

Abstract
Nicorandil is a nitric oxide (NO) donor used in the treatment of angina symptoms. It has also been reported to protect cells and affect the proliferation and death of cells in some tissues. The molecules that interfere with these processes can cause dysfunction in healthy tissues but can also assist in the therapy of some disorders. In this study we examined the effect of nicorandil and of the molecular precursor that does not have the NO radical (N-(beta-hydroxyethyl) nicotinamide) on the cell proliferation and death of human renal carcinoma cells (786-O) under normal oxygenation conditions. The molecular precursor was used in order to analyze the effects independents of NO. In the cytotoxicity test, nicorandil was shown to be cytotoxic at very high concentrations and it was more cytotoxic than its precursor (cytotoxic at concentrations of 2,000 and 3,000 μg/mL, respectively). We propose that the lower cytotoxicity of the precursor is due to the absence of the NO radical. In this study, the cells exposed to nicorandil showed neither statistically significant changes in cell proliferation nor increases in apoptosis or genotoxicity. The precursor generated similar results to those of nicorandil. We conclude that nicorandil causes no changes in the proliferation or apoptosis of the cell 786-O in normal oxygenation conditions. Moreover, the lack of NO radical in the precursor molecule did not show a different result, except in the cell cytotoxicity.
Keywords: Nicorandil, N-(beta-hydroxyethyl) nicotinamide, 786-O cells, Cell proliferation, Apoptosis, Cytotoxicity
Introduction
Nicorandil (N-(beta-hydroxyethyl) nicotinamide nitrate ester) belongs to the organic nitrates group (RONO2) which includes nitroglycerin (TNG). It is used in the treatment of cardiac dysfunction and symptoms of angina pectoris. Nicorandil has two important characteristics: it is a nitric oxide (NO) donor and an ATP-sensitive potassium (KATP) channel activator (Barreto and Correia 2005; Hiremath et al. 2010; Simpson and Wellington 2004). As a potent vasodilator, nicorandil can hyperpolarize the membrane of muscle cells (Frydman 1992), which allows coronary and peripheral vasodilatation leading to a decrease in the input and output pressure of the blood flow in the heart. Nicorandil has also been shown to produce ischemic preconditioning of the cell, which protects cardiac tissue (Ahmed et al. 2011; Carreira et al. 2008; Eeckhout 2003; Sato et al. 2000).
Organic nitrates, like nicorandil, release NO from their structures through enzymatic processes, such as those involving glutathione S-transferase (GST), xanthine oxidase (XO) or complex enzymatic cytochrome P450, or non-enzymatic processes by reacting chemically with acids, alkalis and thiols (Chong and Fung 1991; Seth and Fung 1993). The released NO is a signaling molecule that readily diffuses across the plasma membrane and binds to the soluble enzyme guanylate cyclase (GC), which catalyzes the conversion of intracellular guanosine-5′-triphosphate (GTP) to cyclic guanosine-3′,5′-monophosphate (cGMP). cGMP, in turn, acts as a second messenger to maintain the tone and motility of the smooth muscle tissue of blood vessels, promoting vasodilatation. cGMP is also involved in several other processes, such as proliferation and cellular differentiation, the homeostasis of fluids and electrolytes, and apoptosis (Krumenacker and Murad 2006; Mujoo et al. 2010). Various studies analyze the effects of NO on the cell proliferation and death, which depend on its concentration in the microenvironment, cell type, exposure period and various other factors (Yim et al. 1993; Dimmeler and Zeiher 1997; Masri 2010).
In addition to its effects on vasodilatation, studies have shown that nicorandil inhibits both the cell death of cardiac tissue and the excessive cellular proliferation of renal tissue. The excessive cell proliferation is observed in the kidney of subject affected by cardiac dysfunction. The administration of nicorandil shows improvement of this situation by reducing the cell proliferation (Segawa et al. 2001; Ishii et al. 2007; Jefferson et al. 2008; Sudo et al. 2009). Recurring ischemia-reperfusion events lead to changes in mitochondrial function, which triggers apoptosis in cardiac cells. Previous studies have shown that the activation of the KATP channel by nicorandil inhibits the depolarization of the mitochondrial membrane and the release of cytochrome c into the cytosol, thus inhibiting apoptosis (Akao et al. 2002; Carreira et al. 2008; Lu 2006; Nagata et al. 2003; Sato et al. 2000).
Lu (2006) showed that isolated rat hearts submitted to ischemia-reperfusion when treatments with nicorandil were protected against post-ischemic damage. Akao et al. (2002) observed the inhibition of cytochrome c release in rat cardiomyocytes that were treated with nicorandil and exposed to oxidative stress. Nishikawa et al. (2006) concluded that under hypoxia nicorandil inhibited apoptosis in myocytes via the cGMP signaling pathway and activation of the KATP channel, which inhibited the release of cytochrome c, and through influencing the activity and expression of proteins involved in apoptosis, such as caspase 3, Bax and Bcl-2. However, some studies like Taimor et al. (2000) shows that even though under hypoxia the apoptosis is inhibited, under normal oxygenation conditions substances that release NO induce apoptosis.
Besides protecting heart tissue against cell death, some studies have shown that nicorandil can inhibit the excessive proliferation of mesangial cells that occurs in some renal disorders which aggravate adverse cardiac conditions (Segawa et al. 2001; Sudo et al. 2009). The exact mechanism by which nicorandil functions must still be clarified, but nicorandil can inhibit cell proliferation by decreasing the expression of transforming growth factor β (TGF-β) and platelet-derived growth factor (PDGF), possibly via cGMP (Kastrati et al. 2010; Peters et al. 2003; Segawa et al. 2001; Sudo et al. 2009). Liou et al. (2011) observed that nicorandil inhibited rat cardiac fibroblast proliferation, with inhibition angiotensin II (Ang II) (cardiac fibroblasts proliferate in response to Ang II), and this effect may involve the activation of KATP channels.
Nicorandil’s capacity to interfere in the processes of cell proliferation and death is interesting for drug research in the therapy of some disorders involving such processes. Thus, the goal of this study was to investigate if the nicorandil can affect the proliferation and death of 786-O cells under normal oxygenation conditions. Also to determine if the lack of radical NO in its molecular precursor is able to produce similar effects.
Materials and methods
Chemicals
Nicorandil (N-(beta-hydroxyethyl) nicotinamide nitrate ester) and its precursor molecule (N-(beta-hydroxyethyl)nicotinamide) used in this study were synthesized from nicotinic acid in the chemistry laboratory of the Universidade Federal de Minas Gerais and provided by Dr. Ângelo de Fátima. The precursor (N-(beta-hydroxyethyl) nicotinamide) has a similar structure to nicorandil, but it lacks the nitrite radical (–NO2). The molecules were dissolved at 200 mg/mL in dimethyl sulfoxide (DMSO) (Mallinckrodt, St. Luois, MO, USA) and diluted to working concentrations in the culture medium Dulbecco’s modified Eagle’s medium (DMEM) (Gibco—Life Technologies, Carlsbad, CA, USA).
Doxorubicin (Adriblastina®, Pharmacia) was used as the positive control for the induction of damage in cytotoxicity (0.5 μg/mL), cell proliferation (0.1 μg/mL) and genotoxicity (0.1 μg/mL) assays. Camptothecin (Acros Organics—Fisher Scientific Latin America Headquarters, Suwanee, GA, USA) (20 μg/mL) assays was used as the positive control to induce apoptosis.
Cell culture
The cell line used in this study was the human renal carcinoma cell line (786-O), which was kindly provided by Prof. João Ernesto de Carvalho, CPQBA/UNICAMP. The cells were grown in DMEM (Gibco) supplemented with 10 % fetal bovine serum (FBS) (Gibco) at 37 °C and 5 % CO2.
Cytotoxicity assay
A cytotoxicity assay was performed with MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) (Invitrogen—Life Technologies) in accordance with the protocol described by Mosmann (1983), with some modifications. We seeded 5 × 103 cells in 500 μL of DMEM medium with 10 % FBS in each well of a 24-well culture plate. The plate was incubated for 24 h to stabilize the cells. After this incubation, the medium was replaced with fresh medium plus the treatment solution, and the plates were incubated for 24, 48 or 72 h. We tested both molecules at concentrations of 50, 100, 250, 500, 1,000, 1,500, 2,000 and 3,000 μg/mL. Doxorubicin was used at 0.5 μg/mL as a positive control for cytotoxicity, and the negative control was culture medium with 1.5 % DMSO. After the cells were treated for the indicated times, the medium was withdrawn, and serum-free medium containing 0.167 mg/mL MTT salt was added. After 4 h, the supernatant was removed, and the formazan crystal products were diluted in 500 μL DMSO. The absorbance at 550 nm was converted to a percentage to calculate cell survival using the following formula (Huang et al. 2005):
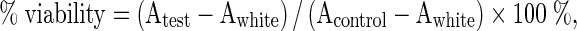 |
where A = absorbance average.
Determination of nitric oxide in the cell culture
NO was quantified through the detection of nitrate and nitrite, which are the products of its decomposition, using the methods of Griess (1879), with previously published modifications (Panis et al. 2010). Two 24-well plates were used for the experiment. The first plate, were seeded with 104 cells in 300 μL medium with FBS, per well; the second plate, received only medium with FBS. Both plates were incubated for 24 h, and 300 μL medium containing the treatment was added as follows: negative control (0.25 % DMSO); doxorubicin (0.1 μg/mL); 20, 100 or 500 μg/mL nicorandil; and 20, 100, or 500 μg/mL N-(beta-hydroxyethyl) nicotinamide. The treatments were added in duplicate, to wells of each plate (with/without cells).
After incubation for 1, 12, 24 or 48 h, 60 μL of the supernatant was collected, deproteinized by adding 75 mM ZnSO4 solution, homogenized, and centrifuged at 10,000 rpm for 2 min at 4 °C. Then 70 μL of a 55 mM NaOH solution was added, and the samples were vortexed and centrifuged at 10,000 rpm for 5 min at 4 °C. Subsequently, 250 μL of the supernatant was diluted in 50 μL of glycine buffer solution (45 g/L, pH 9.7).
Cadmium granules (stored in 100 mM H2SO4) were rinsed in distilled water and maintained for 5 min in a 5 mM CuSO4 solution in a glycine-NaOH buffer (15 g/L, pH 9.7). Subsequently, the copper-coated cadmium granules were added to the sample and suspended with gentle stirring for 10 min while the nitrate from the sample was converted to nitrite.
After 10 min, the samples were transferred to another tube, and the same volume of Griess reagent (reagent I: 50 mg of N-naphthyl ethylenediamine in 250 mL of distilled water; reagent II: 5 g of sulphanilamide in 500 mL of 5 % phosphoric acid) was added to determine the nitrite concentration, then after 10 min they were centrifuged at 10,000 rpm for 2 min. To determine the absorbance of the samples, 100 μL was transferred from each tube to a 96-well plate.
A calibration curve was prepared from a stock solution of 250 μM NaNO2 that was serially diluted to a final concentration of 7.8 μM. The absorbance was determined at 550 nm, and the final results are presented as the concentration of nitrite in μM.
Cellular proliferation
The cellular proliferation was measured following each treatment using the cell count values. The counting was performed after treatment for 24, 48, 72 and 96 h. The cells were seeded at a density of 2.6 × 104 cells per culture tube (10 cm2), contained 2.6 mL of culture medium with FBS plus the following treatments: negative control (0.25 % DMSO); positive control of inhibition of proliferation (0.1 μg/mL doxorubicin); 20, 100, or 500 μg/mL nicorandil; and 20, 100, or 500 μg/mL N-(beta-hydroxyethyl) nicotinamide (concentrations determined from MTT assay results). After the appropriate incubation time, a tube from each treatment condition was trypsinized, and the cells were counted in a Neubauer chamber. The same cell suspension was used to measure cell viability with trypan blue staining. The experiment was repeated three times.
Genotoxicity (comet assay)
A total of 5 mL of culture medium with FBS was added to the culture flasks (25 cm2) in which 5 × 105 cells were grown for 24 h. The following treatments were then added: negative control (0.25 % ×DMSO); positive control for DNA damage induction (0.1 μg/mL doxorubicin); nicorandil (20, 100 or 500 μg/mL); and N-(beta-hydroxyethyl) nicotinamide (20, 100 or 500 μg/mL).
The SCGE assay (single-cell gel electrophoresis, also known as the comet assay) allows the evaluation of primary DNA damage; therefore, we evaluated the cells after 3 h of treatment. The assay was performed under alkaline conditions as described by Singh et al. (1988) and according to Tice et al. (2000). The cells were trypsinized, and 20 μL of the cell suspension was homogenized with 120 μL of 0.5 % LMP agarose (low melting point; Gibco), distributed onto slides pre-treated with agarose (Invitrogen) and covered with a coverslip. After 20 min at 4 °C, the coverslips were removed, and the slides were placed in a lysis solution (1 mL Triton and 10 mL DMSO in 89 mL lysis buffer stock [14.61 g NaCl, 3.22 g EDTA, 0.12 g Tris, and 89 mL deionized H2O], pH 10, adjusted using NaOH) for 60 min at 4°C. After lysis, the slides were placed in an electrophoresis apparatus with an alkaline buffer solution (pH 13) (5 mL EDTA and 30 mL NaOH 10M in 1,000 mL deionized H2O). Denaturing was performed for 20 min before electrophoresis was started.
Electrophoresis was performed at pH 13 at 25 V and 300 mA for 20 min. After electrophoresis, the slides were neutralized with a pH 7.5 buffer (48.5 g 0.4 M Tris in 1,000 mL deionized H2O, pH 7.5, adjusted using HCl), fixed with ethyl alcohol and stained with ethidium bromide (20 μg/mL).
One hundred cells were examined for each treatment condition under a fluorescence microscope with a 40× objective. The cells were classified on the basis of damage into classes 0–3 based on the length and intensity of the comet tail: class 0, absence of a tail and no visible damage; class 1, tail of a size up to the diameter of the nucleoid and little damage visible; class 2, medium-sized tail (up to 2 times the diameter of the nucleoid) and moderate damage visible; and class 3, long tail (length greater than 2 times the diameter of the nucleoid) with major damage visible (Kobayashi 1995).
Cell viability was ascertained with trypan blue exclusion staining. A satisfactory viability for the comet assay was considered to be greater than 80 %. The experiment was repeated three times.
Induction of apoptosis
A total of 4.3 × 104 cells per well were grown on coverslips in 6-well plates in 3 mL of medium with FBS. After 24 h, the treatments were added to the wells as follows: nicorandil (20, 100 and 500 μg/mL); N-(beta-hydroxyethyl) nicotinamide (20, 100 and 500 μg/mL); 20 μg/mL camptothecin (to induce apoptosis); and 0.25 % DMSO (as a negative control).
After 24 h, the coverslips were collected as described by Rovozzo and Burke (1973) and in accordance with the modifications proposed by Tsuboy et al. (2010). The coverslips were washed in saline (PBS), fixed in the Carnoy fixative (methanol:glacial acetic acid ratio of 3:1), and hydrated in a gradual series of alcohol washes (95–25 %). The slides were then transferred to McIlvaine buffer (0.1 M citric acid [1.92 g citric acid in 100 mL 25 % methanol] and 0.2 M disodium phosphate [2.84 g dibasic sodium phosphate in 100 mL 25 % methanol]) for 5 min, transferred to 0.01 % acridine orange (diluted in McIlvaine buffer) for 5 min and then transferred again into McIlvaine buffer for 5 min. The coverslips were placed on a slide and sealed with enamel. The morphological characteristics of a cell that was considered to have undergone apoptosis were a nucleus with condensed chromatin and apoptotic bodies. The characteristic of a normal cell was an intact and uniform nucleus. We analyzed 500 cells for each treatment condition using a fluorescence microscope with a 40× objective.
Real-time RT-PCR (gene expression)
We assessed the gene expression of four protein: caspase 8, protein initiator of the extrinsic pathway of apoptosis (CASP 8); caspase 9, initiator of the intrinsic pathway of apoptosis (CASP 9); survivin, protein inhibitor of caspase (BIRC5); mitochondrial transmembrane protein, anti-apoptotic (BCL-XL).
Real-time RT-PCR was performed according to the MIQE guidelines (Bustin et al. 2009). First, 5 × 105 cells were grown in 25 cm2 flasks with 5 mL of DMEM containing 10 % FBS. After 24 h, the following treatments were added: 0.25 % DMSO (negative control), 500 μg/mL nicorandil or 500 μg/mL N-(beta-hydroxyethyl) nicotinamide.
After 12 h of treatment, the cells were trypsinized, and the total RNA was extracted using the TRIzol-LS reagent (Invitrogen) in accordance with the manufacturer’s instructions. After the extraction, the RNA was resuspended in 30 μL DEPC water, treated with 0.3 μL DNase I (Invitrogen) and kept on ice. The total RNA extracted from the samples was quantified using a spectrophotometer. Samples with an A260/A280 value between 1.9 and 2.1 were used for the experiment. The integrity of the RNA was confirmed using a 0.8 % agarose gel.
The cDNA was synthesized using 2 μL RNA (1 μg), 2 μL 2.5 μM dNTPs (Invitrogen), 1 μL 10 pM oligo-dT (Invitrogen) and 9.9 μL DEPC water (Invitrogen). This mixture (14.9 μL) was incubated at 65 °C for 5 min in a thermal cycler (TECHNE® TC 412; Bibby Scientific Limited, Stone, U.K.) and then quickly transferred to ice. Subsequently, 4 μL of Mlv 5× buffer (Invitrogen), 0.1 μL ribonuclease inhibitor (RNase Out, Invitrogen) and 1 μL reverse transcriptase (M-Mlv-RT, Invitrogen) were added. To complete the reaction, the samples (20 μL) were incubated at 37 °C in a thermal cycler for 50 min followed by incubation at 70 °C for 15 min. The resulting cDNA was stored in a −80 °C freezer.
The real-time PCR was performed using a PTC 200 DNA Engine Cycler (MJ Research, BioRad, Hercules, CA, USA) with SYBR Green dye (Platinum SYBR Green, Invitrogen). After the initial denaturation step (50 °C for 2 min and 95 °C for 3 min), 39 rounds of a 3-step cycle (denaturation at 95 °C for 20 s, annealing at 60 °C for 30 s and extension at 72 °C for 20 s) were performed. The reaction was terminated with an incubation step of 95 °C for 10 s and another at 40 °C for 1 min. At the end of the reaction, a melting curve was generated from 50 to 95 °C, with an increase of 0.5 °C every 5 s. The reference gene was GAPDH (glyceraldehyde 3-phosphate dehydrogenase). The experiment was repeated three times. The sequences of the oligonucleotide primers are shown in Table 1.
Table 1.
The gene and sequence of forward (F) and reverse (R) primers used in the real-time PCR reactions
| Gene | Primer | References or no of NCBI |
|---|---|---|
| GAPDH | F:5′ACAAGATTGTGAAGG TCG GTG TCA 3′ R:5′AGCTTCCCATTCTCAGCCTTGACT 3′ |
Sugaya et al. (2005) (with modification) |
| CASP8 | F:5′ GCAAAAGCACGGGAGAAAGT 3′ R:5′ TGCATCCAAGTGTGTTCCATT3′ |
Castaneda and Rosin-Steiner (2006) |
| CASP9 | F:5′ GCTCTTCCTTTGTTCATCTCC 3′ R:5′ GTTTTCTAGGGTTGGCTTCG 3′ |
Chen et al. (2009) |
| BIRC5 | F:5′ AGCCCTTTCTCAAGGACCAC 3′ R:5′ TGGCTCGTTCTCAGTGGGGCAGT 3′ |
Zhang et at. (2001) (with modification) |
| BCL-XL | F:5′ TGGGCTCACTCTTCAGTCGGAAAT 3′ R:5′ ATGTAG TGGTTCTCCTGGTGGCAA 3′ |
NCBI ID: NM_138578.1 (designera) |
aSequence of forward and reverse primers was designed according to IDT software http://www.idtdna.com/Scitools/Applications/Primerquest
Statistics
The data obtained in the study were analyzed using the GraphPad InStat statistical program. A value of p < 0.05 was accepted as significant for the cytotoxicity, cell proliferation, apoptosis induction and genotoxicity tests using an ANOVA followed by the Dunnett test to compare the treatments with the control. The NO concentration was analyzed using an ANOVA followed by Tukey’s test. The gene expression levels were determined as in Pfaffl (2001), and the statistical analysis was performed using REST-384 software (Pfaffl et al. 2002).
Results
Cytotoxicity assay
The MTT assay results showed that nicorandil started to be cytotoxic at 2,000 μg/mL (57 % after 24 h). The 1,000 μg/mL concentration induced a slight decrease in viability (81 % after 24 h), but the decrease was not statistically significant. N-(beta-hydroxyethyl) nicotinamide was only cytotoxic at the highest concentration tested, 3,000 μg/mL, at all three treatment times (24, 48 and 72 h), the average reduction in cell viability at these concentrations was 60 %. The cells treated with lower concentrations showed cell viability similar to that of the control (Fig. 1).
Fig. 1.

Cytotoxicity. The average cell viability (%) obtained using the MTT assay. The 786-O cells were treated with a nicorandil (2-NN) or b N-(beta-hydroxyethyl) nicotinamide (L1) for 24, 48 and 72 h. The positive control was doxorubicin (Doxo). Asterisk The difference was statistically significant (p < 0.05) compared to the control (DMSO) of 100 % viability
Determination of nitric oxide in the cell culture
The evaluation of NO concentrations showed that the treatment with nicorandil resulted in an increase in the NO concentration in the culture medium compared with the control. The treatment with 500 μg/mL nicorandil induced a statistically significant difference in NO concentration for all of the treatment times (1, 12, 24 and 48 h), with a concentration up to eight times higher than that of the control (325.8 vs. 42.4 μM, respectively; p = 0.0053). The treatment with N-(beta-hydroxyethyl) nicotinamide did not result in a significantly increased or decreased concentration of NO, the values were similar to that of control. Figure 2 shows the average NO concentration generated for each treatment condition.
Fig. 2.

Nitric oxide concentrations in the medium. The average concentration of nitric oxide (NO) in the culture medium of 786-O cells that were treated with nicorandil (2-NN) or N-(beta-hydroxyethyl) nicotinamide (L1). The concentration was measured at one of four exposure times (1, 12, 24 and 48 h). The negative control was 0.25 % DMSO. Asterisk The difference was statistically significant compared to the control (p < 0.05)
Cell proliferation
Compared with the control, there was no statistically significant difference in the proliferation of 786-O cells treated with nicorandil or N-(beta-hydroxyethyl) nicotinamide at any of the concentrations tested. Trypan blue staining revealed no decrease in cell viability in response to any of the treatments (cell viabilities over 90 %). The proliferation curve is shown in Fig. 3.
Fig. 3.

Cellular proliferation. The kinetic curve (exponential curve) of the proliferation of 786-O cells that were treated with a nicorandil (2-NN) or b N-(beta-hydroxyethyl) nicotinamide (L1) for 24, 48, 72 or 96 h. (Control: 0.25 % DMSO). Asterisk The difference was statistically significant (p < 0.05) compared to the control
Genotoxicity, induction of apoptosis, gene expression
None of the tested concentrations of nicorandil or N-(beta-hydroxyethyl) nicotinamide significantly increased the migration of DNA fragments compared with the control cells. The cell viability was greater than 93 % based on the trypan blue staining results.
The condensation of chromatin and the presence of apoptotic bodies were only observed following treatment with camptothecin. The treatment with nicorandil or N-(beta-hydroxyethyl) nicotinamide did not result in a difference in the number of apoptotic cells compared to the control after 24 h of treatment.
The expression levels of the four genes evaluated (CASP8, CASP9, BRIC5 e BCL XL) in cells treated with nicorandil or N-(beta-hydroxyethyl) nicotinamide were unchanged compared to the control.
Discussion
At low concentrations NO functions as a signaling molecule in normal physiology. An increased synthesis of NO is associated with physiopathological conditions, such as degenerative neural diseases (e.g., Parkinson’s and Alzheimer’s) or infection and inflammation (Dimmeler and Zeiher 1997). The beneficial effect of nicorandil on angina symptoms is due to the release of NO from the structure of the drug. According to the literature, nicorandil can affect cellular proliferation and apoptosis in tissues exposed to hypoxia, an effect that can be attributed to the ability of nicorandil to activate mitochondrial KATP channels and cGMP-dependent mechanisms (Sato et al. 2000; Serizawa et al. 2011). In this study we analyzed nicorandil’s capacity to interfere in the processes of cell proliferation and death in cultures under normal oxygenation conditions, in addition, we observed the effect of the structure independent of NO radical.
The structural difference between the nicorandil and N-(beta-hydroxyethyl) nicotinamide is the presence of the radical NO2. Therefore only nicorandil released NO into the culture and the effects observed by the treatment with N-(beta-hydroxyethyl) nicotinamide are independent of NO. In agreement with this, our experiments showed that only the cultures that received nicorandil had NO concentrations higher than the control. We observed that after treatment with 500 μg/mL of nicorandil the concentration of NO in the culture medium was eight times higher than the NO concentration in the control, whereas in the culture treated with 500 μg/mL of N-(beta-hydroxyethyl) nicotinamide neither significantly increased or decreased concentration of NO compared to control was observed. This showed that the N-(beta-hydroxyethyl) nicotinamide besides not releasing NO also does not change the synthesis of NO.
In order to verify the cytotoxicity of the two molecules, we evaluated various concentrations by MTT assay. The 786-O renal cell exhibited reduced cell viability only when exposed to 2,000 μg/mL nicorandil, which reduced the cell viability to 57 %, and the cytotoxicity of N-(beta-hydroxyethyl) nicotinamide was observed only at the tested concentration extreme (3,000 μg/mL, with a reduction in cell viability to 58 %). The high concentration of the N-(beta-hydroxyethyl) nicotinamide required to achieve toxicity, compared to the nicorandil, is possibly due the absence of the NO radical, therefore, the NO radical in the nicorandil structure makes it relatively more cytotoxic for 786-O cells than its precursor. The chosen concentrations of nicorandil (20, 100 and 500 μg/mL) for other trials, were not cytotoxic for cells, although higher concentrations of NO than in the control had been shown in culture.
The sensitivity to NO varies from cell to cell and according to microenvironmental conditions. The unpaired electron of the NO2 radical reacts readily with oxygen, to become a superoxide radical, or reacts with transition metals, such as iron, cobalt, manganese or copper, the relative importance of enzymes with transition metal-containing groups, such as iron-sulfur (heme), in different cells can influence the sensitivity of each cell type to the NO concentration (Moncada et al. 1991; Wink et al. 1998).
Hwang et al. (2009) exposed two different lineages of neural cells (astrocytes and microglia) to NO (35 μM), although the astrocytes did not show any decrease in cell viability, the microglial cells had a 90 % reduction in viability. In our study, the amount of NO observed after treatment with 20 and 100 μg/ nicorandil was similar to that used by Hwang et al. (2009), just as astrocytes, the kidney cells exposed to these conditions did not show altered cell viability, the 786-O renal cell only exhibited reduced cell viability when exposed to 2,000 μg/mL nicorandil.
There are no reports regarding the genotoxicity of nicorandil. However, the reported genotoxicity of NO is dependent on its concentration and the microenvironmental conditions. The activity of NO as a genotoxic agent is primarily attributed to indirect reactions, in which NO interacts with other molecules (e.g., a superoxide anion radical) to generate radicals and other molecules (RNOS, such as peroxide, nitrite, and carcinogenic nitrosamines) that are capable of modifying proteins, acting directly on DNA nucleotides and inhibiting the mechanisms necessary for the repair of DNA lesions (Masri 2010; Nguyen et al. 1992). In this work, the level of DNA damage observed in the cells following exposure to nicorandil or N-(beta-hydroxyethyl) nicotinamide using a comet assay was not statistically significant. Neither nicorandil nor N-(beta-hydroxyethyl) nicotinamide led to DNA damage in 786-O cells, regardless of the presence of NO.
Sudo et al. (2009) showed that in rats with glomerulonephritis the excessive proliferation of the cells of the glomerulus was decreased upon treatment with nicorandil (30 mg/kg), via the inhibition of TGF-β and PDGF expression. Segawa et al. (2001) observed the same effect in the cultured mesangial cells of rats that were exposed to nicorandil (at concentrations of 1 μM to 1 mM), and in rat cardiac fibroblasts, Liou et al. (2011) observed that nicorandil decreased cell proliferation via a mechanism that involved the activation of KATP channels. Even though these studies showed that nicorandil inhibits the proliferation of cells involved in renal injuries, in our study both the nicorandil as N-(beta-hydroxyethyl) nicotinamide, showed no statistically significant reduction in the proliferation of 786-O cells, while at the same time it is possible to observe a trend of decreased proliferation in stages after the treatment (Fig. 3). However, the different models used in these studies (in vivo model, primary culture and permanent cell culture) may explain the divergent results, because different cell types can respond differently to the same stimulus (Moncada et al. 1991).
The inhibitory effect of nicorandil on apoptosis of cardiac tissue under conditions of hypoxia and oxidative stress, alleviates the damage caused in the tissue, and has been attributed to the release of NO, the activation of mitochondrial KATP channels, the interruption of the apoptotic signaling cascade by cGMP-dependent mechanisms and the inhibition of caspase-3 activity (Ahmed et al. 2011; Carreira et al. 2008; Nagata et al. 2003). NO may induce cell death through apoptosis and necrosis, depending on the NO concentration and the interaction of NO with the specific cell type. Taimor et al. (2000) showed that in cardiomyocytes under normal oxygenation conditions, the NO released by S-nitroso-N-acetylpenicillamine (SNAP) induced apoptosis, however, under post-ischemic conditions, apoptosis was inhibited, perhaps because the oxygen radicals formed during reoxygenation scavenged the NO.
Under the normal oxygenation conditions of our experiments, nicorandil did not induce apoptosis in 786-O cells at any of the concentrations tested. All of the treatments with nicorandil and N-(beta-hydroxyethyl) nicotinamide resulted in similar levels of apoptotic cells compared to the control. In some studies the nicorandil appeared to be involved in pre-conditioning the cell that leads to overcome the stress and the stimulation of apoptosis, via the activation of KATP (Nagata et al. 2003).
In our study, we did not observe any change in the gene expression of the pro-apoptotic genes CASP8 or CASP9, which was in agreement with the morphological analysis of apoptosis induction. These reports, combined with the literature, suggest that different conditions (the amount of NO, oxygenation and cell type) can influence the different effects of nicorandil on apoptosis.
In conclusion, nicorandil and N-(beta-hydroxyethyl) nicotinamide had different cytotoxic effects both at very high concentrations. The difference was likely caused by the presence of NO in nicorandil. The evaluation of other parameters at the non-cytotoxic concentrations tested demonstrated that the two molecules functioned similarly. These results indicated that the chemical structure, regardless of the presence of NO, did not affect the induction of apoptosis or cellular proliferation nor have genotoxic effects in 786-O cells. Apoptosis was not induced following treatment, and contrary to the expected results, we observed no inhibition of cellular proliferation in 786-O cells. Although it already had been demonstrated that the treatment with nicorandil can influence the cell proliferation and death in cardiac and renal tissue under specific conditions (hypoxia) (Sudo et al. 2009), the present results suggest that the nicorandil and its precursor molecule are not able to influence cell proliferation and death of cell 786-O under normal oxygenation. Its effects may be related to ischemia-reperfusion condition. However, the molecules can better be studied for their inhibitory effect on cell proliferation, since in our results there was a slight tendency to reduced cell proliferation, whereas in the case of the renal injuries, as well as in other studies of the control of cell growth disorder, the effect is very interesting considering the possible effect independent of NO.
Acknowledgments
We would like to thank the Coordination of Improvement of Higher Education Personnel (CAPES), National Council for Scientific and Technological Development (CNPq) and Araucaria Foundation for their financial support.
Contributor Information
Natália Aparecida de Paula, Phone: +55-43-33714000, Phone: +55-43-33714977, FAX: 55-43-33284440, Email: npbiomed@yahoo.com.br.
Andressa Megumi Niwa, Email: andressamn@yahoo.com.br.
Diogo Campos Vesenick, Email: dcvesenick@hotmail.com.
Carolina Panis, Email: carolpanis@sercomtel.com.br.
Rubens Cecchini, Email: cecchini@uel.br.
Ângelo de Fátima, Email: adefatima@qui.ufmg.br.
Lúcia Regina Ribeiro, Email: lribeiro@fmb.unesp.br.
Mário Sérgio Mantovani, Email: biomsm@uel.br.
References
- Ahmed LA, Salem HA, Attia AS, Agha AM. Pharmacological preconditioning with nicorandil and pioglitazone attenuates myocardial ischemia/reperfusion injury in rats. Eur J Pharmacol. 2011;663:51–58. doi: 10.1016/j.ejphar.2011.04.038. [DOI] [PubMed] [Google Scholar]
- Akao M, Teshima Y, Marbán E. Antiapoptotic effect of nicorandil mediated by mitochondrial ATP-sensitive potassium channels in cultured cardiac myocytes. J Am Coll Cardiol. 2002;40:803–810. doi: 10.1016/S0735-1097(02)02007-7. [DOI] [PubMed] [Google Scholar]
- Barreto RL, Correia CRD. Óxido nítrico: propriedades e potenciais usos terapêuticos. Quim Nova. 2005;28:1046–1054. doi: 10.1590/S0100-40422005000600020. [DOI] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Carreira RS, Monteiro P, Kowaltowski AJ, Gonçalves LM, Providência LA. Nicorandil protects cardiac mitochondria against permeability transition induced by ischemia-reperfusion. J Bioeng Biomembr. 2008;40:95–102. doi: 10.1007/s10863-008-9133-2. [DOI] [PubMed] [Google Scholar]
- Castaneda F, Rosin-Steiner S. Low concentration of ethanol induce apoptosis in HepG2 cells: role of various signal transduction pathways. Int J Med Sci. 2006;3:160–167. doi: 10.7150/ijms.3.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XY, Liu J, Xu KS. Apoptosis of human hepatocellular carcinoma cell (HepG2) induced by cardiotoxin III through S-phase arrest. Exp Toxicol Pathol. 2009;61:307–315. doi: 10.1016/j.etp.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Chong S, Fung HL. Biochemical and pharmacological interactions between nitroglycerin and thiols. Effects of thiol structure on nitric oxide generation and tolerance reversal. Biochem Pharmacol. 1991;42:1433–1439. doi: 10.1016/0006-2952(91)90456-F. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Zeiher AM. Nitric oxide and apoptosis: another paradigm for the double-edged role of nitric oxide. Nitric Oxide. 1997;1:5–281. doi: 10.1006/niox.1997.0133. [DOI] [PubMed] [Google Scholar]
- Eeckhout E. Nicorandil: a drug for many purposes: too good to be true? Eur Heart J. 2003;24:1282–1284. doi: 10.1016/S0195-668X(03)00318-X. [DOI] [PubMed] [Google Scholar]
- Frydman A. Pharmacokinetic profile of nicorandil in humans: an overview. J Cardiovasc Pharmacol. 1992;20:34–44. doi: 10.1097/00005344-199206203-00008. [DOI] [PubMed] [Google Scholar]
- Griess JP. Bemerkungen zu der abhandlung der H.H. Weselsky und Benedikt “ueber einige azoverbindugen”. Chem Ber. 1879;12:426–428. doi: 10.1002/cber.187901201117. [DOI] [Google Scholar]
- Hiremath JG, Valluru R, Jaiprakash N, Katta SA, Matad PP. Pharmaceutical aspects of Nicorandil. Int J Pharm Pharm Sci. 2010;2:24–29. [Google Scholar]
- Huang Y-H, Shang B-Y, Zhen Y-S. Antitumor efficacy of lidamycin on hepatoma and active moiety of its molecule. World J Gastroenterol. 2005;11:3980–3984. doi: 10.3748/wjg.v11.i26.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S-Y, Yoo B-C, Jung J-W, Oh E-S, Hwang J-S, Shin J-A, Kim S-Y, Cha S-H, Han I-O. Induction of glioma apoptosis by microglia-secreted molecules: the role of nitric oxide and cathepsin B. Biochim Biophys Acta. 2009;1793:1656–1668. doi: 10.1016/j.bbamcr.2009.08.011. [DOI] [PubMed] [Google Scholar]
- Ishii H, Toriyama T, Aoyama T, Takahashi H, Yamada S, Kasuga H, Ichimiya S, Kanashiro M, Mitsuhashi H, Maruyama S, Matsuo S, Naruse K, Matsubara T, Murohara T. Efficacy of oral nicorandil in patients with end-stage renal disease: a retrospective chart review after coronary angioplasty in Japanese patients receiving hemodialysis. Clin Ther. 2007;29–1:110–122. doi: 10.1016/j.clinthera.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74–1:22–36. doi: 10.1038/ki.2008.128. [DOI] [PubMed] [Google Scholar]
- Kastrati I, Edirisinghe PD, Wijewickrama GT, Thatcher GR. Estrogen-induced apoptosis of breast epithelial cells is blocked by NO/cGMP and mediated by extranuclear estrogen receptors. Endocrinology. 2010;151:5206–5216. doi: 10.1210/en.2010-0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H. A comparison between manual microscopic analysis and computerized image analysis in the single cell gel electrophoresis assay. MMS Commun. 1995;3:103–115. [Google Scholar]
- Krumenacker JS, Murad F. NO-cGMP signaling in development and stem cells. Mol Genet Metab. 2006;87:311–314. doi: 10.1016/j.ymgme.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Liou JY, Hong HJ, Sung LC, Chao HH, Chen PY, Cheng TH, Chan P, Liu JC. Nicorandil inhibits angiotensin-II-induced proliferation of cultured rat cardiac fibroblasts. Pharmacology. 2011;87:144–151. doi: 10.1159/000323555. [DOI] [PubMed] [Google Scholar]
- Lu C. Nicorandil improves post-ischemic myocardial dysfunction in association with opening the mitochondrial KATP channels and decreasing hydroxyl radicals in isolated rat hearts. Circ J. 2006;70:1650–1654. doi: 10.1253/circj.70.1650. [DOI] [PubMed] [Google Scholar]
- Masri F. Role of nitric oxide and its metabolites as potential markers in lung cancer. Ann Thorac Med. 2010;5:123–127. doi: 10.4103/1817-1737.65036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and citotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Mujoo K, Sharin VG, Martin E, Choi BK, Sloan C, Nikonoff LE, Kots AY, Murad F. Role of soluble guanylyl cyclase–cyclic GMP signaling in tumor cell proliferation. Nitric Oxide. 2010;22:43–50. doi: 10.1016/j.niox.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Obata K, Odashima M, Yamada A, Somura F, Nishizawa T, Ichihara S, Izawa H, Iwase M, Hayakawa A, Murohara T, Yokota M. Nicorandil inhibits oxidative stress-induced apoptosis in cardiac myocytes through activation of mitochondrial ATP-sensitive potassium channels and a nitrate-like effect. J Mol Cell Cardiol. 2003;35:1505–1512. doi: 10.1016/j.yjmcc.2003.09.018. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Brunson D, Crespi CL, Penman BW, Wishnok JS, Tannenbaum SR. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc Natl Acad Sci USA. 1992;89:3030–3034. doi: 10.1073/pnas.89.7.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa S, Tatsumi T, Shiraishi J, Matsunaga S, Takeda M, Mano A, Kobara M, Keira N, Okigaki M, Takahashi T, Matsubara H. Nicorandil regulates Bcl-2 family proteins and protects cardiac myocytes against hypoxia-induced apoptosis. J Mol Cell Cardiol. 2006;40:510–519. doi: 10.1016/j.yjmcc.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Panis C, Mazzuco TL, Costa CZF, Victorino VJ, Tatakihara VLH, Yamauchi LM, Yamada-Ogatta SF, Cecchini R, Rizzo LV, Pinge-Filho P. Trypanosoma cruzi: effect of the absence of 5-lipoxygenase (5-LO)-derived leukotrienes on levels of cytokines, nitric oxide and iNOS expression in cardiac tissue in the acute phase of infection in mice. Exp Parasitol. 2010;127:58–65. doi: 10.1016/j.exppara.2010.06.030. [DOI] [PubMed] [Google Scholar]
- Peters H, Daig U, Martini S, Rückert M, Schäper F, Liefeldt L, Krämer S, Neumayer HH. NO mediates antifibrotic actions of l-arginine supplementation following induction of anti-thy1 glomerulonephritis. Kidney Int. 2003;64:509–518. doi: 10.1046/j.1523-1755.2003.00112.x. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2002–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST©) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:1–10. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovozzo GC, Burke CN. A manual of basic virological techniques. Englewood Cliffs: Prentice-Hall; 1973. [Google Scholar]
- Sato T, Sasaki N, O’Rourke B, Marbán E. Nicorandil, a potent cardioprotective agent, acts by opening mitochondrial ATP—dependent potassium. Channels J Am Coll Cardiol. 2000;35:514–518. doi: 10.1016/S0735-1097(99)00552-5. [DOI] [PubMed] [Google Scholar]
- Segawa K, Minami K, Shiga Y, Shiraishi M, Sata T, Nakashima Y, Shigematsu A. Inhibitory effects of nicorandil on rat mesangial cell proliferation via the protein kinase G pathway. Nephron. 2001;87:263–268. doi: 10.1159/000045924. [DOI] [PubMed] [Google Scholar]
- Serizawa K, Yogo K, Aizawa K, Tashiro Y, Ishizuka N. Nicorandil prevents endothelial dysfunction due to antioxidative effects via normalisation of NADPH oxidase and nitric oxide synthase in streptozotocin diabetic rats. Cardiovasc Diabetol. 2011;10:105. doi: 10.1186/1475-2840-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth P, Fung HL. Biochemical characterization of a membrane-bound enzyme responsible for generating nitric oxide from nitroglycerin in vascular smooth muscle cells. Biochem Pharmacol. 1993;46:1481–1486. doi: 10.1016/0006-2952(93)90115-D. [DOI] [PubMed] [Google Scholar]
- Simpson D, Wellington K. Nicorandil: a review of its use in the management of stable angina pectoris, including high-risk patients. Drugs. 2004;64:1941–1955. doi: 10.2165/00003495-200464170-00012. [DOI] [PubMed] [Google Scholar]
- Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- Sudo H, Hirata M, Kanada H, Yorozu K, Tashiro Y, Serizawa K-I, Yogo K, Kataoka M, Moriguchi Y, Ishizuka N. Nicorandil improves glomerular injury in rats with mesangioproliferative glomerulonephritis via inhibition of proproliferative and profibrotic growth factors. J Pharmacol Sci. 2009;111:53–59. doi: 10.1254/jphs.09072FP. [DOI] [PubMed] [Google Scholar]
- Sugaya S, Nakanishi H, Tanzawa H. Down-regulation of SMT3A gene expression in association with DNA synthesis induction after X-ray irradiation in nevoid basal cell carcinoma syndrome (NBCCS) cells. Mutat Res. 2005;578:327–332. doi: 10.1016/j.mrfmmm.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Taimor G, Hofstaetter B, Piper HM. Apoptosis induction by nitric oxide in adult cardiomyocytes via cGMP-signaling and its impairment after simulated ischemia. Cardiovasc Res. 2000;45:588–594. doi: 10.1016/S0008-6363(99)00272-2. [DOI] [PubMed] [Google Scholar]
- Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu J-C, Sasaki YF. Single cell gel/comet assay: guideline for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(SICI)1098-2280(2000)35:3<206::AID-EM8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Tsuboy MS, Marcarini JC, Luiz RC, Barros IB, Ferreira DT, Ribeiro LR, Mantovani MS. In vitro evaluation of the genotoxic activity and apoptosis induction of the extracts of roots and leaves from the medicinal plant Coccoloba mollis (Polygonaceae) J Med Food. 2010;13:503–508. doi: 10.1089/jmf.2009.0119. [DOI] [PubMed] [Google Scholar]
- Wink DA, Vodovotz Y, Laval J, Laval F, Dewhirst MW, Mitchell JB. The multifaceted roles of nitric oxide in cancer. Carcinogenesis. 1998;19:711–721. doi: 10.1093/carcin/19.5.711. [DOI] [PubMed] [Google Scholar]
- Yim CY, et al. Macrophage nitric oxide synthesis delays progression of ultraviolet light induced murine Skin Cancers. Cancer Res. 1993;53:5507–5511. [PubMed] [Google Scholar]
- Zhang T, Otevrel T, Gao Z, Gao Z, Ehrlich SM, Fields JZ, Boman BM. Evidence that APC regulates survivin expression: a possible mechanism contributing to the stem cell origin of colon cancer. Cancer Res. 2001;61:8664–8667. [PubMed] [Google Scholar]