

Abstract
Multidrug-resistance is a major cause of cancer chemotherapy failure in clinical treatment. Evidence shows that multidrug-resistant cancer cells are as sensitive as corresponding regular cancer cells under the exposure to anticancer ceramide analogs. In this work we designed five new ceramide analogs with different backbones, in order to test the hypothesis that extending the conjugated system in ceramide analogs would lead to an increase of their anticancer activity and selectivity towards resistant cancer cells. The analogs with the 3-ketone-4,6-diene backbone show the highest apoptosis-inducing efficacy. The most potent compound, analog 406, possesses higher pro-apoptotic activity in chemo-resistant cell lines MCF-7TN-R and NCI/ADR-RES than the corresponding chemo-sensitive cell lines MCF-7 and OVCAR-8, respectively. However, this compound shows the same potency in inhibiting the growth of another pair of chemo-sensitive and chemo-resistant cancer cells, MCF-7 and MCF-7/Dox. Mechanism investigations indicate that analog 406 can induce apoptosis in chemo-resistant cancer cells through the mitochondrial pathway. Cellular glucosylceramide synthase assay shows that analog 406 does not interrupt glucosylcer-amide synthase in chemo-resistant cancer cell NCI/ADR-RES. These findings suggest that due to certain intrinsic properties, ceramide analogs’ pro-apoptotic activity is not disrupted by the normal drug-resistance mechanisms, leading to their potential use for overcoming cancer multidrug-resistance.
Keywords: Ceramide, Glucosylceramide synthase (GCS), P-glycoprotein, Multidrug resistance, Anti-cancer drugs
1. Introduction
Ceramides, known as ‘tumor suppressor lipids’, regulate the anti-cancer signals implicated in apoptosis, cell cycle arrest, and autophagic responses.1,2 Exposure to chemotherapeutic agents and radiation therapy lead to increased levels of endogenous ceramide, thereby inducing apoptosis through mitochondrial or non-mitochondrial pathways.3 Similarly, synthetic ceramide analogs have been shown toimitate ceramide action on its downstream targets, leading to apoptosis and cell cycle arrest.2 However, ceramide-metabolizing enzymes can decrease cellular ceramide levels, resulting in cell proliferation, migration, and survival.4,5 One such enzyme, glucosylceramide synthase (GCS), converts ceramide into glucosylceramide on the cytosolic surface of the Golgi apparatus. Evidence shows a close relationship between GCS and P-glycoprotein (P-gp), an important multidrug-resistance (MDR) protein. GCS inhibitor treatment in multidrug-resistant cancer cells down-regulates the expression of MDR1, the P-gp-encoding gene.6 Furthermore, drug-resistant cancer cells exposed to GCS inhibitors become sensitive to anticancer agents.6–8 Ceramide analogs have the potential to act as inhibitors of these ceramide-metabolizing enzymes and thus maintain cellular ceramide levels necessary to induce cell death.9 Therefore, ceramide analogs can exert their anti-cancer activities through two main approaches: (1) acting on ceramide downstream targets to promote cell death, and (2) inhibiting ceramide-metabolizing enzymes to increase cellular ceramide levels.
Due to the crucial role of ceramide in cell death regulation, hundreds of anticancer ceramide analogs have been synthesized and investigated in recent years.10–16 Our previous work and that of many other research groups have shown that certain ceramide analogs preferentially inhibit the growth of chemo-resistant cancer cells in comparison to regular cancer cells.17–22 Because of the relationship between GCS and multi-drug resistance, a hypothesis was formed that manipulation of glucosylceramide levels by inhibiting GCS is a useful way of inducing preferential killing of MDR cells.18 In this work, we have tested this hypothesis by determining GCS activity in the chemo-sensitive and -resistant cancer cells.
In this study, we designed 3-ketone-4,6-diene-containing ceramide analogs to determine whether extending the conjugated system in the backbone of the analogs would lead to an increase in the anticancer activity and selectivity towards chemo-resistant cancer cells. This design was based on the lead compounds 4,6-diene-ceramide and analog 3 as previously reported (Fig. 1).22,23 The α-ketone–diene on the ceramide backbone and phenylacetyl functional group on the amide side chain are expected to increase the molecular rigidity of analog 406, which in turn is expected to enhance its interaction with ceramide downstream targets and enhance cell death. We also seek to elucidate the mechanism of the cell death elicited by our compounds.
Figure 1.
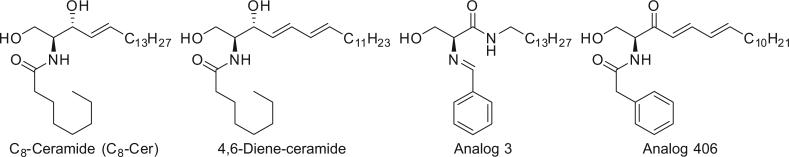
The structure of C8-ceramide, 4,6-diene-ceramide, analogs 3 and 406. C8-ceramide is a short-chain ceramide widely used as a positive control in the studies of ceramide analogs.
2. Results and discussion
2.1. Synthesis of ceramide analogs 401, 402, 403, 404, and 406
The syntheses of analogs 401–406 were achieved according to Chun's method as reported previously.24 Condensation of serine methyl ester derivative 1 with methyl phenyl sulfoxide gave bketosulfoxide 3. In Chun's paper, a diastereomeric mixture of 3 was mentioned. Since the sulfur atom of the sulfinyl functional group is a chiral center, theoretically, compound 3 consists of two diastereomers, 3R and 3S (Scheme 1). These two isomers (3-up and 3-down, a higher spot and a lower spot observed by TLC) were successfully separated in this work through silica column chromatograph with petroleum ether/ethyl acetate 1.5:1 as the eluent. The ratio of 3-up and 3-down depends on the starting material (methyl phenyl sulfoxide) rather than the reaction. We could not designate the absolute configurations of 3-up and 3-down, because each compound has a very complex NMR spectrum, suggesting presence of conformational isomerism, which will be discussed in the followed section. Fortunately, as synthetic intermediates, 3R and 3S possess the same reactivity with the allylic bromide 2, which was derived from the allylic alcohol ((E)-tridec-2-en-1-ol), to give 3-keto-4,6-diene 401, thus isolation of 3R and 3S was not necessary in the actual synthetic procedure. Employing mild reaction conditions (K2CO3 in dimethylformamide (DMF) at room temperature), an all-trans diene 401 was produced executively. The all-trans configuration could not be directly confirmed by the 1H NMR spectrum of 401 since the H-signals overlap in 6.2–6.1 ppm region. However, in its derivatives, 1H NMR, COSY, and NOE spectra can determine the all-trans configuration. Selective reduction of 3-keto-4,6-diene (401) by diisobutylaluminum hydride (DIBAL-H) in tetrahydrofuran (THF) gave (S)-3-hydroxy-4,6-diene (402) as the predominant product in 50% yield. After treatment of 401 with trifluoroacetic acid in dry dichloromethane (DCM), 3-keto-4,6-diene-sphingosine 405 was obtained. d-Sphingosine (purchased from CNH Technologies, Inc., Woburn, MA), (S)-2-amino-3-hydroxy-N-tetradecylpropanamide (synthesis was described previously23), and compound 405 were amidated with phenyl acetyl chloride to give the ceramide analogs, 403, 404, and 406, respectively.
Scheme 1.

Synthetic routes for analogs 401, 402, 403, 404, and 406. Reagents and conditions: (a) methyl phenyl sulfoxide, LDA/THF; (b) triphenylphosphine, NBS/CH2Cl2; (c) K2CO3/DMF; (d) DIBAL-H/THF; (e) trifluoroacetic acid/CH2Cl2; (f) phenylacetyl chloride, NaHCO3/CH2Cl2.
2.2. Isomerism and NMR spectral properties of compound 3
As mentioned above, compound 3 was obtained as a mixture of 3-up and 3-down diastereomers. However, we could not assign R or S designations to the two. Each compound shows complex NMR spectral behaviors, which are fully displayed in the Supplementary Materials. A section of the 13C NMR spectra for these diastereomersis is shown in Figure 2. Generally, two sets of peaks are seen for each single pure compound. The ratio of these two sets of peaks is 0.55:0.45 for both 3-up and 3-down. It is obvious that the two sets of peaks represent two relatively stable conformations of each compound. The hypothesized models of conformational isomerism are shown in Figure 3. The sp2 hybridization of the nitrogen atom in the carbamate group results in a planar carbamate functional group (pink residue). Due to the presence of a bulky group (–COCH2SOPh) on the carbon next to the N in the five-membered oxazoline ring, the steric effects lead to the – COCH2SOPh group locating in the space above or below the carbamate plane. Since these two poses cannot freely interchange, two relatively stable conformations are formed which are reflected by the two sets of signals in the NMR spectra. Figure 3 shows the two possible conformations of 3 in half-chair and envelope forms, respectively. In both conformations the α-H (in blue) is axial, which is consistent with the observations in 1H NMR spectra. The starting material 1, and ceramide analogs 401, and 402 all show the same dualistic signals in NMR spectra with different ratios. Once the five-membered ring is opened, the compounds produced (405 and 406) lose this characteristic, suggesting that the ring is critical in restricting the conformational interchange. In our previous work, we observed similar dualistic signals in the six-membered ring compounds, N-Boc-carboline-3-carboxylic acid and its derivatives.25 These observations indicate that the in-ring nitrogen amidation and the presence of a bulky group on the neighboring ring carbon play important roles in the formation of the two restricted conformations.
Figure 2.

13C NMR spectrum sections for the two diastereomers3-up and 3-down. In each spectrum, two sets of carbon peaks are observed, suggesting the presence of two conformational isomers for each diastereomer. The complete spectral data are provided in the Supplementary Materials.
Figure 3.

Hypothesized conformations for the conformational isomers of compound 3. For easy observation two methyl groups in 2-position of the oxazolidineare not shown up in the projections. The bulky group –COCH2SOPh is located above the carbamate group plane in the half-chair form, while below the amide plane in the envelope form. In both conformations α-H is in an axial position.
2.3. Ceramide analogs inhibiting cellular viability and clonogenic survival in MCF-7, MDA-MB-231, and MCF-7TN-R cells
In order to determine the cytotoxic effects of the ceramide analogs on MCF-7, MDA-MB-231, and MCF-7TN-R cells, MTT cell viability assays were performed using various concentrations of each analog ranging from 1 to 100 μM. The results are shown in Table 1. Compounds 401 and 406 were the most effective compounds across all cell lines with IC50 values of 4.05 ± 1.30 μM and 4.26 ± 1.48 μM, respectively, in the chemo-resistant MCF-7TN-R cell line. Interestingly, IC50 values for all analogs except analog 401 were lower in the chemo-resistant MCF-7TN-R cells compared to the sensitive MCF-7 cells, indicating that these compounds exhibit increased therapeutic potential in drug-resistant cancers (Table 1).
Table 1.
IC50 values for ceramide analogs in MTT viability assay (μM)
| Compound | MCF-7 | MDA-MB-231 | MCF-7TN-R |
|---|---|---|---|
|
IC50 (μM) |
|||
| 401 | 3.91 ± 1.51 | 17.23 ± 0.94 | 4.05 ± 1.30 |
| 402 | 26.44 ± 1.00 | 75.15 ± 0.88 | 9.91 ± 0.65 |
| 403 | 37.28 ± 2.45 | 51.23 ± 0.76 | 4.74 ± 1.07 |
| 404 | 233.6 ± 4.87 | N.D. | 28.96 ± 0.78 |
| 406 | 22.03 ± 1.52 | 161.4 ± 2.28 | 4.26 ± 1.48 |
MCF-7, MDA-MB-231, and MCF-7TN-R cells were treated with increasing concentrations of the analogs and studied for 24 h. The values are the mean ± SE of three independent experiments. N.D., not determined.
There is a debate in the literature as to the reliability of short-term viability assays in predicting clinical response. Therefore, we performed long-term clonogenic survival assays to better determine the efficacy of these analogs. As shown in Table 2, compounds 401 and 406 were again the most effective with IC50 values of 1.85 ± 1.22 μM and 1.81 ± 1.18 μM, respectively, in the MCF-7TN-R cell line. Similar to what was observed in the short-term viability assays, analogs 401 and 406 exhibited the most potent anti-survival effects in the drug-resistant cell lines. Taken together, these results provide proof of principle that these analogs exhibit anti-cancer properties.
Table 2.
IC50 values of ceramide analogs in clonogenic survival assay (μM)
| Compound | MCF-7 | MDA-MB-231 | MCF-7TN-R |
|---|---|---|---|
|
IC50 (μM) |
|||
| 401 | 5.07 ± 0.17 | 1.45 ± 0.06 | 1.85 ± 1.22 |
| 402 | 5.69 ± 0.65 | 3.17 ± 0.10 | 5.19 ± 0.11 |
| 403 | 4.18 ± 0.52 | 1.59 ± 0.10 | 5.62 ± 0.44 |
| 404 | 10.16 ± 0.14 | 17.55 ± 0.10 | 10.05 ± 0.59 |
| 406 | 3.40 ± 0.37 | 1.40 ± 0.13 | 1.81 ± 1.18 |
MCF-7, MDA-MB-231, and MCF-7TN-R cells were treated with increasing concentrations of the analogs studied and allowed to grow until colony formation was noted (generally 10–12 days). The values are the mean ± SE of three independent experiments.
When comparing analogs with different backbones, it is obvious that analog 406 with 3-ketone-4,6-diene moiety shows higher efficacy than its counterparts with 3-hydroxy-4-ene (403) or serine amide (404) backbones in both short-term and long-term assays. Analog 406 is even more potent than 4,6-diene-ceramide (structure shown in Fig. 1), activity of which was previously reported in the chemo-resistant MCF-7TN-R cell line (IC50 11.3 μM in cell viability assay).22 The fact that 3-ketone-4,6-diene backbone is more effective than 3-hydroxy-4,6-diene backbone in killing cancer cells was also proven through comparing the activities of analogs 401 and 402, indicating that rigid modification of ceramide backbone improves anti-cancer activities. On the other hand, 3-ketone-4,6-diene backbone is also a reactive residue, a so-called Michael acceptor, which can react with endogenous nucleophiles, suggesting that these compounds may be a pro-drug in inducing cell death. The selective killing of chemo-resistant cells was observed with certain analogs, especially analog 406. Thus, more cell lines were studied to evaluate the effectiveness of analog 406 towards chemo-resistant cells as described in the following sections.
2.4. Ceramide analogs 401 and 406 induce apoptosis through the intrinsic cell death pathway
The mechanism of ceramide-induced cell death through the induction of apoptosis is well-established.21,22 In this work, we were interested in determining whether our analogs inhibited cell survival through enhancement of programmed cell death. Given that analogs 401 and 406 were the most effective in the viability and clonogenic survival assays, we further determined the ability of these analogs in inducing apoptosis in chemo-resistant breast cancer cells. Fragmented oligonucleotides were determined as a measure of programmed cell death. As seen in Figure 4A, treatment with analog 406 resulted in a 4.30 ± 1.10 fold (p <0.05) increase in apoptosis compared to the control. Similarly, analog 401 increased programmed cell death by 3.09 ± 0.56 fold (p <0.05). Both analogs exhibited increased apoptotic activity compared to parental C8-Cer (structure shown in Fig. 1).
Figure 4.

Effects of ceramide analogs on breast cancer intrinsic cell death. MCF-7TN-R cells were treated with double IC50 concentrations (the IC50 values determined from MTT viability assay) for 24 h. (A) Treatment with analog 406 induced a 4.30 ± 1.10 fold (*p <0.05) increase in apoptosis compared to vehicle control. (B) Treatment with analog 406 induced a 3.59 ± 0.45 fold (*p <0.05) increase in caspase-9 activity compared to vehicle control. DMSO, vehicle control; Taxol and C8-Cer, positive control. The values are the mean ± SE of three independent experiments.
Apoptosis is initiated through either the extrinsic or intrinsic cell death pathways. We further determined whether these analogs utilized the intrinsic pathway through the determination of cellular caspase-9 levels. Caspase-9 is known to be activated in breast cancer cells exclusively in the intrinsic cell death. As shown in Figure 4B, analog 406 increased caspase-9 activity 3.59 ± 0.45 fold (p <0.05), while analog 401 induced caspase-9 activity 1.86 ± 0.75 folds compared to the vehicle control. These results were greater than parental C8-Cer (structure included in Fig. 1), which demonstrated only a 1.18 ± 0.09 fold (p <0.05) increase in caspase-9 activity, thus correlating with our apoptosis findings.
2.5. Resistant cancer cells NCI/ADR-RES and MCF-7/Doxsensitive to analog 406
To clarify the capability of analog 406 for selectively killing chemo-resistant cancer cell lines, anti-viability activities of analog 406 were evaluated independently in pairs of sensitive-resistant lines, OVCAR-8 to NCI/ADR-RES ovarian cancer cells and MCF-7 to MCF-7/Dox breast cancer cells. As it was observed above, analog 406 exhibits a lower IC50 (4.92 μM, Fig. 5B) towards chemo-resistant NCI/ADR-RES cells than towards chemo-sensitive OVCAR-8 cells (7.82 μM, Fig. 5A), indicating its preferentially killing of chemo-resistant cells. On the other hand, analog 406 equally inhibits the viability of MCF-7 and MCF-7/Dox cells (Fig. 5C and D), suggesting that the selectivity towards chemo-resistant cells varied in different cell lines developed by different drugs. Nevertheless, chemo-resistant MCF-7/Dox cells are still sensitive to analog 406 at the same degree as chemo-sensitive MCF-7 cells, proving that analog 406's activity is not interrupted by multi-drug resistance mechanism.
Figure 5.

Ceramide analog 406 effectively eliminates drug-resistant cancer cells in ovarian and breast cancers. Error bars represent the standard errors of three independent experiments. Cells were treated with ceramide analogs for 72 h. *p <0.01 compared with in cells treated with analog 3. The IC50 values of analogs in each cell line are indicated. (A) Drug-sensitive OVCAR-8 human ovarian cancer cells. (B) Drug-resistant NCI/ADR-RES human ovarian cancer cells. (C) Drug-sensitive MCF-7 human breast cancer cells. (D) Drug-resistant MCF-7/Dox human breast cancer cells.
2.6. Effect of analog 406 on glucosylceramide synthase (GCS)
Since glucosylceramide synthase (GCS) is an important target for inhibiting P-gp and consequently reversing or overcoming multi-drug resistance, the effect of analog 406 on glucosylceramide synthase (GCS) was studied in both OVCAR-8 and NCI/ADR-RES cell lines. Based on modification of a previously described protocol, the activity of GCS in cells was determined using the ratio of glucosylceramide to ceramide concentrations.26 The ratio of glucosylceramide to ceramide spots’ intensity as observed on thin layer chromatography (TLC) plates (Fig. 6) shows that the reference analog 3 has a weak inhibitory activity toward GCS, while analog 406 significantly reduces GCS activity in chemo-sensitive OVCAR-8 cells but does not influence GCS activity in chemo-resistant NCI/ADR-RES cells, suggesting that sensitivity of resistant cells to analog 406 is not attributed to GCS inhibition. Thus, the capacity to sensitize resistant cells is expected to be a common characteristic of ceramide analogs regardless of the action on GCS/P-gp pathway. A possible explanation is that due to their lipid solubility, ceramide analogs can freely pass through the plasma membrane. Another explanation given by Shirahama et al. is that P-glycoprotein acts as a transporter for most chemotherapeutic agents but not for ceramide analogs.20 Therefore, ceramide analogs ignore the high expression of MDR proteins in resistant cancer cells. While more data on ceramide derivatives and their anti-cancer activities are needed to provide clarification, ceramide analogs should continue to be studied as potential chemotherapeutic agents for overcoming cancer multi-drug resistance.
Figure 6.

Fluorescent chromatograms of lipid components in OVCAR-8 (A) and NCI/ADR-RES (B) cells. Cells were grown for 24 h in 6-well plates with a 10% FBS RPMI-1640 medium, and then exposed to blank control (DMSO), analog 3, or analog 406 in 5% FBS RPMI-1640 medium for 4 h at 37 °C. Cells were switched to 1% BSA RPMI-1640 medium containing NBD C6-Cer complexed to BSA. After 2 h of incubation at 37 °C, lipids were extracted using chloroform. Fluorescent chromatograms were captured by the AlphaImager, and were inverted using the Adobe Photoshop CS2 Program. Cer, ceramide; GluCer, glucosylceramide.
3. Conclusions
For many cancer patients, resistance to chemo and endocrine therapy (especially multidrug-resistance) is a major cause of treatment failure. We have observed that certain ceramide analogs have the capability to selectively kill resistant cancer cells in comparison to regular cancer cells. Discovering chemotherapeutic agents capable of reversing or ignoring resistance is a valid and important approach for prolonging the lives of advance stage cancer patients. Thus, for the studies presented here, we designed and synthesized five new ceramide analogs, and evaluated their activity on various sensitive and resistant cancer cell lines. Among all of the ceramide analogs with 3-hydroxy-4-ene, 3-hydroxy-4,6-diene, 3-ketone-4,6-diene, and serine amide backbones, which we have designed, synthesized, and studied in this and previous work, analogs 401 and 406 with the 3-ketone-4,6-diene backbone are the most effective in the chemo-resistant MCF-7TN-R cell line. Analog 406 selectively kills TNF-induced resistant breast cancer cells MCF-7TN-R and doxorubicin-induced resistant ovarian cancer cells NCI/ADR-RES, compared with regular MCF-7 and OVCAR-8 cells, respectively. However, this compound has comparable inhibitory activity towards MCF-7 and its resistant cell line MCF-7/Dox. Mechanism investigations show that analogs 401 and 406 can induce cell apoptosis via the intrinsic pathway. These findings suggest that these compounds activate ceramide downstream effectors, supporting our hypothesis that prolonging the conjugated system of the ceramide backbone increases the pro-apoptotic effects. Cellular GCS assays have shown that analog 406 does not interfere with the activity of glucosylceramide synthase in chemo-resistant cancer cells. This indicates that the selectivity of analog 406 towards resistant cancer cells does not correlate with GCS inhibition. Together, our results show that pharmacological intervention via alteration of ceramide signaling and metabolism has promising therapeutic potential for the treatment of chemo-resistant cancers.
4. Experimental section
4.1. Chemistry
All chemicals were purchased from Sigma–Aldrich Corporation (St. Louis, MO) and Fisher Scientific International Inc (Hampton, NH), except d-sphingosine/HCl, which was purchased from CNH Technologies Inc (Woburn, MA). 1H NMR and 13C NMR spectra were recorded on a Bruker Fourier 300 MHz FT-NMR spectrometer. 2D NMR experiments were performed on a Bruker Avance III 400 MHz NMR spectrometer, and the data was processed using the MestReNova NMR software (School of Chemistry, University of Bristol, Bristol, UK). All of the tested compounds were confirmed to be >95% pure via RP-HPLC method. RP-HPLC was performed on an hp Hewlett Packard Series 1050 (Column: Phenomenex Gemini-NX 5u C18 110A). The HPLC conditions were methanol/water (85:15) at a 1.0 mL/min flow rate with a UV detector (Sig = 254.4 nm, Ref = 360.1 nm). Mass spectral data were obtained using an Agilent 6890 GC with a 5973 MS. High-resolution mass spectra (HRMS) were performed on a Bruker Spectrospin APEX TM 47e FT-IRC instrument.
4.1.1. (E)-1-Bromotridec-2-ene (2)
At 0 °C, to a solution of 1.89 g (10 mmol) of (E)-tridec-2-en-1-ol and 2.75 g (10.5 mmol) of triphenylphosphine (Ph3P) in 30 mL of dry CH2Cl2 (DCM), 1.96 g (11 mmol) of N-bromosuccinimide (NBS) was added. The reaction mixture was stirred at 0 °C for 1 h, and then stirred at room temperature for 1 h. The mixture was diluted with 120 mL of hexane and passed through a pad of silica gel (2 cm). The filtrate was concentrated to give 2.06 g (79%) of (E)-1-bromotridec-2-ene as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 6.11 (m, 2H), 4.37 (d, J = 7.5 Hz, 2H), 1.96 (m, 2H), 1.29 (br, 16H), 0.96 (t, J = 6.0 Hz, 3H).13C NMR (CDCl3, 75 MHz) δ 138.79, 129.72, 35.34, 35.07, 32.78, 32.64, 32.52, 32.27, 32.01, 25.80, 17.12.
4.1.2. (4S)-tert-Butyl 2,2-dimethyl-4-(2-(phenylsulfinyl)acetyl)oxazolidine-3-carboxylate (3)
At –15 °C, to a solution of 1.00 g of methyl phenyl sulfoxide (7.1 mmol) in 5 mL of anhydrous tetrahydrofuran (THF), 4.0 mL of lithium diisopropylamide (LDA, 2 M solution in THF/n-heptane/ethylbenzene, 8.0 mmol) was added. The mixture was stirred at –15 °C for 30 min and then chilled to –78 °C. A solution of 1.00 g of (S)-3-tert-butyl 4-methyl 2,2-dimethyloxazolidine-3,4-dicarboxylate (3.8 mmol) in 5 mL of THF was added dropwise. The solution was stirred at –78 °C for 2 h and allowed to warm to room temperature overnight. After saturated aqueous NH4Cl (15 mL) solution was added, the product was extracted with EtOAc, washed with brine, and dried over with MgSO4. Purification by column chromatography (petroleum ether/EtOAc, 1.5:1) gave 1.20 g (86%) of (4S)-tert-butyl 2,2-dimethyl-4-(2-(phenylsulfinyl)acetyl)oxazolidine-3-carboxylate as a colorless oil. This is a mixture of two diastereomers. After a further column chromatography (50 g of silica gel) with petroleum ether/EtOAc (1.5:1), two pure diastereomers (3-up as a colorless oil and 3-down as colorless crystals) were isolated.
3-up 1H NMR (CDCl3, 300 MHz) δ 7.69 (m, 2H), 7.53 (m, 3H), 4.52 (m, 0.45H), 4.41 (m, 0.55H), 4.16–4.03 (m, 2.55H), 3.94 (dd, J = 3.5 Hz, J = 9.8 Hz, 1H), 3.80 (d, J = 14.7 Hz, 0.45H), 1.65–1.35 (m, 15H). 13C NMR (CDCl3, 75 MHz) δ 200.04, 152.53 (151.19), 143.56 (143.26), 131.77 (131.60), 129.56 (129.40), 124.15, 95.43 (94.72), 81.52 (81.33), 66.17 (65.99), 65.86 (65.29), 64.38 (64.14), 28.23, 26.11 (25.56), 24.87 (23.49).ESI-MS (m/e) 390 [M+Na]+, 406 [M+K]+, 575 [2M+Na]+.
3-down mp 115–117 °C. 1H NMR (CDCl3, 300 MHz) δ 7.71 (m, 2H), 7.53 (m, 3H), 4.41 (m, 0.55H), 4.30 (m, 0.45H), 4.19–3.70 (m, 4H), 1.64–1.40 (m, 15H).13C NMR (CDCl3, 75 MHz) δ 200.82 (199.94), 152.55 (151.14), 143.67 (143.58), 131.78 (131.69), 129.49 (129.41), 124.32, 95.48 (94.80), 81.58, 66.81 (66.08), 65.62, 64.50 (63.97), 28.29, 26.17 (25.62), 24.80 (23.34).ESI-MS (m/e) 390 [M+Na]+, 406 [M+K]+, 575 [2 M+Na]+.
4.1.3. (S)-tert-Butyl 2,2-dimethyl-4-((2E,4E)-pentadeca-2,4-dienoyl)oxazolidine-3-carboxylate (401)
To a solution of 0.60 g of (4S)-tert-butyl 2,2-dimethyl-4-(2-(phenylsulfinyl)acetyl) oxazolidine-3-carboxylate (1.6 mmol) in 5 mL of DMF, K2CO3 (170 mg, 1.2 mmol) was added. After the mixture was stirred at room temperature for 1 h under nitrogen, a solution of 0.42 g of (E)-1-bromotridec-2-ene (1.6 mmol) in 4 mL of DMF was added. The mixture was stirred for 4-5 days. Deionized water (5 mL) was added, and the product was extracted with diethyl ether (3 × 15 mL), washed with brine (3 × 15 mL) and water (3 × 15 mL), and dried over with MgSO4. Concentration and purification by flash column chromatography (petroleum ether/EtOAc 10:1) gave 0.34 g (50%) of (S)-tert-butyl 2,2-dimethyl-4-((2E,4E)-pentadeca-2,4-dienoyl)oxazolidine-3-carboxylate as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 7.28 (m, 1H), 6.29–6.14 (m, 3H), 4.67 (dd, J = 3.0 Hz, J = 7.6 Hz, 0.36H), 4.47 (dd, J = 3.8 Hz, J = 7.7 Hz, 0.64H), 4.16 (m, 1H), 3.96 (dd, J = 3.0 Hz, J = 9.3 Hz, 0.36H), 3.91 (dd, J = 3.9 Hz, J = 9.3 Hz, 0.64H), 2.18 (q, J = 6.6 Hz, 2H), 1.71–1.25 (m, 31H), 0.87 (t, J = 6.3 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 197.21 (196.33), 151.51 (152.29), 147.60 (147.23), 144.91, 128.76 (128.90), 122.34 (123.30), 95.09 (94.45), 80.53 (80.79), 66.03 (65.62), 64.46 (64.06), 33.22, 31.90, 29.58, 29.55, 29.42, 29.32, 29.19, 28.62, 28.37, 28.24, 25.29 (26.08), 24.11 (25.14), 22.68, 14.12. HRMS calcd for C25H43NNaO4 (M+Na)+, 444.3090; found, 444.3088.
4.1.4. (S)-tert-Butyl-4-((R,2E,4E)-1-hydroxypentadeca-2,4-dienyl)-2,2-dimethyloxazolidine-3-carboxylate (402)
At –15 °C, to a solution of (S)-tert-butyl 2,2-dimethyl-4-((2E,4E)-pentadeca-2,4-dienoyl) oxazolidine-3-carboxylate (0.25 g, 0.59 mmol) in 10 mL of dry THF, diisobutylaluminium hydride (DIBAL-H, 0.70 mmol) was added. The temperature was gradually raised to 0 °C. After 2 h, the reaction solution was quenched with 20 mL of water, extracted with diethyl ether (3 × 25 mL), washed with brine, and dried over with MgSO4. Concentration and purification by column chromatography (petroleum ether/EtOAc 2:1) gave 180 mg (72%) of (S)-tert-butyl 4-((R,2E,4E)-1-hydroxypentadeca-2,4-dienyl)-2,2-dimethyloxazolidine-3-carboxylate as a colorless oil. 1H NMR (CDCl3, 300 MHz) δ 6.25 (dd, J = 10.5 Hz, J = 14.7 Hz, 1H), 6.01 (dd, J = 10.2 Hz, J = 15.0 Hz, 1H), 5.66 (m, 1H), 5.53 (dd, J = 6.0 Hz, J = 15.0 Hz, 1H), 4.28–3.86 (m, 5H), 2.04 (q, J = 6.9 Hz, 2H), 1.48–1.46 (m, 15H), 1.24 (br, 16H), 0.86 (t, J = 6.6 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 154.12, 135.41, 131.93, 129.45, 128.94, 94.43, 81.06, 73.80, 64.87, 62.33, 32.62, 31.90, 29.61, 29.50, 29.33, 29.21, 29.17, 28.36, 26.33, 24.54, 22.67, 14.11. HRMS calcd for C25H45NNaO4 (M+Na)+, 446.3246; found, 446.3247.
4.1.5. (S,4E,6E)-2-Amino-1-hydroxyheptadeca-4,6-dien-3-one trifluoroacetate (405)
At 0 °C, to a solution of 0.25 g (0.59 mmol) of (S)-tert-butyl 2,2-dimethyl-4-((2E,4E)-pentadeca-2,4-dienoyl)oxazolidine-3-carboxylate in 5 mL of dichloromethane (DCM), 0.5 mL of trifluoroacetic acid (TFA) was added. After the mixture was stirred at 0 °C for 4 h, the solvent was removed under vacuum, and the residue was crystallized in diethyl ether and petroleum ether to give 0.15 g (64%) of (S,4E,6E)-2-amino-1-hydroxyheptadeca-4,6-dien-3-one trifluoroacetate as a colorless crystal. 1H NMR (DMSO-d6, 300 MHz) δ 8.18 (br, 1H), 7.36 (dd, J = 10.5 Hz, J = 15.6 Hz, 1H), 6.44–6.30 (m, 3H), 5.51 (br, 1H), 4.39 (s, 1H), 3.86 (m, 2H), 2.17 (q, J = 6.6 Hz, 2H), 1.39–1.23 (m, 16H), 0.84 (t, J = 6.3 Hz, 3H).13C NMR (DMSO-d6, 75 MHz) δ 194.05, 148.79, 146.01, 129.44, 124.22, 60.28, 59.61, 33.21, 31.95, 29.63, 29.47, 29.38, 29.26, 28.73, 22.73, 14.58. HRMS calcd for C17H32NO2 (M+H)+, 282.2433; found, 282.2437.
4.1.6. N-((S,4E,6E)-1-Hydroxy-3-oxoheptadeca-4,6-dien-2-yl)-2-phenylacetamide (406)
At 0 °C, to a solution of 0.10 g (0.25 mmol) of (S,4E,6E)-2-amino-1-hydroxyheptadeca-4,6-dien-3-one trifluoroacetate in 5 mL of DCM, 5 mL of saturated NaHCO3 was added. The mixture was stirred vigorously for 5 min before the addition of 80 μL (0.50 mmol) phenylacetyl chloride. The reaction mixture was then left to stir at room temperature for 16 h. The organic phase was removed under vacuum, and a white precipitate was formed. 50 mL of EtOAc was then added, and the organic phase was washed with KHSO4 (5%), saturated NaCO3, and saturated NaCl, and dried over by MgSO4. After removal of the solvent, the residue was recrystallized using petroleum ether and ethyl ether to give 70 mg (70%) of N-((S,4E,6E)-1-hydroxy-3-oxoheptadeca-4,6-dien-2-yl)-2-phenyl-acetamide as a colorless powder. 1H NMR (CDCl3, 300 MHz) δ 7.32–7.26 (m, 6H), 6.81 (d, J = 6.0 Hz, 1H), 6.28 (dt, J = 6.5 Hz, J = 15.0 Hz, 1H), 6.19 (d, J = 15.6 Hz, 1H), 6.18 (dd, J = 9.9 Hz, J = 15.0 Hz, 1H), 4.85 (m, 1H), 3.92 (d, J = 11.4 Hz, 1H), 3.79 (dd, J = 5.0 Hz, J = 11.4 Hz, 1H), 3.64 (s, 2H), 2.19 (q, J = 6.8 Hz, 2H), 1.43 (m, 2H), 1.26 (br, 14H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 195.17, 172.15, 148.94, 146.36, 134.33, 129.30, 129.04, 128.56, 127.49, 123.43, 64.51, 60.02, 43.56, 33.30, 31.90, 29.59, 29.55, 29.42, 29.32, 29.20, 28.54, 22.69, 14.13. HRMS calcd for C25H37-NNaO3 (M+Na)+, 422.2671; found, 422.2676.
4.1.7.N-((2S,3R,E)-1,3-Dihydroxyoctadec-4-en-2-yl)-2-phenylacetamide (403)
To a solution of 0.10 g (0.29 mmol) of sphingosine HCl in 5 mL of DCM, 5 mL of saturated NaHCO3 was added under argon atmosphere at 0 °C. The mixture was stirred vigorously for 5 min before the addition of 80 μL (0.50 mmol) phenylacetyl chloride. After stirring at room temperature for 16 h, 25 mL of water was added. The organic phase was removed under vacuum, and white precipitate was formed. After filtration, the precipitate was dried, and washed with a mixture solvent (petroleum ether/EtOAc, 4:1, 20 mL) to give 70 mg (58%) N-((2S,3R,E)-1,3-dihydroxyoctadec-4-en-2-yl)-2-phenylacetamide as a colorless powder. GC/MS (m/e) 399 [M–H2-O]+, 381 [M–2H2O]+.1H NMR (CDCl3, 300 MHz) δ 7.34–7.27 (m, 5H), 6.24 (d, J = 7.0 Hz, 1H), 5.70 (dt, J = 6.8 Hz, J = 15.4 Hz, 1H), 5.44 (dd, J = 6.4 Hz, J = 15.4 Hz, 1H), 4.24 (br, 1H), 3.86 (m, 2H), 3.66 (d, J = 8.7 Hz, 1H), 3.60 (s, 2H), 2.98 (br, 1H), 2.88 (br, 1H), 1.99 (q, J = 6.6 Hz, 1H), 1.27 (br, 22H), 0.89 (t, J = 6.8 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 171.83, 134.65, 134.35, 129.33, 128.98, 128.49, 127.39, 74.07, 62.31, 54.93, 43.77, 32.24, 31.93, 29.70, 29.67, 29.64, 29.50, 29.37, 29.25, 29.06, 22.70, 14.14. HRMS calcd for C26H43NNaO3 (M+Na)+, 440.3141; found, 440.3146.
4.1.8. (S)-3-Hydroxy-2-(2-phenylacetamido)-N-tetradecylpropanamide (404)
To a solution of 0.20 g (0.48 mmol) of (S)-2-amino-3-hydroxy-N-tetradecylpropanamide in 15 mL of DCM, 12 mL of saturated NaHCO3 was added under argon atmosphere at 0 °C. The mixture was stirred vigorously for 5 min before the addition of 160 μL (1.00 mmol) phenylacetyl chloride. After stirring at room temperature for 16 h, 40 mL of water was added. The organic phase was removed under vacuum, and a white precipitate was formed. After filtration, the precipitate was dried, and washed with a mixture solvent (petroleum ether/EtOAc, 4:1, 20 mL) to give 165 mg (82%) (S)-3-hydroxy-2-(2-phenylacetamido)-N-tetradecylpropanamide as a colorless powder. GC/MS (m/e) 399 [M–H2O–H]+, 442 [M+Na+H]+.1H NMR (CDCl3, 300 MHz) δ 7.36–7.24 (m, 5H), 6.91 (t, J = 5.1 Hz, 1H), 6.85 (d, J = 6.9 Hz, 1H), 4.41 (m, 1H), 4.00 (dd, J = 2.5 Hz, J = 11.0 Hz, 1H), 3.83 (br, 1H), 3.59 (s, 2H), 3.53 (m, 1H), 3.16 (q, J = 6.8, 2H), 1.41 (m, 2H), 1.25 (br, 22H), 0.88 (t, J = 6.8 Hz, 3H). 13C NMR (CDCl3, 75 MHz) δ 172.41, 170.85, 134.53, 129.44, 129.25, 127.74, 63.08, 54.18, 43.65, 39.82, 32.19, 29.93, 29.89, 29.82, 29.64, 29.58, 29.54, 27.11, 22.97, 14.40. HRMS calcd for C25H42N2NaO3 (M+Na)+, 441.3093; found, 441.3110.
4.2. Bioassays
4.2.1. Cell culture
MCF-7, MDA-MB-231, MCF-7TN-R, MCF-7/Dox, OVCAR-8, and NCI/ADR-RES cells were cultured as previously described.27–30 Briefly, cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Invitrogen, Carlsbad, CA) enriched with 10% fetal bovine serum (FBS), 1% MEM amino acids, 1% MEM non-essential amino acids, 1% Anti/Anti, human recombinant insulin, 1% sodium pyruvate and plasmocin (10% DMEM) (all from Life Technologies, Gaithersburg, MD). MCF-7/Dox, OVCAR-8, and NCI/ADR-RES cells were culture in RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin and 584 mg/liter l-glutamine. Cells were cultured in 75 cm2 and 125 cm2 tissue culture flasks (Sarsdedt, Newton, North Carolina) in 37 °C humidified atmosphere of 5% CO2 and 95% air.
4.2.2. Viability assays
Viability assays in MCF-7, MDA-MB-231, and MCF-7TN-R cells were performed according to previously published protocols.27,28 Cell lines were plated at 7500–10,000 cells in 200 μL of media per well in 96-well plates using phenol-free DMEM. Cells were incubated and given 24 h to adhere to plates. Cells were then treated with gradient concentrations of ceramide analogs, and incubated for an additional 24-hour period to allow proliferation. MTT (20 μL) was added to each well and allowed to incubate for 3–4 h. Cells were then suspended in 20% SDS buffer and 50% DMF for 24 h. The plates’ absorbance values were read using a spectrophotometer at 550 nm. For analysis, viability values were set to percent control of dimethyl sulfoxide (DMSO) vehicle, and carried out in quadruplicate on three separate occasions at least 24 h apart.
Cell viability of MCF-7, MCF-7/Dox, OVCAR-8, and NCI/ADR-RES was determined by quantitation of ATP, an indicator of live cells, using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI) kit, as described previously.31 Briefly, cells (4000 cells/well) were grown in 96-well plates with 10% FBS RPMI-1640 medium overnight. Cells were treated with ceramide analogs for 72 h. Cell viability was determined by the measurement of luminescent ATP in a Synergy HT microplate reader (BioTek, Winnooski, VT, USA) following incubation with CellTiter-GloReagent.
4.2.3. Clonogenic survival assays
Colony formation assays were performed as previously described.21,23 MCF-7, MDA-MB-231, and MCF-7TN-R cells were plated in 6-well plates at 1000 cells per well in 2 mL of 10% DMEM. After allowing 24 h for cell adhesion, treatment with gradient dilutions of analogs was initiated. Colony growth was assessed each day until suitable colony formation was noted (generally 10–12 days after treatment). Cells were incubated with 3% glutaraldehyde for 30 min in order to fix cells to the plate. Media was then removed in a water bath, and plates were allowed to air dry. After fixation, plates were treated with a 0.2% solution of crystal violet and 20% methanol in deionized water for a period of 40 min. Plates were then washed in distilled water and again air dried. Colonies were hand counted on a light plate with formations of greater than 30 cells quantifying as sufficient size for a positive colony count. Clonogenic survival was normalized to non-analog-treated DMSO control.
4.2.4. Apoptosis assays
As previously described, MCF-7TN-R cells were plated at 7500–10,000 cells in 200 μL of media per well in 96-well plates using phenol-free DMEM.32,33 Cells were incubated and given 24 h to adhere to plates. Cells were then treated with twice the IC50 concentrations of ceramide analogs obtained through MTT viability assay, and incubated for an additional 24-hour period to allow proliferation. Plates were analyzed using a cell death detection ELISA kit purchased from Roche Applied Sciences. Assays were performed in duplicate on three separate occasions at least 24 h apart. For analysis, values were normalized to an MTT viability assay performed on the same plate.
4.2.5. Caspase-9 assays
Caspase activity assays were performed as previously described.33 Briefly, MCF-7TN-R cells were plated at 7500–10,000 cells in 200 μL of media per well in 96-well plates using phenol-free DMEM. Cells were incubated and given 24 h to adhere to plates. Cells were then treated with twice the IC50 concentrations of ceramide analogs obtained by means of MTT viability assays, and incubated for an additional 24-hour period to allow proliferation. Plates were then analyzed using a Caspase Glo 9 assay purchased from Promega. Assays were performed in triplicate on three separate occasions at least 24 h apart.
4.2.6. Statistical analysis
Statistical analysis of IC50 values were calculated from dose–response curves using GraphPad Prism 5.0 (GraphPad Software), using the equation: Y = Bottom + (Top – bottom)/[1 + 10(Log IC50–X)], assuming a standard slope, where the response goes from 10% to 90% of maximal as X increases over two log units. Differences in IC50 and other activity data were compared using Student's unpaired t-test with p <0.05 as the limit of statistical significance. Experiments comparing multiple concentrations to the control were tested with oneway ANOVA with Bonferroni post-test to compare individual concentrations.
4.2.7. Cellular GCS assay
Cells (1 × 106 cells/35-mm dish) were grown in 10% FBS RPMI-1640 medium for 24 h, and then treated with ceramide analog at its IC50 concentration for additional 24 h in 5% RPMI-1640 medium. Cells were switched to 1% BSA RPMI-1640 medium containing 50 μM NBD C6-ceramide complexed to BSA. After 2 h of incubation at 37 °C, lipids were extracted, and resolved on high performance TLC plates (Partisil, Piscataway, NJ, USA) in a solvent system containing chloroform/methanol/3.5 N ammonium hydroxide (85:15:1, v/v/v), as described previously.26,34 NBD C6-glucosylceramide (GlcCer) and NBD C6-ceramide (Cer) were identified using AlphaImager HP imaging system (Alpha Innotech, Santa Clara, CA). For quantitation of GCS activity, ratio of fluorescence of GlcCer to fluorescence of Cer was determined using Synergy HT multi-detection microplate reader (BioTek, Winooski, VT, USA). NBD C6-ceramide (Invitrogen, Carlsbad, CA, USA) and NBD C6-glucosylceramide (N-hexanol-NBD-glucosylceramide; Matreya LLC, Pleasant Gap, PA) were used as standards.
Supplementary Material
Acknowledgements
We acknowledge NIH AREA grant number 1R15CA159059-01 (Multi-Target, Mechanism-Based Drug Design for the Treatment of Breast Cancer: Synthesis, Screening, and Mechanism Investigation of a Ceramide Analogue Library), and DoD Breast Cancer Research Award number W81XWH-11-1-0105 (BC102922) (A Drug Discovery Partnership for Personalized Breast Cancer Therapy). We also like to acknowledge the support of Louisiana Cancer Research Consortium and NIH-RCMI (grant number G12RR026260) for support of the Core Facilities at Xavier University of Louisiana.
Footnotes
Supplementary data
Supplementary data (1H and 13C NMR spectra, 2D NMR spectra, and activity data of all the tested compounds) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.12.065.
References and notes
- 1.Hannun YA, Obeid LM. Nat. Rev. Mol. Cell Biol. 2008;9:139. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 2.Saddoughi SA, Song P, Ogretmen B. Subcell. Biochem. 2008;49:413. doi: 10.1007/978-1-4020-8831-5_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morad SA, Levin JC, Shanmugavelandy SS, Kester M, Fabrias G, Bedia C, Cabot MC. Mol. Cancer Ther. 2012;11:2352. doi: 10.1158/1535-7163.MCT-12-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pyne NJ, Pyne S. Nat. Rev. Cancer. 2010;10:489. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 5.Gouaze-Andersson V, Cabot MC. Biochim. Biophys. Acta. 2006;1758:2096. doi: 10.1016/j.bbamem.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Gouazé V, Liu YY, Prickett CS, Yu JY, Giuliano AE, Cabot MC. Cancer Res. 2005;65:3861. doi: 10.1158/0008-5472.CAN-04-2329. [DOI] [PubMed] [Google Scholar]
- 7.Sietsma H, Veldman RJ, Kolk D, Ausema B, Nijhof W, Kamps W, Vellenga E, Kok JW. Clin. Cancer Res. 2000;6:942. [PubMed] [Google Scholar]
- 8.Dijkhuis AJ, Klappe K, Jacobs S, Kroesen BJ, Kamps W, Sietsma H, Kok JW. Mol. Cancer Ther. 2006;5:593. doi: 10.1158/1535-7163.MCT-05-0457. [DOI] [PubMed] [Google Scholar]
- 9.Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G. Biochim. Biophys. Acta. 2006;1758:1957. doi: 10.1016/j.bbamem.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Szulc ZM, Bai A, Bielawski J, Mayroo N, Miller DE, Gracz H, Hannun YA, Bielawska A. Bioorg. Med. Chem. 2010;18:7565. doi: 10.1016/j.bmc.2010.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong L, Tan SS, Lam Y, Melendez AJ. J. Med. Chem. 2009;52:3618. doi: 10.1021/jm900121d. [DOI] [PubMed] [Google Scholar]
- 12.Bhabak KP, Arenz C. Bioorg. Med. Chem. 2012;20:6162. doi: 10.1016/j.bmc.2012.08.031. [DOI] [PubMed] [Google Scholar]
- 13.Bhabak KP, Kleuser B, Huwiler A, Arenz C. Bioorg. Med. Chem. 2013;21:874. doi: 10.1016/j.bmc.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 14.Singh A, Ha HJ, Park J, Kim JH, Lee WK. Bioorg. Med. Chem. 2011;19:6174. doi: 10.1016/j.bmc.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 15.Camacho L, Simbari F, Garrido M, Abad JL, Casas J, Delgado A, Fabriàs G. Bioorg. Med. Chem. 2012;20:3173. doi: 10.1016/j.bmc.2012.03.073. [DOI] [PubMed] [Google Scholar]
- 16.Liu J, Antoon JW, Ponnapakkam A, Beckman BS, Foroozesh M. Bioorg. Med. Chem. 2010;18:5316. doi: 10.1016/j.bmc.2010.05.044. [DOI] [PubMed] [Google Scholar]
- 17.Crawford KW, Bittman R, Chun J, Byun HS, Bowen WD. Cell Mol. Biol (Noisy-le-grand) 2003;49:1017. [PubMed] [Google Scholar]
- 18.Nicholson KM, Quinn DM, Kellett GL, Warr JR. Br. J. Cancer. 1999;81:423. doi: 10.1038/sj.bjc.6690711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beckham TH, Lu P, Jones EE, Marrison T, Lewis CS, Cheng JC, Ramshesh VK, Beeson G, Beeson CC, Drake RR, Bielawska A, Bielawski J, Szulc ZM, Ogretmen B, Norris JS, Liu XJ. Pharmacol. Exp. Ther. 2013;344:167. doi: 10.1124/jpet.112.199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shirahama T, Sweeney EA, Sakakura C, Singhal AK, Nishiyama K, Akiyama S, Hakomori S, Igarashi Y. Clin. Cancer Res. 1997;3:257. [PubMed] [Google Scholar]
- 21.Antoon JW, Liu J, Ponnapakkam AP, Gestaut MM, Foroozesh M, Beckman BS. Cancer Chemother. Pharmacol. 2010;65:1191. doi: 10.1007/s00280-009-1233-0. [DOI] [PubMed] [Google Scholar]
- 22.Struckhoff AP, Bittman R, Burow ME, Clejan S, Elliott S, Hammond T, Tang Y, Beckman BS. J. Pharmacol. Exp. Ther. 2004;309:523. doi: 10.1124/jpet.103.062760. [DOI] [PubMed] [Google Scholar]
- 23.Antoon JW, Liu J, Gestaut MM, Burow ME, Beckman BS, Foroozesh M. J. Med. Chem. 2009;52:5748. doi: 10.1021/jm9009668. [DOI] [PubMed] [Google Scholar]
- 24.Chun J, Li G, Byun HS, Bittman R. J. Org. Chem. 2002;67:2600. doi: 10.1021/jo0162639. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Cui G, Zhao M, Cui C, Ju J, Peng S. Bioorg. Med. Chem. 2007;15:7773. doi: 10.1016/j.bmc.2007.08.061. [DOI] [PubMed] [Google Scholar]
- 26.Gupta V, Patwardhan GA, Zhang QJ, Cabot MC, Jazwinski SM, Liu YY. J. Lipid Res. 2010;51:866. doi: 10.1194/jlr.D002949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Antoon JW, Meacham WD, Bratton MR, Slaughter EM, Rhodes LV, Ashe HB, Wiese TE, Burow ME, Beckman BS. J. Mol. Endocrinol. 2011;46:205. doi: 10.1530/JME-10-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LV, Ashe HB, Wiese TE, Smith CD, Burow ME, Beckman BS. Endocrinology. 2010;151:5124. doi: 10.1210/en.2010-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu YY, Gupta V, Patwardhan GA, Bhinge K, Zhao Y, Bao J, Mehendale H, Cabot MC, Li YT, Jazwinski SM. Mol. Cancer. 2010;9:145. doi: 10.1186/1476-4598-9-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gupta V, Bhinge KN, Hosain SB, Xiong K, Gu X, Shi R, Ho MY, Khoo KH, Li SC, Li YT, Ambudkar SV, Jazwinski SM, Liu YY. J. Biol. Chem. 2012;287:37195. doi: 10.1074/jbc.M112.396390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patwardhan GA, Zhang QJ, Yin D, Gupta V, Bao J, Senkal CE, Ogretmen B, Cabot MC, Shah GV, Sylvester PW, Jazwinski SM, Liu YY. PLoS One. 2009;4:e6938. doi: 10.1371/journal.pone.0006938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antoon JW, Beckman BS. Bioorg. Med. Chem. Lett. 2012;22:2624. doi: 10.1016/j.bmcl.2012.01.087. [DOI] [PubMed] [Google Scholar]
- 33.Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S, Smith CD, Burow ME, Beckman BS. Cancer Biol. Ther. 2011;11:678. doi: 10.4161/cbt.11.7.14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu YY, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, Holleran WM, Giuliano AE, Jazwinski SM, Gouaze-Andersson V, Consoli DP, Cabot MC. FASEB J. 2008;22:2541. doi: 10.1096/fj.07-092981. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.