

Abstract
Nucleic acid quantification is a relevant issue for the characterization of mammalian recombinant cell lines and also for the registration of producer clones. Quantitative real-time PCR is a powerful tool to investigate nucleic acid levels but numerous different quantification strategies exist, which sometimes lead to misinterpretation of obtained qPCR data. In contrast to absolute quantification using amplicon- or plasmid standard curves, relative quantification strategies relate the gene of interest to an endogenous reference gene. The relative quantification methods also consider the amplification efficiency for the calculation of the gene copy number and thus more accurate results compared to absolute quantification methods are generated. In this study two recombinant Chinese hamster ovary cell lines were analysed for their transgene copy number using different relative quantification strategies. The individual calculation methods resulted in differences of relative gene copy numbers because efficiency calculations have strong impact on gene copy numbers. However, in context of comparing transgene copy numbers of two individual clones the influence of the calculation method is marginal. Therefore especially for the comparison of two cell lines with the identical transgene any of the relative qPCR methods was proven as powerful tool.
Keywords: CHO, Cellline development, Gene copy number, qPCR, LinReg
Introduction
Recombinant mammalian cell lines play an important role in the production of biopharmaceuticals (Walsh 2005). Current transfection strategies in cell line development are based on the random integration of the gene of interest into the host genome with a varying number of gene-copies depending on gene amplification strategies (Kim et al. 2012). The cell clones obtained from these methods are quite heterogeneous and need to be screened thoroughly. Thus, cell line characterization is becoming more and more important and besides specific productivity and growth rates, accurate quantification of nucleic acids needs to be established. The most powerful research tool to quantify nucleic acids is quantitative real-time PCR (qPCR). This method is widely used in biological and medical research, although qPCR data interpretation and quantification remains a controversial topic. Bustin and colleagues proposed a standardization of qPCR analyses by introducing the MIQE Guidelines (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) (Bustin et al. 2009). However, there is still a large variety of quantification methods used in different laboratories rendering interpretation and comparison of published qPCR data difficult. The normalization strategy is an essential part of qPCR derived data processing and relative quantification methods, as mentioned in the MIQE guidelines, appear to be the most progressive. This type of quantification is done using one or more internal reference genes, depending on the research question. Normalization of samples against a standard curve generated from a decimal dilution series of quantified target DNA is also still in use, although this method is error prone (Ginzinger 2002) and sometimes leads to difficulties when comparing obtained results with others found in literature.
In this work we demonstrate how to obtain reproducible qPCR raw data from genomic DNA isolates and compare different relative quantification calculation strategies with and without qPCR efficiency correction using β-actin as reference gene. We analyzed individual recombinant Chinese hamster ovary (CHO) cell lines that were all generated from the same host cell line, producing single-chain fragment crystallisable (scFc) antibody derivates. We point out critical steps of relative qPCR quantification and corresponding qPCR efficiency calculation, showing examples for each efficiency calculation method used. Furthermore, we show that the calculation of qPCR efficiencies has only a marginal impact on the calculated transgene difference between two recombinant cell lines carrying the same transgene and are analyzed under the same experimental conditions.
Materials and methods
Cell culture and genomic DNA isolation
Recombinant CHO clones producing a single chain antibody fragment variable fragment crystallisable fusion proteins (scFc) were described elsewhere (Mader et al. 2013). Cells were cultivated in 125 ml spinner flasks (Techne, Stone, UK) in a volume of 50 mL chemically defined ProCHO5 (Lonza, Visp, Switzerland) culture medium supplemented with 4 mM l-glutamine (PAA, Pasching, Austria), 0.5 mg/ml G418 (PAA) and phenol-red (Sigma-Aldrich, St. Louis, MO, USA). The cultures were stirred (50 rpm) at 37 °C and split twice a week to a seeding density of 2.5 × 105 cells per mL to assure permanent exponential growth. 2 × 106 CHO cells per clone were harvested from the suspension cultures via centrifugation (130 g, 10 min). Cell pellets were snap-frozen in liquid nitrogen and stored at −80 °C. Genomic DNA (gDNA) was isolated from cell pellets using the Blood Mini kit (QIAGEN, Venlo, Netherlands) according to the manufacturer’s protocol (protocol for cultured cells with RNAse treatment). Concentration and purity of gDNA preparations were analyzed photometrically using a Nanophotometer P-300 (Implen, Munich, Germany). The gDNA isolates were diluted to a concentration of 1 ng/μL and heated to 99 °C for 10 min for proper denaturation. Denatured samples were stored at +4 °C.
Primers and hydrolysis probes
Primers and FAM/TAM conjugated hydrolysis probes (Table 1) were designed using the Primer 3 web application version 4.0 (Rozen and Skaletsky 2000) Oligonucleotide properties were evaluated with the web application OligoCalc version 3.26 (Kibbe 2007). All primers and probes were synthesized by Sigma-Aldrich.
Table 1.
List of primers used for qPCR analysis
| Gene | Primer sense Primer antisense |
Hydrolysis probe | Amplicon size (bp) |
|---|---|---|---|
| scFc | ACGAGGACCCTGAAGTGAAG CGGTAGGTGGAGTTGTACTGTTC | [6FAM]AAGTGCACAACGCCAAGACCAAGC[TAM] | 100 |
| β-actin | TGAGCGCAAGTACTCTGTG TTGCTGATCCACATCTCCTG | [6FAM]CCATCCTGGCCTCACTGTCCACCT[TAM] | 78 |
Quantitative real-time PCR
To quantify the transgene copies qPCR was performed on a MiniOpticon™ real-time PCR system with 48-well low white PCR plates and microseal B film sealer (all from BioRad, Hercules, CA, USA) using the designed primers and hydrolysis probes (Table 1). Each qPCR run was performed in duplicates and each sample in triplicate. The untransfected CHO host cell line as negative control (NC) and no template controls (NTC) were included in each qPCR run. Each reaction mix contained 10 μL 2× iQ Supermix (BioRad), 6 pmol of each primer, 4 pmol hydrolysis probe and 3 ng pre-denatured gDNA adjusted with double distilled water to a final volume of 20 μL. The qPCR run was performed as a 2-step protocol with an initial denaturation step at 95 °C for 5 min followed by 40 cycles of annealing and extension at 55 °C for 60 s and denaturation at 95 °C for 15 s. Raw data were processed by the CFX manager 2.1 software application (BioRad) using the settings baseline subtraction and linear regression, to obtain the Cq (Cycle Quantification) (Bustin et al. 2009) values.
Relative quantification methods
qPCR raw data were analysed using different relative quantification approaches with and without efficiency correction. The internal reference gene, β-actin, was measured in addition to the transgenes for each sample. Ratios between the gene of interest and the reference gene were evaluated and compared.
The choice and number of internal reference genes is an essential topic and depends on the research question. Especially if relative quantification methods are used to compare different tissues, cell types, differentially prepared samples or even more when levels of transcripts are quantified, the choice and number of internal references is of extreme importance to obtain valuable data (Bahr et al. 2009). For relative quantification of transgenes theoretically any host DNA sequence could be used as a reference, preferable a unique one that is very stable within the genome. Since β-actin is one of the most used reference gene for qPCR measurements (Heid et al. 1996) it was also decided to use β-actin in this study. Primers and probe were designed using the β-actin CDS of the Chinese Hamster (U20114.1).
2−∆∆Cq Method
The basic calculation method of relative quantification assumes a doubling of the number of amplicons during each qPCR cycle. The efficiency of the amplification is thereby assumed to be 100 % and the calculations are only based on the differences of the respective Cq values between the gene of interest and the reference gene (∆Cq values) (Livak and Schmittgen 2001). To calculate the relative transgene copies within one sample the formula shown in Eq. 1 is used. The equation to calculate the relative difference between two samples (also called ratio) is displayed in Eq. 2.
Relative quantification with efficiency correction
The relative quantification method including efficiency (E) correction was published by Pfaffl and it is similar to the 2−∆∆Cq method but additionally considers the qPCR efficiencies (Pfaffl 2001). To calculate the qPCR efficiencies we used the two following methods.
1:10 Dilution method
In theory, the 1:10 dilution of a template sample results in a ∆Cq value of 3.32 (log2 of 10) if 100 % efficiency is assumed. By plotting the Cq values of a 1:10 dilution series against the logarithmically scaled concentrations of the template, the slope of the generated curve equals the mean −∆Cq value of the dilution series. This value is used to calculate the efficiency using Eq. 3. The slope of the linear regression is therefore directly correlated to the qPCR efficiency; the steeper the curve the lower the efficiency of the PCR (Bustin et al. 2009). According to the MIQE guidelines, the means of calibration curves generated from decimal diluted gDNA was used to calculate the mean efficiencies per amplicon.
Mathematical approach: LinRegPCR
A different way to calculate qPCR efficiencies is based on mathematical operations that calculate the efficiency for each single fluorescence curve. In this work the mean qPCR efficiencies were calculated from all single fluorescence curves in one amplicon group using the open access software application LinRegPCR V12.17 (Ruijter et al. 2009; Tuomi et al. 2010)). The underlying mathematical algorithm calculates real-time PCR efficiencies via linear regression in the exponential part of the fluorescence curve. To calculate the ratio between reference and target gene including the efficiency correction a modified formula derived from the equations published by Pfaffl (2001) was used (Eq. 4).
Equations
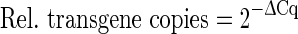 |
1 |
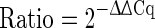 |
2 |
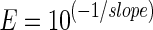 |
3 |
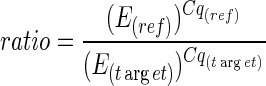 |
4 |
Results
Signal optimization via pre-denaturation
Initial qPCR experiments were done without a pre-denaturation step after gDNA isolation and resulted in relative fluorescence curves with a larger distribution of Cq-values. Therefore, the protocol was modified by adding a 10 min denaturation step at 99 °C before start of qPCR cycles. The improvement of this additional denaturation step is depicted in Fig. 1. gDNA samples from two different recombinant CHO clones (blue and green) were run in nine replicates without or with a pre-denaturation step. With this modified protocol the resolution of the differences in mean cq values (∆Cq) was improved from 1.3 to 2.3 cycles and the standard deviation was decreased from initially 0.8 to 0.3 cycles. The signal improvement through the gDNA pre-denaturation step was reproducible for other tested gDNA targets (data not shown).
Fig. 1.

Effect of pre-denaturing of gDNA before qPCR run. The graph shows relative fluorescence curves plotted against the qPCR cycles. Samples from two different CHO clones (blue and green) were analysed in nine replicates a without and b with gDNA pre-denaturation. (Color figure online)
Relative quantifications
Relative quantification was done with two recombinant cell lines expressing antibody scFc derivates. Prior to relative quantification the copy number of β-actin reference gene was analyzed in both clones to ensure equal amounts (data not shown). Three ng of gDNA preparations of both cell lines were analyzed for relative scFc transgene copies to the β-actin reference gene. Presented results are mean values from three biological samples of the same cell clone analysed in triplicates by two technical runs (n = 18). The obtained mean β-actin Cq values were 27.41 SD ± 0.13 for Clone 1 and 26.10 SD ± 0.10 for Clone 2 while the obtained scFc Cq values were 26.77 SD ± 0.06 and 26.36 SD ± 0.13 for Clone 1 and Clone 2, respectively.
Calculation of amplification efficiency
For relative quantification we either assumed 100 % efficiency for all qPCR runs or calculated the actual efficiency individually. One approach to calculate the efficiency of amplification facilitates the use of a decimal dilution series from a gDNA preparation. For this purpose, a four step 1:10 dilution series from Clone 1 was prepared and each dilution step was measured in triplicate in two technical replicates (n = 6). The calculated mean efficiency was 2.10 for β-actin and 2.07 for scFc (Table 2).
Table 2.
scFc and β-actin amplification efficiency (E) calculation using means (n = 6) of calibration curves generated via 1:10 dilution series of gDNA
| scFc | β-actin | |||
|---|---|---|---|---|
| Dilution | Cqa | SD | Cqa | SD |
| 0 | 19.51 | 0.10 | 20.47 | 0.30 |
| 10−1 | 22.77 | 0.14 | 23.86 | 0.03 |
| 10−2 | 25.91 | 0.11 | 26.99 | 0.10 |
| 10−3 | 29.03 | 0.17 | 29.79 | 0.18 |
| Slope | E | Slope | E | |
|---|---|---|---|---|
| −3.169 | 2.07 | −3.108 | 2.10 |
amean values (n = 6)
In a second approach the qPCR efficiency was calculated via a mathematical algorithm out of the raw fluorescence data from all curves of the same amplicon group by the LinRegPCR (Ruijter et al. 2009; Tuomi et al. 2010) software application. The data were processed using the setting individual fit. The software performs specific quality checks for each fluorescence curve (e.g. no plateau, noisy sample or PCR efficiency outside 5 %) and provides a statistically analyzed mean amplification efficiency value. For the β-actin and the scFc amplicon a total of 36 fluorescence curves for each amplicon were analysed. The range of the curves that passed the quality checks of the LinRegPCR program was between 20 and 40 %. The calculated mean efficiency was 1.85 for β-actin (n = 8) and 1.86 for scFc (n = 14).
Relative trangene copy numbers
The data set used for calculations of relative gene copy numbers (gcn) is summarized in Table 3. The transgene copies were related relative to β-actin using the analysed Cq values and the calculated qPCR efficiencies. To calculate the relative gcn assuming 100 % qPCR efficiency we used the 2−∆Cq method. The obtained gcn using this method were 1.57 SD ± 0.15 for Clone 1 and 0.83 SD ± 0.10 for Clone 2.
Table 3.
Calculation of gene copies relative to β-actin for two different cell clones
| Cellline | Clone 1 | Clone 2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transgene | scFc | β-actin | scFc | β-actin | ||||||||
| Cqa | 26.77 | 27.41 | 26.36 | 26.1 | ||||||||
| SD | 0.06 | 0.13 | 0.13 | 0.1 | ||||||||
| Calculation method | 100% | 1:10 | LinRegPCR | 100% | 1:10 | LinRegPCR | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transgene | scFc | β-actin | scFc | β-actin | scFc | β-actin | scFc | β-actin | scFc | β-actin | scFc | β-actin |
| Efficiency (E) | 2 | 2.07 | 2.1 | 1.86 | 1.85 | 2 | 2.07 | 2.1 | 1.86 | 1.85 | ||
| Relative gcn | 1.57 | 2.38 | 1.19 | 0.83 | 1.2 | 0.68 | ||||||
| SD | 0.15 | 0.24 | 0.1 | 0.1 | 0.15 | 0.07 | ||||||
aMean values (n = 18)
The 1:10 dilution method for qPCR efficiency correction resulted in relative gcn of 2.38 SD ± 0.24 and 1.20 SD ± 0.15 for Clone 1 and Clone 2 respectively.
The usage of the LinRegPCR software application for efficiency correction resulted in 1.19 SD ± 0.10 relative scFc copies for Clone 1 and 0.68 SD ± 0.07 for Clone 2. Independent of the used calculation method there was a two-fold difference in gcn between the two analysed clones.
The obtained results from the different calculation methods for the two recombinant clones are summarized in Fig. 2 and show that the 1:10 dilution method for efficiency correction resulted in the highest relative transgene numbers followed by the 2−∆Cq method (100 % real-time efficiency) and LinRegPCR. However, it should be mentioned that the qPCR efficiency calculated by the 1:10 dilution method has a value higher than two which would mean more than 100 % qPCR efficiency.
Fig. 2.

Relative quantification of two recombinant CHO clones using different qPCR efficiencies. The graph shows the scFc transgene copies of Clone 1 and Clone 2 relative to β-actin for each used relative quantification method
Relative gene copy numbers differ more than 100 % from the lowest to the highest value by application of various efficiency calculation methods. The huge observed differences between the efficiency calculations methods were caused by the extreme differences of efficiencies obtained from each method for each analyzed gene. However, this example illustrates that efficiency correction has a very high impact on the relative gcn of each individual clone, but only little influence when comparing two clones. Comparison of two individual clones will always result in a ratio of relative gcn, because efficiencies are always calculated for the same amplicon and are not specific for a clone. Therefore the influence on the ratio between two clones is rather moderate (Fig. 3).
Fig. 3.

Gene copy number ratio between Clone 1 and Clone 2 using different qPCR efficiencies. The graph shows the ratios between the two scFc clones using different qPCR efficiencies for calculations
Discussion
Optimisation of qPCR by pre-denaturation of gDNA improved the resolution of our qPCR experiments by less scattered fluorescence curves in a multi-parallel experiment (Fig. 1). We could improve the discrimination between two samples and decrease standard deviations. The impact of such a pre-denaturation step was reported before (Wilhelm et al. 2000) for a different PCR system and was attributed to slight temperature variances for each reaction mix inside the qPCR machine. Temperature heterogeneities may cause varying gDNA denaturation from well to well during the first qPCR denaturation step and incomplete denaturation leads to reduced accessibility of the template DNA for the DNA polymerase in the first cycles. Our observation confirmed the advantage of gDNA pre-denaturation to obtain more precise and less scattered data.
To evaluate qPCR quantification methods, three different relative quantification approaches were applied leading to deviating relative transgene copies for each scFc clone (Fig. 2). There was more than a factor of two between the relative quantification approaches. This was caused by the differences of the calculated efficiencies. The presented data show an example with rather high variation in amplification efficiency. However, when comparing two clones with the same transgene, the method of relative quantification was negligible, because all methods lead to comparable fold differences between the two clones (Fig. 3). This statement is of course just valid when the same sets of primers and probes are used on the same qPCR run for the transgene as well as for the reference gene to generate the Cq values. If this is the case and the whole preparation was equal for all biological replicates of both clones, the efficiencies for the transgene as well as for the reference gene are equal for each sample because they are calculated per amplicon group. When the efficiencies are the same for both clones, the result depends mostly on the obtained Cq values. In this presented case, Clone 1 had the transgene approximately twice as often incorporated in its genome compared to Clone 2.
If the research question focuses on the transgene copies relative to the reference gene, the result highly depends on the qPCR efficiency used for calculations. In our view the 2−∆∆Cq Method, which is widely used in research, is not the best choice to determine transgene copy numbers relative to a reference gene because it assumes 100 % qPCR efficiency. Pfaffl’s method with efficiency correction is a very suitable choice for quantification relative to a reference gene but it opens the question of how to determine the qPCR efficiency. In literature, two main concepts of efficiency calculation methods can be found, either via a 1:10 dilution series or via mathematical operations, that calculate mean efficiencies out of the single sample fluorescence curves (Bustin 2009). The concept of the 1:10 dilution method is the most widely used to calculate qPCR efficiencies and is also accepted as gold standard and suggested by the MIQE guidelines (Bustin et al. 2009). The 1:10 dilution method worked well in most cases but sometimes resulted in bad reproducibility from replicate runs (data not shown). Additionally, some experiments, such as the one represented, resulted in very high efficiencies above 100 %, which is theoretically impossible. These facts were attributed to the circumstance that a 1:10 dilution series is error-prone, mostly because of the operator error while pipetting. However, as the MIQE guidelines suggest to work with mean amplification efficiencies it is possible to statistically minimize the failure through the inclusion of sufficient replicates. Overall the 1:10 dilution method works quite well but it is a time and material consuming way to obtain qPCR efficiencies.
Efficiency calculation out of single fluorescence curves using the LinRegPCR software application showed good reproducibility in most of the replicate runs. The advantage of this method is on the one hand, that there is no need to add standards and on the other hand, that the application automatically calculates a mean efficiency value per amplicon group for each plate including a statistical verification by the LinReg software package. In this work we used mean amplification efficiency for all runs as suggested from the MIQE guidelines for 1:10 efficiency calculation. However, in some cases of our analyses, like the presented results, the mean LinRegPCR efficiency was much lower than the efficiency generated by the 1:10 dilution method. Another consideration is that LinRegPCR performs quality checks on each fluorescence curve and only accepts that ones that pass. During our studies we could observe that sometimes more than 50 % of the curves did not pass the quality checks and additional runs were needed to obtain a statistically significant efficiency value.
Conclusion
Relative quantification approaches appear to be more robust than absolute quantification approaches though small changes in qPCR efficiency may cause huge relative quantification differences. In the presented study, all used methods resulted in different values for each transgene relative to β-actin. However, when comparing two cell lines with the same transgene, qPCR efficiencies can be neglected when using the same experimental conditions. Concerning the applied efficiency correction method, we experienced that the 1:10 dilution method as well as the software application LinRegPCR has some drawbacks and the operator should be aware that although the two methods lead to comparable efficiency results in many cases, sometimes, as in the presented example, they result in rather diverse gcn values. Therefore, we propose to cross-check both methods in preliminary qPCR runs when evaluating a new target gene. If the methods result in similar efficiencies, the use of LinRegPCR would safe time and material.
Acknowledgments
Part of this study was partly funded by Polymun Scientific Immunbiologische Forschung GmbH, Donaustraße 99, 3400 Klosterneuburg, Austria.
Conflict of interest
None
References
- Bahr SM, Borgschulte T, Kayser KJ, Lin N. Using microarray technology to select housekeeping genes in Chinese hamster ovary cells. Biotechnol Bioeng. 2009;104:1041–1046. doi: 10.1002/bit.22452. [DOI] [PubMed] [Google Scholar]
- Bustin SA. A-Z of quantitative PCR. La Jolla, CA: International University Line; 2009. [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- Ginzinger DG. Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol. 2002;30:503–512. doi: 10.1016/S0301-472X(02)00806-8. [DOI] [PubMed] [Google Scholar]
- Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- Kibbe WA. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res. 2007;35:W43–W46. doi: 10.1093/nar/gkm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kim YG, Lee GM. CHO cells in biotechnology for production of recombinant proteins: current state and further potential. Appl Microbiol Biotechnol. 2012;93:917–930. doi: 10.1007/s00253-011-3758-5. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mader A, Prewein B, Zboray K, Casanova E, Kunert R (2013) Exploration of BAC versus plasmid expression vectors in recombinant CHO cells. Appl Microbiol Biotechnol 97(9):4049–4054 [DOI] [PubMed]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, Moorman AF. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009;37:e45. doi: 10.1093/nar/gkp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuomi JM, Voorbraak F, Jones DL, Ruijter JM. Bias in the Cq value observed with hydrolysis probe based quantitative PCR can be corrected with the estimated PCR efficiency value. Methods. 2010;50:313–322. doi: 10.1016/j.ymeth.2010.02.003. [DOI] [PubMed] [Google Scholar]
- Walsh G. Biopharmaceuticals: recent approvals and likely directions. Trends Biotechnol. 2005;23:553–558. doi: 10.1016/j.tibtech.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Wilhelm J, Hahn M, Pingoud A. Influence of DNA target melting behavior on real-time PCR quantification. Clin Chem. 2000;46:1738–1743. [PubMed] [Google Scholar]