIB in WT and mutant mice with germline or conditional knockout of Fcgr2b, blood cells and splenic single-cell suspensions were prepared and depleted for erythrocytes, and stained with fluorescent-conjugated anti-CD19 (1D3), anti-NK1.1(PK136), anti-CD11b (M1/70), anti-CD11c (HL3), anti-Gr1 (RB6-8C5), and biotin-conjugated anti-FcγRIIB (Ly17.2) or mouse IgG2a isotype control (MG2a15, Invitrogen) antibodies in the first step and streptavidin-APC in the second step. DAPI was added to exclude dead cells before samples were analyzed using a BD LSRII (BD Biosciences). Acquired data were analyzed using Flowjo (Version 7.05 for windows). In order to analyze FcγRIIB expression in IgG1+ and IgG1− germinal center (GC) B cells, mice were treated with 100 μg of NP-CGG in alum and analyzed 12 days later. Splenic single-cell suspensions depleted for erythrocytes were stained with fluorescent-conjugated anti-B220 (RA3-6B2), anti-Fas (Jo2), anti-IgG1 (A85-1), and anti-FcγRIIB/III (2.4G2). In order to analyze FcγRIIB levels in thioglycollate-elicited macrophages, mice were i.p. injected with 2 ml of thioglycolate. Seven days later, peritoneal cavity cells were harvested by gently flushing with cold PBS, stained with fluorescent-conjugated F4/80 (BM8), CD11b (M1/70) and anti-FcγRIIB (Ly17.2) antibodies. Macrophages were defined CD11b+F4/80+ cells.
Antibody response
Mice were immunized i.p. with 100 μg of NP48-CGG emulsified in complete Freund’s adjuvant on day 0 and boosted with 100 μg of NP48-CGG emulsified in incomplete Freund’s adjuvant on day 28. Levels of NP-specific IgG on day 0, 14, and 42 were determined by ELISA. ELISA plates were coated with 10 μg/ml NP26-BSA; 1:106 diluted serum samples were analyzed; IgG were detected by HRP conjugated goat anti-mouse IgG-Fc (1:5000, Jackson ImmunoResearch) and TMB substrates.
Analysis of autoimmune phenotypes
Mice were monitored for survival for 10 months. Proteinuria levels were monitored using Chemstrip® 2 GP strips (Roche) monthly and mice with proteinuria levels at or above 100 mg/dL were considered “sick”. To analyze levels of anti-nuclear antibodies of IgG class, 1:200 diluted serum samples were applied to REAADS ANA Test plates (Corgenix, Inc.) and incubated at room temperature for one hour, washed 5 times with PBS with 0.05% tween-20. IgG antibodies were detected with HRP-conjugated goat anti-mouse IgG-Fc (1:5000, Jackson ImmunoResearch) and TMB substrate. In order to analyze the levels of total IgM and IgG in sera by ELISA, 1 μg/ml goat anti-mouse Ig (H+L) antibodies (BETHYL laboratories) were used as capture antibodies; 1:100,000 diluted serum samples were used for the analysis of IgM levels; 1:1,000,000 diluted serum samples were used for the analysis of IgG levels; 1:5000 diluted HRP-conjugated goat anti-mouse IgM (Southern biotech) or IgG-Fc antibodies (Jackson ImmunoResearch) plus TMB substrates were used to detect IgM or IgG, respectively. Immunofluorescence staining of Hep-2 cells (MBL International Corporation) was performed following manufacturer’s instructions. Diluted serum samples (1:100) collected from 8~9 month old mice were analyzed. IgG antibodies were detected using FITC conjugated goat anti-mouse IgG-Fc (1:200, Jackson ImmunoResearch). Collagen induced-arthritis experiments were performed as described (28). Briefly, mice were immunized intradermly with 100 μg of bovine or chicken type II collagen emulsified in adjuvant (incomplete Freund’s adjuvant with 4 mg/ml Mycobacterium tuberculosis H37 Ra) to induce arthritis. Arthritis incidences were monitored weekly for 11 weeks. In order to analyze mouse collagen-specific IgG response, serum samples were diluted for 5000 fold and analyzed with mouse IgG anti-mouse collagen type II ELISA kit (MD bioproducts).
K/BxN arthritis model
Experiments using the K/BxN arthritis model were performed as described previously (29). Mice were injected with 200 μl of K/BxN sera intravenously and monitored for the development of arthritis for 6 days. Arthritis clinical scores (0–3) were assigned to each paw of individual mice depending on the severity of arthritis, and the summed scores of four paws of individual mice (0–12) were recorded as their arthritis clinical scores.
OT-I T cell expansion
OT-I T cell expansion experiments were performed using a modified protocol (30). Briefly, on day -1, two million CFSE labeled CD45.1+ OT-I T cells enriched by MACS negative selection were adoptively transferred through i.v. injection into wild-type, Fcgr2b−/−, or Fcgr2b conditional knockout mice with CD11cCre. On day 0, each mouse received 150 μg of rabbit anti-OVA IgG through intravenous injection, followed by 2.5 μg of OVA 4 hours later. The concentration of CD45.1+CD8+Va2+ cells in blood was analyzed 3 days later by FACS.
Statistics
All statistical analyses were performed in Prism 5 for Windows (version 5.04). one-way ANOVA with Dunnett’s post hoc test was used in Figures 2, 4A, 7 to compare all groups to the “WT” or “Fcgr2bfl/fl” control group; one-way ANOVA with Tukey’s post hoc test was used in Figure 3 and Supplementary Figure 3; Chi-square test was used in Figures 4B, 5, 6B and Supplementary Figure 2 to compare every group to the “Fcgr2bfl/fl” control group.
Figure 2.
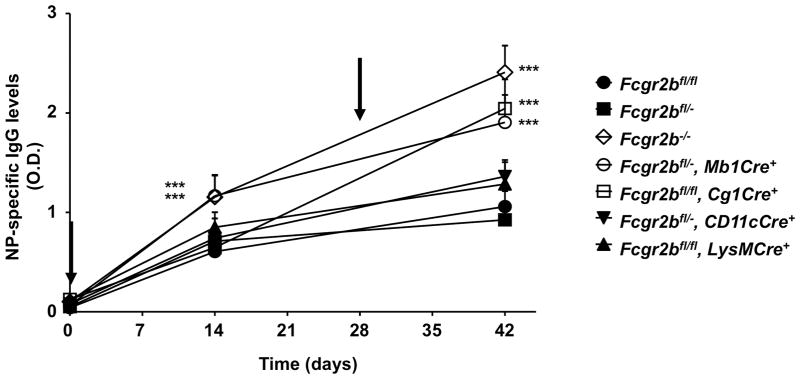
T-dependent IgG responses in WT and mutant mice with germline, partial or conditional knockout of Fcgr2b. Mice were immunized with NP-CGG in complete Freund’s adjuvant on day 0 and boosted with NP-CGG in incomplete adjuvant on day 28 (indicated by arrows). NP-specific IgG levels were analyzed on day 0, 14 and 42 by ELISA, and presented as O.D. values (mean±s.d.). *** p < 0.001, ANOVA with Dunnett’s post hoc. Representative of two independent experiments (5 mice per group).
Figure 4.

Spontaneous anti-nuclear antibodies in WT and mutant mice with germline or conditional knockout of Fcgr2b. (A) Levels of anti-nuclear antibodies of IgG classes in 10 month old WT and mutant mice with germline or conditional knockout of Fcgr2b (16~27 mice per group) were analyzed by ELISA, and presented as O.D. values (symbols represent O.D. values of individual mice and thick horizontal lines represent the means). ** p < 0.01, *** p < 0.001, ANOVA with Dunnett’s post hoc comparing each group to the “WT” group. (B) Increased anti-nuclear antibodies levels in some Fcgr2b−/− mice. Hep-2 human epithelial cells were stained with 1:100 diluted sera from 8–9 month old WT and Fcgr2b−/− mice, followed by FITC conjugated goat anti-mouse IgG. Anti-nuclear IgG antibodies were detected in about half (9/18) Fcgr2b−/− mice, whereas none of 5 wild-type mice were positive in this analysis (p < 0.05, Chi-square test).
Figure 7.
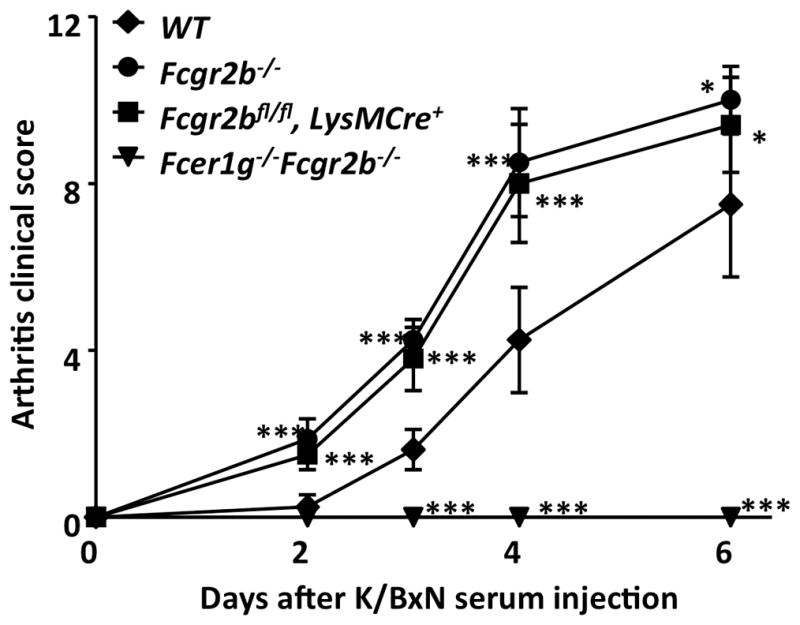
Accelerated development of K/BxN serum induced arthritis in Fcgr2bfl/fl mice with LysMCre. WT, Fcgr2b−/−, Fcgr2bfl/fl mice with LysMCre, and FcγR-deficient (Fcer1g−/−Fcgr2b−/−) mice were treated with K/BxN sera and monitored for the development of arthritis. Arthritis clinical scores (mean±s.d) are presented. * p < 0.05, *** p < 0.001, ANOVA with Dunnett’s post hoc comparing each group to the “WT” group. Representative of two independent experiments.
Figure 3.

Immune complex induced CD8 T cell responses in WT and mutant mice with germline or conditional knockout of Fcgr2b. The expansion of OT-I T cells in mice of the indicated genotypes in response to OVA immune complex was analyzed in blood 3 days after immunization and presented as OT-I T cell concentrations. Symbols represent values from individual mouse and horizontal lines represent the means. * p < 0.05, ** p < 0.01, *** p < 0.001, ANOVA with Tukey’s post hoc. Representative of two independent experiments (5–6 mice per group).
Figure 5.

Susceptibilities of mice with germline or conditional knockout of Fcgr2b to bovine type II collagen induced arthritis (bCIA). Accumulative bCIA incidences in WT (Fcgr2bfl/fl) and the indicated mutant male mice with germline or conditional knockout of Fcgr2b are presented. “n” values are the numbers of mice in each group. ** p < 0.01, *** p < 0.001, **** p < 0.0001, Chi-square test (vs the “Fcgr2bfl/fl” mice). Data are combined from three independent experiments with similar results.
Figure 6.
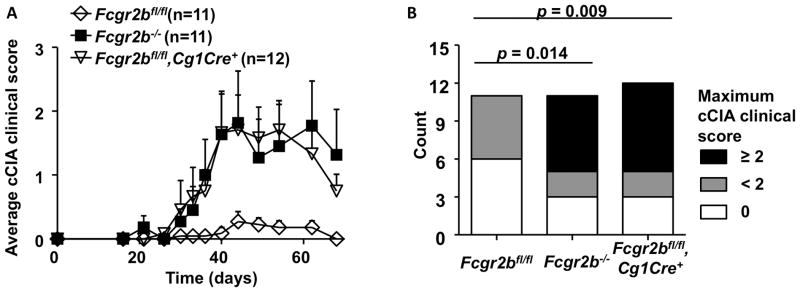
Susceptibilities of mice with germline or conditional knockout of Fcgr2b to the chicken type II collagen induced arthritis (cCIA). (A) The development of cCIA in WT (Fcgr2bfl/fl) and the indicated mutant mice with germline or conditional knockout of Fcgr2b (11–12 mice per group), expressed as average arthritis clinical scores (mean±sem), is presented. (B) The distribution of the maximum arthritis clinical scores observed in the mice in (A) is presented. P values were calculated by Chi-square test.
RESULTS
Generation of Fcgr2b germline and conditional knockout mice from B6 ES cells
Fcgr2b germline and conditional knockout mice were generated by crossing a mouse strain with loxP-flanked Fcgr2b alleles (Fcgr2bfl) derived from B6 ES cells (Supplementary Figure 1) to CagCre (22), Mb1Cre (23), Cg1Cre (24), CD11cCre (25), and LysMCre (26) B6 mice. Mb1Cre is expressed in all B cells while Cg1Cre expression is restricted to germinal center (GC) and post-GC B cells. CD11cCre is primarily expressed in dendritic cells (DCs) and LysMCre is expressed in most myeloid effector cells. As shown in Figure 1A, homozygous Fcgr2bfl/fl mice have equivalent FcγRIIB expression as WT C57BL/6 mice, confirming that the inserted loxP sites in the Fcgr2bfl/fl allele have no effect on the expression of FcγRIIB. Mb1Cre/Fcgr2bfl/fl mediated specific and efficient deletion of Fcgr2b in all B cells examined; in contrast Cg1Cre/Fcgr2bfl/fl did not delete Fcgr2b in resting B cells. To determine which B cell populations delete Fcgr2b in Cg1Cre/Fcgr2bfl/fl mice, Fcgr2bfl/fl mice with Cg1Cre were immunized with NP-CGG in alum and examined for FcγRIIB expression in IgG1+ and IgG1− GC B cells, as defined by B220+Fas+IgG1+ and B220+Fas+IgG1− cells, respectively. As shown in Figure 1B, FcγRIIB expression was reduced in the majority of both IgG1+ and IgG1− GC B cell subsets, consistent with previous studies showing that Cg1Cre expression is restricted to GC and post-GC B cells (24). CD11cCre/Fcgr2bfl/fl mediated efficient deletion of Fcgr2b in dendritic cells (CD11chigh), as well as in some monocytes (CD11cint). LysMCre/Fcgr2bfl/fl mediated deletion of Fcgr2b was detectable in monocyte and thioglycolate-elicited macrophages (Figures 1A and 1C), but not in B cells or DCs. However, the deletion efficiency of LysMCre was estimated to be only 20~60%, as has been reported previously for some other genetic systems (31, 32). These data indicate that we have generated B6 mice with a conditional knockout of Fcgr2b in B cells (Mb1Cre), GC and post GC B cells (Cg1Cre), DCs and some monocytes (CD11cCre), and monocytes and macrophages (LysMCre).
Figure 1.

Expression profiles of FcγRIIB in WT and mutant mice with germline or conditional knockout of Fcgr2b. (A) Histogram profiles showing the expression of FcγRIIB in the indicated cell types of the indicated mice. FcγRIIB levels were analyzed in B cells (CD19+), monocytes (CD11b+NK1.1-Gr1low/−SSClow) and neutrophils (CD11b+NK1.1−Gr1highSSCint) in the peripheral blood, and dendritic cells (DC, CD11chigh) in the spleen in WT C57BL/6 mice, and mice with germline or conditional knockout of Fcgr2b. (B) The gating strategy for non-germinal center B cells, IgG1+ and IgG1− GC B cells, and histogram profiles showing Cg1Cre-mediated deletion of Fcgr2b in these cells. Mice with the indicated genotypes were treated with NP-CGG in alum and analyzed 12 days later for the expression of FcγRIIB in splenic non-GC B cells (B220+Fas−), IgG1+ and IgG1− GC B cells (B220+Fas+IgG1+ and B220+Fas+IgG1−, respectively). (C) Histogram profiles showing FcγRIIB levels in thioglycollate-elicited macrophages (CD11b+F4/80+) isolated from mice of the indicated genotypes. Representative of two independent experiments with 3 mice per group.
FcγRIIB expression in B lineage cells regulates antibody responses
To determine the effect of germline and conditional knockouts of Fcgr2b in different immune cells on the primary and secondary thymic-dependent antibody response, levels of NP-specific IgG were analyzed in mice immunized and boosted with the model antigen NP-CGG. As shown in Figure 2, wild-type (Fcgr2bfl/fl) and Fcgr2b heterozygous (Fcgr2bfl/−) mice had comparable primary and secondary IgG responses, whereas these responses in Fcgr2b−/− mice were significantly enhanced (p < 0.001), consistent with previous studies (8, 33). Analyses of Fcgr2b conditional knockout lines showed that only mice with Mb1Cre have an increase of primary IgG antibody response equivalent to that of Fcgr2b−/− mice. Significant increase of secondary IgG responses were observed in both the Mb1Cre and Cg1Cre lines, whereas CD11cCre and LysMCre mediated deletion of Fcgr2b had no significant effect on either primary or secondary IgG responses. These results suggested that the effect of FcγRIIB-deficiency on IgG antibody responses is B cell intrinsic and FcγRIIB functions as a negative regulator in both primary and secondary IgG responses. While FcγRIIB expressed in resting B cells is important to limit the primary antibody response, FcγRIIB expressed in B lineage cells at the GC or post-GC stages is important to inhibit the secondary response.
FcγRIIB expression in dendritic cell regulates T cell responses
FcγRIIB expression in dendritic cells has been reported to regulate T cell response, and we confirmed these reports (30). As shown in Figure 3, in response to OVA immune complexes, significantly enhanced CD8 T cell response was observed in mice with either germline knockout or dendritic cell specific knockout of Fcgr2b, suggesting that FcγRIIB expression in dendritic cells could contributes to the maintenance of tolerance through regulating T cell responses.
Increased spontaneous ANA antibodies in mice with GC/post-GC B cell specific deletion of FcγRIIB
Since different autoimmune phenotypes have been reported in Fcgr2b knockout mice of different genetic backgrounds in previous studies (5, 8), we analyzed the Fcgr2b-knockout mice generated from B6 ES cells in this study. FcγRIIB-deficiency resulted in modest but significant increases in IgG anti-nuclear antibody titers in 10 month old B6 mice (p < 0.01), with approximately 25% displaying relatively high titers of ANAs (Figure 4A). Anti-nuclear IgG antibodies were detectable in some Fcgr2b−/− mice by Immunofluorescence staining of Hep-2 human epithelial cells (Figure 4B). When Fcgr2b conditional knockout mice were analyzed for ANA IgG antibodies, we found that conditional knockout of Fcgr2b by Cg1Cre in GC/post-GC B cells recapitulated the Fcgr2b germline knockout phenotype, in contrast to CD11cCre or LysMCre mediated conditional knockouts (Figure 4A), suggesting the FcγRIIB expression in GC or post GC B cells is responsible for inhibiting the development of spontaneous autoantibodies.
Increased arthritis incidence in mice with B cell or dendritic cell specific deletion of Fcgr2b on a nonpermissive background
Previous studies have shown that Fcgr2b knockout mice are more susceptible to induced autoimmune diseases (6–8). In order to study the impact of germline or conditional knockout of Fcgr2b on the maintenance of tolerance, mice were analyzed in bovine type II collagen induced arthritis (bCIA) model, which is normally nonpermissive in B6 mice because their H-2b background does not support sustained T cell and subsequent antibody responses against bovine type II collagen required to initiate and perpetuate bCIA (34–36). We found that FcγRIIB-deficiency can sensitize B6 mice in this otherwise resistant model. As shown in Figure 5, Fcgr2b−/− B6 mice are highly susceptible to bCIA (13/19, p < 0.0001), in contrast to the resistant wild-type B6 mice (0/23). The contribution of cell-specific FcγRIIB-expression to the maintenance of tolerance was evaluated in this model using Fcgr2b conditional knockout mice. As shown in Figure 5, 9/20 Fcgr2bflfl mice with Mb1Cre-mediated selective deletion of Fcgr2b in B cells developed arthritis (p < 0.001), as did 3/10 Fcgr2bflfl mice with CD11cCre (p < 0.01). As heterozygous Fcgr2b mice (Fcgr2bfl/−) mice generally did not develop arthritis, although a trend towards disease is suggested (2/17 mice, not statistically significant different from WT mice; Supplementary Figure 2), heterozygous conditional knockout mice with Mb1Cre or CD11cCre were also analyzed and similar results were obtained (Supplementary Figure 2). In contrast, Fcgr2bflfl mice with LysMCre did not develop statistically significant disease, excluding the contribution of CD11cCre activity in monotypes to the phenotype observed in Fcgr2bflfl mice with CD11cCre (Figures 1A and 5). The significantly increased bCIA incidence in Fcgr2b conditional knockout mice with either Mb1Cre or CD11cCre demonstrated that multiple cell compartments, including B cells and dendritic cells, are involved in the development of collagen-induced arthritis and the regulation of these cells by FcγRIIB is critical to the maintenance of tolerance.
Increased arthritis severity in mice with GC/post-GC B cell specific deletion of Fcgr2b on a permissive background
We also studied the impact of germline and conditional knockout of Fcgr2b on the maintenance of tolerance in the chicken type II collagen induced arthritis (cCIA) model. In contrast to the bovine type II collagen induced arthritis model, cCIA is permissive in B6 mice because a robust and sustained T cell response can be mounted against chicken type II collagen (34–36). As shown in Figures 6A–B, while wild-type mice developed only mild arthritis, Fcgr2b−/− mice developed significantly more severe arthritis, consistent with previous reports (10). Analysis of conditional knockout lines in this model showed that selective deletion of Fcgr2b in GC/post-GC B cells is sufficient to recapitulate the exacerbated arthritis phenotype in Fcgr2b−/−mice (Figures 6A–B), which is in sharp contrast to the resistance of Fcgr2bfl/fl mice with Cg1Cre to bCIA (Figure 5). Therefore, FcγRIIB expression in GC and/or post-GC B cells plays an important role in inhibiting autoimmunity in permissive models, but not in non-permissive models.
Exacerbated arthritis in mice with selective deletion of Fcgr2b in myeloid effector cells in response to adoptively transferred arthritic sera
In previous studies, FcγRIIB has been shown to play an important role in modulating antibody-medicated effector functions by setting thresholds for immune complex activation of myeloid effector cells, which has been hypothesized to contributes to the maintenance of tolerance (8). We tested this hypothesis in a passive autoimmune model in which K/BxN autoreactive sera are adoptively transferred into mice with germline deletion of Fcgr2b or conditional deletion of Fcgr2b in myeloid effector cells by LysMCre. As shown in Figure 7, administration of K/BxN sera leads to the development of arthritis in wild-type, but not FcγR-deficient (Fcer1g−/−Fcgr2b−/−) mice, whereas in Fcgr2b−/− mice the development of arthritis was accelerated and exacerbated, consistent with the notion that while activating FcγRs are required for the development of K/BxN serum induced arthritis, FcγRIIB negatively regulates antibody-triggered inflammation. LysMCre-mediated deletion of Fcgr2b, although not complete (Figures 1A, 1C), resulted in near total recapitulation of the effect of Fcgr2b germline deletion (Figure 7), suggesting that the myeloid effector cells responsible for joint inflammation are very sensitive to Fcgr2b levels in this passive autoantibody transfer model of inflammation.
DISCUSSION
The development of autoimmunity has been studied in several FcγRIIB-deficient mouse models, initially in Fcgr2b−/− mice derived from 129/Sv ES cells and backcrossed to either the B6 (B6.Fcgr2b129−/−) or BALB/c background (5), and more recently in Fcgr2b−/− mice derived from B6 ES cells (8). The Fcgr2b−/− mice we independently generated from B6 ES cells (Fcgr2bB6−/−) showed significantly attenuated lupus-like phenotypes (proteinuria and premature mortality) as compared to the backcrossed B6.Fcgr2b129−/−(N12) mice (Supplementary Table), consistent with the report of Boross et al (5). This is also consistent with the finding that in addition to FcγRIIB-deficiency, the 129/Sv derived Sle16 locus may be involved in the autoimmune phenotype in B6.Fcgr2b129−/− mice based on the analysis of B6.Fcgr2b129−/− mice with different lengths of 129/Sv DNA segments around the targeted Fcgr2b gene in a spontaneous arthritis model and an induced tolerance model (37, 38). While these studies supported the conclusion from the early studies that Fcgr2b is an epistatic modifier of autoimmunity, it also demonstrated additional susceptibility factors contributed by 129/Sv genes may have contributed to the severe proteinuria and premature mortality phenotypes observed in B6.Fcgr2b129−/− mice (5). At the same time, we also observed several autoimmune phenotypes in Fcgr2bB6−/− mice, including the increased spontaneous ANA antibody levels and susceptibility to bovine or chicken type II collagen induced arthritis. These results, together with the previously reported moderate glomerulonephritis phenotype and increased incidence of anti-GBM diseases in Fcgr2b−/− mice derived from B6 ES cells (8, 39), confirmed that in B6 mice FcγRIIB plays an important role in the maintenance of tolerance.
FcγRIIB is the most widely expressed of all FcγRs and is found on essentially all lymphoid and myeloid subsets with the exception of T and NK cells. This wide expression pattern has made the assignment of specific phenotypes of Fcgr2b deficient mice to defined cellular populations difficult. The collection of Fcgr2b conditional knockout strains generated in this study has provided us an opportunity to dissect the contribution of cell-specific FcγRIIB expression to a long list of FcγRIIB functions proposed based on the studies using Fcgr2b germline knockout mice. In this study, we focused on the function of cell-specific FcγRIIB and its impact on the maintenance of tolerance. It has been hypothesized in a recent study that the contribution of FcγRIIB-deficiency to autoimmunity is mainly through the regulation of antibody effector pathways, such as immune complex mediated inflammation (5). This seems to be true in models that involve adoptive transfer of autoimmune antibodies, such as the serum transfer K/BxN arthritis model in this study, or the NTN model used in a previous study (40), as immune complex mediated inflammation was enhanced in mice with selective deletion of Fcgr2b in myeloid effector cells by LysMCre or CEBPαCre. However, our analysis of Fcgr2bfl/fl mice with LysMCre in collagen induced arthritis models, in which autoreactive antibodies are actively induced, does not support this hypothesis as these mice are not more susceptible than WT mice to either bovine or chicken (data not shown) type II collagen induced arthritis. In contrast, we demonstrated that FcγRIIB expression in both B cells and dendritic cells are important for the maintenance of tolerance in the bovine type II collagen induced arthritis model, and that the contribution of B cell and dendritic FcγRIIB expression to the maintenance of tolerance might be based on different mechanisms.
We found that the effect of FcγRIIB-deficiency on IgG antibody responses is B cell intrinsic, and FcγRIIB functions as a negative regulator in both primary and secondary IgG responses, suggesting that the activation of both resting B cells and memory B cells, in the primary and secondary responses, respectively, are both regulated by FcγRIIB. The deletion of Fcgr2b by B cell specific Mb1Cre leads to significantly enhanced primary and secondary antibody responses, and the deletion of Fcgr2b by GC and post-GC B cell-specific Cg1Cre specifically enhanced secondary antibody response, consistent with the timing when these Cres become active. FcγRIIB-deficiency may contribute to increased antibody response by promoting B cell activation during the early stage and plasma cell survival during the late stage of B cell differentiation (3, 4, 33). The increased antibody responses in Fcgr2bfl/fl mice with Mb1Cre or Cg1Cre could contribute to the increased autoimmune phenotypes. These studies are consistent with the previous study showing that overexpression of a B cell specific Fcgr2b transgene suppressed T-dependent IgG responses, spontaneous lupus and chicken collagen induced arthritis phenotypes (41) and a more recent study showing that FcγRIIB expression from the autoimmunity-associated polymorphic allele was specifically reduced in GC B cells and resulted in a number of autoimmune phenotypes including the development of more severe chicken collagen-induced arthritis (10).
Interestingly, selective deletion of Fcgr2b by Mb1Cre and Cg1Cre, respectively, resulted in different autoimmune phenotypes. Although hypersensitive to chicken type II collagen induced arthritis, Fcgr2bfl/fl mice with Cg1Cre are not susceptible to bovine type II collagen induced arthritis, in contrast to Fcgr2bfl/fl mice with Mb1Cre. While this could be due to the different impact of Mb1Cre and Cg1Cre mediated Fcgr2b deletion on immune responses, it might be also related to the difference in these two different arthritis models. While both collagen induced arthritis models require robust T cell and antibody responses to initiate and perpetuate arthritis, the H-2b background of B6 mice only support such responses against chicken, not bovine type II collagen (34–36). The fact that conditional knockout of Fcgr2b in antigen presenting cells (dendritic cells and B cells) resulted in the break of tolerance in the bovine type II collagen induced arthritis model suggests that these conditional knockouts of Fcgr2b might result in enhanced antigen presentation, which may lead to the observed increase in T cell and primary antibody responses in these mice and autoimmunity. On the other hand, Cg1Cre mediated deletion of Fcgr2b after B cell activation only results in increased secondary antibody response that is sufficient to enhance autoimmune response in the permissive chicken type II collagen induced arthritis model where T cell tolerance is already broken. This notion is supported by increased anti-mouse type II collagen IgG levels in Fcgr2bfl/fl mice with Cg1Cre (Supplementary Figure 3).
We also confirmed previous studies showing that FcγRIIB expression in dendritic cells can inhibit T cell response (42), presumably by regulating dendritic cell maturation and antigen presentation. This is consistent with other studies showing that selective blockade of FcγRIIB can promote dendritic cell maturation and T cell responses (43–45). Previous studies using B6.Fcgr2b129−/− mice in the EAE model suggested the impact of FcγRIIB expression on T cell response could contribute to the maintenance of tolerance (46). Our study, together with that of van Montfoort et al., established that the increased T cell response in FcγRIIB-deficient B6 mice may contributes to autoimmunity (30). In addition, FcγRIIB has been previously shown to set thresholds for immune complex triggered inflammation in a number of animal models presumably by regulating myeloid effector cells. In this study, partial deletion of FcγRIIB on myeloid effector cells leads to significantly exacerbated arthritis triggered by K/BxN sera, suggesting that myeloid effector cells are very sensitive to the regulation by FcγRIIB levels, in agreement with our finding that increased FcγRIIB expression in myeloid effector cells in response to IVIG is responsible for its significant anti-inflammatory effects in vivo(29, 47, 48).
Our data also showed that quantitative changes in immune responses due to selective deletion of Fcgr2b can result in significant difference in autoimmune models. In corroboration with this notion, previous studies have shown a quantitative increase in TLR7 expression due to gene duplication can accelerate the development of autoimmune diseases (49, 50). These findings suggest that the maintenance of tolerance involves many checkpoints that do not qualitatively but quantitatively regulate immune system at various levels, highlighting the importance of the balance in Immunoregulatory networks.
Taken together, through the analysis of a collection of novel FcγRIIB conditional knockout strains with specific deletion of Fcgr2b in defined cellular compartments, we demonstrated that FcγRIIB expression in multiple cellular compartment is required for the maintenance of peripheral tolerance through different mechanisms, and FcγRIIB expression in the same cell lineage (B cells) but at different differentiation stages also have different impact on the maintenance of tolerance. This collection of FcγRIIB conditional knockout strains are likely useful to investigate other functions assigned to FcγRIIB. For instance, FcγRIIB co-engagement has been recently found to be necessary for the in vivo activities of agonistic antibodies to the TNF receptor family members, such as CD40 and DR5 (51, 52), and these conditional Fcgr2b knockout mice might be also useful to dissect the contribution of cell-specific FcγRIIB to the activities of these antibodies.
Supplementary Material
Acknowledgments
We are grateful to Dr. Michel C. Nussenzweig for helpful discussions. We acknowledge the assistance of the Rockefeller University Gene Targeting Facility in the generation of Fcgr2b−/− and Fcgr2bfl/fl mice; and K. Horiuchi, R. Peraza, I. Londono, M. Kibe, and J. Carroll for expert technical assistance. The authors have no conflict of interest. F.L., P.S., and J.V.R. designed the experiments; F.L. and P.S. performed the experiments; F.L. and J.V.R. wrote the paper.
Footnotes
This work was supported by grants from NIH to J.V.R. F.L. is supported in part by NNSFC project No. 31370934, 973 program 2014CB943600, the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and Grant no. 2757 from the Paralyzed Veterans of America research foundation.
References
- 1.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nature reviews Immunology. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 2.Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nature reviews Immunology. 2010;10:328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fukuyama H, Nimmerjahn F, Ravetch JV. The inhibitory Fcgamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nature immunology. 2005;6:99–106. doi: 10.1038/ni1151. [DOI] [PubMed] [Google Scholar]
- 4.Xiang Z, Cutler AJ, Brownlie RJ, Fairfax K, Lawlor KE, Severinson E, Walker EU, Manz RA, Tarlinton DM, Smith KG. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nature immunology. 2007;8:419–429. doi: 10.1038/ni1440. [DOI] [PubMed] [Google Scholar]
- 5.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13:277–285. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- 6.Yuasa T, Kubo S, Yoshino T, Ujike A, Matsumura K, Ono M, Ravetch JV, Takai T. Deletion of fcgamma receptor IIB renders H-2(b) mice susceptible to collagen-induced arthritis. The Journal of experimental medicine. 1999;189:187–194. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakamura A, Yuasa T, Ujike A, Ono M, Nukiwa T, Ravetch JV, Takai T. Fcγ receptor IIB-deficient mice develop Goodpasture’s syndrome upon immunization with type IV collagen: a novel murine model for autoimmune glomerular basement membrane disease. J Exp Med. 2000;191:899–906. doi: 10.1084/jem.191.5.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boross P, V, Arandhara L, Martin-Ramirez J, Santiago-Raber ML, Carlucci F, Flierman R, van der Kaa J, Breukel C, Claassens JW, Camps M, Lubberts E, Salvatori D, Rastaldi MP, Ossendorp F, Daha MR, Cook HT, Izui S, Botto M, Verbeek JS. The inhibiting Fc receptor for IgG, FcgammaRIIB, is a modifier of autoimmune susceptibility. Journal of immunology. 2011;187:1304–1313. doi: 10.4049/jimmunol.1101194. [DOI] [PubMed] [Google Scholar]
- 9.Pritchard NR, Cutler AJ, Uribe S, Chadban SJ, Morley BJ, Smith KG. Autoimmune-prone mice share a promoter haplotype associated with reduced expression and function of the Fc receptor FcgammaRII. Curr Biol. 2000;10:227–230. doi: 10.1016/s0960-9822(00)00344-4. [DOI] [PubMed] [Google Scholar]
- 10.Espeli M, Clatworthy MR, Bokers S, Lawlor KE, Cutler AJ, Kontgen F, Lyons PA, Smith KG. Analysis of a wild mouse promoter variant reveals a novel role for FcgammaRIIb in the control of the germinal center and autoimmunity. The Journal of experimental medicine. 2012 doi: 10.1084/jem.20121752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGaha TL, Sorrentino B, Ravetch JV. Restoration of tolerance in lupus by targeted inhibitory receptor expression. Science. 2005;307:590–593. doi: 10.1126/science.1105160. [DOI] [PubMed] [Google Scholar]
- 12.Bolland S, Yim YS, Tus K, Wakeland EK, Ravetch JV. Genetic modifiers of systemic lupus erythematosus in FcgammaRIIB(−/−) mice. The Journal of experimental medicine. 2002;195:1167–1174. doi: 10.1084/jem.20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crampton SP, Deane JA, Feigenbaum L, Bolland S. Ifih1 gene dose effect reveals MDA5-mediated chronic type I IFN gene signature, viral resistance, and accelerated autoimmunity. Journal of immunology. 2012;188:1451–1459. doi: 10.4049/jimmunol.1102705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGaha TL, Karlsson MC, Ravetch JV. FcgammaRIIB deficiency leads to autoimmunity and a defective response to apoptosis in Mrl-MpJ mice. Journal of immunology. 2008;180:5670–5679. doi: 10.4049/jimmunol.180.8.5670. [DOI] [PubMed] [Google Scholar]
- 15.Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, Ravetch JV, Diamond B. Selective dysregulation of the FcgammaIIB receptor on memory B cells in SLE. The Journal of experimental medicine. 2006;203:2157–2164. doi: 10.1084/jem.20051503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su K, Wu J, Edberg JC, Li X, Ferguson P, Cooper GS, Langefeld CD, Kimberly RP. A promoter haplotype of the immunoreceptor tyrosine-based inhibitory motif-bearing FcgammaRIIb alters receptor expression and associates with autoimmunity. I. Regulatory FCGR2B polymorphisms and their association with systemic lupus erythematosus. Journal of immunology. 2004;172:7186–7191. doi: 10.4049/jimmunol.172.11.7186. [DOI] [PubMed] [Google Scholar]
- 17.Kyogoku C, Dijstelbloem HM, Tsuchiya N, Hatta Y, Kato H, Yamaguchi A, Fukazawa T, Jansen MD, Hashimoto H, van de Winkel JG, Kallenberg CG, Tokunaga K. Fcgamma receptor gene polymorphisms in Japanese patients with systemic lupus erythematosus: contribution of FCGR2B to genetic susceptibility. Arthritis Rheum. 2002;46:1242–1254. doi: 10.1002/art.10257. [DOI] [PubMed] [Google Scholar]
- 18.Li X, Wu J, Carter RH, Edberg JC, Su K, Cooper GS, Kimberly RP. A novel polymorphism in the Fcgamma receptor IIB (CD32B) transmembrane region alters receptor signaling. Arthritis Rheum. 2003;48:3242–3252. doi: 10.1002/art.11313. [DOI] [PubMed] [Google Scholar]
- 19.Kono H, Kyogoku C, Suzuki T, Tsuchiya N, Honda H, Yamamoto K, Tokunaga K, Honda Z. FcgammaRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet. 2005;14:2881–2892. doi: 10.1093/hmg/ddi320. [DOI] [PubMed] [Google Scholar]
- 20.Floto RA, Clatworthy MR, Heilbronn KR, Rosner DR, MacAry PA, Rankin A, Lehner PJ, Ouwehand WH, Allen JM, Watkins NA, Smith KG. Loss of function of a lupus-associated FcgammaRIIb polymorphism through exclusion from lipid rafts. Nature medicine. 2005;11:1056–1058. doi: 10.1038/nm1288. [DOI] [PubMed] [Google Scholar]
- 21.Baerenwaldt A, Lux A, Danzer H, Spriewald BM, Ullrich E, Heidkamp G, Dudziak D, Nimmerjahn F. Fcgamma receptor IIB (FcgammaRIIB) maintains humoral tolerance in the human immune system in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18772–18777. doi: 10.1073/pnas.1111810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochemical and biophysical research communications. 1997;237:318–324. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- 23.Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M. Testing gene function early in the B cell lineage in mb1-cre mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, Waisman A, Egert A, Ghitza D, Rajewsky K. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7396–7401. doi: 10.1073/pnas.0602353103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. The Journal of experimental medicine. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic research. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 27.Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030–1034. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brand DD, Latham KA, Rosloniec EF. Collagen-induced arthritis. Nature protocols. 2007;2:1269–1275. doi: 10.1038/nprot.2007.173. [DOI] [PubMed] [Google Scholar]
- 29.Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature. 2011;475:110–113. doi: 10.1038/nature10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Montfoort N, t Hoen PA, Mangsbo SM, Camps MG, Boross P, Melief CJ, Ossendorp F, Verbeek JS. Fcgamma receptor IIb strongly regulates Fcgamma receptor-facilitated T cell activation by dendritic cells. Journal of immunology. 2012;189:92–101. doi: 10.4049/jimmunol.1103703. [DOI] [PubMed] [Google Scholar]
- 31.Eftychi C, Karagianni N, Alexiou M, Apostolaki M, Kollias G. Myeloid TAKL acts as a negative regulator of the LPS response and mediates resistance to endotoxemia. PloS one. 2012;7:e31550. doi: 10.1371/journal.pone.0031550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho IH, Hong J, Suh EC, Kim JH, Lee H, Lee JE, Lee S, Kim CH, Kim DW, Jo EK, Lee KE, Karin M, Lee SJ. Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death. Brain : a journal of neurology. 2008;131:3019–3033. doi: 10.1093/brain/awn230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- 34.Pan M, Kang I, Craft J, Yin Z. Resistance to development of collagen-induced arthritis in C57BL/6 mice is due to a defect in secondary, but not in primary, immune response. Journal of clinical immunology. 2004;24:481–491. doi: 10.1023/B:JOCI.0000040919.16739.44. [DOI] [PubMed] [Google Scholar]
- 35.Seki N, Sudo Y, Yoshioka T, Sugihara S, Fujitsu T, Sakuma S, Ogawa T, Hamaoka T, Senoh H, Fujiwara H. Type II collagen-induced murine arthritis. I. Induction and perpetuation of arthritis require synergy between humoral and cell-mediated immunity. Journal of immunology. 1988;140:1477–1484. [PubMed] [Google Scholar]
- 36.Inglis JJ, Criado G, Medghalchi M, Andrews M, Sandison A, Feldmann M, Williams RO. Collagen-induced arthritis in C57BL/6 mice is associated with a robust and sustained T-cell response to type II collagen. Arthritis research & therapy. 2007;9:R113. doi: 10.1186/ar2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato-Hayashizaki A, Ohtsuji M, Lin Q, Hou R, Ohtsuji N, Nishikawa K, Tsurui H, Sudo K, Ono M, Izui S, Shirai T, Takai T, Nishimura H, Hirose S. Presumptive role of 129 strain-derived Sle16 locus in rheumatoid arthritis in a new mouse model with Fcgamma receptor type IIb-deficient C57BL/6 genetic background. Arthritis Rheum. 2011;63:2930–2938. doi: 10.1002/art.30485. [DOI] [PubMed] [Google Scholar]
- 38.Fujii T, Hou R, Sato-Hayashizaki A, Obata M, Ohtsuji M, Ikeda K, Mitsui K, Kodera Y, Shirai T, Hirose S, Nishimura H. Susceptibility loci for the defective foreign protein-induced tolerance in New Zealand Black mice: implication of epistatic effects of Fcgr2b and Slam family genes. European journal of immunology. 2011;41:2333–2340. doi: 10.1002/eji.201141552. [DOI] [PubMed] [Google Scholar]
- 39.Sharp PE, Martin-Ramirez J, Boross P, Mangsbo SM, Reynolds J, Moss J, Pusey CD, Cook HT, Tarzi RM, Verbeek JS. Increased incidence of anti-GBM disease in Fcgamma receptor 2b deficient mice, but not mice with conditional deletion of Fcgr2b on either B cells or myeloid cells alone. Molecular immunology. 2012;50:49–56. doi: 10.1016/j.molimm.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 40.Sharp PE, Martin-Ramirez J, Mangsbo SM, Boross P, Pusey CD, Touw IP, Cook HT, Verbeek JS, Tarzi RM. FcgammaRIIb on myeloid cells and intrinsic renal cells rather than B cells protects from nephrotoxic nephritis. Journal of immunology. 2013;190:340–348. doi: 10.4049/jimmunol.1202250. [DOI] [PubMed] [Google Scholar]
- 41.Brownlie RJ, Lawlor KE, Niederer HA, Cutler AJ, Xiang Z, Clatworthy MR, Floto RA, Greaves DR, Lyons PA, Smith KG. Distinct cell-specific control of autoimmunity and infection by FcgammaRIIb. The Journal of experimental medicine. 2008;205:883–895. doi: 10.1084/jem.20072565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Montfoort ML, Stephan F, Lauw MN, Hutten BA, Van Mierlo GJ, Solati S, Middeldorp S, Meijers JC, Zeerleder S. Circulating nucleosomes and neutrophil activation as risk factors for deep vein thrombosis. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:147–151. doi: 10.1161/ATVBAHA.112.300498. [DOI] [PubMed] [Google Scholar]
- 43.Dhodapkar KM, Banerjee D, Connolly J, Kukreja A, Matayeva E, Veri MC, Ravetch JV, Steinman RM, Dhodapkar MV. Selective blockade of the inhibitory Fcgamma receptor (FcgammaRIIB) in human dendritic cells and monocytes induces a type I interferon response program. The Journal of experimental medicine. 2007;204:1359–1369. doi: 10.1084/jem.20062545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, Steinman RM, Ravetch JV, Dhodapkar MV. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2910–2915. doi: 10.1073/pnas.0500014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells. The Journal of experimental medicine. 2002;195:1653–1659. doi: 10.1084/jem.20020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iruretagoyena MI, Riedel CA, Leiva ED, Gutierrez MA, Jacobelli SH, Kalergis AM. Activating and inhibitory Fcgamma receptors can differentially modulate T cell-mediated autoimmunity. European journal of immunology. 2008;38:2241–2250. doi: 10.1002/eji.200838197. [DOI] [PubMed] [Google Scholar]
- 47.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science. 2001;291:484–486. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 48.Kaneko Y, Nimmerjahn F, Madaio MP, Ravetch JV. Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. The Journal of experimental medicine. 2006;203:789–797. doi: 10.1084/jem.20051900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 50.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, Schultz RA, Wakeland EK. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li F, Ravetch JV. A general requirement for FcgammaRIIB co-engagement of agonistic anti-TNFR antibodies. Cell cycle. 2012:11. doi: 10.4161/cc.21842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.White AL, Chan HT, French RR, Beers SA, Cragg MS, Johnson PW, Glennie MJ. FcgammaRIIB controls the potency of agonistic anti-TNFR mAbs. Cancer immunology, immunotherapy : CII. 2013;62:941–948. doi: 10.1007/s00262-013-1398-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.