

Abstract
Populations encompassing extremes of age, including neonates and elderly, have greater mortality from sepsis. We propose that the increased mortality observed in the neonatal and elderly populations after sepsis is due to fundamental differences in host protective immunity, and are manifested at the level of the leukocyte transcriptome. Neonatal (5–7 days), young adult (6–12 weeks), or elderly (20–24 months) mice underwent a cecal slurry model of intra-abdominal sepsis. Both neonatal and elderly mice exhibited significantly greater mortality to sepsis (p<0.05). Neonates in particular exhibited significant attenuation of their inflammatory response (p<0.05), as well as reductions in cell recruitment and reactive oxygen species production (both p<0.05), all of which could be confirmed at the level of the leukocyte transcriptome. In contrast elderly mice were also more susceptible to abdominal peritonitis, but this was associated with no significant differences in the magnitude of the inflammatory response, reduced bacterial killing (p<0.05), reduced early myeloid cell activation (p<0.05) and a persistent inflammatory response that failed to resolve. Interestingly, elderly mice expressed a persistent inflammatory and immunosuppressive response at the level of the leukocyte transcriptome, with failure to return to baseline by three days. This study reveals that neonatal and elderly mice have profoundly different responses to sepsis that are manifested at the level of their circulating leukocyte transcriptome, although the net result of increased mortality, is similar. Considering these differences are fundamental aspects of the genomic response to sepsis, interventional therapies will require individualization based on the age of the population.
Introduction
Despite the evolution in our understanding of the human response to sepsis, mortality from severe sepsis and organ failure has only modestly improved over the last several decades (1, 2). Additionally, populations encompassing the extremes of age, including neonates and the elderly, are more vulnerable (3). Neonatal sepsis is the leading cause of mortality in infants, with greater than 1 million deaths per year worldwide (4). Likewise, 60% of septic patients are older than 65 years of age, and age is an independent risk factor for poor outcomes in sepsis (5, 6). The economic burdens of caring for these two groups of patients are not insignificant, as estimates place the annual cost of caring for septic neonatal and elderly patients in the United States at $700 million and $17 billion, respectively (5, 7).
The incidence of pediatric sepsis is highest in children less than one year of age, and the neonatal population, specifically premature and very low birth weight (VLBW) infants are the most susceptible (4, 8). Despite improvements in the quality of neonatal intensive care, the mortality from neonatal sepsis over the last 20 years remains high, with infection being the leading cause of mortality in the first days of life (9, 10). Previously, we have shown that the neonatal murine population is more susceptible to sepsis than their young adult counterparts (11) and that neonatal mice have attenuated inflammatory responses following sepsis and functional defects in their peritoneal myeloid cell populations (12). Despite a modest understanding of the neonatal immune system, there are no currently approved immune modulating therapies in use to date (8), and research continues to focus on immune responses in neonates, particularly of the innate immune system as the search for therapeutic strategies continues.
The elderly population also has significantly greater morbidity and mortality following sepsis, and with over 80% of adult deaths from sepsis occurring in those over the age of 65; sepsis is becoming known as a disease of the aged (3, 13). Likewise, the elderly are known to be in a state of immunosenescence, where protective immunity is less able to mount an effective response to invading organisms than the young (14).
In this report we explored the differences in host protective immunity in response to sepsis in the neonatal and aged murine populations compared to juvenile or young, adult mice. We demonstrate that indeed there are fundamental differences in protective immunity which manifest themselves at the level of the leukocyte transcriptome and contribute to the increased neonatal and elderly mortality after sepsis.
Materials and Methods
Murine Models
Male mice were purchased from either Jackson Laboratory (Bar Harbor, ME, USA) or the National Institute of Aging, or were bred at the University of Florida, Animal Care Services. Mixed gender neonatal C57BL/6J mice aged 5–7 days, or male mice age 6–12 weeks (young adult), or 20–24 months (elderly) were used in experiments approved by the University of Florida Institutional Animal Care and Use Committee. Mice were housed in pathogen-free facilities and acclimated at least one week prior to use. Neonatal, young adult, and elderly mice underwent the cecal slurry (CS) model of intra-abdominal sepsis revised for mice (11) and were followed for survival. The CS model of polymicrobial sepsis is used in neonatal mice as the “gold-standard”. In contrast, cecal-ligation and puncture (CLP) is technically challenging in the neonatal mouse due to its small size and risk for cannibalization by the mother after survival surgery. For a matter of uniformity, we chose to use the CS model of intra-abdominal sepsis across all three of the age groups studied. Briefly, when performing CS, cecal contents are harvested from adult C57BL/6j mice and suspended in 5% dextrose in water to make a cecal slurry at a concentration of 80 mg/ml, as previously published. 1.1 mg/gm BW was then injected intraperitoneally. Mouse whole blood was collected via intra-cardiac puncture two hours, one day and three days after injection. Blood was either used for complete blood count (CBC) with differential determination, RNA isolation, determination of plasma cytokine response, or plated on sheep’s blood agar plates for bacterial count determination. The plasma cytokine and chemokine response was measured by multiplex Luminex™ assay (Austin, TX, USA). Additional animals were followed for seven days to judge long-term survival.
Harvest of Peritoneal Cells
Peritoneal cell isolation from peritoneal lavage specimens were performed as previously described (12). Peritoneal lavage samples from individual neonatal mice contain small numbers of cells, so peritoneal wash samples from three to five neonatal mice were pooled for analysis and considered to be one sample. Three to four peritoneal wash samples were collected per experiment. Erythrocytes were lysed with ammonium chloride lysis buffer and washed with PBS. Peritoneal bacterial counts were determined by culturing 100 μl of serially diluted peritoneal lavage sample of sheep’s blood agar plates (Thermo Fisher Scientific) at 37°C. Plates were counted after 24 hours of culture.
Flow Cytometry for Characterization of Cell Phenotype
Single cell suspensions were stained with anti-Ly6G APC (Becton-Dickinson (BD) (Franklin Lakes, NJ), anti-CD11b-PECy7 (BD, Franklin Lakes, NJ), and anti-F4/80-APC (Ebioscience, San Diego, CA). Sytox Blue (Invitrogen, Carlsbad, CA) was used for cell viability analysis. Samples were acquired and analyzed on an LSR II flow cytometer (BD) and analyzed via FACSDiva™ (BD) software. At least 1 × 104 live cells (SYTOX Blue−; Invitrogen, Carlsbad, CA) were collected for analysis. The absolute numbers of innate immune effector cells were determined by multiplying the percentage of neutrophils (CD11b+, Ly6G+) and macrophages)(CD11b+, Ly6G−, F4/80+) within the total sample population by the total sample cell number. In neonates, these were divided by the total number of mice in the pooled sample. Absolute numbers represent total macrophages or neutrophils per mouse.
Functional Analysis of Peritoneal Macrophages or Neutrophils
To assay for reactive oxygen species (ROS) production, cell suspensions containing 2 × 106 cells were stained for cell surface markers, washed with PBS and then labeled with dihydrorhodamine 123 (DHR 123; Invitrogen) to determine ROS production. Cells were then stimulated with 1 μM of phorbol-12-myristate-13-acetate (PMA) (Sigma-Aldrich, St Louis, MO) at 37°C and aliquots were evaluated by flow cytometry every ten minutes for 30 minutes using a LSR II flow cytometer (BD). A minimum of 1 × 104 live, non-debris cells were collected for analysis.
Ex Vivo Whole Blood Cytokine Stimulation
Human anti-coagulated whole blood was also obtained from three healthy control subjects, age range (35–41 years). Fresh neonatal cord blood samples were obtained from the New York Blood Center National Cord Blood Program (Long Island, NY, USA). Both fresh adult and neonatal cord blood were stimulated ex vivo with 100 ng/ml lipopolysaccharide (LPS) (ultrapure via ion exchange chromatography Escherichia coli O26:B6)(Sigma-Aldrich, St Louis, MO) in 12 well microtiter plates in triplicate and incubated at 37° C. After 18 hours, the plasma was collected for cytokine/chemokine determination by multiplex Luminex™ assay (Austin, TX, USA). Blood sampling from healthy control subjects and the purchase of fresh cord blood was reviewed and approved by the Institutional Review Board, at the University of Florida.
Gene Expression Profiling
Leukocytes were isolated from whole blood by lysing erythrocytes with EL buffer™ (Qiagen, Germantown, MD) and centrifugation. Cells were lysed with RLT buffer™ and genome-wide expression analysis was performed on 100 ng of total RNA (RNeasy™, Qiagen) that were labeled and hybridized to GeneChip™ Mouse Genome 430 2.0 Arrays (Affymetrix, Santa Clara, CA) according to the manufacturer’s recommendations. A log2 transformed expression matrix was calculated using RMA™ as implemented in the Partek Genomic Suite 6.6, and gene expression patterns were compared between neonatal, young adult, and elderly mice following CS sepsis using sepsis-responsive genes that were significant at p<0.001 (F-test). Leave-one out cross validation was performed to compute the misclassification rate, and Monte Carlo simulation was used to determine if the miscalculation rate was significantly better than predicted by chance. Once significant genes were identified, fold changes were calculated between each murine age group (neonate, young adult, and elderly) and a control group (neonate or adult).
Functional pathway analysis was performed using Ingenuity Pathway Analysis (IPA, Redwood City, CA). IPA Systems performs functional pathways analysis as part of its tools available to researchers. IPA identifies those pathways that are over-represented, indicating that their expression is affected by the intervention. Significance was determined using a Z-score. Values of 2 < Z < −2 are considered significant and correspond to a 95% confidence interval.
Additional Statistical Analysis
Continuous non-genomic variables were tested for normality and equality of variances. Differences in survival were determined by Fisher’s exact test. Differences among groups were evaluated by either one-way ANOVA with either Dunn’s or Tukey post-hoc analysis, two-way ANOVA with Bonferroni’s post-hoc analysis, or Student’s t-test. Significance was determined at the 95% confidence level.
Results
Elderly and Neonatal Mice Have Impaired Ability to Clear Bacteria from the Peritoneum and the Inability to Activate Immune Cells after CS Leading to Increased Mortality Compared to Young Adult Mice
Murine studies have demonstrated that neonatal and elderly mice are more susceptible to the same insult of polymicrobial abdominal sepsis compared to their young adult counterparts (11, 15), but a direct comparison between neonates and the elderly is lacking. We simultaneously evaluated survival in neonatal, young adult, and elderly mice using an intraperitoneal CS dose of 1.1 mg/gm, known to produce an LD30–40 lethality in young adult mice. As expected, both neonatal and elderly mice had increased mortality compared to young adult mice with the same insult (Figure 1A) (p<0.05). The elderly mice had the greatest susceptibility, with 1.8 times greater mortality than young adult mice, followed by neonatal mice which had a 1.4 times greater mortality. To confirm that this was due to a failure of protective immunity, we examined bacterial counts in peritoneal washes one day after CS sepsis, and we found that elderly mice had significant impairments in the ability to clear bacteria from the site of infection compared to juvenice mice followed by neonatal mice (p<0.0001, One-way ANOVA)(Figure 1B). These findings were substantiated at the level of the leukocyte transcriptome, as elderly mice actually had down-regulation of genes involved in the the activation of leukocytes, whereas young adult mice had significant upregulation of these same genes (Figure 1C). Interestingly, neonatal mice completely failed to exhibit any significant change in a number of genes involved in leukocyte activation at a significance level of p<0.001, thus confirming their attenuated immune response (Figure 1C).
Figure 1. Neonatal and elderly mice have increased mortality compared to young adult mice with impaired control of bacteria at the site of infection and decreased activation of leukocytes.
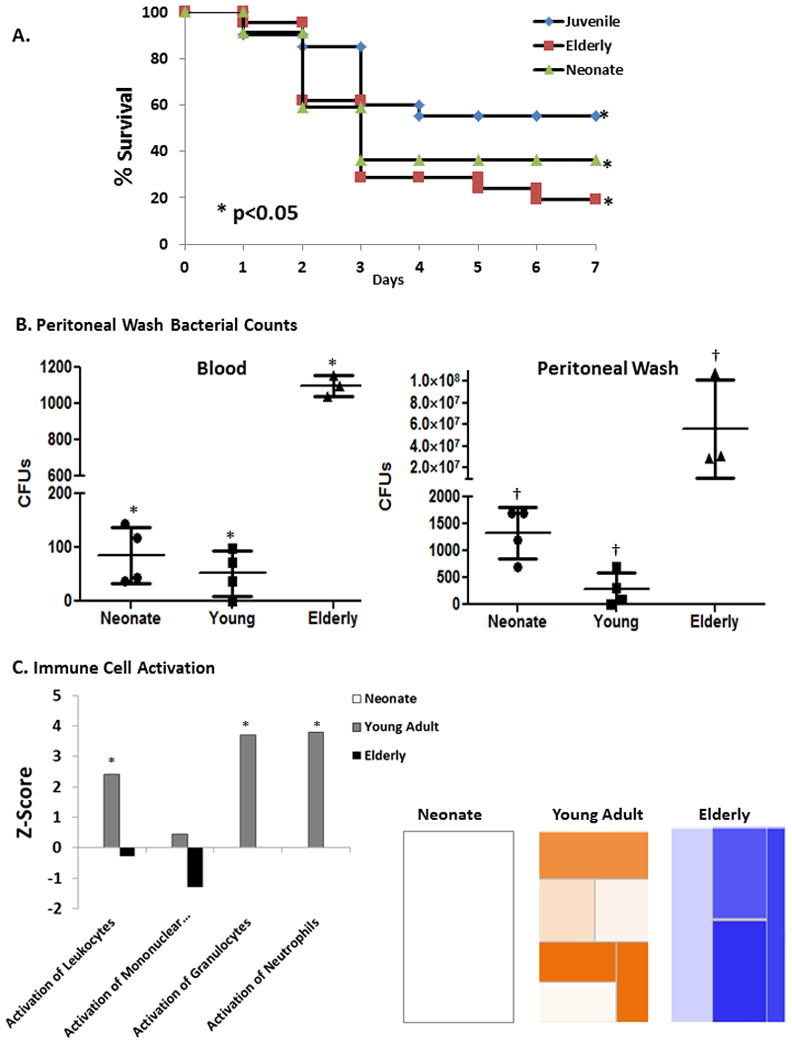
A. Neonatal and elderly mice had increased mortality after CS sepsis compared to young adult mice with the same insult (P<0.05) over 7 days. Neonate (n=22), Young Adult (n=20), Elderly (n=21). B. One day after sepsis both neonatal and elderly mice had significantly increased bacterial counts in the blood (* p<0.0001, One-way ANOVA), and in the peritoneum †p=0.019, One-way ANOVA); with elderly mice having the most severe impairments in clearing bacteria (Blood-p<0.001, Peritoneal wash-p<0.05; Tukey’s Multiple Comparisons post-hoc test) (n=3–4, results shown are from two or more independent experiments). C. Functional pathway analysis showed that elderly mice had down-regulation of genes involved in the the activation of luekocytes, whereas young adult mice had significant upregulation of these same genes (Supplemental Table 1). Neonatal mice niether up or down regulated a large enough number of genes involved in leukocyte activation at a significance level of p<0.001. Heatmaps show the of gene expression of the functional category “Immune Cell Trafficking-Activation” one day after sepsis in neonatal, young adult, and elderly mice. Orange represents pathways with an over-expression of genes leading to the activation of the pathway, whereas blue represents pathways with an over-expression of genes whose activation will lead to down-regulation of the pathway. White represents genes that are neither up or down regulated to significance level of p<0.001. Graph shows the various subcategories and their corresponding significance level (Z-score) of up or down regulation. From left to right, categories include “activation of leukocytes,” “activation of mononuclear leukocytes,” “activation of granulocytes,” and “activation of neutrophils.” (* Z-score >2). See Supplemental Table 2 for a complete list of genes in presented pathways.
Significant Differences in the Phenotypic and Functional Responses by the Neonatal and Elderly Mouse to an Identical Sepsis Challenge
Although both neonatal and elderly mice have increased mortality compared to young adult mice, the mechanisms of this increased mortality, as exhibited both functionally, and at the level of the leukocyte transcriptome, is unique. For example, when examining the magnitude of the inflammatory response to a similar sepsis challenge using the plasma cytokine response, we found that one day after sepsis neonatal mice had significantly attenuated production of the inflammatory cytokines IL-10, IL-6, and TNFα (p<0.01)(Figure 2A), and had significantly decreased production of chemokines MCP-1 and MIP-1alpha at all time points after sepsis (p<0.0001). (Figure 2A). Similar results were seen when human cord blood was stimulated ex vivo with LPS and compared to LPS stimulated whole blood from young adults. Proinflammatory cytokine production was dramatically attenuated in the cord blood (Figure 2B).
Figure 2. Inflammatory and anti-inflammatory chemokine and cytokine responses.
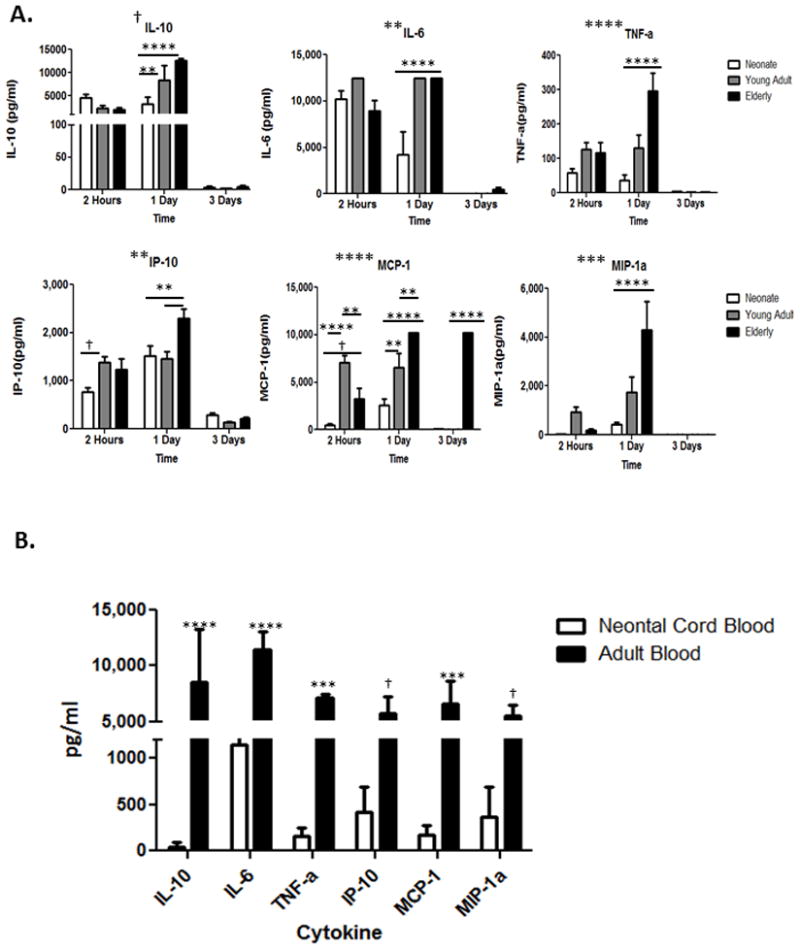
A. Neonatal, young adult, and elderly mice have significant differences in the production of inflammatory and anti-inflammatory cytokine and chemokine responses across all time points after CS sepsis. Significance determined using Two-way ANOVA with Bonferroni multiple comparisons post-hoc test. Symbols placed adjacent to graph titles indicate that age was a significant interaction accounting for a source of variation. Symbols placed within the graph show all significant differences between the age groups for specific cytokine. † = p<0.05. ** = P<0.01. *** = p<0.001. **** = p<0.0001. B. Neonatal cord blood had an attenuated cytokine response compared to adult blood following ex vivo stimulation with LPS across all cytokines studied. † = p<0.05. ** = P<0.01. *** = p<0.001. **** = p<0.0001.
Similarily, we found that recruitment of neonatal peritoneal mouse CD11b+Ly6G+ neutrophils and CD11b+Ly6G− F480+ macrophages to the peritoneum was significantly diminished (p=0.001, Tukey’s Multiple Comparison)(Figure 3), with the recruited cells having decreased ability to produce reactive oxygen species (ROS) compared to adult mice one day after sepsis (p<0.0001, 2-way ANOVA)(Figure 4A).
Figure 3. Elderly mice have increased ability to recruit innate immune effector cells to the peritoneum, whereas neonatal mice have a significantly decreased ability to recruit those same cells.
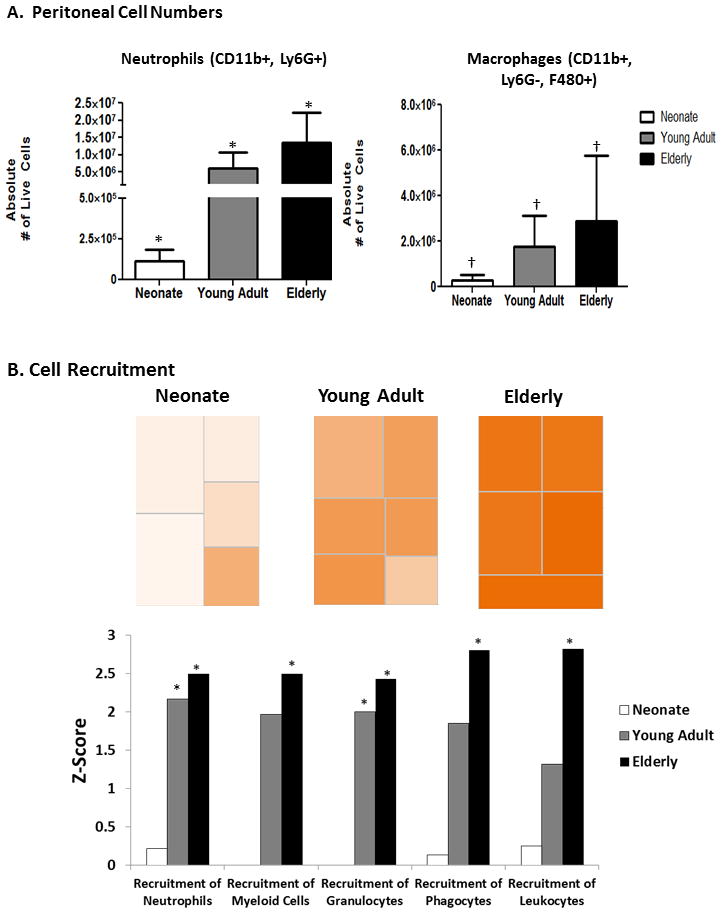
A. Elderly mice have increased absolute numbers of neutrophils (CD11b+, Ly6G+)(* p<0.0001, One-way ANOVA) and macrophages († P<0.05, One-way ANOVA)(CD11b+, Ly6G−, F480+) in the peritoneum via flow cytometry one day after sepsis, whereas neonates had significantly decreased numbers. (n=7–8)(p=0.001, Tukey’s Multiple Comparisons). Data shown are from two or more independent experiments. B. IPA functional pathway analysis revealed that elderly mice have significantly more upregulation of pathways involved in the recruitment of neutrophils, myeloid cells, and phagocytes, while neonates have minimal upregulation of these same pathways (* Z-score >2). Heat maps show the of gene expression of the functional category “Immune Cell Trafficking-Recruitment” 24 hours following sepsis. Orange represents pathways with an over-expression of genes leading to the activation of the pathway, whereas white represents genes that are neither up or down regulated to significance level of p<0.001.
Figure 4. Neontal neutrophils and macrophages have decreased ROS production compared to adult mice.
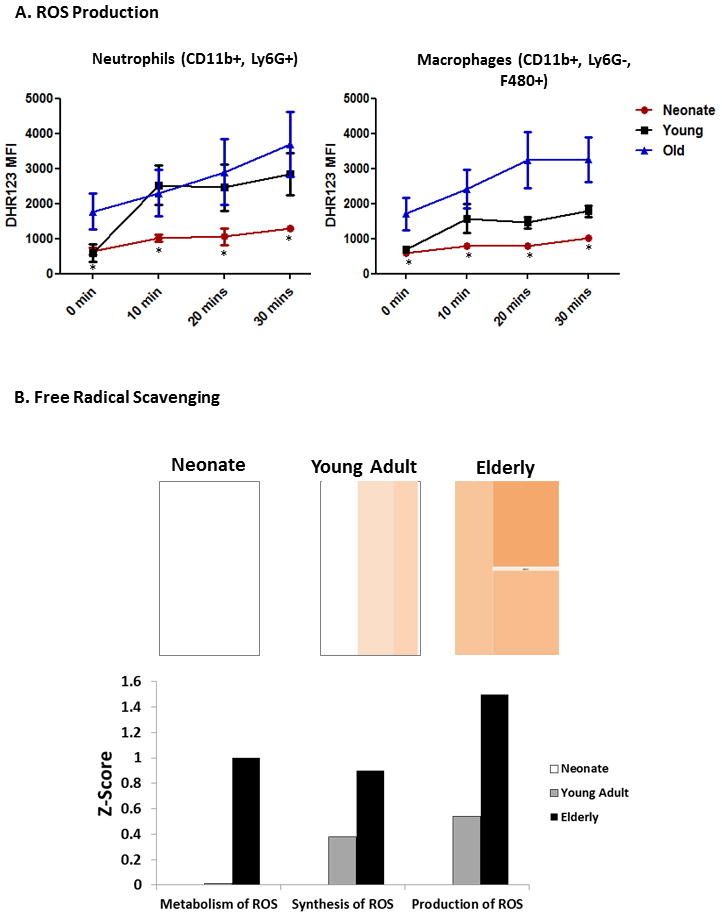
A. Neonatal mouse neutrophils (CD11b+, Ly6G+) and macrophages (CD11b+, Ly6G−, F480+) one day after sepsis had decreased ability to produce reactive oxygen species (ROS) in response to PMA stimulation compared to adult mice one day after sepsis (p<0.0001, 2-way ANOVA). B. Functional pathway analysis showed that elderly mice had increased up regulation of genes involved in the synthesis, production, and metabolism of ROS compared to neonatal and/or young adult mice, which is consistent with in vivo results. Heatmaps show the of gene expression of the functional category “Free Radical Scavenging” 2 hours after sepsis in neonatal, young adult, and elderly mice. Orange represents pathways with an over-expression of genes leading to the activation of the pathway and white represents genes that are neither up or down regulated to significance level of p<0.001. Graph shows the various subcategories and their corresponding significance level (Z-score) of up or down regulation.
In contrast, the response by the elderly mice was fundamentally different. First, their early inflammatory cytokine response was not different than seen in juvenile or young adult animals (Figure 2). We also examined the absolute numbers of CD11b+Ly6G+neutrophils and CD11b+Ly6G−F4/80+ macrophages found in the peritoneum one day after sepsis, and found that elderly mice had an increased ability to recruit innate immune effector cells to the peritoneum, whereas, neonatal mice had a significantly decreased ability to recruit these same cells (p<0.0001, p=0.001, One-way ANOVA)(Figure 3).
Differences in Baseline Gene Expression Compared Among Healthy Neonate, Young Adult or Elderly Mice
We compared the baseline gene expression of naïve neonatal, young adult, and elderly mice to help determine our control populations for genomic comparison after sepsis. Suprisingly, we found that expression patterns from healthy, young adult and elderly naïve mice could not be distinguished from one another by cluster analysis using Pearson correlations (data not shown), despite significant differences in baseline neutrophil and lymphocyte differential counts (p<0.05) (Supplemental Figure 1A). Therefore, it was concluded that the leukocyte transcriptome from healthy elderly and young adult mice could be combined and used as a single control group for comparison to the septic elderly and young adult mice. However, when we compared the transcriptome from healthy, neonatal mice to healthy adult mice, we found that there were 5,798 probesets representing 3987 genes differentially expressed between neonatal and adult mice that were significant at p<0.001. In addition, the the overall pattern of gene expression was significantly different as determined by leave-one-out cross validation (Figure 5A). This difference was unlikely to be caused by the murine WBC differential counts as neonatal WBC differential counts were not significantly different than in young adult or elderly mice. Additionally, when examining the differential make-up of the circulating leukocytes in response to sepsis, it is easy to see that the transcriptomic differences cannot be easily explained by differences or similarities in the prevalent type of circulating leukocyte after injury (Supplemental Figure 1B).
Figure 5. The neonatal transcriptome is fundamentally different at baseline compared to the murine adult.
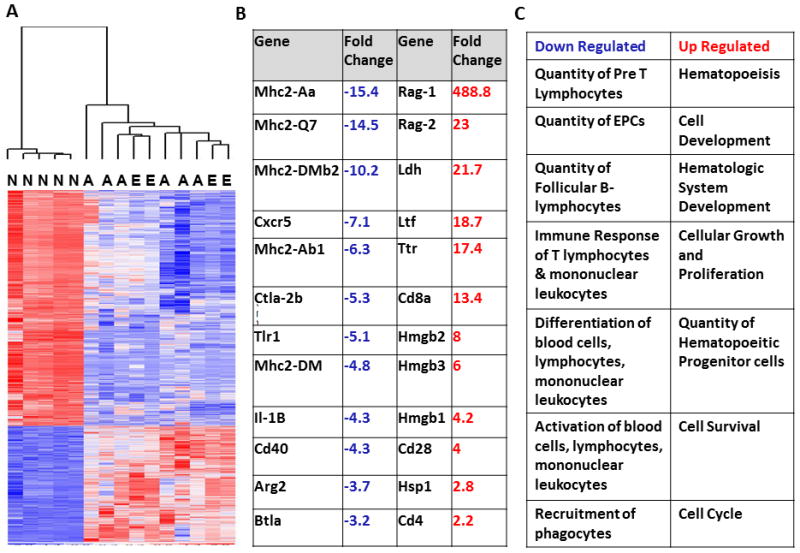
A. Heat map showing the gene expression patterns of naïve neonatal (N), young adult (A), and elderly (E) control mice. Gene expression patterns from young adult and elderly mice could not be differentiated genomically at baseline by an unsupervised analysis. There were 5,798 probe sets (representing 3987 genes) differentially expressed between neonatal and adult mice (including young adult and elderly) that were significant at p<0.001. The overall pattern of gene expression was significantly different as determined by leave-one-out cross-validation. B. Fold changes of selected immune related genes from neonatal control mice compared to adult control mice. Neonatal mice have increased suppression of genes involved in adaptive immunity including MHC II, and decreased expression of genes involving innate immunity and inflammation (p<0.001). C. Categories of functional pathways containing genes that are either up or down regulated in naïve neonatal control mice as compared to adult control mice from Ingenuity Pathway Analysis (IPA).
We examined individual genes that were significantly different between neonatal and healthy adult mice at p<0.001. We found that healthy neonatal mice have decreased expression of genes involved in adaptive immunity, including MhcII, Cd40, and Btla, as well as genes involved in inflammation, such as Il1B and Arg2, which were significantly down regulated compared to adult controls (Figure 5B). The two most highly expressed genes in the naïve neonatal mouse compared to adults were Rag1 and Rag2, which are only expressed in developing lymphocytes, and are indicative that the neonatal adaptive immune system is likely in an active state of development.
We then performed functional analysis on the leukocyte transcriptome from healthy mice using IPA and examined the most highly expressed categories of genes. We found that leukocytes from healthy neonatal mice had over-represented pathways involving hematopoeisis, hematologic systems development, cellular growth and proliferation as well as cellular development, that were all significantly upregulated compared to naïve adults (Z-score>2) (Figure 5C). We also found that neonatal mice had significant down regulation of genes in pathways involved with the quantities, differentiation, and activation of immune cells (Z<−2) (Figure 5C).
Phenotypic Changes are Manifested at the Level of the Blood Leukocyte Transcriptome
We compared the genomic responses between neonatal, young adult, elderly, and control mice at each individual timepoint. There were 10,876, 7,231, and 11,539 probesets, representing 7,012, 4,990, and 7,479 unique genes whose expression changed between neonates, young adults and elderly mice at two hours, one day, and three days respectively (p<0.001). Besides having significant differences in baseline gene expression compared to their adult counterparts (Figure 5), neonatal mice failed to upregulate many genes important in both the innate and adaptive immune resonses, and remained in a predominantly down-regulated state throughout the entire timecourse (Supplemental Figure 2, Supplemental Table 1). The same was true of elderly mice; however, it occurs in a delayed fashion and to a lesser extent.
When reviewing the global gene expression patterns of the neonatal resonse to polymicrobial sepsis over time compared to the adult response, one can see that at two hours following sepsis, neonatal mice only upregulate a very small portion of their genome compared to the patterns of adult mice over time, with only 1,183 probesets representing 884 unique genes that are signficantly altered following sepsis to greater than 4,893 and 4,520 unique genes which are changed in the young adult and elderly populations, respectively (Supplemental Figure 2). Over the time course of sepsis, neonatal mice remain in a mostly unstimulated state, appearing more similar to control mice with minimal up regulation of genes important in the immune response associated with reduced survival. (Supplemental Table 1). Thus, the neonatal response to sepsis is markedly attenuated, and remains unaffected over time compared to both young adult and elderly animals. These results mirrored the results from the IPA functional pathway analysis in which we found that elderly mice had increased up regulation of genes involved in the synthesis, production, and metabolism of ROS compared to neonatal or young adult mice (Figure 4B).
Functional pathway analysis 24 hours after sepsis revealed that neonatal and eldery mice fail to significantly upregulate immune related functional pathways 24 hours after sepsis. In fact, the functional analysis at this time point revealed that neonatal mice have significantly downregulated genes in pathways having to do with proliferation of immune cells, quantity of T-lymphocytes, development of blood cells, leukocytes, and mononuclear leukocytes, and the proliferation of immune related cells (Z-score<−2) (Figure 6). Alternatively, young adult mice upregulate pathways involved in, activation of leukocytes, cell movement and migration of cells, chemotaxis, phagocytosis, and cell homing (Z-score >2), which are not significantly upregulated or are down regulated in either neonataes or elderly mice (Figure 6). Although elderly mice do upregulate some immune related pathways such as recruitment and quantities of phagocytes and neutrophils, they do not upregulate or have down regulated pathways involving immune cell activation, chemotaxis, and leukocyte homing.
Figure 6. Neonatal and elderly mice do not upregulate important immune related pathways 24 hours following sepsis.
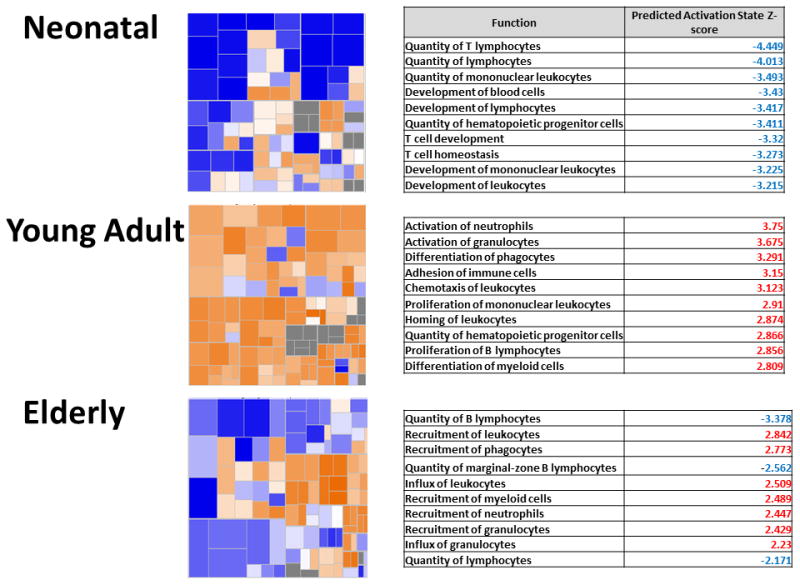
Heatmaps show the of gene expression of the functional category “Hematological Systems Development and Function” 24 hours following sepsis. Orange represents pathways with an over-expression of genes leading to the activation of the pathway, whereas blue represents pathways with an over-expression of genes whose activation will lead to down-regulation of the pathway. Neonatal mice have a large proportion of their pathways which are predominantly down regulated 24 hours after sepsis, which young the majority of young adult pathways are upregulated. Elderly mice fall in between. The corresponding tables show the top 10 pathways and their corresponding Z-score within the functional category 24 hours after sepsis. Red indicates that the pathway is significantly upregulated, whereas blue indicates that the pathway is significantly down regulated.
This functional genomic data at the level of the leukocyte transcriptome, mirrors the results we found when performing functional experiments in vivo. First, we examined the absolute numbers of CD11b+Ly6G+neutrophils and CD11b+Ly6G−F4/80+ macrophages found in the peritoneum one day after sepsis, and found that elderly mice had an increased ability to recruit innate immune effector cells to the peritoneum, whereas, neonatal mice had a significantly decreased ability to recruit these same cells (p<0.0001, p<0.05, One-way ANOVA)(Figure 3A). Likewise, IPA functional pathway analysis revealed that indeed, elderly mice have significantly more upregulation of pathways involved in the recruitment of neutrophils, myeloid cells, and phagocytes (Z-Score >2), whereas, neonates have minimal upregulation of these same pathways (Figure 3B).
Additionally, elderly mice have gene expression changes consistent with the down regulation of pathways involved with the quantity and function of the adaptive immune system, with specific increased down-regulation of humoral immune related pathways involving decreased quanitities of B lymphocytes, IgG, IgM, and immunoglobulin (Z-score <−2).
Three days following sepsis, functional analysis in surviving animals reveals that both neonatal and young adult mice have gene expression patterns consistent with the down regulation of immune related pathways, including T cell development and homeostasis, and quantity and differentiation of leukocytes (Z-score <2), whereas the gene expression of elderly mice three days after sepsis is more consistent with the upregulation of pathways involved with cell movement and homing, recruitment of myeloid cells, granulocytes, and neutrophil transcription, proliferation of blood cells, and the quantities of leukocytes and HSCs (Z-score >2), hence it may be the continued expression of inflammatory mediators that ultimately leads to their increased mortality.
Discussion
We can confirm using a murine model of polymicrobial sepsis that both the neonate and the elderly fare poorly when compared to the young adult. This is the first report comparing the functional and genomic responses to the exact same model of sepsis in the neonate and the elderly, two populations who experience increased mortality. At the most fundamental level of the leukocyte transcriptome, the genomic response to polymicrobial sepsis is markedly different between the aged and the neonatal mouse, despite a similar increased mortality response.
Here, we have demonstrated that the genomic expression of leukocytes from healthy, neonatal mice is fundementally different than that of adult mice, thus likely a strong contributing factor the the neonate’s subsequent dysfunctional response to polymicrobial sepsis. We have also shown that despite aged mice having increased mortality from sepsis, there are minimal gene expression differences at baseline that can help distinguish them, thus indicating that there is something inherent within the elderly murine host response to sepsis that is reponsible for their icreased mortality. Regardless, both the neonatal and the elderly murine genomic reponse to sepsis is strikingly different from the young adult mouse, and this response is associated with adverse outcomes. This difference in genomic response is not a result of the differences in the WBC differntial counts (Supplemental Figure 1). In naïve animals, elderly mice have a higher percentage of neutrophils and a lower percentage of lymphocytes than young adult mice; however, there are no differences between neonatal mice and young or elderly mice, which is where there are significant genomic differences. Additionally, the differences in baseline neutrophil and lymphocyte percentages between elderly and young adult mice are significant (p<0.05) (Supplemental Figure 1A), but these two groups of mice were unable to be distinguished from each other genomically (Figure 5).
Mortality from sepsis remains a significant healthcare problem. For the neonatal population, the mortality from sepsis is highest in VLBW infants that were born prematurely, and has remained relatively stable throughout the years, despite improvements in neonatal intensive care and evolution of ventilatory strategies (4, 8). It is well known that immunity in neonates is distinct from that in adults, and it has been shown that in mice, the adaptive immune system does not play a protective role in neonatal sepsis, and instead, neonatal mice rely on the innate immune system for protection from severe infection(12). The neonatal innate immune response has been found to be “dysfunctional” or “immature” as studies have found murine neonates to have impaired phagocytic ability of innate immune effector cells and poor production of neutrophil extracellular traps (NETs) compared to adults (12, 16, 17). Additionally, genomic studies examine the response to sepsis in neonates through school aged children have found that neonates have impaired innate immunity compared to older children (18).
For the elderly population, the incidence of sepsis continues to increase, right along with the increase in the aging population, and mortality remains high (3). For years, it was thought that the elderly’s defense against pathogens was compromised mainly because of changes in the adaptive immune system mediated by defective T and B lymphocytes; however, it has since been discovered that aging has a profound impact on innate immunity as well (19) with a chronic pro-inflammatory state as well as a predilection towards myelopoiesis. One can easily see the common thread underlying the immune related deficits in both the neonatal and elderly populations, and similar to the neonatal murine population, it has been shown that elderly mice have a higher mortality than young adult mice following polymicrobial sepsis (14, 19, 20), and current evidence for the mechanism of this increased mortality is limited.
There have been conflicting reports implicating the role of serum cytokines, with some studies citing increased levels of serum cytokines as being responsible for the elderly’s higher mortality following sepsis (3), whereas others found a lack of cytokine up-regulation following infection (21, 22). Also, it is known that elderly mice have a predisposition toward myeloid responses following an infectious insult (23); however, these myeloid cells have been shown to exhibit phenotypic differences from those found in young adult mice (24–26). This was shown in our functional analysis of the elderly response to sepsis when 24 hours after sepsis elderly mice exhibited significant upregulation of pathways involving the recruitment and quanties of myeloid cells, however, did not significantly upregulate or had down regulatation of pathways leading to their chemotaxis, homing, or activation, all of which were highly expressed in the young adult mice (Z-score>2). Simlarily, we have shown in vivo that elderly mice preserve the ability to recruit myeloid cells to the peritoneum after sepsis, however, fail to activate those cells leading to the inability to clear bacteria from the site of infection, likely contributing to their increased mortality.
From a functional standpoint, at baseline, neonatal mice upregulate such systems as Hematologic Systems Development, Cellular Development, Growth and Proliferation, Hematopoeisis, and Tissue Morphlogy, as would be expected (Z-score>2). They have down regulation of systems involoved with cell death and organismal survival along with decreased quantities of both B and T lymphocytes (Z-score<−2). This baseline difference is not surprising as neonates are in a transition period from a state in utero where they need a less reactive immune system in order to not mount a response against the mother’s antigens, to an independent, fully functional immune system after they are born. Some evidence of this is that the in murine mice aged 5–7 days, the splenic architecture is different than that of the adult spleen. We have found that the neonatal spleen had poor marginal zone development, increased numbers of myleoid cells, and resembles more of a hematopoeitic organ as it does in fetal life rather than a clearance organ as it is in adult life (11). When this “switch” occurs and is complete is unknown and will be key in developing appropriate therapies to modulate the neonatal immune system so that it responds appropriately to an infectious insult.
Although murine neonates do appear to upregulate some important inflammatory genes after sepsis (i.e. CXCL10, TNFα related genes), they do so to a much lesser extent than adult mice (Supplemental Table 1). These findings were also evident when we performed ex vivo whole blood stimulation of human adult and neontal cord blood with endotoxin. We found that the neontal cord blood inflammatory cytokine and chemokine responoses were markedly attenuated compared to stimulated adult blood (p<0.001) (Figure 2B). These findings are also in accordance with human genomic data published that showed that compared to school aged children, neonates had a larger number of downregulated genes following sepsis compared to healthy neonatal controls (18). Similarily, two hours after polymicrobial sepsis 63% of the genome is downregulated and significantly different from adults (p<0.05) in neonatal mice.
Additionally, although some individual genes may be upregulated in response to sepsis, both functional in vivo and genomic analysis reveals that overall, neonates do not generate an immune response great enough to significantly upregulate important immune related pathways after sepsis, leading to decreased immune cell recruitment, activation, and function. In fact a majority of their immune related pathways remain in a signficantly down regulated state (Figure 6).
The elderly mice also exhibit a suboptimal response to CS sepsis, although the pattern is quite different than the suboptimal response found in the neonatal population. Like neonates, the overall genomic response is attenuated compared to young adult mice. However, although elderly mice do preserve the ability to recruit myeloid cells to the peritoneum, they do not fully activate these cells and they remain partially dysfunctional. Additionally, elderly mice do not appear to be as able to return toward baseline levels of gene expression as is observed in young adult mice, and have a significant number of inflammatory genes whose expression remains increased to a greater extent than young adult mice three days after sepsis (Supplemental Table 1). This elderly murine response with an inability to return to baseline is similar to published data from the Glue Grant “Inflammation and Host Response to Injury Large Scale Collaborative Research Program,” in which we reported that patients with complicated outcomes exhibited prolonged, altered gene expression and a failure to return to baseline gene expression levels within 28 days (27).
In summary, the murine response to polymicrobial sepsis varies greatly over the spectrum of age from the neonatal period through young adulthood, and into the elderly population. Both neonatal and elderly mice have increased mortality following a murine model of polymicrobial sepsis, however, the underlying mechanisms for this increased mortality appear to be fundementally distinct when examining their transcriptomic responses to sepsis. The neonatal transcriptome appears to be distinct from the adult transcriptome not only at baseline, where adaptive immune gene expression is suppressed, but also in response to polymicrobial sepsis where their innate immune responses are markedly attenuated. Elderly mice exhibit a dysfunctional response to sepsis as well, also characterized by attenuated innate inflammatory response with continued systemic inflammation and adpative immune suppression, with the inability to return toward baseline homeostasis. Considering these genomic differences, sepsis therapies aimed at the neonatal and elderly populations will have to overcome many hurdles.
Supplementary Material
Acknowledgments
AGC and LFG were supported by a T32 training grant (T32 GM-008721-13) in burns and trauma from the NIGMS. Other support was provided by R01 GM-40586-24 and R01 GM-80576-06, awarded by the NIGMS, and the Claude D. Pepper Older Americans Independence Center (NIH/NIA P30AG028740).
Footnotes
Authorship Contributions
LFG contributed to the analysis of data, conception, design, composition, drafting, and editing of the manuscript.
MCP contributed to the analysis of data and editing of the manuscript.
DCN performed animal experiments, data collection, and contributed to the editing of the manuscript.
EV, AGC, SL, and AC contributed to the manuscript editing.
RU contributed animal experiments and laboratory data collection as well as to the editing of the manuscript.
CL contributed to the design and editing of the manuscript.
BAM contributed to the manuscript and editing.
FAM contributed to the editing of the manuscript.
HVB contributed to the conception and design of the manuscript, data analysis and interpretation, and the composition/editing of the manuscript.
LLM contributed to the conception and design of the manuscript, data analysis and interpretation, and the composition/editing/writing of the manuscript.
PAE contributed to the analysis of data, conception, design, composition, drafting, and editing of the manuscript
Conflict of Interest Disclosures None of the authors have disclosed any conflict of interest.
References
- 1.Carlet J, Cohen J, Calandra T, Opal SM, Masur H. Sepsis: time to reconsider the concept. Crit Care Med. 2008;36:964–966. doi: 10.1097/CCM.0B013E318165B886. [DOI] [PubMed] [Google Scholar]
- 2.Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001;16:83–96. doi: 10.1097/00024382-200116020-00001. [DOI] [PubMed] [Google Scholar]
- 3.Turnbull IR, Clark AT, Stromberg PE, Dixon DJ, Woolsey CA, Davis CG, Hotchkiss RS, Buchman TG, Coopersmith CM. Effects of aging on the immunopathologic response to sepsis. Crit Care Med. 2009;37:1018–1023. doi: 10.1097/CCM.0b013e3181968f3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watson RS, Carcillo JA, Linde-Zwirble WT, Clermont G, Lidicker J, Angus DC. The epidemiology of severe sepsis in children in the United States. Am J Respir Crit Care Med. 2003;167:695–701. doi: 10.1164/rccm.200207-682OC. [DOI] [PubMed] [Google Scholar]
- 5.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29:1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 6.Martin GS, Mannino DM, Moss M. The effect of age on the development and outcome of adult sepsis. Crit Care Med. 2006;34:15–21. doi: 10.1097/01.ccm.0000194535.82812.ba. [DOI] [PubMed] [Google Scholar]
- 7.PrabhuDas M, Adkins B, Gans H, King C, Levy O, Ramilo O, Siegrist CA. Challenges in infant immunity: implications for responses to infection and vaccines. Nature immunology. 2011;12:189–194. doi: 10.1038/ni0311-189. [DOI] [PubMed] [Google Scholar]
- 8.Wynn JL, Wong HR. Pathophysiology and treatment of septic shock in neonates. Clinics in perinatology. 2010;37:439–479. doi: 10.1016/j.clp.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lukacs SL, Schoendorf KC, Schuchat A. Trends in sepsis-related neonatal mortality in the United States, 1985–1998. The Pediatric infectious disease journal. 2004;23:599–603. doi: 10.1097/01.inf.0000131633.74921.90. [DOI] [PubMed] [Google Scholar]
- 10.Lawn JE, Cousens S, Zupan J T Lancet Neonatal Survival Steering. 4 million neonatal deaths: when? Where? Why? Lancet. 2005;365:891–900. doi: 10.1016/S0140-6736(05)71048-5. [DOI] [PubMed] [Google Scholar]
- 11.Wynn JL, Scumpia PO, Delano MJ, O’Malley KA, Ungaro R, Abouhamze A, Moldawer KL. Increased mortality and altered immunity in neonatal sepsis produced by generalized peritonitis. Shock. 2007;28:675–683. doi: 10.1097/SHK.0b013e3180556d09. [DOI] [PubMed] [Google Scholar]
- 12.Wynn JL, Scumpia PO, Winfield RD, Delano MJ, Kelly-Scumpia K, Barker T, Ungaro R, Levy O, Moldawer LL. Defective innate immunity predisposes murine neonates to poor sepsis outcome but is reversed by TLR agonists. Blood. 2008;112:1750–1758. doi: 10.1182/blood-2008-01-130500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sands KE, Bates DW, Lanken PN, Graman PS, Hibberd PL, Kahn KL, Parsonnet J, Panzer R, Orav EJ, Snydman DR, Black E, Schwartz JS, Moore R, Johnson BL, Jr, Platt R G Academic Medical Center Consortium Sepsis Project Working. Epidemiology of sepsis syndrome in 8 academic medical centers. JAMA. 1997;278:234–240. [PubMed] [Google Scholar]
- 14.Weng NP. Aging of the immune system: how much can the adaptive immune system adapt? Immunity. 2006;24:495–499. doi: 10.1016/j.immuni.2006.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turnbull IR, Wlzorek JJ, Osborne D, Hotchkiss RS, Coopersmith CM, Buchman TG. Effects of age on mortality and antibiotic efficacy in cecal ligation and puncture. Shock. 2003;19:310–313. doi: 10.1097/00024382-200304000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, Chandler NB, Rodesch CK, Albertine KH, Petti CA, Weyrich AS, Zimmerman GA. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood. 2009;113:6419–6427. doi: 10.1182/blood-2008-07-171629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuenca AG, Wynn JL, Moldawer LL, Levy O. Role of Innate Immunity in Neonatal Infection. American journal of perinatology. 2013 doi: 10.1055/s-0032-1333412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wynn JL, Cvijanovich NZ, Allen GL, Thomas NJ, Freishtat RJ, Anas N, Meyer K, Checchia PA, Lin R, Shanley TP, Bigham MT, Banschbach S, Beckman E, Wong HR. The influence of developmental age on the early transcriptomic response of children with septic shock. Mol Med. 2011;17:1146–1156. doi: 10.2119/molmed.2011.00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Solana R, Pawelec G, Tarazona R. Aging and innate immunity. Immunity. 2006;24:491–494. doi: 10.1016/j.immuni.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 20.Schneider CP, Schwacha MG, Chaudry IH. Impact of sex and age on bone marrow immune responses in a murine model of trauma-hemorrhage. Journal of applied physiology. 2007;102:113–121. doi: 10.1152/japplphysiol.00848.2006. [DOI] [PubMed] [Google Scholar]
- 21.Mares CA, Sharma J, Ojeda SS, Li Q, Campos JA, Morris EG, Coalson JJ, Teale JM. Attenuated response of aged mice to respiratory Francisella novicida is characterized by reduced cell death and absence of subsequent hypercytokinemia. PloS one. 2010;5:e14088. doi: 10.1371/journal.pone.0014088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hinojosa E, Boyd AR, Orihuela CJ. Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. The Journal of infectious diseases. 2009;200:546–554. doi: 10.1086/600870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dykstra B, Olthof S, Schreuder J, Ritsema M, de Haan G. Clonal analysis reveals multiple functional defects of aged murine hematopoietic stem cells. J Exp Med. 2011;208:2691–2703. doi: 10.1084/jem.20111490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyd AR, Shivshankar P, Jiang S, Berton MT, Orihuela CJ. Age-related defects in TLR2 signaling diminish the cytokine response by alveolar macrophages during murine pneumococcal pneumonia. Experimental gerontology. 2012;47:507–518. doi: 10.1016/j.exger.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McConnell KW, Fox AC, Clark AT, Chang NY, Dominguez JA, Farris AB, Buchman TG, Hunt CR, Coopersmith CM. The role of heat shock protein 70 in mediating age-dependent mortality in sepsis. J Immunol. 2011;186:3718–3725. doi: 10.4049/jimmunol.1003652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang SC, Matsutani T, Choudhry MA, Schwacha MG, Rue LW, Bland KI, Chaudry IH. Are the immune responses different in middle-aged and young mice following bone fracture, tissue trauma and hemorrhage? Cytokine. 2004;26:223–230. doi: 10.1016/j.cyto.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, Hayden DL, Hennessy L, Moore EE, Minei JP, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Brownstein BH, Mason PH, Baker HV, Finnerty CC, Jeschke MG, Lopez MC, Klein MB, Gamelli RL, Gibran NS, Arnoldo B, Xu W, Zhang Y, Calvano SE, McDonald-Smith GP, Schoenfeld DA, Storey JD, Cobb JP, Warren HS, Moldawer LL, Herndon DN, Lowry SF, Maier RV, Davis RW, Tompkins RG. A genomic storm in critically injured humans. J Exp Med. 2011;208:2581–2590. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.