

Abstract
Cell cultures from reef-building scleractinian corals are being developed to study the response of these ecologically important organisms to environmental stress and diseases. Despite the importance of cell division to support propagation, cell proliferation in polyps and in vitro is under-investigated. In this study, suspended multicellular aggregates (tissue balls) were obtained after collagenase dissociation of Pocillopora damicornis coral, with varying yields between enzyme types and brands. Ultrastructure and cell type distribution were characterized in the tissue balls (TBs) compared to the polyp. Morphological evidence of cellular metabolic activity in their ciliated cortex and autophagy in their central mass suggests involvement of active tissue reorganization processes. DNA synthesis was evaluated in the forming multicellular aggregates and in the four cell layers of the polyp, using BrdU labeling of nuclei over a 24 h period. The distribution of BrdU-labeled coral cells was spatially heterogeneous and their proportion was very low in tissue balls (0.2 ± 0.1 %), indicating that suspended multicellular aggregate formation does not involve significant cell division. In polyps, DNA synthesis was significantly lower in the calicoderm (<1 %) compared to both oral and aboral gastroderm (about 10 %) and to the pseudostratified oral epithelium (15–25 % at tip of tentacle). DNA synthesis in the endosymbiotic dinoflagellates dropped in the forming tissue balls (2.7 ± 1.2 %) compared to the polyp (14 ± 3.4 %) where it was not different from the host gastroderm (10.3 ± 1.2 %). A transient (24 h) increase was observed in the cell-specific density of dinoflagellates in individually dissociated coral cell cultures. These results suggest disruption of coral cell proliferation processes upon establishment in primary culture.
Keywords: Primary culture, DNA synthesis, BrdU, Coral, Scleractinia, Dinoflagellate, Cell proliferation
Introduction
Since the 1960s there have been multiple efforts to develop cell cultures from the scleractinian coral organisms, which build the structural framework of marine reefs. Coral cell cultures are potentially alternative tools to study the response of corals to environmental stress and diseases. In vitro experiments may complement and extend in vivo experiments in the aquarium or the natural reef environment. However, to date no coral cell lines have been established and isolated scleractinian coral cells can only be maintained in primary cultures on timescales from a few days to a few weeks, without continuous propagation.
Development of the methodology for coral cell cultures has been sporadic. Difficulties stem from a limited knowledge about cell nutritional requirements and physiology in vitro that lead to short-term functional viability (Domart-Coulon et al. 2004a). Limited tools for characterization of cell types, frequent overgrowth of contaminants (especially chytrid protists) (Rinkevich 1999, 2005), and a lack of characterization of the proliferating stem cell niches (Rinkevich 2011) add more complexities. Moreover, most unsuccessful strategies have remained unpublished.
To isolate cells, methods include mechanical detachment of soft tissue from the skeleton, either by pinching off tissue fragments with tweezers (Kramarsky-Winter and Loya 1996; Vizel et al. 2011) or by blasting the tissue with a jet of pressurized seawater (Nesa and Hidaka 2009). Alternately, coral soft tissue is chemically induced to detach, in response to removal of divalent cations (Gates and Muscatine 1992; Frank et al. 1994; Kopecky and Ostrander 1999; Domart-Coulon et al. 2001, 2004a; Mass et al. 2012) or exposure to a reducing agent, such as N-acetylcysteine (Peng et al. 2008). Enzymatic digestion has also been widely used to dissociate the tissue into single cells (Gates and Muscatine 1992; Frank et al. 1994; Helman et al. 2008; Downs et al. 2010). Types and brand of enzymes used in dissociation studies vary between laboratories, and are often not detailed by authors, making it difficult to standardize protocols for coral cell isolation. Spontaneous dissociation has also been reported after freeze–thaw cycles (Frank et al. 1994) or after exposure of larvae to antibiotics (Reyes-Bermudez et al. 2009).
These methods yield either tissue (explant) cultures, or mixed cell type single dissociated cultures, which may be further enriched in specific cell types and for example applied to ecotoxicology testing (Downs et al. 2010). Suspended multicellular isolates (explants) are formed in cell culture medium within 3 days of culture initiation after divalent cation removal (Kopecky and Ostrander 1999; Domart-Coulon et al. 2001, 2004a). Similar structures (called tissue balls) are also formed in seawater via individual cell aggregation after mechanical dissociation (Nesa and Hidaka 2009) or rounding up and closure of soft tissue spheroids microdissected from the skeleton (Kramarsky-Winter and Loya 1996; Vizel et al. 2011).
Suspension cultures of multicellular aggregates have been applied to the study of symbiosis, the response to thermal stress, and interactions between coral and fungi (Kopecky and Ostrander 1999; Nesa and Hidaka 2009; Domart-Coulon et al. 2004b). Adherent cultures of multicellular aggregates have been used to develop models of in vitro biomineralization (Domart-Coulon et al. 2001; Helman et al. 2008; Mass et al. 2012). Decreasing in vitro viability usually limits the timescale of experiments using such primary cultures from a few days to a few weeks, enabling only short-term study of physiological mechanisms.
A major problem is the current lack of characterization of in vitro proliferation. Once the tissue is dissociated from the initial fragment, the biomass of primary cultures does not increase over time (in terms of cell number or protein content), so it is assumed that coral cells do not divide significantly. Clusters of cells have been reported to form in suspension in several models, including embryonic cells (Reyes-Bermudez et al. 2009) as well as adult cells (Kopecky and Ostrander 1999; Nesa and Hidaka 2009) or in adherent cultures on Primaria substrate (Domart-Coulon et al. 2001; Helman et al. 2008; Mass et al. 2012). In the soft coral Sinularia flexibilis, these aggregates were interpreted as a sign of cell proliferation (Khalesi et al. 2008). Recent encouraging results have reported regeneration of polyps from suspended spheroid tissue cultured in seawater (Vizel et al. 2011). However cell division activity has not been investigated within these coral primary cultures and especially during formation of the multicellular aggregates.
Incorporation of BrdU (5-Bromo-2′-deoxy-uridine analogue of deoxythymidine) in the DNA of cell nuclei is an indicator of DNA synthesis and is widely used to estimate rates of cell division (Dolbeare and Selden 1994), although its interpretation requires caution as it may also detect DNA repair events not linked to S-phase of the cell cycle (Taupin 2007). Within the phylum Cnidaria, this method is commonly used in hydrozoans, for example in the marine species Hydractinia echinata and Clytiahemisphaerica, in short-term pulse (5 mM BrdU for 30 min to a few hours)—chase assays to detect mitotic activity and cellular self-renewal (Müller et al. 2004; Denker et al. 2008). In adult colonies of scleractinian corals the BrdU labeling method has recently been used, at much lower concentrations and longer exposure periods, to investigate cell proliferation activity in aquarium-based studies. DNA synthesis was detected in 2–4 % of the Symbiodinium dinoflagellate endosymbionts of Montipora capitata, via BrdU (50 μM) labeling during 48 h (Santos et al. 2009). In another study, mitotic activity was assessed by expression of the conserved Proliferating Cell Nuclear Antigen (PCNA) protein in the species Montipora foliosa and Acropora pulchra, following creation of a lesion (D’Angelo et al. 2012). But in both cases, the spatial distribution of the proliferation activity was determined only at the level of the colony, comparing top versus underside and margin versus inner area of plate-forming Montipora corals, and comparing apex versus branch of branching Acropora corals. A very recent study reported differential cellular kinetics in the oral and aboral tissue layers of healthy and diseased corals (Porites australiensis and Montipora informis) exposed to BrdU (100 μM) for 3 days, showing an increase in cell division and a decrease in apoptosis in growth anomaly lesions versus control tissue (Yasuda and Hidaka 2012).
In this study, we have evaluated the efficiency of different brands and types of collagenase to obtain cells and tissue balls (TBs) from adult colonies of the Indo-Pacific scleractinian coral Pocillopora damicornis. The structure and cell type distribution of TBs were characterized and compared to polyps. The DNA synthesis activity was investigated via BrdU incorporation into nuclei over a 24 h period, at the onset of primary cultures in the forming TBs, and in vivo in the polyps. BrdU-labeled cells were localized and quantified in the cortex and the center of TBs, and in the four tissue-layers characteristic of coral polyps. The cell-specific density (CSD) of dinoflagellate symbionts was also monitored in vitro, during the first 2 days after establishment of single dissociated cell cultures. Implications for the proliferation activity of coral cells and their dinoflagellate endosymbionts in the polyp and in the TBs at the onset of primary cultures are discussed.
Materials and methods
Biological material
Small nubbins (~5 cm height) of the scleractinian coral P. damicornis (Linnaeus, 1758) were prepared by fragmentation from one large adult colony grown at the Aquarium Tropical, Palais de la Porte Dorée (ATPD), Paris, France. They were allowed to recover more than 3 weeks until completely covered with tissue, then used for cell isolation and BrdU labeling of DNA synthesis activity. Colony nubbins were cultured under a 12/12 h light/dark cycle (5000 Lux, provided by 6 fluorescent tubes, including 3 white light 10,000 K and 3 blue light 20,000 K) in equilibrated closed-circuit artificial seawater at 25 °C, salinity 35°/°°, pH 8.1 ± 0.2 (daily pH fluctuations due to photosynthesis and respiration activity). During the 24 h BrdU labeling experiment, the light cycle was conserved, seaweater was renewed every 6 h, and air was gently bubbled in for gaz equilibration. Temperature and pH were monitored to remain within the recorded growing range (25–27.5 °C and pH 8.0–8.3).
Coral cell isolation
The apex of branches (~5 mm height) from P. damicornis nubbins were sampled with scissors at the Aquarium Tropical, Palais de la Porte Dorée (ATPD, Paris) and placed for ~2–3 h at room temperature in 0.2 μm filtered sea water (FSW)—sampled from a 1,500 L tank with artificial seawater (closed system) equilibrated for a live stony coral exhibit. This seawater was supplemented with (v:v) 3 % antibiotics-antimycotics 100× solution (AB-AM, Gibco/Life Technologies, Carlsbad, CA, USA) corresponding to final concentrations of Amphotericin B <0.3 %, Penicillin 1.5–4.5 %, Streptomycin 1.5–4.5 % .
After three successive rinsing steps to remove loosely attached surface contaminants, the coral fragments were incubated at 24 °C in 5 mL collagenase 0.05–0.15 % (weight/volume) solution in FSW, stirred at 75 rpm with a magnetic stir bar, for periods ranging 10–30 min. Different types and commercial brands of collagenase were tested, with collagenase activity given either in Collagen Degrading Units (CDU) or in Wünsch units/mg of product. For comparison of the efficiency of collagenase digestion between brands, the Wünsch unit was converted in CDU (1 Wünsch unit/mg = 1,000 CDU/mg, after Roche manufacturer’s manual). The enzyme percent concentration (weight/volume) traditionally used was standardized a posteriori per mg of wet tissue digested: this was calculated from the final weight of enzyme in a defined volume (5 mL) standardized to 100 mg of wet tissue. Here, wet tissue is defined by the difference between wet weight of coral fragments before (t = 0) and after complete tissue dissociation (t = 3 days), once the skeleton is bare. The range of activity for each enzyme was derived from repeated experiments with the same enzyme, on different batches of coral fragments.
The exact composition of collagenase blends is not specified by the provider(s). Collagenase type IV Sigma-Aldrich (C5138) from Clostridium histolyticum has a collagenase activity of 125 CDU per mg of product and also contains clostripain, a non-specific neutral protease and tryptic activities. Collagenase type IV Gibco (17104-019) from Clostridium histolyticum is prepared with low amount of tryptic activity and guaranteed to have at least 160 CDU per mg of product. Collagenase type I Sigma-Aldrich (C0130) from Clostridium histolyticum has 125 CDU per mg of product, and contains non-specific protease and clostripain activity. Two Liberase purified enzyme blends from Roche Diagnostics (Mannheim, Germany) were also used: Liberase DL (05401160001) and Liberase TM (05401119001). Both contain the same amount of collagenase activity (26 Wünsch units/5 mg of product, i.e. 5,200 CDU/mg of product) but differ in the type of combined neutral protease, its amount and its aggressiveness. According to the manufacturer, Liberase DL contains low amount of dispase with weak aggressiveness and Liberase TM contains medium amount of thermolysin with high aggressiveness.
Tissue digestion was stopped by rinsing fragments 15 min with FSW at 75 rpm and 24 °C. Then each collagenase-treated apex was placed individually into separate wells of a 6-well plate (Nunclon/Thermo Fisher, Illkirch, France) in 5 mL of cell culture medium. These primary explant cultures were incubated at 24 °C under air, with a 10/14 h day/night light cycle under very low white light, 2–4 photosynthetic photon flux density (PAR) μmol per m2 per second (measured with LI-COR LI-250A light meter Quantum/Radiometer/Photometer). Tissue dissociation was monitored using an inverse phase contrast light microscope Olympus CK40 (Tokyo, Japan) and micrographs were acquired with Canon camera (Tokyo, Japan). Cell viability was determined by the trypan blue exclusion assay and cell counting on a Malassez hemacytometer.
The cell culture medium was modified from Domart-Coulon et al. (2001) and Helman et al. (2008). Its composition was based on filtered seawater (FSW) supplemented with commercial formula for vertebrate cell culture, with added salts to adjust osmolarity, and various amino-acid supplements, as well as trace amount of fetal calf serum to provide undetermined growth factors Antibiotics-antimycotics mix (AB-AM, Gibco) was added to limit overgrowth of microbial contaminants. Prepared in FSW, this complex medium contained (v/v) 12.5 % modified DMEM (Gibco 11880 without phenol red and with 1 g L−1 glucose and 100 mg L−1 sodium pyruvate, to which were added NaCl 18.1 g L−1, KCl 0.35 g L−1, CaCl2 1.1 g L−1, Na2SO4 1 g L−1, MgCl2–6H2O 10.2 g L−1, aspartic acid 20 mg L−1, taurine 52 mg L−1, SrCl2 26 mg L−1) buffered with Hepes 5.96 g L−1, and pH adjusted to 7.9 and (v/v) 1.25 % Fetal Calf Serum (Sigma-Aldrich, St. Louis, MO, USA), (v/v) 1 % AB-AM (Gibco), (v/v) 1 % Glutamax (Gibco), and 50 μg/mL ascorbic acid (Sigma-Aldrich) freshly prepared from frozen 5 mg/mL stock solution. Unless otherwise specified, all chemicals were from Sigma-Aldrich. Final concentration of antibiotics and antimycotics were Penicillin 0.5–1.5 %, Streptomycin 0.5–1.5 %, Amphotericin B <0.1 %. The culture medium was renewed every week by 50 %, with weekly addition of 50 μg/mL ascorbic acid as antioxidant compound.
Isolated cells or tissue which detached from the skeleton were transferred into fresh culture medium in separate wells of a 6 or 24 well microplate (Nunclon), in order to limit contamination by ciliates, bacteria or chytrid-like protists associated with the original coral fragments. Substrate of culture dishes was plastic (Nunclon) or glass (autoclaved coverslips or Labtek chambers).
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) of coral polyps and multicellular aggregates formed in primary cultures
For ultrastructural observations, polyps at the apex of colony branches and cell aggregates formed in primary cultures were fixed overnight at 4 °C in 2.5 % glutaraldehyde and 0.2 % saturated picric acid in 0.1 M Sörensen-phosphate buffer at pH 7.9 (made with NaH2PO4–2H2O and Na2HPO4–2H2O) containing 0.6 M sucrose. The skeleton of fixed colony fragments was decalcified in EDTA 0.5 M in 0.1 M Sörensen-phosphate buffer at pH 7.9 for 2 days at 4 °C. Polyps were then microdissected under a binocular. Fixed samples (polyps or cell aggregates) were rinsed three times in Sörensen-phosphate-sucrose buffer, post-fixed in 1 % osmium tetroxide (OsO4) in 0.1 M Sörensen-phosphate-0.6 M sucrose buffer for 1 h at room temperature, and dehydrated in a graded series of increasing ethanol concentrations.
For scanning electron microscopy (SEM), ethanol was substituted with liquid CO2 and samples were critical point dried, mounted on SEM stubs, fractured with a scalpel, and gold-coated. Observations were performed at 15 kV with a JEOL 840A scanning electron microscope (Tokyo, Japan) equipped with SEMAFORE image acquisition software at the electron microscopy platform (PIME) of the Muséum National d’Histoire Naturelle.
For transmission electron microscopy (TEM), dehydrated samples were embedded at 60 °C in Spürr resin. Semi-thin sections (0.5–1 μm) were cut with a 35° diamond blade (Diatome) on an ULTRACUT E Reichert-Jung microtom (Buffalo, NY, USA) and stained with 1 % toluidine blue containing sodium tetraborate (EMS, Fort Washington, PA, USA) followed by 0.1 % basic fuchsin (EMS). Ultra-thin sections (50–70 nm) were contrasted with saturated uranyl acetate 50 % in ethanol at room temperature, and observed at 75 kV with a Hitachi H-7100 transmission electron microscope (Tokyo, Japan) equipped with a CCD Hamamatsu camera (Hamamatsu Photonics, Hamamatsu, Japan). Photos were taken with AMTV 542 image acquisition software, at the electron microscopy platform (PIME) of the Muséum National d’Histoire Naturelle.
Estimation of DNA synthesis via immunodetection of BrdU labeled cell nuclei
For primary cultures and coral colonies incubated, respectively, in cell culture medium and seawater, the BrdU labeling solution was renewed every 4–6 h, due to the reported instability of BrdU beyond 6 h after dilution in seawater (Santos et al. 2009). Reagents from the commercial 5-Bromo-2′-deoxy-uridine Labeling and Detection Kit II (Roche) were used, except that the colorimetric detection system was replaced by an epifluorescence detection system after preliminary assays revealed significant endogenous alkaline phosphatase activity in coral tissue, creating false positive labeling. Therefore a secondary (goat) antimouse antibody coupled to Alexa594 fluorochrome (Invitrogen/Life Technologies, Carlsbad, CA, USA) was used for detection of the primary antiBrdU mouse antibody (Roche).
In order to evaluate DNA synthesis at the onset of primary cultures, coral cells were incubated with 50 μM BrdU labeling reagent in cell culture medium (5 mL), between Day 1 and Day 2 from collagenase treatment. Labeling was carried out during one diurnal cycle (24 h), with two successive changes of BrdU solution, first after 4 h then after 8 h, for an estimated exposure time of at least 14 h to active BrdU, covering the last 5 h of the dark photoperiod and 9 h of the light photoperiod. The experiment was repeated three times independently.
In order to evaluate DNA synthesis in situ in the polyps, coral nubbins were incubated with 25 μM BrdU in seawater (250 mL) during one diurnal cycle (24 h), with four successive changes of the BrdU-labeled seawater, i.e. every 6 h starting with the 12 h light photoperiod and finishing with the 12 h dark photoperiod.
For immunohistological detection of BrdU incorporation, the apex of colony branches (four replicate nubbins exposed to BrdU and one unexposed control) and the multicellular aggregates were fixed overnight at 4 °C with 0.5 % glutaraldehyde (Sigma-Aldrich), 2 % paraformaldehyde (Fluka/Sigma-Aldrich), and 0.2 % saturated picric acid (Sigma-Aldrich) in 0.1 M Sörensen-phosphate—0.6 M sucrose buffer at pH 7.9. Fixed tissue was rinsed in 0.1 M Sörensen-phosphate—0.6 M sucrose buffer, and the skeleton of colony fragments was decalcified in EDTA 0.5 M in 0.1 M Sörensen-phosphate buffer at pH 7.9 for 2 days at 4 °C, before microdissection of individual polyps under a binocular.
Samples were dehydrated in graded ethanol series and embedded at 37 °C under vacuum in LR-White medium grade resin (Fluka/Sigma-Aldrich). Serial semi-thin sections (1 μm) were cut with a 35° diamond knife (Diatome) on an ULTRACUT microtome and placed on Superfrost glass slides in five sets of five serial sections. One set was stained with toluidine blue—basic fuchsin for orientation in the tissue (cf. above for detailed stain composition), two sets were used for immunodetection of BrdU and the two other sets were used for internal immunolabeling negative controls, without antibodies (for autofluorescence) and without primary antibody. Finally, for each slide, the total tissue area examined to detect BrdU-positive nuclei corresponded to a surface of 15 × 104 μm2 by 5–10 μm depth, and consecutive areas were spaced by at least 30–40 μm. So, within an individual polyp or TB, each area corresponded in fact to a specific depth in the polyp or the TB.
Sections were permeabilized with PBS, 10 mM, pH 7.4, containing (v:v) 0.1 % Triton X100 (PBST), treated 10 min with HCl 2 M, washed with PBST, pre-incubated for 30 min in saturation solution (PBST with (w:v) 1 % BSA and (v:v) 2 % Normal Goat Serum), then incubated for 3 h at room temperature with the primary anti-BrdU mouse antibody (Roche kit no. 11299964001) diluted 1/50 in the saturation solution. Sections were washed three times in PBST then incubated 1 h at RT with the secondary goat anti-mouse antibody labeled with Alexa-594 (Invitrogen) diluted 1/100 in PBS. Sections were washed three times in PBS and slides were coverslipped in the mounting medium Prolong Gold Antifade with DAPI (Invitrogen).
Labeling was detected at ×20 and ×40 at the Centre de Microscopie de fluorescence et d’IMagerie numérique (PIME) of the Muséum National d’Histoire Naturelle with a Nikon Eclipse TE 300 inverse wide-field fluorescence microscope, equipped with a mercury lamp, a z acquisition design, and a CDD camera. Images and image stacks were acquired using Metamorph software with MonoD excitation-emission settings (λex: 340–380 nm, λem: 445–465 nm) for DAPI, with MonoR excitation-emission settings (λex: 540–565 nm, λem: 580–620 nm) for dinoflagellates autofluorescence, and with HQ-APC excitation-emission settings (λex: 563–617 nm λem: 632–697 nm) for Alexa-594. Selected optical planes were stacked to visualize BrdU-labeled structures. Localization of dinoflagellate cells was confirmed by overlay of chlorophyll autofluorescence images (MonoR settings) with DAPI images (MonoD settings). Specificity of BrdU signal was checked by colocalization of Alexa-594 (HQ-APC settings) with DAPI-counterstained nuclei (MonoD settings).
Evaluation of the cell specific density of dinoflagellate endosymbionts
The cell-specific density (CSD) is an estimation of the number of dinoflagellates contained in one host cell. CSDs were estimated at the onset of coral primary cultures in five independent collagenase experiments, with the method of Muscatine et al. (1998). The number of dinoflagellates per host cell was counted with an Olympus CK40 inverse phase contrast microscope (providing enhanced contrast of the host cell cytoplasmic membrane), in three to five hundred coral gastrodermal cells per well, in four to five replicate wells per culture. The cell-specific density data were expressed in terms of the frequency or percent distribution of host cells (fi) with a given number of dinoflagellates per cell (ri).
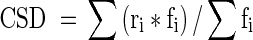 |
Data and statistical analysis
For quantification of the relative abundance of cells having engaged in DNA synthesis during the 24 h labeling period, BrdU-positive and DAPI-positive nuclei were counted using ImageJ image analysis software (U.S. National Institutes of Health, Bethesda, Maryland, USA, http://www.rsb.info.nih.gov/ij/) using the cell counting tool and checking for colocalization of Alexa-594 and DAPI signals with the colocalization plugin. Each slide was considered to represent one area in the depth of the polyp or the TB, as it corresponded to 5–10 serial semi-thin (1 μm) sections, for which the total numbers of counted nuclei were pooled. Percentages of BrdU-labeled nuclei were compared after arcsine transformation and the data distribution was tested. Non-parametric statistical analyses of the data were performed with either Kruskal–Wallis test for comparison between more than two groups, or Mann–Whitney test for comparison between two non-paired groups, or signed Wilcoxon test between two paired groups (e.g. DNA synthesis data between tissue types from the same series of serial sections—same slide).
For CSDs of dinoflagellates measured over time in primary culture, data were transformed using arcsine. Data coherence between the five independent collagenase experiments was checked with a non-parametric Kruskal–Wallis test. When no significant differences were found, data were pooled. Differences between CSD at each time point were evaluated with non-parametric Mann–Whitney test, after at least one difference was found between timepoints with a Kruskal–Wallis test. In all statistical analyses, significance was associated with p values <0.05.
Results
Effect of collagenase type and commercial brand on tissue dissociation efficiency
Within 3 days from collagenase treatment, cells continued to detach from the polyp tissue, coming off the skeleton of the colony (Fig. 1a) as single dissociated individual cells and as tissue fragments. Derived primary cultures displayed a mixed cell type composition (Fig. 1b), including cnidocytes, brown-pigmented dinoflagellate endosymbionts of coral cells (about 8 μm in diameter), and undetermined rounded clear or granulous coral cells (4–10 μm in diameter).
Fig. 1.

In vitro cell aggregation and the formation of tissue balls in primary cultures from P. damicornis scleractinian coral. a Polyps at the apex of a colony branch (scale bar 500 μm). b Mixed morphological cell types in 1 day cell culture from collagenase type IV (Sigma) dissociated tissue (scale bar 15 μm); brown spherical dinoflagellates (dino); spindle cells corresponding to various subtypes of cnidocytes (cni); clear rounded unidentified coral cells. c Irregular cluster formed by aggregation of mixed cell types in 1 day cell culture (scale bar 100 μm). d Cell aggregate formed by fusion of irregular cell clusters between first and second day of primary culture (scale bar 500 μm). e Tissue balls are spheroid cell aggregates with a smooth surface, rotating in suspension in a 2 day primary culture (scale bar 500 μm). f Fusion of suspended tissue balls in a 2 day primary culture (scale bar 500 μm)
Spontaneous aggregation of individual cells was observed in vitro, forming irregular cell clusters about 100 μm in diameter (Fig. 1c) which fused on the second day (Fig. 1d) and developed a smooth surface on the third day of culture (Fig. 1e). Both these multicellular aggregates and the detached tissue fragments rounded up and closed into spherical or ellipsoidal tissue balls (TBs) delimited by a smooth surface and rotating in suspension. Fusion of TBs over the first 5 days of the primary cultures (Fig. 1f) led to increase in TB size, reaching final diameters in the range of 250–900 μm, and averaging 520 ± 230 μm (n = 133, pooled from all experiments), generally below the average diameter of apical polyps (620 ± 70 μm, n = 18) and of branch lateral polyps (900 ± 130 μm, n = 27). Dissociation of the initial colony fragment tissue was usually complete within 3–5 days, leaving the skeleton bare.
Results presented in Table 1 indicate that the dissociation efficiency varied depending on the type and brand of commercial collagenase enzyme used. Each collagenase type targets a specific type of collagen molecule, and commercial brands include varying amounts of other proteases (detailed in “Material and methods”). Standardized to collagenase digestive units (CDU) per mg wet coral tissue, the tissue dissociation yield varied depending on treatment, in terms of number of individual cells, and number and size of tissue balls. The viability of dissociated cells measured by the trypan blue exclusion assay was, however, similar between all treatments, in the range of 70–80 %. (The trypan blue assay has low precision, due to the differential staining reaction for different coral cell types and dinoflagellates.) The quantity of single dissociated cells obtained right after the incubation was low with collagenase Sigma type I and collagenase Gibco type IV and higher with collagenase Sigma type IV (for similar activity range), and was also high with Liberase TM and Liberase DL. Nevertheless, the three latter enzymes had distinct effects on the capacity of individual cells to reaggregate over time in primary culture. Aggregation of single dissociated cells into TBs was not observed for Liberase Roche TM, which contains high concentrations of additional proteases. Collagenase Sigma type IV and Liberase DL demonstrated the highest efficiency in terms of single cell numbers right after incubation (at t = 0) but reproducibility in obtaining TBs in primary culture (at t = 2 days) was better with collagenase Sigma type IV compared to Liberase DL. The average size of tissue balls did not vary with enzyme treatment, but their number was reproducibly highest after treatment with 0.15 % collagenase type IV from Sigma. As a conclusion, the treatment with (weight/vol) 0.15 % collagenase Sigma type IV had a good yield of single cell isolation at culture initiation and was the most efficient to obtain the formation of tissue balls in primary culture.
Table 1.
Primary culture yield and viability, depending on collagenase type and commercial origin
| Enzyme | Activity (Collagen degrading units per mg wet tissue) | Digestion protocol | Number of single dissociated cells at t = 0 (106 viable cells per 100 mg wet tissue) | Cell viability (%) | Number of tissue balls (TB) at t = 2 days (per 100 mg wet tissue) | Diameter (μm) of tissue balls (mean ± SD) |
|---|---|---|---|---|---|---|
| Collagenase Sigma type I (0.15 %) | 7–8 | 1 × 30 min (+1 × 15 min rinse) | 0.02–0.25 | 50–75 | 3–7 | 554 ± 195 (n = 14) |
| Collagenase Sigma type IV (0.15 %) | 5–15 | 1 × 15 min (+1 × 15 min rinse) | 3 | 75–80 | 5–15 | 336 ± 140 (n = 28) |
| Collagenase Gibco type IV (0.15 %) | 5–15 | 1 × 30 min (+1 × 15 min rinse) | 0.15–1.5 | 70 | <1 | 500 (n = 1) |
| Liberase Roche DL (0.05 %) | 120–130 | 1 × 30 min (+1 × 15 min rinse) | 5–6 | 70–75 | 0–10 | 509 ± 79 (n = 10) |
| Liberase Roche TM (0.05 %) | 70–150 | 1 × 30 min (+1 × 15 min rinse) | 2–3 | 75–80 | 0 | 0 |
Morphology and structure of the tissue balls formed in vitro
The structure of tissue balls formed in vitro was compared to the structure of the polyps, visualized in scanning electron microscopy (SEM) after decalcification of the skeleton (Fig. 2). Typical of scleractinians, the polyp (Fig. 2a) is characterized by four cell layers organized in two tissues: the oral tissue facing seawater, composed of the pseudostratified epithelium and the oral gastroderm containing dinoflagellate endosymbionts; the aboral tissue facing the skeleton, composed of the aboral gastroderm and the calicoderm. The gastroderm lines the internal gastric cavity (Fig. 2c). Comparatively, the organization of tissue balls formed in primary culture (Fig. 2b) was simpler, with only two distinct zones, described as a cortex and a central mass of cells. A small internal cavity was sometimes present inside the central mass (Fig. 2d, white hatched line). Both polyps and tissue balls displayed ciliae on their outer surface, facing respectively seawater or cell culture medium (Fig. 2e, f). The ciliary activity of the cells forming the cortex created TB rotation in suspension.
Fig. 2.

Structure of tissue ball (TB) compared to polyp. Scanning electron micrographs of a Fractured polyp of P. damicornis and b Fractured set of 2 fused tissue balls. Insets are higher magnifications showing (c) the polyp’s four cell layers, including pse.: pseudo-stratified epithelium, o.g.: oral gastroderm, ab.g.: aboral gastroderm, cal.: calicoderm, and (d) the TB’s two zones, including co.: cortex, and c.m.: central mass, with a small internal cavity (white dots). Close-up on the ciliated external surface of respectively the polyp e and the TB f, facing respectively sea water or cell culture medium. Scale bars: a, b 100 μm, c, d 10 μm, and e, f 1 μm
Histological observations (Fig. 3) confirmed differences in organization of the tissue balls as compared to the polyps (Fig. 3a), with an outer cortex differentiated from the central mass of cells, as visualized in semi-thin sections after aggregation of tissue fragments (Fig. 3b) or individual cells (Fig. 3e). The cortex (~15 μm thick) was almost continuous, with the exception of an occasional small opening in TBs formed from tissue fragments (top in Fig. 3b). The spatial distribution of cell types was significantly different between polyp (Fig. 3a) and TBs (Fig. 3c, d, f). There was no recognizable pseudostratified epithelium in the TB. The cortex was rich in cells containing intracellular vesicles stained red with basic fuchsin and blue with toluidine blue borax, indicating basophile content. The localization of dinoflagellate endosymbionts was restricted to the central mass, and the dinoflagellate-hosting gastrodermal cells were dispersed throughout the TB central mass instead of being grouped in specific layers (as in the gastrodermal layer of polyps). Cnidocytes were located underneath the cortex, in clusters at the periphery of the central mass. Mucocytes stained purple with basic fuchsin were distributed heterogeneously inside the central mass.
Fig. 3.

Histological organization of coral tissue balls compared to polyp. Optical micrographs of semi-thin sections (0.5–1 μm) stained with toluidine blue borax then basic fuchsin. Tangential section through (a) a polyp of P. damicornis and (b–f) a TB formed in vitro, either (b, c, d) via fusion and reorganization of detached tissue fragments or (e, f) via aggregation of single dissociated cells; g, h Single dissociated coral cell types featuring a cnidocyte g and a gastrodermal host cell containing a doublet of symbiotic dinoflagellate (h) SW sea water, sk. decalcified skeleton, pse. pseudo-stratified epithelium, t. tentacle, o.g. oral gastroderm, ab.g. aboral gastroderm, g.c. gastric cavity, cal. calicoderm, co. cortex, c.m. central mass. Scale bars: a, b, e 100 μm and c, d, f, g, h 10 μm
Ultrastructural observations (Fig. 4) confirmed the spatially differentiated cell type distribution inside tissue balls, with distinct cell types in the cortex and the central mass. The cortex was composed of interdigitated ciliated cells with apical microvilli and numerous mitochondria, joined together by apical intercellular septate junctions (Fig. 4a, b). Abundant electron-dense vesicles and fibrillar material were observed in proximity to Golgi structures in the cortex cells, indicating high secretory activity (Fig. 4c, d). In the central mass, few cnidocytes were detected underneath the cortex cells, mostly grouped in small clusters, with intact or partially lysed cnidocyst capsule (Fig. 4e). The central mass also contained coral cells hosting dinoflagellate symbionts (holobiont cells) (Fig. 4f). Intercellular spaces were abundant (Fig. 4g) and various types of autophagosomes were detected throughout the central mass (Fig. 4h), providing morphological evidence of autophagy processes.
Fig. 4.

Ultrastructure of tissue balls. Transmission electron micrographs of TBs formed in vitro by a Aggregation of tissue fragments or b Aggregation of single dissociated cells; c, d Higher magnification of the cortex epithelium, composed of interdigitated ciliated cells joined by apical septate junctions (black arrows), and containing abundant mitochondria (m) and electron-dense fibrillar material (white arrows) in proximity to Golgi structures (G.) Higher magnification of the central mass, with e Cnidocytes (cni) grouped in clusters, f a doublet of dinoflagellates (dino) in their coral host cell, g cnidocytes, dinoflagellate and intercellular spaces. h Autophagosome figures (black stars) are abundant inside cells of the central mass, and a few pigmented cells (pi) are observed. Scale bars a, e, f, g 2 μm and b, c, d, h 500 nm
Autofluorescence observations in epifluorescence microscopy of live material (Fig. 5) revealed differences between tissue balls and polyps in the spatial distribution of chlorophyll-containing dinoflagellates (in red) and of the coral cells containing Green Fluorescent Protein (GFP)-like molecules (in green). In the polyp, dinoflagellate density was highest in the oral tissue and tentacles (Fig. 5a), and GFP-like molecules were most concentrated at the apex of tentacles and at the periphery of the oral disk (Fig. 5c and overlay Fig. 5e). In the tissue balls, the distribution of dinoflagellates (Fig. 5b) and of GFP-like molecules (Fig. 5d and overlay Fig. 5f) was spatially highly heterogeneous, corresponding to individual cells dispersed throughout the tissue.
Fig. 5.

Compared auto-fluorescence of a live polyp and a tissue ball. Epifluorescence images of chlorophyll (in red), GFP-like molecules (in green) and their image overlay in a polyp of P. damicornis (a, c, e), and a tissue ball formed in primary cell culture (b, d, f), showing differences in the distribution of chlorophyll and GFP between the polyp and the TB. Scale bars 500 μm
DNA synthesis in vitro in the tissue balls compared to in situ in the polyps
Detection of BrdU incorporation into cell nuclei in semi-thin sections of the polyps and of the tissue balls (Fig. 6) allowed localization of DNA synthesis both in situ in the polyp and in vitro, during formation of the tissue balls. Specific labeling of DAPI stained nuclei (Fig. 6a, b) with Alexa-594-conjugated anti-BrdU secondary antibody (Fig. 6c, d) was confirmed by co-localization of BrdU with DAPI (Fig. 6e, f) and absence of labeling in internal negative controls, and in tissue unexposed to BrdU (data not shown).
Fig. 6.

BrdU-labeled nuclei in polyp and in tissue ball formed in 2 days primary cell culture. Semi-thin (1 μm) sections stained with toluidine blue and basic fuchsin of a polyp and b TB. Insets are light micrographs indicating the orientation of the sections in respectively the P. damicornis polyp or the TB. Fluorescence of nuclei stained with DAPI (in blue) (c, d), immunolabeled with Alexa-594 (in red) (e, f) and their image overlay (g, h) in semi-thin sections of a polyp (a, c, e, g) and a tissue ball (b, d, f, h). sk. skeleton, pse. pseudo-stratified epithelium, o.g. oral gastroderm, ab.g. aboral gastroderm, cal. calicoderm, g.c. gastric cavity. Scale bars: 100 μm
The percentage of cells having incorporated BrdU during 24 h was quantified in apical polyps from four replicate colonies, and in three replicate TBs. Several areas corresponding each to data pooled from 5 to 10 serial sections were analyzed, corresponding each to a specific depth in the polyp or TB (see “Materials and methods”). Average relative abundance of BrdU-positive nuclei are reported in Table 2 for each cell layer and each zones of the polyp and the TB. Differences in average BrdU-labeling index between polyp cell layers or zones in the TBs, and between coral cells (oral gastroderm) and dinoflagellates are illustrated in Fig. 7. The percentage of BrdU-positive coral cell nuclei was counted separately for each cell layer of the polyp and each zone (cortex or central mass) of the TB (Fig. 7a). The BrdU-labeling index of dinoflagellate endosymbionts was also evaluated, to compare DNA synthesis rate of symbionts and host cells, in the polyp and in the TBs (Fig. 7b).
Table 2.
BrdU labeling index of nuclei of coral cells and dinoflagellate symbionts in the different tissue layers of the polyp and in vitro, during (2d) formation of the tissue balls
| % BrdU-positive nuclei | Polyp | Tissue ball | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Pseudo-stratified epithelium | Oral gastroderm | Aboral gastroderm | Calicoderm | Cortex | Central mass | ||||
| Oral disk, coenosarc, tentacle | Tip of tentacle | Dinoflagellate endosymbionts | Coral cells | Coral cells | Dinoflagellate endosymbionts | ||||
| Average | 15.9 % | 21.4 % | 14.0 % | 10.3 % | 10.3 % | 0.8 % | 0.2 % | 0.2 % | 2.7 % |
| Standard error | 4.2 % | 5.6 % | 3.4 % | 2.2 % | 3.3 % | 0.4 % | 0.1 % | 0.1 % | 1.2 % |
| Min–max | 6.1–31.5 % | 5.3–40.7 % | 5–21 % | 5.1–15.9 % | 4.0–23.7 % | 0–1.5 % | 0–0.7 % | 0–1.2 % | 0–10 % |
| Standard deviation | 10.3 % | 13.6 % | 6.8 % | 4.9 % | 8.2 % | 0.9 % | 0.3 % | 0.4 % | 3.4 % |
| Number of replicate tissue areas | 6 | 6 | 4 | 5 | 6 | 6 | 8 | 8 | 8 |
| Total counted nuclei | 10,207 | 4,760 | 317 | 1,919 | 1,418 | 866 | 11,054 | 37,567 | 2,910 |
Fig. 7.

Compared BrdU incorporation rates of coral cells and dinoflagellates in polyps and in tissue balls obtained in 2 days primary culture. Data indicate mean ± standard error (SE) for a coral cells in the different polyp tissue layers and the TB zones, and b for dinoflagellates in the polyp oral gastroderm and in the TB (central mass). pse pseudo-stratified epithelium. Black star indicates significant difference between calicoderm and other tissue layers, and between TB and polyps
For TBs, no statistical differences in BrdU incorporation rates were found within similar zones (cortex or central mass) between the replicate TBs, so data were pooled. For comparison between zones, a similar very low rate of BrdU incorporation (0.2 ± 0.1 %, mean ± standard error) was detected in nuclei of the coral cells in the cortex and the central mass (Signed Wilcoxon, p value = 0.205, n = 40 total sections). In the central mass, BrdU labeling index of dinoflagellate symbionts was significantly higher than BrdU labeling index of coral cells (Signed Wilcoxon, paired data, p value = 0.000, n = 38 total sections), indicating that inside the tissue balls DNA synthesis activity was higher in dinoflagellates compared to coral cells.
For polyps, the average percentage of BrdU-positive nuclei was evaluated per area in the tissue depth and, within each area, the BrdU incorporation rates were collected separately for each cell layer. The BrdU labeling index of each cell layer varied between areas. All results are summarized in Table 2. For the pseudo-stratified oral epithelium (pse), BrdU incorporation rates ranged from 6.1 to 31.5 %, reaching 5.3 to 40.7 % at the tip of tentacles. In the oral gastroderm (o.g.), BrdU incorporation rates were similar in coral cells (from 5.1 to 15.9 %) and in dinoflagellate symbionts (from 5 to 21 %). In the aboral gastroderm (ab.g.), BrdU incorporation rates in coral cells ranged from 4 to 23.7 %. The calicoderm was the only cell layer with low variations in BrdU incorporation rates between areas (from 0 to 1.5 %). In order to evaluate differences in DNA synthesis activity between the four cell layers, arcsine transformed data averaged from all areas in the polyp replicates were compared with non-parametric tests. For coral cells, the calicoderm was the only cell layer statistically different from all others, with lowest BrdU labeling (0.8 ± 0.4 % mean ± standard error, n = 6 areas). In the pseudostratified epithelium, BrdU labeling was 15.9 ± 4.2 % (n = 6), reaching 21.4 ± 3.4 % (n = 6) at tip of tentacle. BrdU labeling index of coral cells was similar in the oral gastroderm (10.3 ± 2.2 % n = 5), and the aboral gastroderm (10.3 ± 3.3 % n = 6) (Mann–Whitney tests, p value <0.0001). The dinoflagellate symbionts displayed no statistically significant differences in BrdU incorporation rates compared to their host coral cells (oral gastroderm) (Signed Wilcoxon, p value = 0.584 n = 4).
Compared to polyp coral cells, the DNA synthesis activity of coral cells in TBs was significantly reduced (Fig. 7a). The DNA synthesis activity of dinoflagellate endosymbionts was also reduced in TBs compared to polyps, with rates of BrdU incorporation rates estimated at 2.7 ± 1.2 % in vitro inside the central mass of TBs compared with 14.0 ± 3.4 % in situ in the oral gastroderm of polyps (Fig. 7b). (The lower number of counted nuclei from dinoflagellates compared to coral cells was due to their lower relative abundance inside the TB central mass or the polyp, and to the lower number of semi-thin sections (1 μm) passing through their nucleus.)
For polyps as well as for tissue balls, the important standard deviations noted for measured BrdU-labeling indexes were caused by the wide range of data distribution, due to high labeling variability between areas in the tissue depth. This large spatial variability indicates that the distribution of cells engaged in DNA synthesis is spatially highly heterogeneous, depending on their location in the polyp or the TB: within the same cell layer, cell proliferation rates vary locally.
In single dissociated cells, exposed to BrdU in parallel to tissue ball formation at the onset of primary cultures, preliminary assessment of BrdU labeling of nuclei suggests that up to 20 % of the dinoflagellates had incorporated BrdU (14/70 total counted nuclei), but not the coral cells. The low number of counted nuclei (due to low cell yield of immunocytochemistry on suspended cells) will however require future confirmation of this preliminary observation.
Evolution of the cell specific density of dinoflagellate symbionts in primary cultures
The cell-specific density (CSD) of dinoflagellate endosymbionts was quantified in single dissociated gastrodermal cells (after the method of Muscatine et al. 1998) at the onset of primary cultures, over the first 2 days from collagenase treatment. Data were averaged for three to five hundred gastrodermal (holobiont) cells counted per replicate well of a primary culture. No statistical difference was detected between the five independent collagenase experiments for each time point (Kruskal–Wallis test, p value >0.05 at t = 16, 20, 24 h; 40 and 45 h, n = 4–6), so data were pooled. Between time points there were statistically significant differences (Kruskal–Wallis test, p value <0.0001). The CSD at t = 0 h differed from the CSD at t = 16/20/24 h (Mann–Whitney test, p value = 0.001/0.010/0.043). Figure 8 illustrates the increase in average CSD during the first 20 h of primary culture, starting from 1.29 dinoflagellates per host cell at culture initiation and reaching 1.46 dinoflagellates per host cell at t = 16 h. After 40 h, the CSD returned to control level at beginning of culture.
Fig. 8.

Cell Specific Density of dinoflagellate symbionts at initiation of coral primary cultures. For each time point, data were pooled from five independent experiments and are expressed as mean ± standard deviation. A dark star indicates significant difference with control at t = 0 h
Discussion
Cell dissociation methods are critical to the isolation of scleractinian coral cells and affect their in vitro survival and functionality. Enzyme digestion induces dissociation of individual cells and small cell clusters, which remain suspended or adhere to the culture substrate and may aggregate over time (Gates and Muscatine 1992; Frank et al. 1994; Helman et al. 2008; Downs et al. 2010). The mesoglea gel separating the two epithelial layers (epiderm and gastroderm) of cnidaria is an extracellular matrix (ECM) which, in Hydra, has been reported to contain collagen type IV (Fowler et al. 2000) associated to laminin in the subepithelial basal lamina and to fibrillar collagen type I (Shimizu et al. 2008; Sarras 2012). Collagenase is the most widely used enzyme for cnidarian tissue dissociation, with protocols first developed for hydrozoans, jellyfish and sea anemones, then adapted to the octocorals and stony corals (Gates and Muscatine 1992; Frank et al. 1994; Helman et al. 2008). Optimal reported concentrations of collagenase range from 0.05 to 0.15 % (dry weight/volume) with incubation times of 30 min to 4 h. However information on collagenase type and commercial brand is frequently missing from the literature. In this study, various collagenase-based cell dissociation methods were compared for their effects on cell yield and aggregation in primary culture. This study highlights differences in the nature and yield of tissue versus single cell cultures obtained, depending on the collagenase type and commercial brand used. Aggregation of individual cells into suspended multicellular aggregates with a smooth surface (tissue balls) was optimized for P. damicornis after 15 min treatment with collagenase type IV (Sigma) adjusted to 5–15 collagen-degrading units (CDUs) per mg of wet tissue. These results confirm previous reports of the formation of adherent cell aggregates following collagenase (unspecified type) dissociation of tissue from the coral Montipora digitata (Helman et al. 2008). Compared to calcium removal treatment, after which dissociated cells failed to re-aggregate (Domart-Coulon et al. 2004a), this collagenase treatment preserves the coral cell adhesion capacities.
The timing of formation of suspended multicellular aggregates (tissue balls) in scleractinian coral primary cultures has been described (Kopecky and Ostrander 1999; Domart-Coulon et al. 2004a; Nesa and Hidaka 2009). Nevertheless mechanisms underlying formation of these structures are poorly understood, including potential involvement of in vitro proliferation. In this study, detailed morphological and ultrastructural observations of the forming cell aggregates highlight their tissue-grade structure, with a cortex differentiated from the central mass by its cell type composition. Morphologically, these structures are similar to spheroids formed by wound healing of scleractinian soft tissue fragments manually peeled off the skeleton (Kramarsky-Winter and Loya 1996; Vizel et al. 2011), or to bailed out polyps (Sammarco 1982) after a few days incubation in seawater without attachment (data not shown).
Visualization of apical septate junctions between the interdigitated ciliated cells of the cortex indicates that this cell layer constitutes an epithelium, delimitating the surface of cell aggregates. A morphologically distinct underlying basal matrix was however not detected, in contrast to mesoglea observed in spheroids formed from Fungia granulosa tissue (Vizel et al. 2011), possibly reflecting species-specific differences in extra-cellular matrix abundance. Inside the cortex cells, abundance of mitochondria, electron-dense fibrillar material and Golgi apparatus are morphological indications of intense secretory activity. In a previous study, it was reported that the cells located at the surface of multicellular isolates formed from P. damicornis coral were immunoreactive to an antibody raised against the soluble organic matrix from the skeleton of the related Pocilloporid coral Stylophora pistillata (Domart-Coulon et al. 2004b; Puverel et al. 2005). These cells might thus share features of calicoblast cell types.
Abundant autophagosomes in the central mass provide morphological evidence that autophagy events are involved in the formation of TBs. Combined with the spatially restricted distribution of cnidocytes and dinoflagellate endosymbionts to the center of tissue balls, these results suggest that cell sorting activities may exist, which could play a role during in vitro aggregation of scleractinian coral cells. Indeed, in the hydrozoan Hydra sp. homotypic interactions switching to heterotypic interactions have been reported during aggregation of epithelial cells (Hobmayer et al. 2001).
It was shown that a critical step for obtaining polyp regeneration from cultured spheroid explants is the development of a mouth, which can be induced by light and temperature cycling in seawater (Vizel et al. 2011). In the polyps of P. damicornis, we have observed that the oral disk was delimited by a concentrated ring of GFP-like molecules. According to D’Angelo et al. (2012), GFP-like proteins are concentrated in areas of growth and accelerated cell proliferation in several coral species (Porites lobata, Montipora foliosa, Acropora pulchra/polystoma) and could be used as a biomarker of locally accelerated growth and tissue regeneration. In the forming tissue balls of P. damicornis, we did not record any spatially differentiated pattern of distribution of GFP-like molecules. Instead, cells containing GFPs were dispersed throughout the structure of the tissue balls, with a highly heterogeneous spatial distribution. These results indicate disorganization of tissue balls compared to polyps, with lack of an oral disk structure.
It is known that collagenase treatment of the extra-cellular matrix of cnidarian tissue may stimulate reprogramming and transdifferentiation of cell types, a process well described in the jellyfish (Scyphozoa) Podocoryne carnea (Schmid et al. 1999; Schmid and Reber-Müller 1995). In the jellyfish, collagenase treatment of isolated pieces of the umbrella triggers de-differentiation of striated muscle cells, followed by transdifferentiation to smooth muscle cells. In our case, collagenase may induce scleractinian coral tissue re-organization processes and the differentiation of cortex cells. Tissue re-organization may also be a common mechanism in scleractinian corals, triggered by several signals, after artificial or spontaneous detachment from skeleton.
Our results using BrdU labeling of nuclei show a very low level and high spatial heterogeneity of DNA synthesis activity inside the forming tissue balls, in our primary culture conditions. As DNA repair mechanisms in response to stress also involve limited DNA synthesis events before engagement in cellular apoptosis pathways, the BrdU labeling method cannot unambiguously detect cell proliferation activity in the coral primary cultures and should be complemented by other methods targeting proteins regulating progression through the cell cycle, to label cells engaged in mitosis. However, the low DNA synthesis rates measured for coral cells throughout the TB (about 0.2 %) indicate that the formation of these multicellular aggregates does not involve significant proliferation. Further analyses to investigate more precisely cell fate in longer-term cultures of tissue balls will be performed.
In situ labeling with BrdU of the polyps of P. damicornis, over a 24 h period in aquarium settings, revealed that DNA synthesis occurred in all four cellular layers composing the tissue, with a significantly lower rate in the calicoderm layer, which is involved in skeleton formation. No statistical differences of BrdU incorporation were found between the three other cell layers. The pseudostratified oral epithelium was highly labeled with 15.9 ± 4.2 % BrdU-positive cells, reaching 21.4 ± 5.6 % at tips of tentacles. Coral cells of the oral and aboral gastroderm were characterized by respectively 10.3 ± 2.2 % and 10.3 ± 3.3 % BrdU incorporation rates. Our results are in agreement with the values reported for the BrdU labeling index of the gastroderm in polyps of Montipora informis and Porites australiensis healthy colonies (Yasuda and Hidaka 2012) but we report lower rates in the calicoderm and higher rates in the pseudostratified epithelium (oral epidermis). However this recent study used a longer exposure time to BrdU (3 days) than our study (24 h) and did not focus on differences between cell layers within an individual polyp. With long exposure periods, the distribution of BrdU positive cells is the result of combined cell division and migration, the cellular turnover masking potential differences in cell proliferation between cell layers (for example accumulation of BrdU positive cells may occur in regions of low turnover). The high variations of BrdU labeling index we observed from one area to the next within the same tissue layer of the same polyp suggest that there may be some highly proliferative clusters of cells inside each tissue layer. Additional experiments must be done to identify potential niche of stem cells and distinguish migration from division, with varying BrdU exposure times in pulse-chase labeling experiments.
Major differences in proliferation activity of the dinoflagellate endosymbionts were observed between in vitro and in situ conditions. Cell division of dinoflagellates was higher when located inside individually dissociated gastrodermal cells than when located inside the intact polyp or inside the tissue balls: the endosymbiont specific density (CSD) of gastrodermal cells peaked within the first day of individual cell culture and the proportion of dinoflagellate cells having incorporated BrdU seemed to increase from 14 % in intact polyp to 20 % in individually dissociated cells, whereas it dropped to 2.7 % inside the tissue balls. These combined BrdU labeling and CSD data suggest that tissue dissociation into individual gastrodermal cells induces an increase in dinoflagellate proliferation. Tissue dissociation is likely to disrupt the controls exerted by the coral host cell on its dinoflagellate symbiont proliferation, explaining the rapid and transient rise of CSDs. Restrictions of the intracellular availability of nitrogenous nutrients needed to undertake dinoflagellate cell cytokinesis is one of the processes which maintain dinoflagellates in a growth-limited state (Wooldridge 2010). In single dissociated cell cultures, the dissolved amino-acids contained in the complex cell culture medium are likely to be rapidly assimilated by the dinoflagellates (Grover et al. 2008), fueling their cell growth and division. The expulsion or degradation of dinoflagellates by the host cell after the peak of CSD could explain the return to initial CSD. Alternately, the coral host cell may die after the peak of CSD, releasing dividing dinoflagellates into the culture medium.
In this study, a nutrient-rich culture medium (containing (v:v) 12.5 % commercial DMEM) was used to establish primary coral cell cultures, under air and 10/14 h light/dark cycle, with very low white light intensity (2–4 photosynthetic photon flux density micromoles m−2 s−1). These conditions differ from the natural conditions for growth of the polyps and may contribute to limit the in vitro cellular DNA synthesis rate. Tissue-grade cell aggregates can also be obtained in seawater (Vizel et al. 2011; Nesa and Hidaka 2009), and they survive longest at low light intensity and constant low temperature as reported for the spheroid tissue from Fungia granulosa (Vizel et al. 2011). Nutrients and light intensity can have an impact on the survivorship of multicellular isolates from some coral species (e.g. Acropora micropthalma) and not from others (e.g. P. damicornis) (Kopecky and Ostrander 1999). These observations confirm that cell culture conditions have to be adjusted between species, since optimal light- and temperature-range differ between scleractinian coral species.
Understanding processes leading to in vitro coral growth arrest and cellular senescence remains an exciting challenge (Rinkevich 2011). To date, most coral primary cultures are established from differentiated tissue from scleractinian adult colonies. In order to improve in vitro cell proliferation, it would be useful to differentiate more finely which area in the adult tissue layer has the highest proliferation potential, and attempt cultures from selected microdissected tissue fragments. Alternately, cultures could be established from highly proliferative larval tissue, as attempted on embryonic cells from Acropora millepora (Reyes-Bermudez et al. 2009) or hyperplasic tissue from growth anomalies (so-called ‘tumors’) for example of Porites compressa (Domart-Coulon et al. 2006), and Porites australiensis or Montipora informis (Yasuda and Hidaka 2012).
Conclusion
In this study, spatial differences in DNA synthesis activity were detected in the different cell layers of scleractinian coral polyps with the BrdU labeling method (24 h exposure), with significantly lower proliferation in the calicoderm at the surface of skeleton compared to the pseudostratified epithelium facing seawater and the gastroderm lining the digestive cavity. BrdU incorporation rates evaluated during the process of formation of suspended multicellular aggregates in primary cultures initiated with collagenase were found to be very low, in both coral and dinoflagellate cells, indicating that cell aggregation does not imply simultaneous proliferation. Dissociation of the tissue into single cells was followed by a transient rise in the cell specific density of dinoflagellate symbionts of isolated gastrodermal cells. These results suggest that mechanisms controlling proliferation of coral cells and their dinoflagellate endosymbionts are disrupted by current in vitro primary culture conditions.
Acknowledgments
This work was supported by the ATM Biomineralization of the Museum National d’Histoire Naturelle (‘scleractinian coral’ project) to I. Domart-Coulon, and by European Research Council Advanced Grant 246749 (BIOCARB) to A. Meibom. It was presented at the Symposium on ‘Marine Invertebrate Cell Cultures’, August 30–31 2012, at the Station de Biologie Marine du MNHN in Concarneau, France. We especially thank Michel Hignette and all the team from the Aquarium Tropical, Palais de la Porte Dorée, Paris, France, for access to biological material and facilities for aquarium experiments.
Footnotes
I. Domart-Coulon was formerly affiliated with UMR 7208 BOREA Département Milieux et Peuplements Aquatiques, Muséum National d’Histoire Naturelle, Case Postale 26, 43 rue Cuvier, 75005 Paris, France
References
- D’Angelo C, Smith EG, Oswald F, Burt J, Tchernov D, Wiedenmann J (2012) Locally accelerated growth is part of the innate immune response and repair mechanisms in reef-building corals as detected by green fluorescent protein (GFP)-like pigments. Coral Reefs 31(4):1045–1056
- Denker E, Manuel M, Leclère L, Le Guyader H, Rabet N. Ordered progression of nematogenesis from stem cells through differentiation stages in the tentacle bulb of Clytia hemisphaerica (Hydrozoa, Cnidaria) Dev Biol. 2008;315:99–113. doi: 10.1016/j.ydbio.2007.12.023. [DOI] [PubMed] [Google Scholar]
- Dolbeare F, Selden JR (1994) Immunochemical quantitation of bromodeoxyuridine: application to cell-cycle kinetics. Methods Cell Biol 41:297–316 [PubMed]
- Domart-Coulon IJ, Elbert DC, Scully EP, Calimlim PS, Ostrander GK. Aragonite crystallization in primary cell cultures of multicellular isolates from a hard coral, Pocillopora damicornis. Proc Natl Acad Sci USA. 2001;98:11885–11890. doi: 10.1073/pnas.211439698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domart-Coulon IJ, Sinclair CS, Hill RT, Tambutte S, Puverel S, Ostrander GK. A basidiomycete isolated from the skeleton of Pocillopora damicornis (Scleractinia) selectively stimulates short-term survival of coral skeletogenic cells. Mar Biol. 2004;144:583–592. doi: 10.1007/s00227-003-1227-0. [DOI] [Google Scholar]
- Domart-Coulon I, Tambutte S, Tambutte E, Allemand D. Short term viability of soft tissue detached from the skeleton of reef-building corals. J Exp Mar Biol Ecol. 2004;309:199–217. doi: 10.1016/j.jembe.2004.03.021. [DOI] [Google Scholar]
- Domart-Coulon IJ, Traylor-Knowles N, Peters E, Elbert D, Downs CA, Price K, Stubbs J, McLaughlin S, Cox E, Aeby G, Brown PR, Ostrander GK. Comprehensive characterization of skeletal tissue growth anomalies of the finger coral Porites compressa. Coral Reefs. 2006;25:531–543. doi: 10.1007/s00338-006-0133-6. [DOI] [Google Scholar]
- Downs CA, Fauth JE, Downs VD, Ostrander GK. In vitro cell-toxicity screening as an alternative animal model for coral toxicology: effects of heat stress, sulfide, rotenone, cyanide, and cuprous oxide on cell viability and mitochondrial function. Ecotoxicology. 2010;19:171–184. doi: 10.1007/s10646-009-0403-5. [DOI] [PubMed] [Google Scholar]
- Fowler SJ, Jose S, Zhang X, Deutzmann R, Sarras MP, Jr, Boot-Handford RP. Characterization of hydra type IV collagen. Type IV collagen is essential for head regeneration and its expression is up-regulated upon exposure to glucose. J Biol Chem. 2000;275:39589–39599. doi: 10.1074/jbc.M005871200. [DOI] [PubMed] [Google Scholar]
- Frank U, Rabinowitz C, Rinkevich B. In vitro establishment of continuous cell cultures and cell lines from 10 colonial cnidarians. Mar Biol. 1994;120:491–499. doi: 10.1007/BF00680224. [DOI] [Google Scholar]
- Gates RD, Muscatine L. 3 Methods for isolating viable anthozoan endoderm cells with their intracellular symbiotic dinoflagellates. Coral Reefs. 1992;11:143–145. doi: 10.1007/BF00255468. [DOI] [Google Scholar]
- Grover R, Maguer JF, Allemand D, Ferrier-Pages C. Uptake of dissolved free amino acids by the scleractinian coral Stylophora pistillata. J Exp Biol. 2008;211:860–865. doi: 10.1242/jeb.012807. [DOI] [PubMed] [Google Scholar]
- Helman Y, Natale F, Sherrell RM, LaVigne M, Starovoytov V, Gorbunov MY, Falkowski PG. Extracellular matrix production and calcium carbonate precipitation by coral cells in vitro. Proc Natl Acad Sci USA. 2008;105:54–58. doi: 10.1073/pnas.0710604105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobmayer B, Snyder P, Alt D, Happel CM, Holstein TW. Quantitative analysis of epithelial cell aggregation in the simple metazoan Hydra reveals a switch from homotypic to heterotypic cell interactions. Cell Tissue Res. 2001;304:147–157. doi: 10.1007/s004410000344. [DOI] [PubMed] [Google Scholar]
- Khalesi MK, Vera-Jimenez NI, Aanen DK, Beeftink HH, Wijffels RH. Cell cultures from the symbiotic soft coral Sinularia flexibilis. In Vitro Cell Dev Biol Anim. 2008;44:330–338. doi: 10.1007/s11626-008-9128-7. [DOI] [PubMed] [Google Scholar]
- Kopecky EJ, Ostrander GK. Isolation and primary culture of viable multicellular endothelial isolates from hard corals. In Vitro Cell Dev Biol Anim. 1999;35:616–624. doi: 10.1007/s11626-999-0101-x. [DOI] [PubMed] [Google Scholar]
- Kramarsky-Winter E, Loya Y. Regeneration versus budding in fungiid corals: a trade-off. Mar Ecol-Prog Ser. 1996;134:179–185. doi: 10.3354/meps134179. [DOI] [Google Scholar]
- Mass T, Drake JL, Haramaty L, Rosenthal Y, Schofield OM, Sherrell RM, Falkowski PG. Aragonite precipitation by “proto-polyps” in coral cell cultures. PLoS ONE. 2012;7:e35049. doi: 10.1371/journal.pone.0035049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller WA, Teo R, Frank U. Totipotent migratory stem cells in a hydroid. Dev Biol. 2004;275:215–224. doi: 10.1016/j.ydbio.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Muscatine L, Ferrier-Pages C, Blackburn A, Gates RD, Baghdasarian G, Allemand D (1998) Cell specific density of symbiotic dinoflagellates in tropical anthozoans. Coral Reefs 17(4):329–337
- Nesa B, Hidaka M. High zooxanthella density shortens the survival time of coral cell aggregates under thermal stress. J Exp Mar Biol Ecol. 2009;368:81–87. doi: 10.1016/j.jembe.2008.10.018. [DOI] [Google Scholar]
- Peng SE, Luo YJ, Huang HJ, Lee IT, Hou LS, Chen WNU, Fang LS, Chen CS. Isolation of tissue layers in hermatypic corals by N-acetylcysteine: morphological and proteomic examinations. Coral Reefs. 2008;27:133–142. doi: 10.1007/s00338-007-0300-4. [DOI] [Google Scholar]
- Puverel S, Tambutte E, Zoccola D, Domart-Coulon I, Bouchot A, Lotto S, Allemand D, Tambutte S. Antibodies against the organic matrix in scleractinians: a new tool to study coral biomineralization. Coral Reefs. 2005;24:149–156. doi: 10.1007/s00338-004-0456-0. [DOI] [Google Scholar]
- Reyes-Bermudez A, Lin Z, Hayward DC, Miller DJ, Ball EE. Differential expression of three galaxin-related genes during settlement and metamorphosis in the scleractinian coral Acropora millepora. BMC Evol Biol. 2009;9:178. doi: 10.1186/1471-2148-9-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkevich B. Cell cultures from marine invertebrates: obstacles, new approaches and recent improvements. J Biotechnol. 1999;70:133–153. doi: 10.1016/S0168-1656(99)00067-X. [DOI] [Google Scholar]
- Rinkevich B. Marine invertebrate cell cultures: new millennium trends. Mar Biotechnol (NY) 2005;7:429–439. doi: 10.1007/s10126-004-0108-y. [DOI] [PubMed] [Google Scholar]
- Rinkevich B. Cell cultures from marine invertebrates: new insights for capturing endless stemness. Mar Biotechnol (NY) 2011;13:345–354. doi: 10.1007/s10126-010-9354-3. [DOI] [PubMed] [Google Scholar]
- Sammarco PW. Polyp bail-out—an escape response to environmental stress and a new means of reproduction in corals. Mar Ecol Prog Ser. 1982;10:57–65. doi: 10.3354/meps010057. [DOI] [Google Scholar]
- Santos SR, Toyoshima J, Kinzie RA., III Spatial and temporal dynamics of symbiotic dinoflagellates (Symbiodinium: dinophyta) in the perforate coral Montipora capitata, Galaxea. J Coral Reef Stud. 2009;11:139–147. doi: 10.3755/galaxea.11.139. [DOI] [Google Scholar]
- Sarras MP. Components, structure, biogenesis and function of the Hydra extracellular matrix in regeneration, pattern formation and cell differentiation. Int J Dev Biol. 2012;56:567–576. doi: 10.1387/ijdb.113445ms. [DOI] [PubMed] [Google Scholar]
- Schmid V, Reber-Muller S. Transdifferentiation of isolated striated muscle of jellyfish in vitro: the initiation process. Semin Cell Biol. 1995;6:109–116. doi: 10.1006/scel.1995.0016. [DOI] [PubMed] [Google Scholar]
- Schmid V, Ono SI, Reber-Muller S. Cell-substrate interactions in cnidaria. Microsc Res Tech. 1999;44:254–268. doi: 10.1002/(SICI)1097-0029(19990215)44:4<254::AID-JEMT5>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Shimizu H, Aufschnaiter R, Lib L, Sarras MP, Borza D-B, Abrahamson DR, Sadof Y, Zhang X. The extracellular matrix of Hydra is a porous sheet and contains type IV collagen. Zoology (Jena) 2008;111:410–418. doi: 10.1016/j.zool.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taupin P. BrdU immunohistochemistry for studying adult neurogenesis: paradigms, pitfalls, limitations, and validation. Brain Res Rev. 2007;53:198–214. doi: 10.1016/j.brainresrev.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Vizel M, Loya Y, Downs CA, Kramarsky-Winter E. A novel method for coral explant culture and micropropagation. Mar Biotechnol (NY) 2011;13:423–432. doi: 10.1007/s10126-010-9313-z. [DOI] [PubMed] [Google Scholar]
- Wooldridge SA. Is the coral-algae symbiosis really ‘mutually beneficial’ for the partners? BioEssays. 2010;32:615–625. doi: 10.1002/bies.200900182. [DOI] [PubMed] [Google Scholar]
- Yasuda N, Hidaka M. Cellular kinetics in growth anomalies of the scleractinian corals Porites australiensis and Montipora informis. Dis Aquat Organ. 2012;102:1–11. doi: 10.3354/dao02530. [DOI] [PubMed] [Google Scholar]