

Abstract
Mechanosensory hair cells are the receptor cells of hearing and balance. Hair cells are sensitive to death from exposure to therapeutic drugs with ototoxic side effects, including aminoglycoside antibiotics and cisplatin. We recently showed that the induction of heat shock protein 70 (HSP70) inhibits ototoxic drug–induced hair cell death. Here, we examined the mechanisms underlying the protective effect of HSP70. In response to heat shock, HSP70 was induced in glia-like supporting cells but not in hair cells. Adenovirus-mediated infection of supporting cells with Hsp70 inhibited hair cell death. Coculture with heat-shocked utricles protected nonheat-shocked utricles against hair cell death. When heat-shocked utricles from Hsp70–/– mice were used in cocultures, protection was abolished in both the heat-shocked utricles and the nonheat-shocked utricles. HSP70 was detected by ELISA in the media surrounding heat-shocked utricles, and depletion of HSP70 from the media abolished the protective effect of heat shock, suggesting that HSP70 is secreted by supporting cells. Together our data indicate that supporting cells mediate the protective effect of HSP70 against hair cell death, and they suggest a major role for supporting cells in determining the fate of hair cells exposed to stress.
Introduction
Hearing loss is the most common sensory impairment in humans, and it affects over 16% of adults in the United States (1). Hearing loss is often caused by the death of mechanosensory hair cells in the inner ear. Hair cells are the sensory cells of hearing and balance, transducing mechanical stimuli into neural signals. Hair cells are damaged by a variety of stresses including aging, noise trauma, genetic mutations, and exposure to certain therapeutic drugs, including aminoglycoside antibiotics and the antineoplastic agent cisplatin. Hair cell death caused by exposure to ototoxic drugs is a significant health problem that results in hearing loss for an estimated 500,000 Americans each year (2). Aminoglycoside antibiotics remain among the most commonly used antibiotics worldwide, and significant hearing loss or balance impairment (or both) occurs in up to 20% of patients receiving these drugs (3).
The induction of heat shock proteins (HSPs) in response to cellular stress is a ubiquitous and highly conserved response that can significantly inhibit apoptosis in many systems (4). We have shown that HSP induction via heat shock inhibits aminoglycoside-induced hair cell death in organ cultures of utricles from adult mice (5). HSP70 is required for this protective effect, and HSP70 overexpression inhibits ototoxic hair cell death (6). Furthermore, HSP70 is protective against aminoglycoside-induced hearing loss and cochlear hair cell death in vivo (7). Taken together, these data indicate that HSP70 induction is a critical stress response that can promote survival of hair cells exposed to aminoglycosides.
The mechanism(s) underlying the protective effect of HSP70 against aminoglycoside-induced hair cell death are unknown. Stress-induced HSP70 expression occurs in response to a variety of stressors and can inhibit apoptosis, both via its chaperone activity and via direct inhibition of apoptotic signaling (reviewed in refs. 8–10). Here, we have used an in vitro preparation of utricles from adult mice to examine the mechanisms underlying the protective effect of HSP70 against aminoglycoside-induced hair cell death.
Results
HSP levels in control and heat-shocked utricles.
HSP expression levels in control and heat-shocked utricles from CBA/J mice were examined by Western blotting (Figure 1A). Heat shock resulted in a robust (∼14-fold) increase in HSP70. Heat shock also resulted in the induction of HSP40 and HSP27. We observed that the levels of HSP90, HSP60, and HSP32 remained relatively unchanged after heat shock. We examined Hsp mRNA induction in utricles from Hsp70+/+ and Hsp70–/– mice using quantitative RT-PCR (Figure 1B). Heat shock resulted in a comparable induction of HSP27 in utricles from Hsp70+/+ and Hsp70–/– mice. We found that Hsp70 mRNA was induced by heat shock in utricles from Hsp70+/+ mice, but not in utricles from Hsp70–/– mice.
Figure 1. Effects of heat shock on HSP levels.

(A) Control and heat-shocked utricles from CBA/J mice were examined for expression levels of HSPs using Western blotting. Heat shock results in upregulation of HSP70, HSP40, and HSP27. Numbers below each band indicate the fold change relative to the utricles that were not heat shocked. (B) Control and heat-shocked utricles from Hsp70+/+ and Hsp70–/– mice were examined for Hsp mRNA expression using real-time quantitative PCR (RT-qPCR). Heat shock resulted in a robust induction of Hsp70 mRNA in utricles from Hsp70+/+ mice. In heat-shocked utricles from Hsp70–/– mice, Hsp27 mRNA was induced, but Hsp70 mRNA was not.
Heat shock results in HSP70 induction in supporting cells.
In order to examine the cellular localization of HSP70 in response to heat shock, the utricles were heat shocked and then fixed 6 hours later. We sectioned the utricles and stained them for myosin 7a (hair cells) and HSP70 immunoreactivity (Figure 2). The control utricles show the typical tissue architecture with a single row of hair cell nuclei above a single row of supporting cell nuclei. While hair cells contact only the luminal surface of the epithelium, supporting cells contact both the luminal and basal surfaces and extend processes between the hair cells (see schematic diagram in Figure 3). The control utricles show very little HSP70 immunoreactivity (Figure 2A), whereas the heat-shocked utricles show robust HSP70 immunoreactivity, with HSP70 localized to supporting cells and no detectable HSP70 immunoreactivity in the hair cells (Figure 2, B–E). Since heat shock inhibits aminoglycoside-induced hair cell death (5), these data suggest that supporting cells mediate the protective effect of heat shock against aminoglycoside-induced hair cell death.
Figure 2. Heat shock results in HSP70 upregulation in supporting cells.
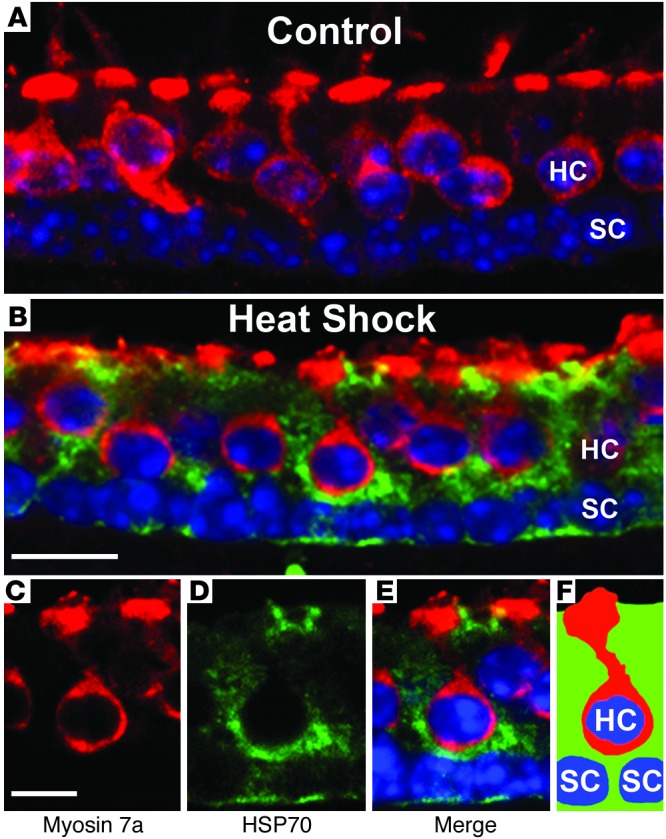
Control and heat-shocked (43°C for 30 minutes) utricles were allowed to recover from heat shock for 6 hours and were sectioned (see Methods). Sections were labeled using antibodies against myosin 7a (red, hair cell marker) and HSP70 (green). Nuclei were labeled with Hoechst dye (blue). Confocal microscopy was performed using identical settings for laser power, gain, offset, and zoom. Shown are a control utricle (A) and a heat-shocked utricle (B–E). Control utricles (A) show very little HSP70 immunoreactivity. Heat-shocked utricles (B–E) show robust upregulation of HSP70 in supporting cells. C–E show a higher-magnification image of a heat-shocked utricle. (C) Myosin 7a labeling of a single hair cell. (D) HSP70 immunoreactivity is present in supporting cells but not in the hair cell. (E) Merged image. (F) Schematic showing locations of the hair cell (HC) and supporting cells (SC) in C–E. Scale bars: 10 μm (A and B); 5 μm (C–E). Images are representative of 3 experiments for a total of 10 to 12 utricles per condition.
Figure 3. Adenovirus specifically infects supporting cells.

Utricles were infected with Ad-RFP for 2 hours and then cultured in control media for an additional 24 hours and fixed. Hair cells and supporting cells were visualized using myosin 7a (blue) and SOX2 (green) immunochemistry, respectively. Schematic shows the structure of the utricular sensory epithelium and the locations of the hair cells and supporting cells. Locations of confocal (optical) sections shown in the upper and lower panels are indicated by lines and arrows in the schematic. Upper panels: in confocal images taken at the level of the hair cell nuclei, the Ad-RFP signal appears in the spaces between hair cells and does not overlap with the hair cell marker myosin 7a. Lower panels: in confocal sections at the level of the supporting cell nuclei, the Ad-RFP signal colocalizes with the supporting cell marker SOX2. Ad-RFP infection results in RFP expression in supporting cells only, and no hair cells are infected. Scale bar: 20 μm.
Adenovirus specifically infects supporting cells in adult mouse utricles in vitro.
To further explore the roles of supporting cells as mediators of the protective effect of HSP70, we sought a method for inducing HSP70 expression specifically in supporting cells. Utricles were infected with Ad-RFP and cultured in virus-free media for 24 hours, fixed, and then processed for myosin 7a (hair cells) and SOX2 (supporting cells) immunoreactivity. As shown in Figure 3, the adenovirus specifically and efficiently infected supporting cells without infecting hair cells. Supporting cell infection efficiency was 47.9 ± 11%, and no RFP+ hair cells were observed (n = 10 utricles). Importantly, we found that adenoviral infection of supporting cells does not result in toxicity to hair cells or supporting cells, as cell counts in infected utricles are equivalent to those in noninfected utricles (11).
Adenoviral infection of supporting cells with HSP70 inhibits neomycin-induced hair cell death.
To examine the role of supporting cells in mediating the protective effect of HSP70 against ototoxic hair cell death, we used an adenovirus to deliver HSP70 to supporting cells. We infected the utricles with Ad-HSP70 (or Ad-GFP as a control) and then cultured them for an additional 24 hours in either control media or media containing neomycin. We then fixed the utricles and visualized the hair cells using myosin 7a immunochemistry (Figure 4). The control utricles show normal numbers of hair cells (Figure 4A). Culture in either Ad-GFP (Figure 4B) or Ad-HSP70 (Figure 4C) did not result in hair cell toxicity. Neomycin exposure resulted in substantial loss of hair cells in uninfected utricles (Figure 4D). Infection of supporting cells with Ad-GFP was not protective against neomycin-induced hair cell death (Figure 4E), while Ad-HSP70 inhibited neomycin-induced hair cell death (Figure 4, F and H). A similar protective effect of Ad-HSP70 was seen in hair cell counts from the striolar region of the utricle (data not shown). Western blot data confirm that infection with Ad-HSP70 resulted in increased HSP70 expression in utricles (Figure 4G). Because hair cells were not infected with the adenovirus (Figure 3), these data indicate that HSP70 in supporting cells protects hair cells against neomycin-induced death and that supporting cells thus act as mediators of hair cell survival.
Figure 4. Adenoviral infection of supporting cells with HSP70 inhibits neomycin-induced hair cell death.

Utricles were infected with Ad-HSP70 (or Ad-GFP). Under these conditions, the adenovirus infects supporting cells, and no hair cells are infected (see Figure 3). Hair cells are labeled with myosin 7a. (A) Control utricles show normal hair cell numbers. (B) Infection with Ad-GFP does not result in hair cell loss (for clarity, the GFP channel is not shown). (C) Infection with Ad-HSP70 does not result in hair cell loss. (D) Neomycin treatment results in significant loss of hair cells. (E) Infection of supporting cells with Ad-GFP is not protective against neomycin-induced hair cell death. (F) Infection of supporting cells with Ad-HSP70 inhibits neomycin-induced hair cell death. (G) Western blot analysis of HSP70 expression in control and Ad-HSP70–infected utricles. (H) Ad-HSP70 is protective against neomycin-induced hair cell death. 1-way ANOVA, F5,46 = 12.03, P < 0.0001. n = 5–12 utricles per condition. *P < 0.001 between neomycin and neomycin plus Ad-HSP70. Ctrl, control; Neo, neomycin.
Coculture with heat-shocked utricles is protective.
The mechanism(s) by which HSP70 in supporting cells results in the protection of hair cells are unknown. We hypothesized that HSP70 may be secreted by supporting cells and internalized by hair cells. This hypothesis makes several specific predictions. First, it predicts that coculture with utricles that have been heat shocked will protect hair cells in utricles that have not undergone heat shock. Second, HSP70 should be detectable in the media surrounding heat-shocked utricles. Third, it predicts that adding exogenous HSP70 to the media should be protective. And fourth, it predicts that HSP70 should translocate from supporting cells to hair cells.
In order to begin to test the hypothesis that HSP70 is secreted by supporting cells and internalized by hair cells, we performed a series of coculture experiments using 24-well tissue culture plates fitted with Transwell permeable membrane inserts (Figure 5). These experiments used utricles from both Hsp70+/+ and Hsp70–/– (12) mice, allowing us to examine the role of HSP70 in any observed protective effect. In the first experiment, utricles from wild-type (Hsp70+/+) mice were heat shocked and then placed in the top chamber of a well containing a Transwell insert. Utricles (wild-type) that had not been heat shocked were placed in the same well beneath the Transwell insert containing the heat-shocked utricles (see schematic, Figure 5A). As shown in Figure 5A, gentamicin resulted in significant loss of hair cells in utricles that were not cocultured with heat-shocked utricles. Heat-shocked utricles (in the top chamber) were partially protected against gentamicin-induced hair cell death, in agreement with our previous results (5). In addition, coculture with heat-shocked utricles protected hair cells in utricles that had not been heat shocked (i.e., in the bottom chamber). Moreover, the protective effect of coculture with heat-shocked utricles was comparable to the protective effect of heat shock itself. These data indicate that heat-shocked utricles secrete factor(s) that are protective against aminoglycoside-induced hair cell death.
Figure 5. Coculture with heat-shocked utricles is protective.

Utricles were cultured in 24-well plates containing Transwell permeable inserts. For each experiment, heat-shocked utricles were placed in the upper chamber of the Transwell device, and nonheat-shocked utricles were placed in the lower chamber. (A) Wild-type (Hsp70+/+) utricles were cocultured. Control utricles and heat-shocked utricles had comparable hair cell densities. Gentamicin caused significant loss of hair cells. Heat-shocked utricles (upper chamber) were partially protected against gentamicin-induced hair cell death. Nonheat-shocked utricles (bottom chamber) were protected in a manner comparable to heat-shocked utricles. (B) When utricles from Hsp70–/– mice were used in both chambers (heat-shocked and nonheat-shocked), protection against gentamicin-induced hair cell death was abolished in both groups of utricles. (C) Hsp70–/– utricles were heat shocked and cultured in the upper chamber of the Transwell device, while Hsp70+/+ utricles (nonheat shocked) were in the lower chamber. No protective effect against gentamicin-induced hair cell death was observed in either group. (D) Hsp70+/+ utricles were heat shocked and cultured in the upper chamber of the Transwell device, while Hsp70–/– utricles (nonheat shocked) were in the lower chamber. Both groups were protected against gentamicin-induced hair cell death. ANOVAs, *P < 0.05. n = 5–11 utricles per condition. Gent, gentamicin; HS, heat shocked.
Next, we used utricles from Hsp70–/– mice to determine whether HSP70 is required for the protective effect of coculture (Figure 5B). When utricles from Hsp70–/– mice were used in both the upper and lower chambers of the Transwell device, the protective effect was abolished in both the heat-shocked utricles (upper chamber) and the nonheat-shocked utricles (lower chamber).
The finding that the protective effect of coculture is abolished when Hsp70–/– utricles are used in both chambers indicates that HSP70 is required for the protective effect observed in coculture. Is HSP70 required in the heat-shocked utricles themselves, in the cocultured (nonheat-shocked) utricles, or both? To address this question, we performed mixed coculture experiments in which Hsp70–/– utricles were in 1 chamber and Hsp70+/+ utricles were in the other chamber. First, Hsp70–/– utricles were heat shocked and placed in the upper chamber over wild-type (Hsp70+/+) utricles (Figure 5C). The use of Hsp70–/– utricles in the top chamber abolished the protective effect in both chambers, indicating that HSP70 is required in the heat-shocked utricles. In the converse experiment, wild-type utricles were heat shocked and placed in the upper chamber over nonheat-shocked utricles from Hsp70–/– mice (Figure 5D). Both the heat-shocked wild-type utricles in the top chamber and the nonheat-shocked Hsp70–/– utricles in the bottom chamber were protected against gentamicin-induced hair cell death relative to utricles that were not heat shocked or cocultured. Taken together, these data indicate that HSP70 was required in the heat-shocked utricles in the upper chamber, but not in the nonheat-shocked utricles in the bottom chamber, suggesting that either HSP70 is itself secreted, or HSP70 is required for the secretion of other protective molecule(s).
HSP70 is secreted into the media.
Our hypothesis that HSP70 is secreted by supporting cells predicts that HSP70 should be present in the media surrounding heat-shocked utricles. We analyzed HSP70 levels in media from control and heat-shocked utricles using an ELISA (Figure 6A). Media from control utricles contained 561 ± 31 ng of HSP70, while media from heat-shocked utricles contained 1,105 ± 31 ng of HSP70, representing a significant increase (P < 0.0005; Figure 6A). When HSP70 levels were examined in the homogenized tissue itself, levels in control utricles (458 ± 141 ng) were comparable to those in the control media, while heat-shocked utricles contained approximately 4-fold more HSP70 (1,814 ± 49 ng). No HSP70 was detected in the control or heat-shocked utricles from Hsp70 knockout mice (data not shown). These data indicate that heat-shocked utricles secrete HSP70 into the surrounding media.
Figure 6. HSP70 is secreted to the medium, and depletion of HSP70 from the medium abolishes the protective effect of coculture.

(A) HSP70 levels in medium from control and heat-shocked utricles were examined by ELISA. Medium surrounding heat-shocked utricles contained significantly more HSP70 than medium surrounding the control (nonheat-shocked) utricles (ANOVA, P < 0.0005). HSP70 levels in control tissue were comparable to those in control medium, while heat-shocked utricles contained significantly higher levels of HSP70. Shown are data from 3 biological replicates processed in parallel. The experiment was repeated 5 times with similar results. (B) Heat-shocked (upper chamber) and nonheat-shocked (lower chamber) utricles from Hsp70+/+ mice were cocultured in a Transwell device containing 5 mM of gentamicin. As in Figure 5, gentamicin resulted in significant hair cell death, and both heat-shocked (upper) and nonheat-shocked (cocultured, lower) utricles were protected. Addition of a control IgG to the medium did not alter the protective effect of either heat shock (upper) or coculture with heat-shocked (lower) utricles. Depletion of HSP70 from the medium using an HSP70 function-blocking antibody (fbAb) abolished the protective effect of both heat-shocked (upper) and nonheat-shocked, cocultured (lower) utricles. ANOVA, *P < 0.05 relative to gentamicin. n = 5–15 utricles per condition.
Extracellular HSP70 is required for coculture protection.
In order to examine whether secreted HSP70 is required for the protective effect of coculture, we used a function-blocking antibody to deplete the media of HSP70 (13, 14). In the absence of the function-blocking antibody, we again observed a protective effect in both the heat-shocked utricles in the upper chamber and the nonheat-shocked utricles in the lower chamber (Figure 6B). However, when the media was depleted of HSP70 using the blocking antibody, the protective effect of heat shock was abolished in both chambers (Figure 6B). Use of a control IgG in the media did not alter the protective effect of heat shock (Figure 6B). These data indicate that extracellular HSP70 is required for both the protective effect of heat shock and the protective effect seen in the nonheat-shocked, cocultured utricles in the bottom chamber.
Ad-HSP70 is protective in coculture.
We next examined whether utricles infected with Ad-HSP70 can protect noninfected utricles in a manner similar to the protection provided by heat-shocked utricles in the coculture paradigm. Utricles from CBA/J mice were infected with an adenovirus carrying HSP70 fused to mCherry (Ad-HSP70mCherry). Ad-HSP70mCherry–infected utricles were placed in the top chamber of a Transwell dish, and noninfected utricles were placed in the bottom chamber. Infection with Ad-HSP70mCherry inhibited neomycin-induced hair cell death in the infected utricles (upper chamber) as well as in the noninfected cocultured utricles (lower chamber) (Figure 7). These data indicate that the expression of HSP70 in supporting cells is sufficient for the protective effect against hair cell death that we observed in cocultured utricles. Taken together, the data from the coculture experiments indicate that HSP70 is secreted by supporting cells and that this secreted HSP70 inhibits hair cell death.
Figure 7. Ad-HSP70-mCherry is protective in coculture.

Utricles were infected with Ad-HSP70-mCherry and placed in the upper chamber of a Transwell device containing 3 mM of neomycin. Noninfected utricles were placed in the lower chamber. No utricles were heat shocked. Infection of supporting cells with Ad-HSP70-mCherry resulted in significant inhibition of neomycin-induced hair cell death in both the infected utricles (upper) and noninfected utricles (lower). ANOVA, *P < 0.05 relative to neomycin alone. n = 6–11 utricles per condition.
Exogenous HSP70 is protective.
The data from the coculture experiments are consistent with the hypothesis that HSP70 is secreted by supporting cells. As mentioned above, the third prediction of this hypothesis is that adding exogenous HSP70 to the media should be protective. In order to test this prediction, we treated utricles with neomycin in the presence (or absence) of exogenous human HSP70 (Figure 8A). Treatment with exogenous HSP70 inhibited neomycin-induced hair cell death, while treatment with a control protein (HSA) was not protective. These data support the idea that extracellular HSP70 alone is protective, and they argue against the idea that HSP70 is merely chaperoning another protective molecule.
Figure 8. Exogenous HSP70 is protective.

(A) Utricles were cultured in neomycin with (or without) recombinant human HSP70. Recombinant HSA was used as a control. Neomycin resulted in significant loss of hair cells (ANOVA, *P < 0.05 relative to control). Addition of exogenous HSP70 inhibited neomycin-induced hair cell death (ANOVA, #P < 0.05 relative to neomycin), while HSA was not protective. n = 5–20 utricles per condition. (B) Exogenous HSP70 is not internalized by hair cells. Neomycin-treated utricles were cultured in the presence of FITC-labeled recombinant human HSP70 (ExHSP70-FITC) or BSA (ExBSA-FITC) and examined for the presence of intracellular HSP70-FITC (or BSA-FITC) (green). Hair cells were labeled with an antibody against myosin 7a (red). Neither ExHSP70-FITC nor ExBSA-FITC was internalized by hair cells, although supporting cells internalized small amounts of both exogenous proteins. Scale bar: 10 μm (applies to all panels). Orth, orthogonal.
HSP70 is not internalized by hair cells.
Our hypothesis that HSP70 is secreted by supporting cells and internalized by hair cells predicts that HSP70 should translocate from supporting cells to hair cells. Our data (Figure 2) demonstrate that 6 hours after heat shock, HSP70 is localized to the supporting cells. We examined later time points after heat shock to determine whether HSP70 eventually appears in hair cells, and we did not detect increased HSP70 immunoreactivity in hair cells at 12, 18, or 24 hours after heat shock (data not shown). We then examined HSP70 internalization by hair cells in 2 additional paradigms. First, we examined the localization of exogenously applied HSP70 by using an FITC-tagged human HSP70 (ExHSP70-FITC). No ExHSP70-FITC was detected in hair cells (Figure 8B). We did detect small amounts of ExHSP70-FITC in supporting cells, however, supporting cells also internalized small amounts of the tagged control protein, BSA-FITC (ExBSA-FITC). Taken together, these data suggest that the protective effect of extracellular HSP70 is probably not mediated by the internalization of HSP70 by hair cells.
We next examined the localization of an HSP70-mCherry fusion protein after infection of supporting cells with Ad-HSP70mCherry. For this experiment, we first confirmed that Ad-HSP70mCherry is protective against neomycin-induced hair cell death (Figure 9A), indicating that fusion with the mCherry tag did not reduce the protective effect of Ad-HSP70. Ad-HSP70mCherry was expressed exclusively in supporting cells 24 hours after infection (Figure 9B). Even 48 hours after infection, we did not detect any HSP70-mCherry in hair cells (data not shown). Using all 3 of these approaches, we found no evidence that HSP70 is internalized by hair cells.
Figure 9. Ad-HSP70-mCherry is protective, and it is not internalized by hair cells.

(A) Supporting cells were infected with Ad-HSP70-mCherry (or Ad-mCherry as a control). Infected utricles were cultured in the presence or absence of neomycin (3 mM). Neither virus resulted in significant hair cell death, while neomycin caused a significant loss of hair cells. Infection of supporting cells with Ad-HSP70-mCherry inhibited neomycin-induced hair cell death (ANOVA, *P < 0.05). Infection of supporting cells with Ad-mCherry was not protective. n = 6–11 utricles per condition. (B) Utricles infected with Ad-HSP70-mCherry were fixed, and hair cells were labeled using antimyosin 7a (green). Utricles were examined using confocal microscopy (original magnification, ×63). Schematic shows the location of each panel. Upper panels show confocal images taken at the level of the hair cell cuticular plates, while the lower panels show confocal images from the same field taken at the level of the hair cell nuclei. Ad-HSP70-mCherry (red) is restricted to supporting cells and does not colocalize with myosin 7a (green, hair cells), indicating that Ad-HSP70-mCherry does not translocate from supporting cells to hair cells. Scale bars: 20 μm.
Discussion
Supporting cells are important mediators of hair cell survival.
Our data indicate that heat shock–induced HSP70 upregulation occurs primarily in supporting cells with no detectable upregulation in hair cells. In addition, adenoviral-mediated infection of supporting cells with HSP70 inhibited aminoglycoside-induced hair cell death. Since hair cells of the mature mouse utricle were not infected with the adenovirus (Figure 3 and ref. 11), it shows that HSP70 in supporting cells protects hair cells against ototoxic death. These findings, therefore, indicate that supporting cells act as critical mediators of hair cell survival. Furthermore, recent evidence suggests that supporting cells are also important mediators of hair cell death (15) and subsequent phagocytosis (16). Hair cell damage caused by either laser ablation or aminoglycoside exposure results in the activation of extracellularly regulated kinases 1 and 2 (ERK1/2) in supporting cells (Deiters’ and other phalangeal cells) in the neonatal rat cochlea (15). ERK1/2 is not activated in hair cells in response to this damage. Pharmacological inhibition of ERK1/2 signaling inhibited aminoglycoside-induced hair cell death (15). Taken together with our data, these findings demonstrate that supporting cells play a major role in determining whether hair cells under stress ultimately live or die.
Potential mechanisms for supporting cell–mediated protection against hair cell death.
We hypothesized that supporting cells secrete HSP70, which is then internalized by hair cells. Under normal physiological conditions, HSP70 is largely an intracellular protein involved in the folding, assembly, and translocation of other proteins. HSP70 expression is strongly induced by a wide variety of environmental stresses, a process known as the stress response (17). These stressful conditions result in extracellular release of HSP70 from a variety of cell types, including neurons, glia, B cells, and PBMCs (18–24). HSP70 secretion does not occur via the classical secretory pathway, since it carries no secretion signal, and inhibitors of Golgi-mediated secretion do not inhibit HSP70 release (20). Instead, HSP70 is released via secretory exosomes (25, 26), which are endosome-derived microvesicles of 50 to 200 nm in size. Extracellular HSP70 can act as a danger signal that increases immune surveillance (reviewed in ref. 27). However, extracellular HSP70 can also protect against stress-induced cell death in a variety of systems (28–31). The protective effects of extracellular HSP70 may be particularly important in the nervous system. The application of exogenous HSP70 inhibits axotomy-induced death of motor and sensory neurons (31), and systemic injections of human HSP70 increased motoneuron survival and lifespan in a mouse model of amyotrophic lateral sclerosis (ALS) (32). The source of stress-induced extracellular HSP70 appears to be glial cells. Glia release HSP70 in response to a variety of stresses, and neurons exposed to extracellular HSP70 are more resistant to stress-induced injury than unexposed neurons (21, 33–35).
Stress-induced HSP70 production by glia and glia-like supporting cells may be a general mechanism by which these cells provide protection to sensory cells. In the CNS, stress-induced HSP70 induction occurs largely in glial cells, with little induction in neurons (36–38). Similarly, in the rat olfactory system, stress-induced HSP70 production occurs primarily in supporting (sustentacular) cells and not in olfactory sensory neurons (39). In addition, olfactory sensory neurons are protected by HSP induction (40). Targeted overexpression of HSP70 in astrocytes protects CA1 hippocampal neurons from death caused by transient forebrain ischemia (41). Consistent with our finding that stress-induced HSP70 induction is specific to supporting cells, a recent report showed that noise trauma resulted in increased HSP70 immunoreactivity in Deiters’ cells (42).
Several lines of evidence support the hypothesis that HSP70 is secreted by supporting cells. First, coculture with heat-shocked utricles protects utricles that have not been heat shocked (Figure 5A). HSP70 is required for both the protective effect of heat shock itself (in the heat-shocked utricles) and for the protective effect on cocultured, nonheat-shocked utricles (Figure 5, B–D). Second, HSP70 is present in the media surrounding heat-shocked utricles (Figure 6A). Third, depletion of HSP70 from the media abolished the protective effect of the coculture (Figure 6B). Together, these findings indicate that HSP70 is secreted by supporting cells in response to heat shock. While our finding that exogenous HSP70 is protective (Figure 8A) suggests that extracellular HSP70 alone is sufficient for the protective effect against hair cell death, our data do not rule out the possibility that secreted HSP70 is chaperoning additional protective molecules.
Although our data indicate that HSP70 is secreted by supporting cells, we found no evidence that it is internalized by hair cells. We examined hair cells at various time points after heat shock and found that HSP70 immunoreactivity is consistently strong in supporting cells but not in hair cells (Figure 2). Fluorescently tagged HSP70 was not internalized by hair cells (Figure 8B), and Ad-HSP70mCherry did not translocate from supporting cells to hair cells (Figure 9B). These data suggest that rather than being internalized by hair cells, extracellular HSP70 may be acting at the cell surface, perhaps by ligating a cell surface receptor and causing signaling downstream of that receptor. Extracellular HSP70 can bind a variety of cell surface receptors on the secreting cell, on adjacent cells, or on distant cells (20). Several classes of cell surface receptors have been identified as HSP70 receptors, including TLRs, scavenger receptors (SRs), and c-type lectins (19, 27, 43). Extracellular HSP70 is an endogenous ligand for TLR2/4. In the human squamous carcinoma cell line A431, ligation of TLR2/4 with HSP70 results in the formation of heterodimers between TLR 2/4 and the EGFR (44). This complex results in transactivation of the EGFR and prosurvival signaling in the EGFR-expressing cell (45). Similarly, HSP70 expressed on the surface of tumor-derived exosomes from mouse cell lines results in the ligation of TLR2 and the subsequent activation of STAT-3 and NFκB (46), both of which can have prosurvival effects. These data suggest that HSP70-mediated activation of prosurvival signaling via TLRs does not require the internalization of HSP70.
Recent studies have found that supporting cells can act as mediators of hair cell death. These studies demonstrated that damage to hair cells results in the activation of ERK1/2 exclusively in supporting cells, and that pharmacological inhibition of ERK1/2 inhibits hair cell death (15). In addition, supporting cells were found to mediate the removal of dead hair cells from the sensory epithelium (16). Our data indicate that supporting cells can act as mediators of hair cell survival. Taken together, these data implicate supporting cells as critical determinants of whether a hair cell under stress ultimately lives or dies. The roles of supporting cells as mediators of hair cell survival and death are only beginning to be explored. In addition, the mechanisms by which hair cells send stress signals and supporting cells sense and respond to these signals are largely unknown. From a basic science perspective, elucidating these intercellular signaling mechanisms in the mature inner ear will be critical to understanding the fundamental roles of supporting cells as determinants of hair cell survival versus death. From a clinical perspective, understanding these intercellular signals will inform the rational design of therapies aimed at preventing hearing loss and balance disturbances resulting from hair cell death.
Methods
Animals
CBA/J mice of both sexes were obtained from either the Jackson Laboratory or Harlan. Hsp70–/– mice (12) were obtained from the Mutant Mouse Regional Resource Center at the UCSD. These mice carry a deletion of an 11-kb region of genomic DNA that contains both Hspa1a and Hspa1b (12). The mouse genome contains 3 inducible Hsp70 genes: Hspa1a (formerly Hsp70.3), Hspa1b (formerly Hsp70.1), and Hspa2 (formerly Hsp70.2). Hspa1a and Hspa1b are strongly induced by heat and other stresses. Hspa2 expression is restricted to spermatogenic cells (47). Young adult mice (4–6 weeks of age) were euthanized by CO2 inhalation and decapitated.
Utricle culture
The adult mouse utricle culture preparation has been described in detail (11, 48). Utricles were dissected under sterile conditions and cultured free floating (5–8 utricles per well) in a 24-well tissue culture plate overnight in a tissue culture incubator at 37°C and 5% CO2. The culture medium consisted of DMEM/F12 (11320; Invitrogen), 5% FBS, and 50 U/ml of penicillin G (Sigma-Aldrich).
Aminoglycoside treatment
Before treatment, neomycin sulfate (Sigma-Aldrich) was equilibrated for at least 30 minutes in a tissue culture incubator at 37°C in a humidified atmosphere of 95% air and 5% CO2. Utricles were exposed to neomycin (2–3 mM in culture medium) for various periods of time at 37°C. The control utricles were not exposed to neomycin.
Because Hsp70–/– mice carry a neomycin resistance gene in place of the HSP70 coding region (12), we used gentamicin C (5 mM for 24 hours) in the experiments on utricles from these mice. The neomycin resistance gene used to develop these knockout mice encodes an aminoglycoside kinase that modifies neomycin, kanamycin, and related aminoglycosides at the 3′-hydroxyl group (49). Other aminoglycosides, including gentamicin C and tobramycin, are not substrates for this enzyme (49), and utricles from Hsp70–/– mice show the same susceptibility to gentamicin-induced hair cell death as utricles from wild-type mice (6).
Heat shock protocol
Heat shock treatment was carried out as previously described (5). Utricles and surrounding culture media were transferred from the 24-well plate into sterile 1.5 ml microcentrifuge tubes. Tubes containing utricles to be heat shocked were placed into a water bath at 43°C for 30 minutes. Control utricles (no heat shock) were replaced in the incubator at 37°C. Following heat shock, all utricles were returned to the tissue culture plates and maintained in culture at 37°C for 6 hours prior to neomycin exposure.
Quantitative RT-PCR
Control and heat-shocked utricles (4 per condition) from Hsp70+/+ and Hsp70–/– mice were homogenized in ground-glass homogenizers, and total RNA was collected using an RNeasy Micro Kit (QIAGEN). RNA was reverse transcribed using random hexamer primers (Life Technologies). cDNA was amplified with SYBR Green qPCR (Applied Biosystems) using the following primer sets (5′–3′): 18S (forward) TTCGGAACTGAGGCCATGATT, (reverse) TTTCGCTCTGGTCCGTCTTG; HSP27 (forward) GAGAACCGAACGACCGTCC, (reverse) CCCAATCCTTTGACCTAACGC; HSP32 (forward) CTCACAGATGGCGTCACTTCGTCA, (reverse) TTGCCAACAGGAAGCTGAGA; HSP70 (forward) AGGCCAGGGCTGGTATTACT, (reverse) AATGACCCGAGTTCAGGATG; HSP90 (forward) GCAATTTCTGCCTGAAAGGC, (reverse) GTGCGTGTTCATTCAGCCAC. Results are reported as fold changes in mRNA levels relative to control (nonheat-shocked) utricles from mice of the same genotype.
Western blot analyses
After treatment, utricles (10 utricles per condition) were homogenized using a ground-glass homogenizer containing RIPA buffer (150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% Na-deoxycholate, 0.1% SDS) supplemented with 1 mM Na3VO4, 1 mM NaF, and a Protease Inhibitor Cocktail Set III (539134; EMD Chemicals) used at 1:100 dilution. Supernatants were resuspended in Laemmli buffer, boiled for 5 minutes, and separated by SDS-PAGE using 4%–15% Tris-HCl minigels (Bio-Rad). After wet transfer to PVDF membranes and incubation in blocking buffer (1X TBS with 0.1% Tween 20 and 5% w/v nonfat dry milk), the membranes were probed with primary antibodies. The following antibodies were obtained from Cell Signaling Technology and diluted 1:1,000 in blocking buffer: anti-human HSP90 (4875), anti-human HSP70 (4872), anti-human HSP60 (4870), and anti-human HSP40 (4868). Other antibodies included anti-human HSP32/HO-1 (MAB3776; R&D Systems; diluted 1:500), anti-hamster HSP27 (06-517; Millipore; diluted 1:200), and anti-pan actin (A-2066; Sigma-Aldrich; diluted 1:200). Antibody binding was visualized by chemiluminescence (SuperSignal West Dura Extended Duration Substrate) using preflashed CL-XPosure Film (Pierce Biotechnology). Each Western blot experiment was performed at least 3 times with similar results. Quantification of Western blots was performed using ImageJ software (NIH). All HSP band intensities were normalized to that of actin. Densitometry data are reported relative to the control (no heat shock, no neomycin) conditions.
Immunofluorescence
Whole mounts.
At the end of the culture period, utricles were fixed for 30 minutes in 4% paraformaldehyde and then washed. Tissue was permeabilized for 1 hour in PBT (PBS plus 0.1% Tween 20). Utricles were blocked for 1 hour in 5% fish gelatin (G7765; Sigma-Aldrich) in PBT. Anti-SOX2 antibody (sc-17320; Santa Cruz Biotechnology) was diluted 1:500 in blocking solution and incubated overnight at 4°C. SOX2 was detected using Alexa 594–conjugated donkey anti-goat IgG (all secondary antibodies were from Invitrogen). Anti-HSP70 (SPA-810; Assay Designs) recognizes inducible HSP70 (HSPA1A and HSPA1B) and does not recognize constitutive HSP70 (HSPA8). This antibody was diluted 1:200 in blocking solution and detected using Alexa 488–conjugated goat anti-mouse IgG. Antimyosin 7a (Proteus BioSciences rabbit polyclonal ab 25-6790, diluted 1:250 in blocking solution; or Developmental Studies Hybridoma Bank mouse monoclonal 138-1, diluted 1:00 in blocking solution) was detected using Alexa 647–conjugated goat anti-rabbit IgG. Axiovision software version 4.8 (Carl Zeiss Microscopy) was used to draw 4–6 boxes (30 μm on a side) in the extrastriolar region of the utricle, and hair cells were counted in each of these boxes. At least 2 regions were sampled from the medial extrastriolar region and 2 from the lateral extrastriolar region (50). We regularly generated a second set of hair cell counts that were performed by a blinded observer, and each time these counts agreed with the original cell counts. Hair cell densities are reported as mean hair cells per unit area.
Sections.
For utricle sections, we modified the cryoembedding protocol described previously for cochlea (51). In brief, utricles were fixed overnight in 4% paraformaldehyde, followed by 3 washes with 1× PBS (pH 7.4) for 15 minutes each. Otoconia were removed using fine forceps (no. 55). Utricles were incubated for 30 minutes in 10% sucrose (Sigma-Aldrich) and then in 15% sucrose with gentle rotation at room temperature. The utricles then remained in 15% sucrose overnight at 4°C with rotation. On the following day, the utricles were transferred into a 1:1 solution of 15% sucrose and Tissue-Tek OCT Compound (4583; Sakura) and incubated overnight with rotation at 4°C. OCT-containing tissue culture plates were placed in a vacuum dessicator for 1 hour to remove air bubbles, and the utricles were then transferred to those wells and incubated at 4°C overnight with rotation. The utricles were then transferred to degassed OCT-containing vinyl cryomolds (4566; Tissue-Tek), oriented, and cryoembedded in a slurry of absolute alcohol and dry ice. Frozen sections (30-μm thickness) were cut on a cryostat (Leica CM), mounted on glass slides (Superfrost Plus; Fisher Scientific), and air dried for 1 to 2 hours before using them for immunostaining.
Sections were treated with 2% PFA for 5 minutes and then washed 3 times (5 minutes each). Sections were kept in blocking solution (0.5% triton X-100 [T-8787; Sigma-Aldrich], 2% BSA [A3294; Sigma-Aldrich], and 0.8% normal goat serum in 1× PBS) for 2 hours. Primary antibodies against myosin 7a and HSP70 (33575, 1:100 in blocking solution; Santa Cruz Biotechnology) were added, and the slides were incubated overnight at 4°C in a humid chamber. The following day, utricles were washed, and secondary antibodies (Alexa-594 goat anti-mouse and Alexa-488 goat anti-rabbit; Invitrogen) were added and incubated for 2 hours at room temperature. Hoechst staining was applied for 30 seconds. The sections were washed and mounted on glass slides using Fluoromount G (SouthernBiotech).
Infection of supporting cells using adenoviruses
For utricle cultures that were to be infected using adenoviruses, otoconia were removed during the dissection using an eyelash tool (no. 113; Ted Pella) (11). We used recombinant adenoviruses to infect supporting cells in the adult mouse utricle. In all cases, adenovirus type 5 (dE1/E3) was used to deliver a transgene under the control of the human CMV promoter (Vector BioLabs). Stock viruses included adenovirus red fluorescent protein (Ad-RFP), adenovirus GFP (Ad-GFP), adenovirus-mCherry (Ad-mCherry), and adenovirus HSP70 (Ad-HSP70). Ad-HSP70-mCherry was generated by PCR amplification of wild-type inducible HSP70 from pDrive-HSP70 (gift from Wolfgang Dillmann, UCSD). This fragment was cloned into the BamHI/EcoRV site of the pDual2CCM-mCherry shuttle vector (Vector BioLabs) to generate a fusion protein in which mCherry was fused to the C-terminal end of HSP70. This expression cassette was then cloned into the pAd viral backbone to generate Ad-HSP70-mCherry. Viral stocks were obtained at titers of 1.0 to 4.0 × 1010 PFU/ml. For infection, utricles were transferred to individual wells of a Nunc MiniTray (136528; Thermo Scientific). Each well contained 15 μl of serum-free culture medium, to which 0.5–2 μl (1.0–4.0 × 107 PFUs) of viral stock was added for 2 hours. After 2 hours, the utricles were transferred back into serum-containing media in the 24-well tissue culture plate. Utricles were cultured for an additional 24 hours before being fixed in 4% paraformaldehyde. Myosin 7a immunochemistry was used to label hair cells, and SOX2 immunochemistry was used to label supporting cells (and a subset of hair cells) (52). Utricles were examined at ×63 magnification by confocal microscopy. Within each experiment, all images were obtained using identical settings for laser power, gain, offset, and zoom. For Ad-HSP70 experiments, utricles were cultured as above in Ad-HSP70, in which the CMV promoter was upstream of HSP70 (1047; Vector BioLabs). Ad-HSP70 was removed from the cultures after 2 hours, and the utricles were cultured for 24 hours before the medium was changed to neomycin-containing (2 mM) medium for an additional 24 hours and fixed. Hair cells were visualized using myosin 7a immunochemistry.
Coculture experiments
The utricle cultures were maintained in an incubator at 37°C in a 5% CO2/95% air environment overnight. Heat shock was performed as described above. Immediately after heat shock, the utricles were transferred to 24-well tissue culture plates containing Transwell inserts (3422; Corning). These inserts have a permeable polycarbonate membrane bottom with an 8.0-μm pore size, thus they allow 2 sets of utricles to be cultured in the same medium without physically contacting one another. Heat-shocked utricles were placed in the upper chamber of the Transwell plate, while nonheat-shocked utricles were placed in the bottom chamber. Using culture medium, gentamicin was added to a final concentration of 5 mM in 1 ml and added to the Transwell dish. The control utricles were cultured in the absence of gentamicin. All utricles were incubated for 24 hours before being fixed and prepared for myosin 7a immunohistochemistry and hair cell counts as described above.
Some cocultures were incubated with an HSP70 function-blocking antibody to deplete the culture medium of HSP70. We tested the specificity of this antibody (SC-33575; Santa Cruz Biotechnology) using control and heat-shocked utricles from HSP70 knockout mice and their wild-type littermates and found that the antibody labeled supporting cells in heat-shocked utricles from Hsp70+/+ mice, but it did not label utricles from heat-shocked (or control) Hsp70–/– mice (data not shown). For these experiments, utricles from wild-type mice were cultured as above. Heat-shocked utricles were placed in the upper chamber of a Transwell plate, and nonheat-shocked utricles were placed in the lower chamber. The medium contained both gentamicin (5 mM) and either the HSP70 function-blocking antibody (SC-33575, 1 μg/ml; Santa Cruz Biotechnology) or a control IgG (AB37361, 1 μg/ml; Abcam). Cocultures were incubated in gentamicin with or without HSP70 function-blocking antibody for 24 hours and then fixed. Hair cells were labeled using antimyosin 7a, and hair cell counts were performed as above.
For cocultures with virus-infected utricles, the utricles were cultured as described above and infected with Ad-HSP70-mCherry in 15 μl of serum-free culture medium for 2 hours. Utricles were then transferred to 1 ml of serum-containing medium and cultured 24 hours in an incubator at 37°C in a 5% CO2/95% air environment. A Transwell insert was placed in control (untreated) or neomycin-containing (3 mM) medium within a 24-well plate as above. The virus-infected utricles were placed in the top chamber of the Transwell plate, while noninfected utricles were placed in the bottom chamber. All utricles were incubated for 24 hours before being fixed and prepared for myosin 7a immunohistochemistry and hair cell counts as described above.
ELISA
Utricles were cultured and heat shocked as described above. Twenty-four hours after heat shock, culture medium was collected from the wells. Utricles (8 per condition) were homogenized and analyzed for HSP70 expression separately from the culture medium. The utricles were homogenized using a mammalian cell lysis kit (MCL-1; Sigma-Aldrich). HSP70 was quantified using an ELISA according to the manufacturer’s instructions (SUV1663; R&D Systems). Using a multimode microplate reader (LB 940; Berthold Technologies Mithras), absorbance levels were read at 450 nm, with a dual wavelength correction at 540 nm. Sample values were calculated from a standard curve generated using recombinant HSP70.
Exogenous HSP 70 experiments
Whole-organ utricle cultures were prepared as described above. The utricles were allowed to recover in culture medium overnight, then 5 μg of purified human recombinant HSP70 (ADI-ESP-555-F; Enzo Life Sciences) or 5 μg of HSA (AB94020; Abcam) was added to the utricle-containing wells for 24 hours. After 24 hours, 3 mM of neomycin (Sigma-Aldrich) was equilibrated for 3 hours in 95% air/5% CO2 and added to the culture wells for 24 hours. The control utricles were maintained in untreated culture medium. The utricles were fixed in 4% paraformaldehyde overnight at 4°C. Specimens were prepared for myosin 7a immunochemistry, and hair cells were counted as described above.
To examine whether exogenous HSP70 is internalized by hair cells or supporting cells, human recombinant HSP70 and BSA were each tagged with FITC according to the manufacturer’s instructions (F6434; Invitrogen). Unbound FITC molecules were removed and FITC-tagged proteins were purified using spin columns provided in the kit. After overnight recovery, the cultured utricles were incubated for an additional 24 hours in 5 μg of FITC-labeled recombinant human exogenous HSP70 (or BSA). Utricles were then fixed in 4% paraformaldehyde at 4°C for 2 hours. Hair cells were labeled with antimyosin 7a as above, and images were taken at ×63 magnification with a Zeiss 780 laser scanning confocal microscope.
Statistics
Each experiment was repeated at least 3 times. Data on hair cell counts are shown in figures as the mean ± SEM hair cell densities (average hair cells per 900 μm2). Data were analyzed by ANOVA with Tukey’s multiple comparison post-hoc tests (IBM SPSS Statistics version 19). Differences were considered significant at P < 0.05.
Study approval
All animal experiments were approved by the IACUC of the Medical University of South Carolina (MUSC) or the National Institute on Deafness and Other Communication Disorders (NIDCD). Experiments using adenoviruses were performed at Biosafety Level II using procedures approved by the MUSC and NIDCD biosafety committees.
Acknowledgments
This work was supported by grants from the NIH (5R01 DC007613 and DC07613-SI) and the NIH/NIDCD Division of Intramural Research (ZIA DC 000079). The authors are grateful to Jonathan Gale for many helpful discussions about supporting cells. Our thanks to Thomas Friedman, Andrew Griffith, and Matthew Kelley for helpful comments on the manuscript.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(8):3577–3587. doi:10.1172/JCI68480.
See the related Commentary beginning on page 3206.
References
- 1.Agrawal Y, Platz EA, Niparko JK. Prevalence of hearing loss and differences by demographic characteristics among US adults: data from the National Health and Nutrition Examination Survey, 1999–2004. Arch Intern Med. 2008;168(14):1522–1530. doi: 10.1001/archinte.168.14.1522. [DOI] [PubMed] [Google Scholar]
- 2.Mukherjea D, et al. Short interfering RNA against transient receptor potential vanilloid 1 attenuates cisplatin-induced hearing loss in the rat. J Neurosci. 2008;28(49):13056–13065. doi: 10.1523/JNEUROSCI.1307-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forge A, Schacht J. Aminoglycoside antibiotics. Audiol Neurootol. 2000;5(1):3–22. doi: 10.1159/000013861. [DOI] [PubMed] [Google Scholar]
- 4.Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 5.Cunningham LL, Brandon CS. Heat shock inhibits both aminoglycoside- and cisplatin-induced sensory hair cell death. J Assoc Res Otolaryngol. 2006;7(3):299–307. doi: 10.1007/s10162-006-0043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Taleb M, Brandon CS, Lee FS, Lomax MI, Dillmann WH, Cunningham LL. Hsp70 inhibits aminoglycoside-induced hair cell death and is necessary for the protective effect of heat shock. J Assoc Res Otolaryngol. 2008;9(3):277–289. doi: 10.1007/s10162-008-0122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taleb M, Brandon CS, Lee FS, Harris KC, Dillmann WH, Cunningham LL. Hsp70 inhibits aminoglycoside-induced hearing loss and cochlear hair cell death. Cell Stress Chaperones. 2009;14(4):427–437. doi: 10.1007/s12192-008-0097-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meriin AB, et al. Protein-damaging stresses activate c-Jun N-terminal kinase via inhibition of its dephosphorylation: a novel pathway controlled by HSP72. Mol Cell Biol. 1999;19(4):2547–2555. doi: 10.1128/mcb.19.4.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mosser DD, et al. The chaperone function of hsp70 is required for protection against stress-induced apoptosis. Mol Cell Biol. 2000;20(19):7146–7159. doi: 10.1128/MCB.20.19.7146-7159.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (hsp70) as an emerging drug target. J Med Chem. 2010;53(12):4585–4602. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brandon CS, Voelkel-Johnson C, May LA, Cunningham LL. Dissection of adult mouse utricle and adenovirus-mediated supporting-cell infection. J Vis Exp. 2012. (61).pii: 3734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt CR, et al. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol. 2004;24(2):899–911. doi: 10.1128/MCB.24.2.899-911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roue G, Pichereau V, Lincet H, Colomer D, Sola B. Cyclin D1 mediates resistance to apoptosis through upregulation of molecular chaperones and consequent redistribution of cell death regulators. Oncogene. 2008;27(36):4909–4920. doi: 10.1038/onc.2008.126. [DOI] [PubMed] [Google Scholar]
- 14.Li CJ, Ning W, Matthay MA, Feghali-Bostwick CA, Choi AM. MAPK pathway mediates EGR-1-HSP70-dependent cigarette smoke-induced chemokine production. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1297–L1303. doi: 10.1152/ajplung.00194.2006. [DOI] [PubMed] [Google Scholar]
- 15.Lahne M, Gale JE. Damage-induced activation of ERK1/2 in cochlear supporting cells is a hair cell death-promoting signal that depends on extracellular ATP and calcium. J Neurosci. 2008;28(19):4918–4928. doi: 10.1523/JNEUROSCI.4914-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bird JE, Daudet N, Warchol ME, Gale JE. Supporting cells eliminate dying sensory hair cells to maintain epithelial integrity in the avian inner ear. J Neurosci. 2010;30(37):12545–12556. doi: 10.1523/JNEUROSCI.3042-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lindquist S. The heat-shock response. Annu Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- 18.Hightower LE, Guidon PT. Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J Cell Physiol. 1989;138(2):257–266. doi: 10.1002/jcp.1041380206. [DOI] [PubMed] [Google Scholar]
- 19.Theriault JR, Adachi H, Calderwood SK. Role of scavenger receptors in the binding and internalization of heat shock protein 70. J Immunol. 2006;177(12):8604–8611. doi: 10.4049/jimmunol.177.12.8604. [DOI] [PubMed] [Google Scholar]
- 20.Calderwood SK, Mambula SS, Gray PJ. Extracellular heat shock proteins in cell signaling and immunity. Ann N Y Acad Sci. 2007;1113:28–39. doi: 10.1196/annals.1391.019. [DOI] [PubMed] [Google Scholar]
- 21.Tytell M, Greenberg SG, Lasek RJ. Heat shock-like protein is transferred from glia to axon. Brain Res. 1986;363(1):161–164. doi: 10.1016/0006-8993(86)90671-2. [DOI] [PubMed] [Google Scholar]
- 22.Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol. 2006;177(11):7849–7857. doi: 10.4049/jimmunol.177.11.7849. [DOI] [PubMed] [Google Scholar]
- 23.Clayton A, Turkes A, Navabi H, Mason MD, Tabi Z. Induction of heat shock proteins in B-cell exosomes. J Cell Sci. 2005;118(pt 16):3631–3638. doi: 10.1242/jcs.02494. [DOI] [PubMed] [Google Scholar]
- 24.Hunter-Lavin C, Davies EL, Bacelar MM, Marshall MJ, Andrew SM, Williams JH. Hsp70 release from peripheral blood mononuclear cells. Biochem Biophys Res Commun. 2004;324(2):511–517. doi: 10.1016/j.bbrc.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 25.Bausero MA, Gastpar R, Multhoff G, Asea A. Alternative mechanism by which IFN-gamma enhances tumor recognition: active release of heat shock protein 72. J Immunol. 2005;175(5):2900–2912. doi: 10.4049/jimmunol.175.5.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lancaster GI, Febbraio MA. Exosome-dependent trafficking of HSP70: a novel secretory pathway for cellular stress proteins. J Biol Chem. 2005;280(24):23349–23355. doi: 10.1074/jbc.M502017200. [DOI] [PubMed] [Google Scholar]
- 27.Asea A, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277(17):15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 28.Yu Q, Kent CR, Tytell M. Retinal uptake of intravitreally injected Hsc/Hsp70 and its effect on susceptibility to light damage. Mol Vis. 2001;7:48–56. [PubMed] [Google Scholar]
- 29.Johnson AD, Berberian PA, Tytell M, Bond MG. Differential distribution of 70-kD heat shock protein in atherosclerosis. Its potential role in arterial SMC survival. Arterioscler Thromb Vasc Biol. 1995;15(1):27–36. doi: 10.1161/01.ATV.15.1.27. [DOI] [PubMed] [Google Scholar]
- 30.Novoselova TV, et al. Treatment with extracellular HSP70/HSC70 protein can reduce polyglutamine toxicity and aggregation. J Neurochem. 2005;94(3):597–606. doi: 10.1111/j.1471-4159.2005.03119.x. [DOI] [PubMed] [Google Scholar]
- 31.Tidwell JL, Houenou LJ, Tytell M. Administration of Hsp70 in vivo inhibits motor and sensory neuron degeneration. Cell Stress Chaperones. 2004;9(1):88–98. doi: 10.1379/CSC-9R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gifondorwa DJ, et al. Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2007;27(48):13173–13180. doi: 10.1523/JNEUROSCI.4057-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guzhova I, et al. In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Res. 2001;914(1–2):66–73. doi: 10.1016/S0006-8993(01)02774-3. [DOI] [PubMed] [Google Scholar]
- 34.Sheller RA, Smyers ME, Grossfeld RM, Ballinger ML, Bittner GD. Heat-shock proteins in axoplasm: high constitutive levels and transfer of inducible isoforms from glia. J Comp Neurol. 1998;396(1):1–11. doi: 10.1002/(SICI)1096-9861(19980622)396:1<1::AID-CNE1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 35.Tytell M. Release of heat shock proteins (Hsps) and the effects of extracellular Hsps on neural cells and tissues. Int J Hyperthermia. 2005;21(5):445–455. doi: 10.1080/02656730500041921. [DOI] [PubMed] [Google Scholar]
- 36.Satoh J, Yamamura T, Kunishita T, Tabira T. Heterogeneous induction of 72-kDa heat shock protein (HSP72) in cultured mouse oligodendrocytes and astrocytes. Brain Res. 1992;573(1):37–43. doi: 10.1016/0006-8993(92)90111-L. [DOI] [PubMed] [Google Scholar]
- 37.Sprang GK, Brown IR. Selective induction of a heat shock gene in fibre tracts and cerebellar neurons of the rabbit brain detected by in situ hybridization. Brain Res. 1987;427(1):89–93. doi: 10.1016/0169-328x(87)90049-0. [DOI] [PubMed] [Google Scholar]
- 38.Pavlik A, Aneja IS. Cerebral neurons and glial cell types inducing heat shock protein Hsp70 following heat stress in the rat. Prog Brain Res. 2007;162:417–431. doi: 10.1016/S0079-6123(06)62020-7. [DOI] [PubMed] [Google Scholar]
- 39.Carr VM. Induced and constitutive heat shock protein expression in the olfactory system — a review, new findings, and some perspectives. J Neurocytol. 2005;34(3–5):269–293. doi: 10.1007/s11068-005-8358-9. [DOI] [PubMed] [Google Scholar]
- 40.Simpson SA, Alexander DJ, Reed CJ. Heat shock protein 70 in the rat nasal cavity: localisation and response to hyperthermia. Arch Toxicol. 2004;78(6):344–350. doi: 10.1007/s00204-004-0541-8. [DOI] [PubMed] [Google Scholar]
- 41.Xu L, Emery JF, Ouyang YB, Voloboueva LA, Giffard RG. Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia. 2010;58(9):1042–1049. doi: 10.1002/glia.20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gratton MA, et al. Noise-induced changes in gene expression in the cochleae of mice differing in their susceptibility to noise damage. Hear Res. 2011;277(1–2):211–226. doi: 10.1016/j.heares.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, et al. CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC-chemokines. Immunity. 2001;15(6):971–983. doi: 10.1016/S1074-7613(01)00242-4. [DOI] [PubMed] [Google Scholar]
- 44.Evdonin AL, Guzhova IV, Margulis BA, Medvedeva ND. Extracellular heat shock protein 70 mediates heat stress-induced epidermal growth factor receptor transactivation in A431 carcinoma cells. FEBS Lett. 2006;580(28–29):6674–6678. doi: 10.1016/j.febslet.2006.11.024. [DOI] [PubMed] [Google Scholar]
- 45.Evdonin A, Kinev A, Tsupkina N, Guerriero V, Raynes DA, Medvedeva N. Extracellular HspBP1 and Hsp72 synergistically activate epidermal growth factor receptor. Biol Cell. 2009;101(6):351–360. doi: 10.1042/BC20080069. [DOI] [PubMed] [Google Scholar]
- 46.Chalmin F, et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest. 2010;120(2):457–471. doi: 10.1172/JCI40483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dix DJ, et al. Targeted gene disruption of Hsp70-2 results in failed meiosis, germ cell apoptosis, and male infertility. Proc Natl Acad Sci U S A. 1996;93(8):3264–3268. doi: 10.1073/pnas.93.8.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cunningham LL. The adult mouse utricle as an in vitro preparation for studies of ototoxic-drug-induced sensory hair cell death. Brain Res. 2006;1091(1):277–281. doi: 10.1016/j.brainres.2006.01.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright GD, Thompson PR. Aminoglycoside phosphotransferases: proteins, structure, and mechanism. Front Biosci. 1999;4:D9–D21. doi: 10.2741/Wright. [DOI] [PubMed] [Google Scholar]
- 50.Desai SS, Ali H, Lysakowski A. Comparative morphology of rodent vestibular periphery. II. Cristae ampullares. J Neurophysiol. 2005;93(1):267–280. doi: 10.1152/jn.00747.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Coleman B, Rickard NA, de Silva MG, Shepherd RK. A protocol for cryoembedding the adult guinea pig cochlea for fluorescence immunohistology. J Neurosci Methods. 2009;176(2):144–151. doi: 10.1016/j.jneumeth.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oesterle EC, Campbell S, Taylor RR, Forge A, Hume CR. Sox2 and JAGGED1 expression in normal and drug-damaged adult mouse inner ear. J Assoc Res Otolaryngol. 2008;9(1):65–89. doi: 10.1007/s10162-007-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]