

Abstract
Background:
Microarrays have been used extensively to profile transcriptome remodeling in failing human heart, although the genomic coverage provided is limited and fails to provide a detailed picture of the myocardial transcriptome landscape. Here, we describe sequencing-based transcriptome profiling, providing comprehensive analysis of myocardial mRNA, microRNA (miRNA) and long non-coding RNA (lncRNA) expression in failing human heart, before and after mechanical support with a left ventricular assisted device (LVAD)
Methods and Results:
Deep sequencing of RNA isolated from paired non-ischemic (NICM,n=8) and ischemic (ICM,n=8) human failing LV samples collected pre- and post-LVAD, as well as from non-failing human LV (n=8), was conducted. These analyses revealed high abundance of mRNA (37%) and lncRNA (71%) of mitochondrial origin. miRNASeq revealed 160 and 147 differentially expressed miRNAs in ICM and NICM, respectively, compared with non-failing LV. Among these only 2 (ICM) and 5 (NICM) miRNAs are normalized with LVAD. RNASeq detected 18480, including 113 novel, lncRNAs in human LV. Among the 679 (ICM) and 570 (NICM) lncRNAs differentially expressed with heart failure, ~10% are improved or normalized with LVAD. In addition, the expression signature of lncRNAs, but not miRNAs or mRNAs, distinguishes cardiomyopathy of ischemic and non-ischemic origins. Further analysis suggests that cis-gene regulation represents a major mechanism of action of human cardiac lncRNAs.
Conclusions:
The myocardial transcriptome is dynamically regulated in advanced heart failure and following LVAD support. The expression profiles of lncRNAs, but not mRNAs or miRNAs, can discriminate failing hearts of different etiologies and are markedly altered in response to LVAD support. These results suggest an important role for lncRNAs in the pathogenesis of heart failure and in reverse remodeling observed with mechanical support.
Keywords: Next-generation sequencing, noncoding RNA, heart failure, mechanical circulatory support
Introduction
Transcriptional profiling has been utilized extensively in human heart failure (HF) to gain new insights into complex disease pathways,1 to identify biomarkers for better diagnostic and prognostic accuracy,2 and to examine the impact of therapeutic treatments, including medications and implanted devices.3-5 Although intriguing results have been generated, the genomic coverage provided in most, if not all, of these studies was limited owing to use of conventional microarray or PCR-based assays. In addition, it has also become increasingly clear that the transcription of the eukaryotic genome is far more pervasive and complex than previously appreciated.6 Indeed, present estimates are that up to 90% of the mammalian genome is transcribed, although messenger RNAs (mRNAs) and microRNAs (miRNAs), the main transcript species targeted in previous myocardial transcriptional profiling studies, account for only ~1% of all transcribed species.6 A large proportion of the mammalian genome is transcribed as long non-coding RNAs (lncRNAs), a heterogeneous group of non-coding transcripts longer than 200 nucleotides, residing within or between (long intergenic RNA, lincRNA) coding genes.7,8 In addition, many lncRNAs have been shown to be functional and involved in specific physiological and pathological processes, through transcriptional or post-transcriptional regulatory mechanisms.9-11 To date, however, lncRNAs have never been included in analyses of the human myocardial transcriptome. We sought to determine whether a more comprehensive myocardial transcript profiling, encompassing cardiac coding (mRNA) and non-coding (miRNA and lncRNA) transcriptomes, would provide a more complete picture of the myocardial transcriptional landscape in heart failure, and whether these changes were dynamically regulated following hemodynamic unloading with a left ventricular assist device (LVAD).
In the studies presented here, a molecular and bioinformatic pipeline optimized for comprehensive analysis and quantification of myocardial mRNA, miRNA and lncRNA expression with next-generation sequencing was developed. The LV myocardial transcriptional signatures of ischemic cardiomyopathy (ICM) and non-ischemic cardiomyopathy (NICM), before and after mechanical circulatory support with an LVAD, were analyzed and compared with those of non-failing human LV samples. These analyses revealed that while the coding and noncoding transcriptomes are each dynamically altered with advanced HF, lncRNA expression profiles are more sensitive to different HF etiologies when compared with mRNA or miRNA expression profiles. In addition, the pathological expression pattern of lncRNAs associated with cardiomyopathy improves in response to LVAD support to a greater extent than that of either mRNAs or miRNAs, suggesting a biological role for lncRNAs in mechanical circulatory support-induced reverse remodeling. Finally, leveraging the unprecedented comprehensive human myocardial transcriptome data obtained, we demonstrate that cis-, rather than trans-, gene regulation likely is the predominant mechanism of action of cardiac lncRNAs. Taken together, the results presented here provide a transcriptome blueprint to identify novel molecular targets and pathways in human heart failure, as well as new insights into the mechanisms involved in the reverse remodeling of myocardial dysfunctions in cardiomyopathy associated with mechanical circulatory support.
Methods
An expanded Methods section is available in the Online Data Supplement.
All high-throughput sequencing data have been submitted to the NCBI gene expression and hybridization array data repository (GEO ID:GSE46224).
All studies were conducted in accordance with protocols approved by the Washington University and Columbia University Institutional Review Boards.
Human Left Ventricular Tissue Acquisition and RNA Extraction
Paired LV samples from patients with severe heart failure of ischemic (ICM, n=8) and non-ischemic (NICM, n=8) origin were collected at the time of LVAD implantation (pre-LVAD) and during cardiac transplant (post-LVAD). Clinical parameters and the medical histories of each subject are summarized in Supplemental Table S1. Non-failing human LV samples (n=8) were collected from donor hearts, deemed not suitable for transplantation for technical or non-cardiac reasons, from Mid-America Transplant Services (Supplemental Table S2). Transmural LV tissue samples were collected from the LV apex, and total RNA was isolated from individual samples and quality controlled using described methods.12,13
RNA Library Preparation, Sequencing and Data Processing
Barcoded RNA libraries were prepared from 3μg of total RNA of each LV sample using TrueSeq RNA Sample Prep Kits (Illumina, CA) in accordance with the manufacturer’s recommendations. To minimize lane and batch effects in RNASeq experiments, barcoded libraries prepared from NF, paired ICM and NICM samples were mixed and pooled in equimolar (10 nmol/L) amounts and diluted to 4 pmol/L for cluster formation on a single flow cell lane, followed by pair-end sequencing on an Illumina HiSeq 2000 sequencer.
After separating the multiplexed sequencing data, adapter sequences were removed and the individual libraries were converted to the FASTQ format. Sequence reads were aligned to the human genome (hg19) with TopHat14 and the resulting alignment files were reconstructed using Cufflinks.15 For mRNA analyses, the RefSeq and Ensembl transcript databases were chosen as the annotation references. For lncRNA analyses, we generated an annotation database that includes lncRNAs from the NONCODE 3.0 human non-coding RNA database,16 the Human Body Map lincRNAs catalog,17 and the 113 novel lncRNAs identified in the human LV samples analyzed here (See Results). The read counts of each transcript were normalized to the length of the individual transcript and to the total mapped read counts in each sample and expressed as RPKM (Reads Per Kilobase of exon per Million mapped reads). Sequence reads mapped to different isoforms of individual genes were pooled together for subsequent comparative analyses.
Small RNA Library Construction, Sequencing and Data Processing
Small RNA libraries were prepared from 1 μg total RNA of each LV sample using TrueSeq Small RNA Sample Prep Kits (Illumina) in accordance with the manufacturer’s instructions. Barcoded libraries prepared from NF, paired ICM and NICM samples were mixed and pooled in equimolar (10 nmol/L) amounts and diluted to 8 pmol/L for cluster formation on a single flow cell lane, followed by single-end sequencing on an Illumina HiSeq 2000 sequencer.
Using miRanalyzer 18 and Bowtie,19 sequence reads were mapped miRBase20v.18 human database to detect known miRNA. The read counts of each known miRNA were then normalized to the total counts of sequence reads mapped to miRBase v.18 database and are presented as PMMR (sequences Per Million Mapped Reads).
Sequencing Data Analyses and Statistical Methods
Cuffdiff 2.0 (mRNA and lncRNA) and miRAnalyzer (miRNA) were utilized for differential expression analyses of the NF, compared with the ICM or NICM samples, where normalized read counts, fold change, and P values (corrected for multiple testing) of each transcript were reported (see Results). The statistical significance of differences between paired heart failure (HF) samples before and after LVAD support was evaluated by paired sample Wilcoxon signed rank test, where a two-tailed P value<0.05 was considered statistically significant. Gene symbols and PMMR and RPKM values were imported into MultiExperiment Viewer (MeV v4.7.4) for preparation of heat-map and hierarchical clustering analyses. Partek Genomics Suite version 6.5 (Partek, St Louis, MO) was used for principal component analyses. Correlation coefficients for comparisons of miRNA and mRNA expression between biological replicates were calculated using Excel (Microsoft).
Results
Deep RNA Sequencing Identified Novel Human Cardiac lncRNAs and Revealed Distinct Expression Signatures of Coding and Non-coding RNAs in Human LV
A total of 40 barcoded RNA and small RNA libraries were prepared from human LV samples, including paired pre-LVAD and post-LVAD LV samples from patients with ICM (n=8) and NICM (n=8); the time between collection of these samples varied from 111 to 690 days, with an average time 305±50 days. Non-failing (NF) human LV samples (n=8) were also obtained from donor hearts. From these samples, a total of 572,026,460 read pairs and 507,195,283 reads were generated from RNASeq and miRNASeq experiments, respectively (Table 1). For RNASeq, more than 500 million read pairs (89.0%) were aligned to the human genome (hg19), where ~287 million (56.6%) mapped within exons, ~19.7 million (3.9%) mapped to introns and ~166 million (32.8%) mapped to intergenic regions. For miRNASeq, ~507 million reads (97.7%) were uniquely aligned to the human genome, ~333 million (66.1%) mapped to known miRNAs, and ~71 million (14.1%) to RNA species other than miRNA.
Table 1.
Summary of RNA- and miRNASeq read counts and mapping results
| RNASeq Read Pairs |
NF (n=8) |
ICM pre-LVAD (n=8) |
ICM post-LVAD (n=8) |
NICM pre-LVAD (n=8) |
NICM post-LVAD (n=8) |
Total |
|---|---|---|---|---|---|---|
| Total | 110618151 | 123220366 | 102603539 | 111812494 | 123771910 | 572026460 |
| Uniquely mapped |
98707139 (89.2%) |
109496189 (88.9%) |
91373026 (89.1%) |
99767663 (89.2%) |
109455159 (88.4%) |
508799176 (89.0%) |
| Mapped within exons |
53202618 (53.9%) |
62803455 (57.4%) |
49216082 (53.9%) |
55862334 (56.0%) |
66813974 (61.0%) |
287898462 (56.6%) |
| Mapped within introns |
3540222 (3.6%) |
3850216 (3.5%) |
3464881 (3.8%) |
3730959 (3.7%) |
5085069 (4.7%) |
19671347 (3.9%) |
| Mapped to intergenic regions |
35841497 (36.3%) |
35437442 (32.4%) |
32790010 (35.9%) |
33596835 (33.7%) |
29284167 (26.8%) |
166949951 (32.8%) |
|
| ||||||
| miRNASeq Read Counts |
NF (n=8) |
ICM pre-LVAD (n=8) |
ICM post-LVAD (n=8) |
NICM pre-LVAD (n=8) |
NICM post-LVAD (n=8) |
Total |
|
| ||||||
| Total | 96645624 | 101702266 | 107449757 | 102917401 | 98480235 | 507195283 |
| Uniquely mapped |
94296940 (98.2%) |
98472955 (97.3%) |
104312124 (97.5%) |
99583887 (97.3%) |
96290979 (98.3%) |
492956885 (97.7%) |
| Known miRNAs |
66756546 (69.5%) |
60103247 (59.4%) |
66245884 (61.9%) |
68378019 (66.8%) |
72042543 (73.6%) |
333526239 (66.1%) |
| Mapped to other RNA species |
9210690 (9.6%) |
17164602 (17.0%) |
15445738 (14.4%) |
17619257 (17.2%) |
11812904 (12.1%) |
71253191 (14.1%) |
NF: non-failing; ICM: ischemic cardiomyopathy; NICM: non-ischemic cardiomyopathy; LVAD: left ventricular assist device
RNASeq reads first underwent transcriptome reconstruction using Cufflinks, which identified 2049 novel transcripts that do not overlap with any known coding or non-coding genes (Figure 1), 158 of which are multi-exon transcripts. Among these novel multi-exon transcripts, 118 were found to have low coding potentials and all but 5 were expressed at ≥0.5 RPKM in ≥2 individual samples. Among these 113 transcripts determined as novel human cardiac lncRNAs, 100 are located in intergenic regions, and 13 intersect with known genes. The genomic loci, transcript lengths and normalized read counts of each of these novel lncRNAs in individual LV samples are summarized in Supplemental Table S3. To explore the possible correlation between epigenetic modification and cardiac lncRNA expression, we examined the epigenetic markers and DNaseI hypersensitivity information reported by the ENCODE project21 at the genomic regions encoding the 18480 lncRNAs detected in human LV samples analyzed here (Figure 1). Most of these genomic regions contain DNaseI hypersensitivity loci (n=13418,72.6%), as well as epigenetic markers signaling active transcription, including trimethylation at H3K4 (H3K4me3, n=18311,99%) and acetylation at H3K27 (H3K27Ac, n=17164,92.9%). These findings are consistent with previous suggestions that lncRNAs are encoded from genomic regions with active transcriptional activity.22
Figure 1.
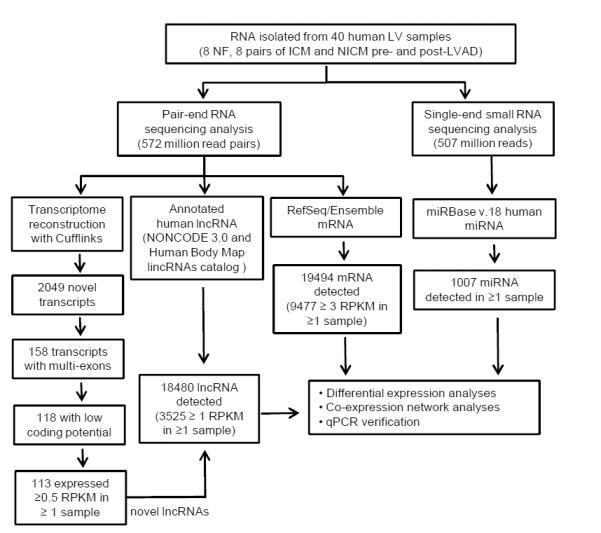
Workflow of comprehensive quantification of miRNAs, mRNAs and lncRNAs in human LV. Total RNA was isolated from non-failing (n=8) LV, and from ICM (n=8) and NICM (n=8) LV, before and after LVAD support; the latter were matched samples from the same patient. Poly-A(+) RNA libraries were constructed for pair-end RNA sequencing, whereas small RNA libraries were prepared for single-end sequencing. Pair-end sequencing reads went through transcriptome reconstruction and computational prediction pipelines to identify novel transcripts; among the 2049 novel transcripts uncovered by RNASeq, 113 were identified as novel lncRNAs. A compiled lncRNA annotation database, including NONCODE3.0, the Human Body Map lincRNAs catalog and the 113 novel lncRNAs identified in this study, was generated and utilized for quantitatve analyses of lncRNAs. RefSeq and the Ensemble database were used for quantification of mRNAs, and miRBase v.18 was used for miRNA quantification analyses. Results from RNASeq were subjected to differential expression and co-expression network analyses. In addition, quantitative PCR analysis of selected transcripts was conducted and compared with the results from RNASeq.
For transcript expression quantification analyses in RNASeq experiments, read counts mapped to different isoforms of individual mRNA or lncRNA were pooled together to calculate the RPKM value of each gene. For miRNASeq, normalized read counts of each of the annotated miRNAs were presented as PMMR (see Methods). Scatter plots of the normalized read counts of all mRNA/lncRNA (β=1.00, R2 >0.99 Supplemental Figure 1) and miRNA (β=1.06, R2 >0.99, Supplemental Figure 2) showed high degrees of correlation between technical replicates, indicating high levels of reproducibility. Similar to the reports from our23 and other24 groups, quantification of transcripts by deep sequencing is highly correlated with the results derived from qPCR analyses (Supplemental Figure 3), reflecting the accuracy and reliability of deep sequencing analyses.
Using the criterion that a miRNA sequence must be detected in ≥2 small RNA libraries, miRNASeq reads were mapped to 1007 mature miRNAs reported in miRBase v.18 (Figure 1 and Supplemental Table S4). All lncRNAs expressed at ≥1 RPKM and all mRNAs expressed at ≥3 RPKM, in at least 2 different samples, are listed in Supplemental Tables S5 and S6, respectively. Strikingly, mRNAs and lncRNAs encoded by mitochondrial DNA constitute the majority of the cardiac mRNAs and lncRNAs, accounting for 37% and 71% of the total cardiac mRNA and lncRNA read counts, respectively (Figure 2A, B). While non-mitochondrial mRNAs are evenly distributed among high (mean RPKM>3, 40%), intermediate (mean RPKM 1-3, 18%) and low (mean RPKM<1, 42%) abundance groups (Figure 2D), the majority (88%) of non-mitochondrial lncRNAs detected in human LV samples are expressed at <1 RPKM, and only 6% of all cardiac non-mitochondrial lncRNAs are expressed at ≥3 RPKM (Figure 2E). In contrast to mRNAs and lncRNAs, all of the annotated cardiac miRNAs identified are non-mitochondrial; 54% of cardiac miRNAs are expressed at low (<1 PMMR) levels and 37% at high (≥3 PMMR) expression levels (Figure 2F). The 20 most abundant non-mitochondrial mRNAs, lncRNAs and mature miRNAs accounted for 24.6%, 48.7% and 81.8% of total non-mitochondrial mRNA, lncRNA and miRNA reads in human LV, respectively (Figure 2G-I). The striking distinctions in the relative abundances, expression patterns and types of RNA species in human LV are consistent with the multi-faceted roles of RNA, as well as with the suggestion that the biological functions of lncRNAs are very different from those of mRNAs and miRNAs (see Discussion).
Figure 2.
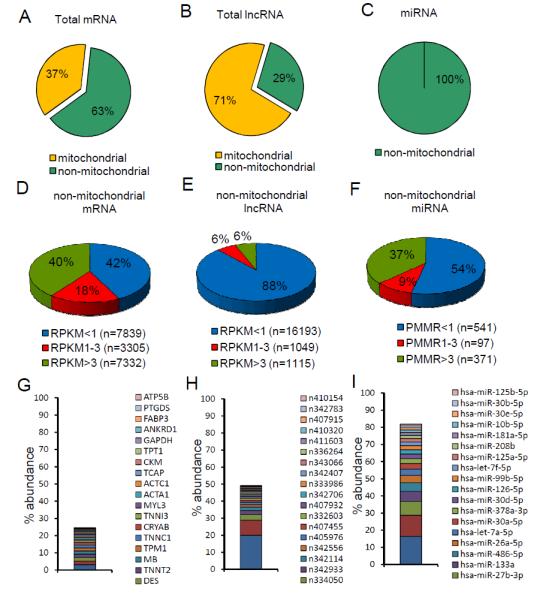
RNASeq reveals distinct expression pattern of different RNA species in human hearts. (A-C)Pie charts showing read count distributions of mitochondrial and non-mitochondrial mRNAs (A), lncRNAs (B) and miRNAs (C) in human LV as percentages of the total read counts. (D-F)Pie charts illustrating the percent distributions of non-mitochondrial mRNAs (D), lncRNAs (E) and miRNAs (F) in human LV based on abundance. (G-H)The top 20 most abundant non-mitochondrial mRNAs (G), lncRNAs (H) and miRNAs (I) account for markedly different percentages of the total mRNA, lncRNA or miRNA species identified in human LV.
Expression Signature of lncRNAs, but not mRNAs or miRNAs, Differentiates Ischemic and non-Ischemic Human Failing Hearts
Unsupervised hierarchical clustering of the expression profiles of cardiac mRNAs, miRNAs and lncRNAs (Figure 3A-C) revealed a distinct expression signature of all three RNA species in the cardiomyopathic, compared to the NF, samples. Principal component analyses also revealed that mRNA, miRNA and lncRNA expression profiles distinguish failing from non-failing LV (Figure 3D-F). Interestingly, lncRNA expression profiles also largely distinguish the cardiomyopathic samples of ischemic and non-ischemic origin, with 7 of the 8 NICM and 7 of the 8 ICM samples classified correctly (Figure 3C). The profiles of miRNA and mRNA expression, in contrast, do not distinguish ischemic and non-ischemic cardiomyopathic samples (Figure 3A,B). Principal component analyses also revealed the overlap in both mRNAs (Figure 3D) and miRNAs (Figure 3E) in the ICM and NICM samples. To exclude the possibility that the marked difference in the discriminatory power between mRNA and lncRNA resulted from the different low-abundance filtering criteria (≥3 RPKM for mRNA and ≥1 RPKM for lncRNA) used, we lowered the filtering criterion for mRNAs to ≥1 RPKM and conducted a similar sample clustering analysis with the mRNA dendrogram. As shown in Supplemental Figure 4, using mRNAs expressed at ≥1 RPKM, the same criterion applied to lncRNAs for sample clustering, did not improve the power to discriminate between ICM and NICM samples. This result indicates that the exquisite sensitivity of lncRNA profiles to distinguish between cardiomyopathic samples cannot be explained by usage of different low-abundance filtering criteria. Taken together, these data suggest that lncRNA expression profiles are more sensitive than those of either miRNA or mRNA in discriminating advanced heart failure of different etiologies (see Discussion). It has previously been reported that the expression profiles of subsets of mRNAs2 or miRNAs25 are useful in discriminating ICM from NICM. To explore the hypothesis that similar discriminatory power would be revealed in our data, we conducted additional analyses on subsets of mRNAs and miRNAs using the panel of 90 genes reported by Kitleson et al,2 as well as the panel of 29 miRNAs reported by Ikeda et al,25 to cluster the samples in a mRNA (Supplemental Figure 5A) or miRNA dendrogram (Supplemental Figure 5B). Analyses of these reduced subsets of mRNA or miRNA, however, did not discriminate between ICM and NICM samples, possibly due to the differences in the platforms of gene profiling (i.e., microarray versus deep sequencing of gene profiling used). Interestingly, using only the 100 most differentially-expressed mRNAs identified here to cluster these samples (Supplemental Figure 6) resulted in improved discriminatory power of mRNA profiles and ICM/NICM samples were more clearly separated (only 2 ICM and 2 NICM samples were misclassified). These results suggest that stringent selection criteria are needed when choosing mRNA/miRNA classifier genes to discriminate between cardiomyopathies of different causes.
Figure 3.
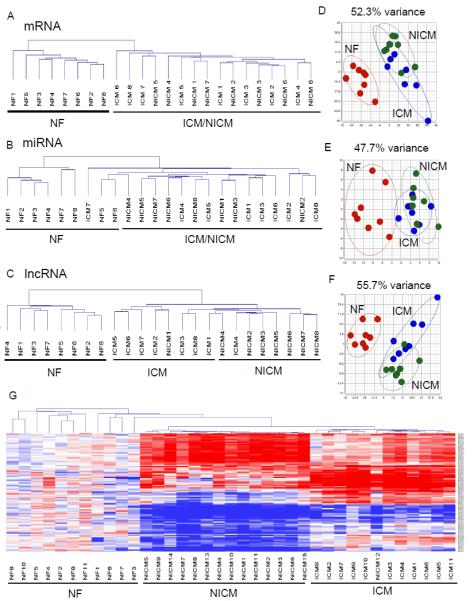
Expression signature of lncRNAs, but not mRNAs or miRNAs, distinguishes failing human hearts of ischemic and non-ischemic origin. (A-C)Unsupervised hierarchical clustering of the expression profiles of human cardiac mRNAs (A), miRNAs (B) and lncRNAs (C) reveals that all three distinguish failing from non-failing LV samples. In addition, the lncRNA, but not the mRNA or miRNA, expression profiles discriminate ICM from NICM LV samples. (D-F)Principal component analyses of mRNA (D), miRNA (E) and lncRNA (F) expression profiles showed similar findings. The variance explained by the principal components chosen is shown on top of each plot. (G)Heat map and unsupervised hierarchical clustering of lncRNA expression profiles derived from the microarray data of an independent cohort of human cardiac samples (11 NF, 11 ICM and 15 NICM) showed that lncRNA expression signature correctly classified all but one sample in all three groups.
To explore further the hypothesis that lncRNA expression profiles can be used to discriminate between ICM and NICM, we analyzed microarray data from an independent cohort of human cardiac samples (11 NF, 11 ICM and 15 NICM) obtained from Harvard Medical School CardioGenomics Programs for Genomic Applications (GSE1145).26 It has been demonstrated previously that 10-30% of the probes in expression microarrays designed for protein-coding gene analyses actually map to lncRNAs,27 and that with proper re-annotation of these probe sets, many of these arrays can provide expression information on lncRNAs.27,28 Using the microarray-reannotation R package provided by Gatexplorer,27 we were able to extract and analyze the expression profiles of 5635 lncRNAs from these conventional microarrays. Hierarchical clustering of the lncRNA expression profiles across these failing and non-failing samples (Supplemental Figure 7A) demonstrated that 97.3% (36/37) of these cardiac samples can be correctly classified by lncRNA expression signature, with only one NICM sample mis-classified as ICM sample. In contrast, the expression profiles of mRNAs from the same microarray data failed to distinguish ICM from NICM samples (Supplemental Figure 7B). The expression heat map and the clustering dendrogram of a subset of the lncRNAs in these failing and non-failing samples were illustrated in Figure 3G. Taken together, these results clearly support the hypothesis (above) that signature of lncRNA expression discriminates between cardiomyopathies of different etiologies, which may provide unique insights into the pathogenesis of heart failure in these different conditions.
Deep Sequencing Reveals Dynamic Regulation of Cardiac lncRNAs with Heart Failure and in response to LVAD Support
To determine the functional impact of LVAD support, echocardiographic (echo) and hemodynamic data, obtained from HF patients before and after LVAD support, were reviewed. As shown in Figure 4A-C, the dilated LV end-diastolic dimensions (LVEDD), high systolic and diastolic pulmonary arterial pressures (PAP) in HF patients before LVAD support were significantly (P<0.01) reduced after LVAD support. Expression of the heart failure marker, atrial natriuretic factor (ANF), which is significantly increased in ICM and NICM samples, is reduced with LVAD support (Figure 4D). The marked reductions in PAP, LV size and myocardial ANF expression after LVAD support are consistent with hemodynamic unloading and reverse remodeling that have been reported following mechanical circulatory support.
Figure 4.
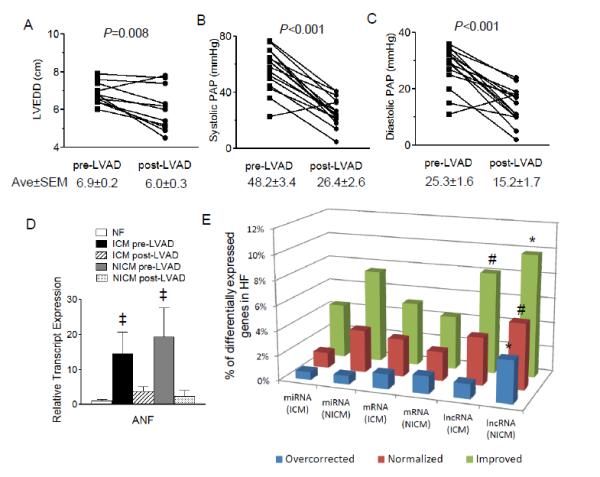
Higher proportion of HF-associated lncRNAs, compared with mRNAs and miRNAs, are improved or normalized by LVAD support. (A-D)The LV end-diastolic dimension (LVEDD) (A), systolic (B) and diastolic (C) pulmonary arterial pressures (PAP), as well as myocardial expression levels of the heart failure marker ANF (D), are all reduced significantly (‡P<0.05) in HF patients with LVAD support. (E)A significantly (#P<0.01,*P<0.001) higher percentage of differentially expressed lncRNAs, compared to mRNAs and miRNAs, are improved or normalized by LVAD in both ICM and NICM LV samples.
Analyses of the changes in expression of miRNAs, mRNAs and lncRNAs following LVAD support suggest that these RNA species are differentially regulated in response to hemodynamic unloading. Comparative analyses of miRNAs in HF samples revealed that 160 (100 up- and 60 down-regulated) and 147 (106 up- and 41 down-regulated) miRNAs are differentially expressed (absolute fold change>1.2 and adjusted P<0.05)3 in ICM and NICM samples, respectively (Figure 4E and Supplemental Table S7,S8 and S9). Among these, 7 (ICM, 4.4%) and 11 (NICM, 7.5%) miRNAs are significantly improved (corrected by at least 25%)4 after LVAD support. In contrast, only 2 (hsa-miR-365a-3p and 378a-3p) miRNAs in ICM and 5 (hsa-miR-548d-5p,760,425-5p, 93-3p and 193b-5p) miRNAs in NICM are normalized to values that are within 10% of the mean levels in NF samples (Supplemental Table S10). RNASeq analyses also revealed that there are 2262 (1945 up- and 317 down-regulated) and 1929 (1508 up- and 421 down-regulated) mRNAs differentially regulated in ICM and NICM hearts, respectively, with 115 (ICM, 5.1%) and 83 (NICM, 4.3%) improved (corrected by at least 25%) and 68 (ICM, 3.0%) and 45 (NICM, 2.3%) normalized (to <10% different from the mean value of NF), by LVAD support (Figure 4E and Supplemental Tables S9,S11,S12 and S13). Among the 679 (ICM, 569 up and 110 down-regulated) and 570 (NICM, 438 up- and 132 down-regulated) differentially expressed lncRNAs in failing hearts (Figure 4E and Supplemental Tables S9,S14,S15 and S16), 55 (ICM, 8.1%) and 56 (NICM, 9.8%) lncRNAs are improved and 26 (ICM, 3.8%) and 30 (NICM, 5.3%) lncRNAs are normalized with LVAD support. Viewed together, these results showed that a significantly higher proportion of differentially expressed lncRNAs in failing hearts are improved (P=0.0092 and 0.0015 for ICM and NICM, respectively; Chi-square test was used to compare 3 RNA groups) and/or normalized (P=0.21 and 0.0015 for ICM and NICM, respectively, by Chi-square test) by LVAD support, when compared to changes in miRNAs or mRNAs.
Interestingly, unsupervised hierarchical clustering of the cardiac lncRNA expression profiles revealed that lncRNA expression signature not only distinguishes the NF from the cardiomyopathic LV samples, it also distinguishes the cardiomyopathic (both ICM and NICM) LV samples before and after LVAD treatment (Figure 5A,B). Principal component analyses also revealed that lncRNA expression profiles differentiate failing (pre-LVAD), from hemodynamically unloaded (post-LVAD), LV samples (Figure 5C,D). In contrast to the lncRNAs, the expression profiles of both mRNAs and miRNAs failed to distinguish pre-LVAD from post-LVAD HF samples (Supplemental Figure 8).
Figure 5.
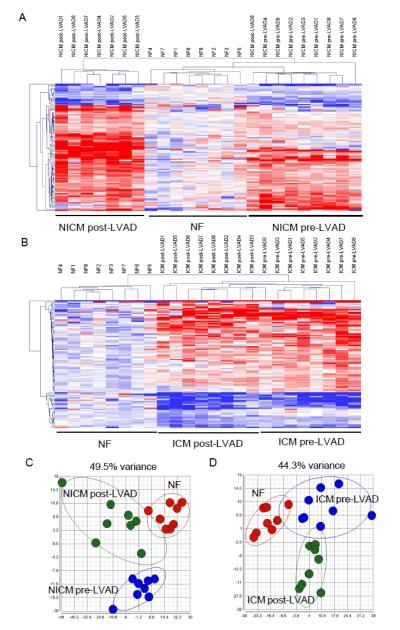
lncRNA expression signature distinguishes HF samples before and after mechanical circulatory support. (A,B)Heat maps and unsupervised hierarchical clustering of lncRNA expression profiles in NF, NICM (A) and ICM (B) samples before and after LVAD support. (C,D)Principal component analyses of lncRNA expression profiles in NF, NICM (C) and ICM (D) before and after LVAD support. Both analyses showed that lncRNA expression signature distinguishes HF samples before and after LVAD support.
It has previously been reported that combining mRNA/miRNA profiles can provide distinctions between HF samples before and after LVAD support.4 Additional analyses were conducted, therefore, using the reported set of 50 mRNAs and 28 miRNAs4 to cluster the NF and HF samples, before and after LVAD support. These analyses revealed that the combined mRNA/miRNA profiles, although sufficient to separate NF from HF samples, did not discriminate pre-LVAD from post-LVAD HF samples of either ischemic (Supplemental Figure 9A) or non-ischemic (Supplemental Figure 10A) origins. Interestingly, by adding the 50 most differentially expressed lncRNAs into the mRNA/miRNA mix, the power was increased and 96% (23 out of 24) of the samples were classified correctly in either ICM (Supplemental 9B) or NICM (Supplemental 10B). These results suggest that lncRNAs can provide an orthogonal and powerful marker for each disease state.
Taken together, these data indicate that lncRNA expression is dynamically regulated in advanced heart failure to a much greater extent than either miRNAs or mRNAs following mechanical circulatory support and that lncRNA expression profiles distinguish failing hearts of different etiologies, and may serve as a useful biomarker for different disease states.
Expression of Cardiac Mitochondrial lncRNAs Are Negatively Associated with Nuclear-Encoded Mitochondrial Regulatory Proteins
The high relative abundance (71%) of lncRNAs encoded from mitochondrial genome (mito-lncRNA) in human LV (Figure 2) was intriguing and additional analyses were completed to explore how these mito-lncRNAs are regulated with HF and in response to LVAD support. We compared the mito-lncRNA abundance across different disease states, and examined the association between mito-lncRNA expression levels and the nuclear-encoded proteins known to regulate mito-lncRNA expression including ELAC2, MRPP3, PTCD1, PTCD2,29 as well as factors known to regulate mitochondrial gene transcription, including PPARG1CA, PPARG1CB, PPRC1, GABPA, NRF1, TFAM, TFB2M, TFB1M,30 across all samples. As shown in Supplemental Figure 11A, mito-lncRNA abundance trended lower in ICM (by 12%, P=0.17) and NICM (by 9%, P=0.27), compared with the NF, LV samples. In addition, LVAD support significantly (P=0.007) reduced mito-lncRNA abundance in NICM (by 22%), but not in ICM, suggesting that the mitochondrial genomic response to mechanical circulatory support varies with the disease state. Interestingly, there was a significant (P<0.0001) negative correlation between the nuclear-encoded mitochondrial regulators PPRC1 (r=−0.59), GABPA (r=−0.63), NRF1 (r=−0.63) and cardiac mito-lncRNA abundance (Supplemental Figure 11B). It is possible that decreased mitochondrial transcript abundance in failing myocardium and in response to LVAD support triggers a compensatory upregulation of these nuclear-encoded mitochondrial regulators. Further experiments are needed to determine the physiological impact of mito-lncRNA regulation with heart failure and with mechanical circulatory support.
Expression of Cardiac lncRNAs and Neighboring Coding Genes Are Highly Correlated
Numerous studies have shown that lncRNAs act through regulating transcriptional expression of nearby mRNA/protein coding genes.17,31 To explore the potential interactions between the cardiac lncRNAs and neighboring coding mRNAs identified here, Pearson’s correlation coefficients between the expression levels of the lncRNAs and nearby coding genes (cis-mRNAs) were computed and compared with that of random gene pairs and mRNA:cis-mRNA pairs. As shown in Figure 6A, there was no significant positive or negative correlation between random gene pairs (0.4% with Pearson’s r>0.5, median=0.04), whereas mRNA:cis-mRNA pairs are positively correlated (Figure 6B, 20.7% with Pearson’s r>0.5, median=0.27), consistent with previous reports suggesting that adjacent genes may be co-regulated through common cis-regulatory elements.32 Interestingly, lncRNA:cis-mRNA pairs show even stronger correlations (Figure 6C, 41.4% with Pearson’s r>0.5, median=0.43) than mRNA:cis-mRNA pairs (P<0.0001 by Fisher’s exact test). The highly coordinated expression between neighboring lncRNAs and mRNAs may be attributed to the presence of common regulatory elements shared by lncRNAs and cis-mRNAs and/or to the positive regulatory potentials of lncRNAs on neighboring mRNAs. Figure 6D illustrates one example of a lncRNA:cis-mRNA pair that is upregulated in heart failure and shows a strong positive correlation: the expression of the retinoic acid receptor alpha (RARA) and lncRNA n340651, which resides in the third intron of RARA; both are upregulated with HF and showed a strong positive correlation (Pearson’s r=0.87, Figure 6E). This finding was confirmed by quantitative PCR analyses (Figure 6F, Pearson’s r=0.80). The strong positive correlation between lncRNA n340651 and the neighboring mRNA RARA suggests a cis regulatory role for lncRNA n340651 on RARA gene expression.
Figure 6.
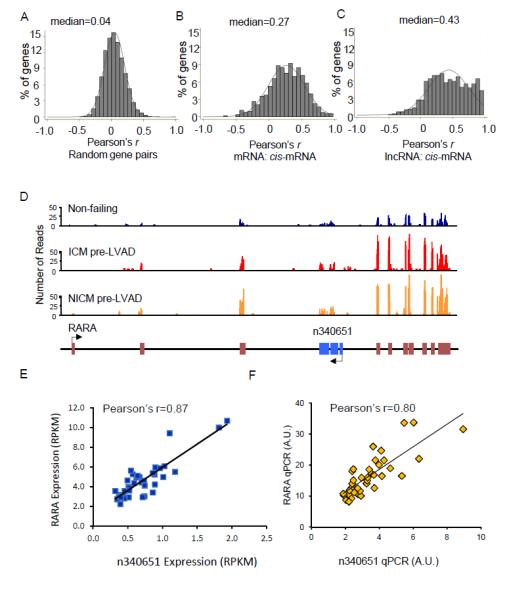
Expression levels of many cardiac lncRNAs are positively correlated with neighboring coding genes. (A-C)Histograms of Pearson correlations between random gene pairs (A) mRNA:cis-mRNA (B) and lncRNA:cis-mRNA gene pairs (C) reveals a strong positive correlation between lncRNAs and cis-mRNAs (C), compared with either mRNA:cis-mRNA (B) or random gene pairs (A). (D)Read distributions of a HF-associated lncRNA, n340651 (blue boxes), and its neighboring coding gene RARA (brown boxes), in NF, ICM and NICM LV samples. (E,F)Scatter plots of n340651 and RARA expression levels in individual human LV samples determined by RNASeq (E) and qPCR (F); both analyses reveal strong correlations for this lncRNA:cis-mRNA gene pair.
The Potential of Cardiac lncRNAs to Interact with Remote Coding Genes Is Not Higher than Background trans mRNA-mRNA Interaction
Although many lncRNAs function through cis regulation, thereby sharing high correlation with nearby coding genes, others have been shown to interact with or to regulate the expression of remote genes in trans.9,33 To explore the trans regulatory potential of cardiac lncRNAs, pairwise correlations encompassing all detectable human cardiac lncRNAs and mRNAs were computed. Pearson’s correlation coefficients of ~1 million lncRNA-mRNA, as well as mRNA-mRNA, gene pairs that are located at a distance >1Mb or on different chromosomes, representing trans-correlation of expression,34 were obtained. As shown in Figure 7A, 7.1% of the tested lncRNA:trans-mRNA gene pairs showed positive correlation (Pearson’s r>0.5), while 0.2% showed negative correlation (Pearson’s r< -0.5). Similarly, mRNA:trans-mRNA gene pairs also showed more frequent positive than negative correlations (Figure 7B, 8.9% vs 0.4%, respectively). However, the percentage (7.3%) of lncRNAs showing a positive or negative correlation with trans-mRNAs are lower than the percentage (9.3%) between mRNA:trans-mRNA gene pairs (P<0.0001 by Fisher’s exact test). These observations suggest that the potential of lncRNAs to regulate/interact with remote coding genes in trans in the human ventricular myocardium is, at most, not higher than the extent of background mRNA:trans-mRNA interactions.
Figure 7.
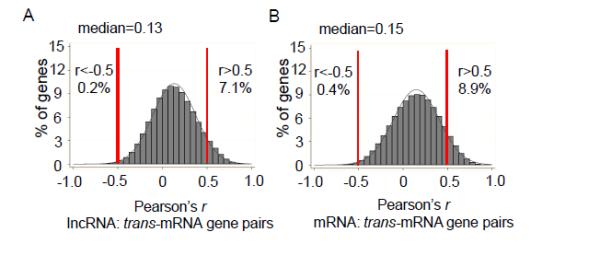
The trans-acting potential of lncRNAs is similar to that of mRNAs. Histograms of Pearson correlations between (A) lncRNA:trans-mRNA and (B) mRNA:trans-mRNA gene pairs reveals that the percentage of lncRNA:trans-mRNA gene pairs that show a significant positive (r>0.5) or negative (r<-0.5) correlation is actually slightly lower than that between mRNA:trans-mRNA gene pairs, suggesting that the potential of lncRNAs co-expressing/interacting with remote coding genes in trans is not higher than that of background trans mRNA-mRNA interaction.
4. Discussion
The present study utilized next-generation sequencing to provide a quantitative and comprehensive analysis of the coding and non-coding transcriptome in non-failing human LV and in failing human LV, before and after LVAD support. These analyses revealed significant differences in the patterns of mRNA, miRNA and lncRNA expression in ischemic and non-ischemic failing hearts, as well as dynamic changes in response to hemodynamic unloading with an LVAD. Here we show for the first time that the expression patterns of lncRNAs discriminate between ischemic vs non-ischemic heart failure, whereas the mRNA or miRNA expression signature does not discriminate between heart failure etiologies. Moreover, this study shows that the changes in the expression levels of lncRNAs are more dynamically regulated following hemodynamic unloading than mRNA or miRNA levels. The observation that the expression pattern of lncRNAs in the failing heart was both etiology-specific, as well as sensitive to hemodynamic loading conditions suggests that the observed changes were not due to random transcriptional noise secondary to low fidelity binding of RNA polymerase to randomly occurring weak promoter sequences in the genome.
Distinct Expression Patterns Reflect the Functional Complexities of Different RNA Species
The discovery of multiple classes of non-coding RNAs that are widely transcribed from the genomes of most complex organisms7 challenges the traditional view that RNA is simply an intermediate between gene and protein. Indeed, the marked differences in the expression patterns and abundances of cardiac mRNAs, lncRNAs and miRNAs reported here highlight the distinct biological roles of these RNAs that have evolved differently over time. Most cardiac mRNAs, for example, are expressed at intermediate (1-3 RPKM,18%) to high (≥3 RPKM,40%) levels, whereas the expression levels of most lncRNAs are low (<1 RPKM,88%) (Figure 2D,E). Because the evolutionary rates of genes are often inversely correlated with their expression levels,35 the low abundance of lncRNAs likely reflects more rapid evolution compared with protein-coding mRNAs, which are largely conserved across organisms of vastly different complexities and ages.6 Consistent with this hypothesis, there is evidence showing that lncRNAs have been subject to purifying selection for phenotypic innovation,36 suggesting that the lncRNAs contribute to complex genome regulatory mechanisms during evolution.
A particularly striking finding here is the high abundance of mRNAs and lncRNAs of mitochondrial origin:13 mitochondrial mRNAs and 9 mitochondrial lncRNAs alone account for 37% and 71% of the total cardiac mRNA and lncRNA read counts, respectively (Figure 2A,B). The high levels of mitochondrial mRNA and lncRNA expression likely reflect the large numbers of mitochondria in ventricular myocytes, owing to the high-energy demands of the working myocardium. Consistent with our findings, one recent report on human mitochondrial transcriptome demonstrated that mitochondrial mRNA and lncRNA content are higher in the heart compared to 16 other tissues.37 Because polyadenylated RNA was used here in the preparation of the RNASeq libraries, it is not possible to determine whether rRNAs or tRNAs of mitochondrial origin also dominate total cardiac rRNAs or tRNAs.
Expression Signature of Myocardial lncRNAs Differentiates Ischemic and Non-ischemic Cardiomyopathy
Ischemic and non-ischemic cardiomyopathy, distinguished by the presence or absence of significant coronary artery disease, have distinct survival outcomes38 and differential responses to therapies.39 Several studies have suggested that the expression profiles of subsets of mRNAs2 or miRNAs25 may be useful in discriminating ICM from NICM; however, the overall expression signatures of mRNAs and miRNAs determined here (Figure 3A,B,D,E) do not provide such distinction.4 It is possible that etiology-specific changes in mRNA and/or miRNA expression develop relatively early in the course of heart failure, and that the mRNA and/or miRNA changes observed with end-stage cardiomyopathy are the consequences of advanced disease, rather than the underlying causes.40 By contrast, the observation that the lncRNA expression signature, determined from our RNASeq data (Figure 3C and 3F), as well as from analyses of an independent cohort of human myocardium expression arrays (Figure 3G), distinguishes ICM from NICM, suggests that much of the etiology-specific changes in lncRNAs with HF may be preserved throughout the course of the disease, even at end-stage heart failure. These data also suggest that distinct populations of lncRNAs are involved in the pathogenesis of ICM and NICM. Exploring the functional roles of lncRNAs in this regard should provide mechanistic insights into the pathophysiology of HF of different etiologies.
Myocardial lncRNA, but not mRNA or miRNA, Expression Profiles Identify Reverse Remodeling with LVAD Support
Previous studies have suggested a limited role for mRNA regulation in LVAD-mediated reverse remodeling.3,4 Indeed, the results presented here revealed that only 2-3% of the mRNAs that are differentially expressed in failing human hearts are normalized with LVAD support (Figure 4E) and that mRNA expression profiles do not distinguish HF samples before and after LVAD treatment (Supplemental Figure 4A,B). Recently, Matkovich and colleagues4 reported that 20 out of the 28 miRNAs differentially expressed in failing human hearts are normalized by LVAD support, leading the authors to conclude that miRNA is more sensitive than mRNA to reflect reverse remodeling in HF by mechanical support. Unlike the present study, however, Matkovich and colleagues4 utilized a case-control study design and they compared samples from HF patients without LVAD to samples from HF patients with LVAD, rather than using paired samples from the same patient, as was done in the present study. The experimental results obtained, therefore, may be confounded by the differences between groups of patients, rather than, or in addition to, reflecting the impact of LVAD support on miRNA regulation. Consistent with this suggestion, another recent study reported that LVAD support had a limited impact on cardiac miRNA expression in failing hearts.5 The longitudinal analysis here of LV samples from HF patients before and after LVAD support revealed only 2 and 5 miRNAs that are abnormally expressed in ICM and NICM, respectively, are normalized by mechanical support (Supplemental Table S9) and that miRNA expression profiles do not discriminate HF samples before and after LVAD support (Supplemental Figure 4C,D). Compared with mRNA and miRNA, a significantly higher proportion of abnormally expressed lncRNAs in failing hearts are normalized fully or partially by LVAD support (Figure 4E). In addition, lncRNA expression profiles readily distinguish LV samples from patients with ischemic and non-ischemic HF, before and after LVAD support. Taken together, our results suggest that cardiac lncRNA expression profiles respond more sensitively to LVAD support than do mRNA and miRNA expression profiles and may serve, therefore, as a useful biomarker to assess either LV reverse remodeling or myocardial recovery in response to mechanical circulatory support.
Cis-Gene Regulation Is an Important Mechanism of Action of Cardiac lncRNAs
Unlike miRNAs or mRNAs, the functions of lncRNAs cannot be inferred from sequence or primary structure alone.7 The apparent lack of sequence conservation across species has also made it difficult to generalize lncRNA functions identified in animal models to likely human counterparts. The studies presented here analyzed lncRNA-mRNA genomic proximity information to explore the potential cis- and trans-regulatory roles of human cardiac lncRNAs on coding mRNAs. While the probability of correlation between lncRNAs and cis-mRNAs is much higher than that between neighboring mRNAs (42.6% vs 20.7%,respectively,P<0.0001), the percentage of correlated expression between lncRNAs and remote coding genes is actually lower than background mRNA:trans-mRNA co-expression (7.3% vs 9.3%,respectively,P<0.0001). These observations suggest that trans-acting lncRNAs may not be as prevalent as cis-acting lncRNAs in human left ventricular myocardium.
The observation that lncRNA profiles segregate biological samples better than mRNA also suggests that lncRNAs function beyond simply regulating mRNA transcription. Indeed, although very little is known with regard to lncRNAs in the heart, lncRNAs are known to control gene expression on multiple levels through diverse mechanisms.10 Functionally, lncRNAs are best known for their roles as regulators of transcription, including epigenetic modification of chromatin structure.31 More recently, lncRNAs have been implicated as regulators of post-transcriptional mechanisms including pre-mRNA splicing,41 mRNA decay,11 and mRNA translation.42 In this regard lncRNAs are unique, insofar as their functions are not dependent solely on sequence (as with miRNAs) or structure (as for RNA-binding proteins). Rather, lncRNAs, appear to function both by sequence homology/complementarity with other nucleic acids, as well as by structure, forming molecular frameworks and scaffolds for the assembly of macromolecular complexes.10
Study Limitations
The number of samples analyzed here was relatively small, and the samples were obtained from a heterogeneous (in terms of age, race, gender, ancestry, etc.) cohort of patients and donors. Each of these factors could have effects on cardiac remodeling and gene expression that cannot be controlled for and may, therefore, confound the results of the analyses completed and presented. In addition, one of the NICM patients (NICM1), although diagnosed dilated cardiomyopathy at presentation, had received myectomy 40 years prior (Table S1), suggesting hypertrophic cardiomyopathy. Although it is possible that including this sample in the NICM transcriptome analysis introduced some bias, additional analyses excluding this one sample from the NICM group (Supplemental Figure 12) did not reveal significant differences from the primary results presented above. Finally, it is important to note that sequencing technologies and data analysis methods are continuing to evolve. Additional studies conducted on larger numbers of well-annotated clinical samples and exploiting improved sequencing technologies and analytical methods will be of considerable interest.
5. Conclusions
The studies presented here utilized next-generation sequencing technologies for comprehensive cardiac mRNA, miRNA and lncRNA expression profiling in non-failing and failing human hearts, before and after LVAD support These experiments revealed distinct relative abundance, expression pattern and genomic origin of each of these RNA species in human heart, highlighting the different biological roles of the individual RNA classes during evolution. Interestingly, the results presented here also revealed that the lncRNA expression signature discriminates between ischemic and non-ischemic cardiomyopathy, whereas mRNA and miRNA expression profiles do not. In addition, and also unlike mRNA and miRNA, the lncRNA expression signature also distinguishes cardiomyopathic samples before and after LVAD support. The observation that lncRNA profiles segregate cardiac samples better than either mRNA or miRNA profiles is consistent with the multiplicity of functional roles for lncRNAs, in addition to the regulation of gene transcription.11,41,42 The analyses presented also suggest that regulatory interactions with nearby genes (cis-acting), rather than with distant genes (trans-acting), could be the major mechanisms of action for cardiac lncRNAs. Although lncRNAs were once referred to as the “dark matter of the genome” because of difficulties detecting expression and/or discerning function, the results of the present study suggest, for the first time, that lncRNAs may play an important functional role in the pathogenesis of cardiomyopathy and well as contribute to reverse LV remodeling following hemodynamic unloading with mechanical circulatory support.
Supplementary Material
Acknowledgements
This publication was made possible by the resources provided the Translational Cardiovascular Biobank and Repository at Washington University Medical School, supported by the Washington University Institute for Clinical and Translational Sciences (ICTS), recipient of a Clinical and Translational Sciences Award (UL1 RR024992) from the NIH National Center for Research Resources, the Barnes-Jewish Hospital Foundation and the Richard J. Wilkinson Trust. The authors also wish to thank Drs. Michael K. Pasque and Scott C. Silvestry for expertise and assistance with cardiac tissue acquisition and the Genome Technology Access Center at Washington University Medical School for assistance with next-generation sequencing experiments.
Funding Sources: The experiments described here were supported directly by the National Heart, Lung and Blood Institute of the National Institutes of Health (R01 HL-034161 and HL-066388 to JMN; R01 HL-111094 to DLM) and by the NIH National Center for Research Resources (UL1 RR024992 to the ICTS). None of these funding sources were involved in study design, data collection, data analyses, data interpretation, manuscript preparation or the decision to submit this article for consideration for publication.
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Hannenhalli S, Putt ME, Gilmore JM, Wang J, Parmacek MS, Epstein JA, Morrisey EE, Margulies KB, Cappola TP. Transcriptional genomics associates FOX transcription factors with human heart failure. Circulation. 2006;114:1269–1276. doi: 10.1161/CIRCULATIONAHA.106.632430. [DOI] [PubMed] [Google Scholar]
- 2.Kittleson MM, Ye SQ, Irizarry RA, Minhas KM, Edness G, Conte JV, Parmigiani G, Miller LW, Chen Y, Hall JL, Garcia JG, Hare JM. Identification of a gene expression profile that differentiates between ischemic and nonischemic cardiomyopathy. Circulation. 2004;110:3444–3451. doi: 10.1161/01.CIR.0000148178.19465.11. [DOI] [PubMed] [Google Scholar]
- 3.Margulies KB, Matiwala S, Cornejo C, Olsen H, Craven WA, Bednarik D. Mixed messages: transcription patterns in failing and recovering human myocardium. Circ Res. 2005;96:592–599. doi: 10.1161/01.RES.0000159390.03503.c3. [DOI] [PubMed] [Google Scholar]
- 4.Matkovich SJ, DJ Van Booven, Youker KA, Torre-Amione G, Diwan A, Eschenbacher WH, Dorn LE, Watson MA, Margulies KB, Dorn GW., 2nd Reciprocal regulation of myocardial microRNAs and messenger RNA in human cardiomyopathy and reversal of the microRNA signature by biomechanical support. Circulation. 2009;119:1263–1271. doi: 10.1161/CIRCULATIONAHA.108.813576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramani R, Vela D, Segura A, McNamara D, Lemster B, Samarendra V, Kormos R, Toyoda Y, Bermudez C, Frazier OH, Moravec CS, Gorcsan J, 3rd, Taegtmeyer H, McTiernan CF. A micro-ribonucleic acid signature associated with recovery from assist device support in 2 groups of patients with severe heart failure. J Am Coll Cardiol. 2011;58:2270–2278. doi: 10.1016/j.jacc.2011.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mattick JS. The central role of RNA in human development and cognition. FEBS Lett. 2011;585:1600–1616. doi: 10.1016/j.febslet.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 8.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 9.Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, Kong B, Langerod A, Borresen-Dale AL, Kim SK, van de Vijver M, Sukumar S, Whitfield ML, Kellis M, Xiong Y, Wong DJ, Chang HY. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621–629. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogawa Y, Sun BK, Lee JT. Intersection of the RNA interference and X-inactivation pathways. Science. 2008;320:1336–1341. doi: 10.1126/science.1157676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marionneau C, Brunet S, Flagg TP, Pilgram TK, Demolombe S, Nerbonne JM. Distinct cellular and molecular mechanisms underlie functional remodeling of repolarizing K+ currents with left ventricular hypertrophy. Circ Res. 2008;102:1406–1415. doi: 10.1161/CIRCRESAHA.107.170050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang KC, Foeger NC, Marionneau C, Jay PY, McMullen JR, Nerbonne JM. Homeostatic regulation of electrical excitability in physiological cardiac hypertrophy. J Physiol. 2010;588:5015–5032. doi: 10.1113/jphysiol.2010.197418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bu D, Yu K, Sun S, Xie C, Skogerbo G, Miao R, Xiao H, Liao Q, Luo H, Zhao G, Zhao H, Liu Z, Liu C, Chen R, Zhao Y. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012;40:D210–215. doi: 10.1093/nar/gkr1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hackenberg M, Sturm M, Langenberger D, Falcon-Perez JM, Aransay AM. miRanalyzer: a microRNA detection and analysis tool for next-generation sequencing experiments. Nucleic Acids Res. 2009;37:W68–76. doi: 10.1093/nar/gkp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol Biol. 2006;342:129–138. doi: 10.1385/1-59745-123-1:129. [DOI] [PubMed] [Google Scholar]
- 21.ENCODE_Project_Consortium A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jia H, Osak M, Bogu GK, Stanton LW, Johnson R, Lipovich L. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA. 2010;16:1478–1487. doi: 10.1261/rna.1951310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang KC, Ku YC, Lovett M, Nerbonne JM. Combined deep microRNA and mRNA sequencing identifies protective transcriptomal signature of enhanced PI3Kalpha signaling in cardiac hypertrophy. J Mol Cell Cardiol. 2012;53:101–112. doi: 10.1016/j.yjmcc.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31:367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 26.Genomics of Cardiovascular Development, Adaptation, and Remodeling NHLBI Program for Genomic Applications, Harvard Medical SChool. URL: http://www.cardiogenomics.org [Mar, 2013 accessed]
- 27.Risueno A, Fontanillo C, Dinger ME, De Las Rivas J. GATExplorer: genomic and transcriptomic explorer; mapping expression probes to gene loci, transcripts, exons and ncRNAs. BMC Bioinformatics. 2010;11:221. doi: 10.1186/1471-2105-11-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michelhaugh SK, Lipovich L, Blythe J, Jia H, Kapatos G, Bannon MJ. Mining Affymetrix microarray data for long non-coding RNAs: altered expression in the nucleus accumbens of heroin abusers. J Neurochem. 2011;116:459–466. doi: 10.1111/j.1471-4159.2010.07126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rackham O, Shearwood AM, Mercer TR, Davies SM, Mattick JS, Filipovska A. Long noncoding RNAs are generated from the mitochondrial genome and regulated by nuclear-encoded proteins. RNA. 2011;17:2085–2093. doi: 10.1261/rna.029405.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- 31.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santini S, Boore JL, Meyer A. Evolutionary conservation of regulatory elements in vertebrate Hox gene clusters. Genome Res. 2003;13:1111–1122. doi: 10.1101/gr.700503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011;146:119–133. doi: 10.1016/j.cell.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigo R. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Managadze D, Rogozin IB, Chernikova D, Shabalina SA, Koonin EV. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome Biol Evol. 2011;3:1390–1404. doi: 10.1093/gbe/evr116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, Cabili MN, Jaenisch R, Mikkelsen TS, Jacks T, Hacohen N, Bernstein BE, Kellis M, Regev A, Rinn JL, Lander ES. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mercer TR, Neph S, Dinger ME, Crawford J, Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O, Stamatoyannopoulos JA, Filipovska A, Mattick JS. The human mitochondrial transcriptome. Cell. 2011;146:645–658. doi: 10.1016/j.cell.2011.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–1084. doi: 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- 39.Follath F, Cleland JG, Klein W, Murphy R. Etiology and response to drug treatment in heart failure. J Am Coll Cardiol. 1998;32:1167–1172. doi: 10.1016/s0735-1097(98)00400-8. [DOI] [PubMed] [Google Scholar]
- 40.Aronow BJ, Toyokawa T, Canning A, Haghighi K, Delling U, Kranias E, Molkentin JD, Dorn GW., 2nd Divergent transcriptional responses to independent genetic causes of cardiac hypertrophy. Physiol Genomics. 2001;6:19–28. doi: 10.1152/physiolgenomics.2001.6.1.19. [DOI] [PubMed] [Google Scholar]
- 41.Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB, Pena R, Bonilla F, de Herreros AG. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev. 2008;22:756–769. doi: 10.1101/gad.455708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, Pesce E, Ferrer I, Collavin L, Santoro C, Forrest AR, Carninci P, Biffo S, Stupka E, Gustincich S. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491:454–457. doi: 10.1038/nature11508. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.