

Abstract
N,N′-((4-(Dimethylamino)phenyl)methylene)bis(2-phenylacetamide) was discovered by using 3D pharmacophore database searches and was biologically confirmed as a new class of CB2 inverse agonists. Subsequently, 52 derivatives were designed and synthesized through lead chemistry optimization by modifying the rings A–C and the core structure in further SAR studies. Five compounds were developed and also confirmed as CB2 inverse agonists with the highest CB2 binding affinity (CB2 Ki of 22–85 nM, EC50 of 4–28 nM) and best selectivity (CB1/CB2 of 235- to 909-fold). Furthermore, osteoclastogenesis bioassay indicated that PAM compounds showed great inhibition of osteoclast formation. Especially, compound 26 showed 72% inhibition activity even at the low concentration of 0.1 µM. The cytotoxicity assay suggested that the inhibition of PAM compounds on osteoclastogenesis did not result from its cytotoxicity. Therefore, these PAM derivatives could be used as potential leads for the development of a new type of antiosteoporosis agent.
INTRODUCTION
In the past decades, cannabinoid research has witnessed significant evolution, including the discoveries of cannabinoid (CB) receptors, their endogenous ligands, the putative anandamide membrane transporter (AMTa)1 for endocannabinoid cellular uptake and inactivation, the fatty acid amide hydrolase (FAAH),2 and the monoacylglycerol lipase (MAGL)3 enzymes responsible for CB ligand metabolisms. Among the discovered cannabinoid receptors, the subtypes CB1 and CB2 share 48% identity at the amino acid level4,5 and belong to the rhodopsin-like family class A of G-protein-coupled receptors (GPCRs). While the location of the CB receptor subtypes is recently being debated,6–8 it is believed that the CB1 receptor is expressed predominantly in the brain (central receptor for cannabinoids)4 and the CB2 receptor in peripheral cells and tissues derived from the immune system (peripheral receptor for cannabinoids).5
Importantly, the discovered endocannabinoid system is known to play a key role in numerous biological processes and exhibits pharmacological effects in a large spectrum of diseases and disorders, such as pain,9 immune and inflammatory disorders,10,11 cancer,12,13 osteoporosis,14 and cardiovascular and gastrointestinal disorders.15–17 While the investigations were aimed at designing new synthetic molecules that target cannabinoid receptors in past years, cannabinoid (CB) drug research is experiencing a challenge, as the CB1 antagonist rimonabant, launched in 2006 as an anorectic/antiobesity drug, was withdrawn from the European market because of complications of suicide and depression side effects. These undesirable central nervous system side effects are thought to be CB1-receptor-mediated.18 Thus, strategic medicinal chemistry design is needed to develop CB2 selective ligands for therapeutic medications without undesirable side effects.
The therapeutic potential of CB2 receptor modulation has prompted the development of CB2 receptor selective ligands, either as agonists or as antagonists/inverse agonists. Several reviews,19–23 including the latest review from our lab,23 summarize the advances of new CB2 ligands from literature and patents (Figure 1). The first CB2 inverse agonist discovered is 1 (SR144528).24 This compound and 2 (AM630)25 have been extensively used as standards to measure the specificity of various cannabinoid agonists for CB2 in animal models. 3 (JTE-907)26 and 4 (Sch225336)27 received much attention for their immunomodulatory properties against inflammatory disorders in which leukocyte recruitment is involved. Recently, the natural product 5 (MH)28 and several derivatives were shown to selectively target CB2 receptors and act as inverse agonists with anti-inflammatory and antiosteoclastogenic properties. In addition, the pyrimidine derivative 6 (GW842166X) was found to be a potentially promising therapeutic agent for the treatment of inflammatory and neuropathic pain.29 More recently, it was reported that 7 (JWH-133) dose-dependently inhibited intravenous cocaine self-administration, cocaine-enhanced locomotion, and cocaine-enhanced accumbens extracellular dopamine in wild-type and CB1 receptor knockout mice.30 This result suggests that brain CB2 receptors may be a drug target for the pharmacotherapy of drug abuse and addiction. Moreover, the natural product 8 ((E)-β-caryophyllene [(E)-BCP]) was identified as a functional nonpsychoactive CB2 receptor ligand and as a macrocyclic anti-inflammatory cannabinoid in Cannabis.31 Taken together, these published studies show that the CB2 receptor is an attractive target for developing potentially therapeutic ligands.
Figure 1.

Representative CB2 receptor-selective compounds with various chemical scaffolds.
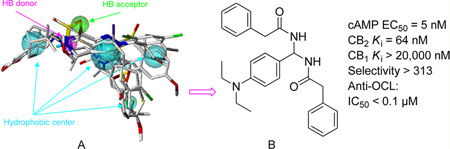
In order to discover novel CB2 selective inverse agonists, we used the genetic algorithm-based pharmacophore alignment (GALAHAD, SYBYL 8.0) approach to derive active pharmacophore models based on the reported CB2 inverse agonists, including 1–4 (Figure 1). A representative pharmacophore model was illustrated in Figure 2, showing one H-bond (HB) acceptor (green), one HB donor (pink), and four hydrophobic (light blue) features. We then performed a 3D database search using the defined pharmacophore query via the UNITY pharmacophore search program (SYBYL 8.0), and identified compound 9 (N,N′-((4-(dimethylamino)phenyl)methylene)-bis(2-phenylacetamide)) (Xie95 or PAM, Figure 2B) as a novel chemotype with selective CB2 activity (CB2 Ki = 777 nM, selectivity index of >26-fold) validated by [3H]CP-55040 radiometric binding assays. On the basis of this promising result, we chose compound 9 as a prototype for further SAR medicinal chemistry studies. In this report, we have designed and synthesized a series of novel PAM derivatives (Scheme 1). Binding activities and effects of these derivatives on the CB2 receptor downstream cAMP production have also been investigated to define their structure–activity relationships and ligand functionality. Our systematic studies led to the identification of five new derivatives (Figure 3) as novel CB2 selective ligands with improved CB2 binding affinity and high selectivity. Importantly, some showed promising inhibition activity to osteoclast cells derived from human bone marrow. The toxicity of PAM compounds on normal human mononuclear cells was also investigated.
Figure 2.

Novel CB2 ligand 9 discovered by 3D pharmacophore database virtual screening search and confirmed by experimental bioassays: (A) pharmacophore query; (B) virtually screened hit 9; (C) 9 validated by [3H]CP-55040 radiometric binding assays showing high CB2 receptor binding affinity, Ki = 777 nM and selectivity (>26-fold).
Scheme 1.

General Synthesis of PAM Derivativesa
aReagents and conditions: (a) concentrated H2SO4, 0 °C, 12 h. (b) Method 1: aldehyde, anhydrous dichloroethane, TMSCl, 70 °C, 3–12 h. (c) Method 2: aldehyde, anhydrous dichloromethane, F3CSO3SiMe3, rt, 12 h. (d) Ethanol, palladium (10%), hydrazine, 70 °C, 3 h. (e) DMF, K2CO3, rt, 12 h.
Figure 3.

Structures of the lead compound 9 and the modified target compounds 18, 26, 27, 30, and 59.
RESULTS AND DISCUSSION
Pharmacophore Modeling and Virtual Screening
A representative 3D pharmacophore model was derived via a genetic algorithm-based pharmacophore alignment method (GALAHAD)32,33 using a set of known CB2 inverse agonists/antagonists including 1, 2, 3, and 4 (Figure 1). The model was then refined and analyzed using our in-house training database that contained non-CB2 ligands and active CB2 ligands. As shown in Figure 2, the final model consisted of one H-bond (HB) acceptor (green), one HB donor (pink), and four hydrophobic (light blue) features. Subsequently, the model was used as a pharmacophore query to screen our in-house structurally diverse chemical database of 540 000 compounds that was constructed from a parent database containing 5.3 million compounds constructed using our published cell-based partition chemistry-space matrix calculation algorithm.34 Out of top ranked 40 compounds, 20 of them were available commercially or via material transfer agreement and then experimentally tested for CB2 binding affinity and selectivity. Among experimentally validated compounds, there were three compounds with good CB2 binding affinities including our lead compound 9.
Pharmacology and SAR Analysis
On the basis of the lead discovered, we have carried out medicinal chemistry modification and synthesized 52 analogues. The binding affinities of these 52 derivatives to CB2 were determined by performing [3H] CP-55,940 radioligand competition binding assays using membrane protein preparations of CHO cells stably expressing human CB2 receptor. The CB1 binding assay was also conducted for those compounds with high CB2 receptor binding potency (Ki < 1000 nM) using membrane proteins harvested from the CHO cells stably transfected with the human CB1 receptor. CB2 selective ligand 1 (SR144528, CB2 inverse agonist) and CB1 ligand 10 (SR141716, CB1 inverse agonist)35 were used as positive controls along with the tested compounds in bioassays experiments. The chemical structures, physiochemical properties, binding activities, and selectivity index are summarized in Tables 1–5.
Table 1.
Radioligand Competition Binding Affinity(Ki)Data of PAM Derivatives
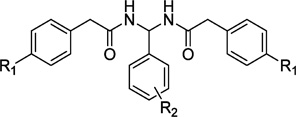 | |||||||
|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | MW | cLogP | Ki (CB2), nMa,b | Ki (CB1, nMa,c | SId |
| 9 | H | p-(CH3)2N- | 401.50 | 4.04 | 777 | >20000 | >26 |
| 11 | H | H- | 358.43 | 3.93 | 9930 | NT | |
| 12 | H | o-F- | 376.42 | 4.08 | 35330 | NT | |
| 13 | H | m-F- | 376.42 | 4.08 | 12670 | NT | |
| 14 | H | p-F- | 376.42 | 4.08 | 10900 | NT | |
| 15 | H | p-Cl- | 392.88 | 4.54 | 3081 | NT | |
| 16 | H | p-Br- | 437.33 | 4.70 | 2226 | NT | |
| 17 | H | p-CH3- | 372.46 | 4.45 | 494 | 109 | |
| 18 | H | p-i-C3H7- | 400.51 | 5.18 | 85 | >20000 | >235 |
| 19 | H | p-CH3O- | 388.46 | 3.78 | 783 | >20000 | >26 |
| 20 | H | p-C2HsO- | 402.49 | 4.13 | 1500 | NT | |
| 21 | H | p-i-C3H7O- | 416.51 | 4.55 | 313 | >20000 | >64 |
| 22 | H | o-CF3- | 426.43 | 4.81 | 11780 | NT | |
| 23 | H | p-CF3- | 426.43 | 4.81 | 596 | >20000 | >34 |
| 24 | H | p-NO2 | 403.43 | 3.87 | NB | NT | |
| 25 | H | p-H2N- | 373.45 | 2.51 | 12550 | NT | |
| 26 | H | p-(C2H5)2N- | 429.55 | 4.76 | 64 | >20000 | >313 |
| 27 | H | p-(C3H7)2N- | 457.61 | 5.80 | 22 | >20000 | >909 |
| 28 | H | p-(C4H9)2N- | 485.66 | 6.69 | 221 | >20000 | >90 |
| 29 | H | p-(benzyl)2N- | 553.69 | 7.33 | 203 | >20000 | >99 |
| 30 | H | p-pyrrolidinyl- | 427.53 | 4.45 | 71 | >20000 | >281 |
| 31 | H | p-piperidyl- | 441.56 | 4.89 | 595 | >20000 | >34 |
| 32 | Cl | H- | 427.32 | 5.14 | NB | NT | |
| 33 | Cl | o-F- | 445.31 | 5.29 | 10850 | NT | |
| 34 | Cl | p-F- | 445.31 | 5.29 | NB | NT | |
| 35 | Cl | p-Cl- | 461.77 | 5.75 | 154 | >20000 | >130 |
| 36 | Cl | p-CH3- | 441.35 | 5.66 | 462 | >20000 | >43 |
| 37 | Cl | p-CH3O- | 457.35 | 4.98 | 310 | >20000 | >65 |
| 38 | Cl | o-CF3- | 495.32 | 6.02 | 158 | >20000 | >127 |
| 39 | Cl | p-CF3- | 495.32 | 6.02 | 101 | >20000 | >198 |
| 40 | Cl | p-NO2- | 472.32 | 5.08 | NB | NT | |
| 41 | CF3 | H- | 494.43 | 5.69 | NB | NT | |
| 42 | CF3 | o-F- | 512.42 | 5.83 | NB | NT | |
| 43 | CF3 | p-F- | 512.42 | 5.83 | NB | NT | |
| 44 | CF3 | p-Cl- | 528.87 | 6.29 | NB | NT | |
| 45 | CF3 | p-CH3- | 508.46 | 6.20 | NB | NT | |
| 46 | CF3 | p-CH3O- | 524.45 | 5.53 | NB | NT | |
| 47 | CF3 | p-CF3- | 562.43 | 6.57 | NB | NT | |
| 1e,f | 2.1 | NT | |||||
| 10e,g | NT | 10.6 | |||||
Binding affinities of compounds for CB1 and CB2 receptor were evaluated using [3H]CP-55,940 radioligand competition binding assay.
NB: no binding, Ki > 20000 nM.
NT: not tested.
SI: selectivity index for CB2, calculated as Ki(CB1)/Ki(CB2) ratio.
The binding affinities of reference compounds were evaluated in parallel with compounds 9, 11–61 under the same conditions.
CB2 reference compound SR144528.
CB1 reference compound SR141716.
Table 5.
Radioligand Competition Binding Affinity(Ki)Data of PAM Derivatives
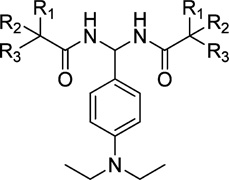 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd | Ri | R2 | R3 | MW | cLog P | Ki (CB2), nMa,b | Ki (CB1), nMa,c | SId |
| 56 | H | CH3 | CH3 | 333.46 | 3.57 | 2636 | NB | |
| 57 | CH3 | CH3 | CH3 | 361.52 | 4.69 | 3553 | NB | |
| 58 | H | H | C3H7 | 361.52 | 4.27 | 182 | >20000 | >109 |
| 59 | H | H | C4H9 | 389.57 | 5.16 | 25 | >20000 | >800 |
| 60 | H | H | C6H13 | 445.68 | 7.9 | 146 | >20000 | >136 |
| 61 | H | H | C8H17 | 501.79 | 10.0 | 160 | >20000 | >125 |
| 1e,f | 2.1 | NT | ||||||
| 1e,g | NT | 10.6 | ||||||
Binding affinities of compounds for CB1 and CB2 receptor were evaluated using [3H]CP-55,940 radioligand competition binding assay.
NB: no binding, Ki > 20000 nM.
NT: not tested.
SI: selectivity index for CB2, calculated as Ki(CB1)/Ki(CB2) ratio.
The binding affinities of reference compounds were evaluated in parallel with compounds 9, 11–61 under the same conditions.
CB2 reference compound SR144528.
CB1 reference compound SR141716.
First, the SAR study was focused on the effect of the side chains on aromatic ring C. Twenty-one compounds were synthesized (11–31, Table 1). The aromatic ring C was modified with substituents that varied in their size, electronic character, and position. Removal of the p-dimethylamino group (compound 11, CB2 Ki = 9930 nM) dramatically decreased the CB2 binding activity. Introducing fluorine atoms to different positions of ring C also lowered the CB2 receptor affinity (compounds 12–14, CB2 Ki of 35 330,12 670, and 10 900 nM, respectively). The CB2 receptor binding affinities of the F-substituted compounds decreased in the order of o-F < m-F < p-F. From these results, we deduced that substitution at the para position of the phenyl ring may play an important role in the CB2 receptor binding activity. This deduction was also confirmed by compounds 22 and 23. Compound 23 bearing p-trifluoromethyl showed improved activity (23, CB2 Ki = 596 nM), while compound 22 bearing o-trifluoromethyl showed dramatically decreased activity (22, CB2 Ki = 11780 nM). Moreover, replacing the p-fluorine with chlorine (compound 15) or bromine (compound 16) relatively increased the activity, but the binding affinities were still weak. While introduction of a methyl group to the para position (compound 17) improved CB2 receptor affinity, unfortunately, compound 17 also had high affinity for the CB1 receptor, the only compound that exhibited significant CB1 receptor binding activity among the 52 compounds (17, CB1 Ki = 109 nM; CB2 Ki = 494 nM). Replacement of p-dimethylamino group with bioisostere isopropyl (compound 18) dramatically improved the binding activity and selectivity (CB2 Ki = 85 nM, selectivity index of >235).
As for the compounds with alkoxy groups (compounds 19–21), the compound bearing methoxy (19) showed similar activity and selectivity compared to the parent compounds, the compound bearing ethoxy (20) showed slightly decreased activity, and the compound bearing isopropoxy (21) showed slightly increased activity. This result indicated that various alkoxy groups were tolerated, but their activity and selectivity for the CB2 receptor were sensitive to the group size. To explore the electronic and steric effects on CB2 binding activity, we introduced a nitro group to the benzene ring (24), but compound 24 completely lost its binding affinity to CB2. Reduction of compound 24 to the corresponding amine resulted in compound 25, which displayed relatively improved activity but was still weak. Replacement of the amine with a diethylamine group, however, resulted in a promising compound (26), which showed much improved activity and selectivity compared to the lead compound (CB2 Ki = 64 nM, selectivity index of >313). When the p-diethylamino group was identified as a better chemical group on ring C, additional substituted amino groups were further studied, resulting in several potent compounds 27–31 with p-dipropylamino, p-dibutylamino, p-pyrrolidinyl, p-piperidyl, and p-dibenzylamino, respectively. Compared with the lead compound 9, these five compounds showed greatly improved activity and selectivity (CB2 Ki of 22–595 nM, selectivity index of 34–909). When compared to compound 26 bearing a diethylamino group, compound 27 with a p-dipropylamino group showed the most potential binding affinity and selectivity (CB2 Ki = 22 nM, selectivity index of >909). Compound 30 with a p-pyrrolidinyl group showed similar activity (CB2 Ki = 71 nM, selectivity index of >28l). The modification result showed that CB2 binding affinity decreased as the size of the functional group at the para position of the benzene ring C increased (compounds 28, 29, and 31). Hence, we conclude that the substituted amino group at the para position plays a significant role in CB2 receptor binding activity and the p-dipropylamino group is optimal.
Subsequently, the SAR was further explored on the variation on aromatic rings A and B by introducing Cl or CF3, resulting in two series of compounds: 32–40 and 41–47. Among the first series compounds bearing Cl on rings A and B (32–40), five compounds (35–39) showed increased CB2 binding affinity and selectivity. All the compounds with CF3 on rings A and B in the second series (41–47) showed no binding activity to CB2 receptors. The results indicated that p-Cl is a better substituent than CF3 and H on rings A and B. Comparison of compound 38 with o-CF3 and compound 39 with p-CF3 further indicates that the para position of the phenyl ring C plays an important role in the CB2 binding activity.
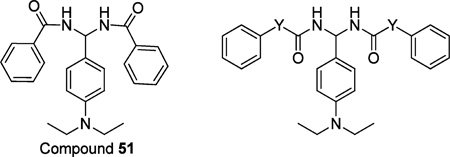
In addition, the distance from ring C to the methylene amide group as well as from rings A and B to the amide group was also explored (compounds 48–50, Table 2; compound 51–53, Table 3). The data indicated that inserting CH2 (compound 48), CH2CH2 (compound 49), or CH=CH double bond (compound 50) between ring C and methylene amide group resulted in a complete loss of activity or weak binding affinity. While removing CH2 from compound 26 or inserting CH2CH2 (compound 52) or CH=CH double bond (compound 53) between rings A/B and the methylene amide group led to a slight decrease in binding affinity, these compounds still showed good CB2 binding affinity and selectivity (167 ≤ CB2 Ki ≤ 688 nM; 29 ≤ selectivity index ≤ 119).
Table 2.
Radioligand Competition Binding Affinity(Ki) Data of PAM Derivatives
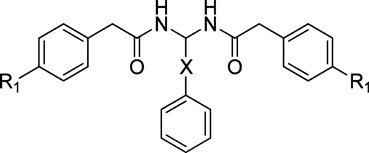 | ||||||
|---|---|---|---|---|---|---|
| compd | Ri | X | MW | cLogP |
Ki(CB2), nMa,b,c |
Ki(CB1), nMa,b,c |
| 48 | H | CH2 | 372.46 | 3.99 | NB | NB |
| 49 | H | CH2CH2 | 386.49 | 4.44 | 9,319 | NB |
| 50 | H | CH=CH | 384.47 | 4.54 | 5,683 | NB |
| 1d,e | 2.1 | NT | ||||
| 10d,f | NT | 10.6 | ||||
Binding affinities of compounds for CB1 and CB2 receptor were evaluated using [3H]CP-55,940 radioligand competition binding assay.
NB: no binding, Ki > 20000 nM.
NT: not tested.
The binding affinities of reference compounds were evaluated in parallel with compounds 9, 11–61 under the same conditions.
CB2 reference compound SR144528.
CB1 reference compound SR141716.
Table 3.
Radioligand Competition Binding Affinity (Ki) Data of PAM Derivatives
 | ||||||
|---|---|---|---|---|---|---|
| compd | Y | MW | cLogP |
Ki (CB2), nMa,b |
Ki (CB1), nMa,c |
SId |
| 51 | 401.50 | 4.80 | 688 | >20000 | >29 | |
| 52 | CH2CH2 | 457.61 | 5.64 | 213 | >20000 | >93 |
| 53 | CH=CH | 453.58 | 5.80 | 167 | >20000 | >119 |
| 1e,f | 2.1 | NT | ||||
| 10e,g | NT | 10.6 | ||||
Binding affinities of compounds for CB1 and CB2 receptor were evaluated using [3H]CP-55,940 radioligand competition binding assay.
NB: no binding, Ki > 20000 nM.
NT: not tested.
SI: selectivity index for CB2, calculated as Ki(CB1)/Ki(CB2) ratio.
The binding affinities of reference compounds were evaluated in parallel with compounds 9, 11–61 under the same conditions.
CB2 reference compound SR144528.
CB1 reference compound SR141716.
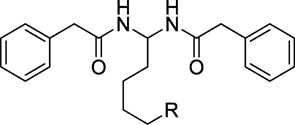
Furthermore, the importance of aromatic ring C in the CB2 binding activity was explored (compounds 54 and 55, Table 4). Replacing ring C with alkyl chain butyl (54) or pentyl (55) led to a complete loss of activity or very weak binding affinity. We conclude that the aromatic ring C plays a significant role in CB2 receptor binding affinity and may be an essential element to retain activity.
Table 4.
Radioligand Competition Binding Affinity (Ki) Data of PAM Derivatives
 | |||||
|---|---|---|---|---|---|
| compd | R | MW | cLogP | Ki (CB2), nMa,b | Ki (CB1, nMa,c |
| 54 | H | 338.44 | 3.75 | 35970 | NT |
| 55 | CH3 | 352.47 | 4.19 | 18200 | NB |
| 1d,e | 2.1 | NT | |||
| 10d,f | NT | 10.6 | |||
Binding affinities of compounds for CB1 and CB2 receptor were evaluated using [3H]CP-55,940 radioligand competition binding assay
NB: no binding, Ki > 20000 nM.
NT: not tested.
The binding affinities of reference compounds were evaluated in parallel with compounds 9, 11–61 under the same conditions.
CB2 reference compound SR144528.
CB1 reference compound SR141716.
After discovering the importance of the aromatic ring C for CB2 binding affinity, we then explored the importance of rings A and B by replacing aromatic rings A and B with different alkyl chains (compounds 56–61, Table 5). The results indicated that replacing the benzyl group with a branched chain isopropyl (compound 56) or tert-butyl group (compound 57) dramatically decreased the CB2 binding affinity, whereas replacing the benzyl group with a long alkyl chain butyl (compound 58) showed slightly decreased affinity. Interestingly, replacement of benzyl with the straight chain pentyl group led to another promising compound 59, which showed greatly improved binding affinity and selectivity (CB2 Ki = 25 nM, selectivity index of >800). To further explore the effect of the alkyl chain, we also replaced aromatic rings A and B with longer chains n-C7H15 (compound 60) and n-C9H19 (compound 61). Compared to compound 59, however, they both showed slightly decreased binding affinity (60, CB2Ki = 146 nM, selectivity index of >136; 61, CB2 Ki = 160 nM, selectivity index of > 125). From these results, we conclude that the aromatic rings A and B may be replaced by an alkyl chain and the pentyl group is optimal.
Cell-Based Functional Bioassay in Vitro
Cellular bioassay was carried out using our published protocol36 to measure the agonistic or antagonistic functional activities of the CB2 selective compounds. Briefly, the cell-based LANCE cAMP assays were performed on 384-well plates using CHO cells stably expressing the CB2 receptors in the presence of phosphodiesterase inhibitor RO20-1724 and adenyl cyclase activator forskolin. Since CB2 is a Gαi-coupled receptor, an agonist inhibits the forskolin-induced cAMP production, resulting in an increase of the LANCE signal. On the other hand, an antagonist or inverse agonist decreases the LANCE signal toward forskolin-induced cAMP accumulation. Therefore, the detected LANCE signal is inversely proportional to cAMP level. As shown in Figure 4, reduction of the LANCE signal occurred with increasing concentrations of compounds 9, 18, 26, 27, 30, 59; and 1. These ligands acted as inverse agonists, indicated by increasing forskolin-induced cAMP production, with EC50 values of 159.1 ± 8.68,4.11 ± 3.66, 5.73 ± 6.37, 28.33 ± 2.54, 17.08 ± 2.1nM, 13.42 ± 2.07, and 13.71 ± 2.81 nM, respectively. Such a phenomenon was not observed with agonsits CP55940 and HU308, which inhibited cAMP production with EC50 values of 23.29 ± 4.17 and 83.81 ± 5.63 nM, respectively. The results clearly indicated that six compounds (9, 18, 26, 27, 30, and 59) indeed behaved as inverse agonists.
Figure 4.

Comparisons of LANCE signal of different CB2 receptor ligands in stably transfected CHO cells expressing human CB2 receptors in a concentration-dependent fashion. EC50 values of compounds 9, 18, 26, 27, 30, 59, and 1 are 159.1 ± 8.68, 4.11 ± 3.66, 5.73 ± 6.37, 28.33 ± 2.54,17.08 ± 2.11, 13.42 ± 2.07, and 13.7 ± 2.81 nM, respectively EC50 for CP-55,940 and HU308 are 23.29 ± 4.17 and 83.81 ± 5.63 nM. Data are the mean ± SEM of one representative experiment of two or more performed in duplicate or triplicate.
Osteoclast Formation Bioactivity
On the basis of binding affinity, selectivity, functionality, and druglikeness studies above, four compounds were selected as top candidates for further biological study. As shown in Figure 5A, we tested the effect of these most promising CB2 ligands on osteoclast (OCL) formation using human nonadherent mononuclear bone marrow cells.37 Each ligand tested induced a concentration-dependent inhibition of osteoclastogenesis. Compared with the known CB2 inverse agonist 1, our compounds exhibited the same or stronger potency in suppressing OCL formation. Especially, compound 26 showed the strongest inhibition activity, with inhibition rates of 72%, 79% and 84% at 0.1, 1, and 10 µM, respectively. Importantly, 26 showed a more potent inhibitory effect than the parent ligand Xie95 (compound 9), suggesting that our medicinal chemistry modification and SAR studies of Xie95 led to overall improved compounds not only for CB2 activity but also for osteoclastogenesis inhibition.
Figure 5.

Inhibition of human osteoclastogenesis by CB2 ligands. (A) Human-bone-marrow-derived mononuclear cells were cultured in a 96-well plate for 3 weeks in the presence of RANKL (50 ng/mL) to form osteoclast-like cells, as described in the Experimental Section. After 3 weeks, the cultures were stained with the 23c6 antibody. 23c6-positive OCLs containing three or more nuclei were scored microscopically. All experiments were performed in triplicate. Results are shown as the mean ± SD. SR = SR144528. The control on the left is vehicle control, and the right one is positive control. (B) Cytotoxic effects of PAM compounds on normal human mononuclear cells. Samples of primary PBMCs (105 cells per well in 96-well plate) from healthy donors were treated in culture for 72 h with the indicated compounds. The viability of cells was determined using trypan blue exclusion assay. The results were presented as the mean ± SD of three assays.
Cytotoxicity Studies Using Normal Human Cells
Our newly discovered compounds showed promising inhibition activity with respect to osteoclastogenesis. To examine whether the impaired osteoclastogenesis in the presence of PAM compounds is due to their cell toxicity, we investigated the cytotoxicity profile of PAM compounds on normal human cells. First, mononuclear cells were isolated from healthy donors. After treatment of these normal cells with the PAM compounds for 3 days, the results indicated that the cell viability was not significantly affected in comparing with the vehicle control group (Figure 5B). The best compound 26 did not show any cytotoxic effects at the concentration (1 µM) of 79% inhibition of osteoclastogenesis, and only slight effects on cell viability were observed at high concentration of 10 µM. These results indicate that our compounds possess favorable therapeutic indexes and the inhibition of human osteoclastogenesis is not a result of their cytotoxicity.
QSAR Pharmacophore Modeling Studies of the New CB2 Ligands
To compare the theoretical SAR models and activity data for further SAR study, 3D QSAR studies were carried out for the PAM analogues to generate CB2 CoMFA SAR models by using our published protocol.38,39 Given its high CB2 affinity, selectivity, and strongest inhibition of osteoclastogenesis, compound 26 was selected as a template compound in our CoMFA studies. To search for preferred conformations of compound 26, molecular dynamic simulations and molecular mechanics (MD/MM) were carried out based on our established computational protocol.40 As described in the Experimental Section, MD simulations were performed with time steps of 1 fs for 300 ps with 1 ps interval recording time, which resulted in 300 conformers sampled after the simulations. All 300 conformations were minimized and converged to four families. Among four representative MD-generated conformers, one conformer had the conformation most similar to the docking pose that resulted from the molecular docking simulation (data not shown) using our refined 3D CB2 receptor model.41 The conformer was then chosen as one of the preferred active templates, and then all compounds from the training and test data sets were aligned to such preferred conformer of compound 26. The final alignments of each set are depicted in Figure 6A,B.
Figure 6.

Overall alignments of training set molecules (A) and test set molecules (B) to compound 26 and CoMFA contour maps of compound 26 showing steric and electrostatic (C) interactions. Sterically (bulk) favored areas are color-coded in green, and sterically unfavored areas are in yellow. Electrostatically (charge) preferred regions are in blue, and red regions are electrostatically unfavored areas.
After molecular alignment, leave-one-out cross-validation (LOOCV) analysis was performed to determine the optimal number of components and to evaluate the predictive ability of the derived CoMFA model which was measured by a cross-validated r2 (rcv2). It is defined as
where SD is the sum of the squared deviations of each biological property value from their mean and PRESS is the sum, over all compounds, of the squared differences between the actual and predicted biological activity values. The LOOCV analysis showed that the optimal number of components was 4 and the rcv2 was 0.52, which was within the range of the generally accepted criterion for statistical validity.
Subsequently, non-cross-validated PLS analysis was performed and an r2 of 0.924 with a standard error of estimate of 0.28 was obtained. Such a result indicates that the trained CoMFA model correlates well between PAM analogue structures and their CB2 receptor affinity values. In order to evaluate the derived CoMFA model’s generalization ability, it was used to predict the CB2 binding activity values of test set compounds that were separated from the training set and hence were not included during the model training. A good correlation coefficient (r2) of 0.76 was obtained from such prediction, and the result demonstrated that the CoMFA model had a good generalization performance on the test set compounds. As shown in Table 6, the predicted pKi values are close to the experimental pKi values for molecules in both training and test sets. Figure 7 shows the relationship between the calculated and experimental pKi values for the non-cross-validated training set predictions and for the test set predictions. The linearity of the plot indicates a very good correlation and the ability of the developed CoMFA model to predict CB2 receptor binding affinities of PAM derivatives.
Table 6.
Experimental (expt) and Predicted (pred) pKi Values of PAM Derivatives in the Training Set and Test Set
| Compd | pKi (expt) | P-Kj (pred) | residual |
|---|---|---|---|
| 9 | 6.109579 | 6.63026 | −0.5207 |
| 11 | 5.003051 | 5.15014 | −0.1471 |
| 12 | 4.451856 | 4.61118 | −0.1593 |
| 13 | 4.897223 | 4.98821 | −0.091 |
| 14 | 4.962574 | 4.85432 | 0.10825 |
| 15a | 5.511308 | 5.58159 | −0.0703 |
| 16a | 5.652475 | 5.72248 | −0.07 |
| 17 | 6.306273 | 5.97563 | 0.33064 |
| 18 | 7.070581 | 6.99702 | 0.07356 |
| 19a | 6.106238 | 5.87445 | 0.23179 |
| 20 | 5.823909 | 6.04419 | −0.2203 |
| 21 | 6.504456 | 6.48389 | 0.02057 |
| 22 | 4.928855 | 4.58176 | 0.3471 |
| 23 | 6.224754 | 6.05444 | 0.17031 |
| 25a | 4.901356 | 5.63905 | −0.7377 |
| 26 | 7.19382 | 6.98267 | 0.21115 |
| 27a | 7.657577 | 7.06706 | 0.59052 |
| 28 | 6.655608 | 7.11863 | −0.463 |
| 29a | 6.692504 | 6.60822 | 0.08428 |
| 30a | 7.148742 | 6.83115 | 0.31759 |
| 31a | 6.225483 | 6.94563 | −0.7201 |
| 33 | 4.96457 | 5.46751 | −0.5029 |
| 35 | 6.812479 | 6.5766 | 0.23588 |
| 36 | 6.335358 | 6.6504 | −0.315 |
| 37a | 6.508638 | 6.86546 | −0.3568 |
| 38 | 6.801343 | 6.47965 | 0.32169 |
| 39 | 6.995679 | 6.92502 | 0.07066 |
| 49 | 5.030631 | 5.18212 | −0.1515 |
| 50 | 5.245422 | 5.10916 | 0.13626 |
| 51 | 6.162412 | 6.11595 | 0.04646 |
| 52 | 6.67162 | 6.78351 | −0.1119 |
| 53a | 6.777284 | 6.84095 | −0.0637 |
| 54 | 4.44406 | 4.26222 | 0.18184 |
| 55 | 4.739929 | 4.82097 | −0.081 |
| 56 | 5.579055 | 5.64843 | −0.0694 |
| 57a | 5.449405 | 5.87713 | −0.4277 |
| 58a | 6.739929 | 6.67326 | 0.06667 |
| 59 | 7.60206 | 7.09237 | 0.50969 |
| 60 | 6.835647 | 6.86835 | −0.0327 |
| 61 | 6.79588 | 6.69406 | 0.10182 |
Molecules from the test set.
Figure 7.

Plots of CoMFA-calculated and experimental binding affinity values (pKi) for the training and test sets.
To further predict favorable and unfavorable regions of PAM derivatives for CB2 receptor binding activity, CoMFA contour maps were derived. In particular, CoMFA contour maps depict the color-coded electrostatic and steric regions around the molecules that associate with ligand biological activities. Green regions indicate favorable steric interactions that enhance binding affinity, whereas yellow regions display unfavorable steric interactions. On the other hand, the blue and red regions show preferred and not-preferred electrostatic interactions, respectively. As shown in Figure 6C, there is a sterically preferred region near the p-dimethylamino group, which means the hydrophobic pharmacophore feature in this part of the molecule is expected to enhance CB2 receptor binding affinity. In fact, such a hydrophobic moiety may interact and fit well in the previously suggested hydrophobic pocket within transmembrane regions 3, 5, 6, and 7.36,42–44 Moreover, this finding is consistent with our previous CoMFA studies,39 which showed that the presence of a steric bulky group enhanced the CB2 receptor binding activity and selectivity. On the other hand, electrostatic interactions are not preferred near the p-dimethylamino group as highlighted by a red region. This is congruent with the chemistry modifications of compounds 25 and 40 with p-NH2 and p-NO2 groups, respectively, which lost CB2 binding activity. Once a hydrophobic feature was reintroduced, however, the CB2 affinity and selectivity were restored, as demonstrated by compounds 17, 18, 21, 26, 27, 28, 29, 30, and 31. Therefore, our CoMFA studies corroborate our SAR hypothesis that aromatic ring C plays an important role in CB2 receptor binding activity and introducing a hydrophobic feature at the para position of ring C is expected to enhance CB2 receptor activity and selectivity.
CONCLUSION
We reported PAM as a novel chemotype with selective CB2 receptor binding activity. In our SAR studies we have synthesized 52 new PAM derivatives designed through variations of the aromatic rings A–C and the substituents of different positions on these three rings. The SAR analyses reveal that (i) the para-substituted amino group on ring C plays a significant role in CB2 receptor binding activity, a variety of functional groups was tolerated, and the p-dipropylamino group is optimal, (ii) p-Cl is a much better substituent than CF3 and H on rings A and B, and aromatic rings A and B may be replaced by alkyl chains with the pentyl group being optimal, and (iii) aromatic ring C is an essential element to retain compound potency to CB2. Among the derivatives, five compounds 18, 26, 27, 30, and 59 were confirmed as CB2 inverse agonists with the strongest CB2 receptor binding affinity and best selectivity. SAR pharmacophoric studies also confirmed our SAR findings that aromatic ring C is important for CB2 receptor activity and a hydrophobic feature at the ring C’s para position is crucial to improve CB2 activity and selectivity of the PAM analogues. The results were congruent by chemistry, bioassay validation, and computer modeling studies. More importantly, osteoclastogenesis assay indicated that PAM compounds have promising inhibition activity to osteoclast cells derived human bone marrow. The most promising compound, 26, showed 72% inhibition activity even at the low concentration of 0.1 µM. The inhibition of human osteoclastogenesis is not due to cytotoxic effects. Therefore, these PAM derivatives could be used as potential leads for the development of a new type of antiosteoporosis agent. Overall, the data presented here show that PAM is a new scaffold different from the existing CB2 ligands and is promising for the design of new selective CB2 receptor inverse agonists for further CB2 signaling and antiosteoclast studies.
EXPERIMENTAL SECTION
Pharmacophore Modeling and Virtual Screening
A genetic algorithm-based pharmacophore alignment (GALAHAD) approach32,33 was used to derive a 3D pharmacophore model based on known CB2 antagonists including 1, 2, 3, and 4 (Figure 2). The pharmacophore model was then examined and refined using our in-house training database which contained a mixture of decoy molecules and known CB2 ligands. The derived pharmacophore model was subsequently used as a query in the UNITY program33 to perform virtual screening on a structurally diverse representative compound database.34 Top ranked screened compounds from the pharmacophore search were obtained commercially or via material transfer agreement (MTA) to be experimentally validated for CB2 binding activity and selectivity.
Chemistry
All reagents were purchased from commercial sources and used without further purification. Analytical thin-layer chromatography (TLC) was performed on SiO2 plates on alumina. Visualization was accomplished by UV irradiation at 254 nm. Preparative TLC was conducted using preparative silica gel TLC plates (1000 µm, 20 cm × 20 cm). Flash column chromatography was performed using the Biotage Isolera flash purification system with SiO2 60 (particle size 0.040–0.055 mm, 230–400 mesh). 1H NMR was recorded on a Bruker 400 MHz spectrometer. Splitting patterns are indicated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; br, broad peak. Purity of all final derivatives for biological testing was confirmed to be >95% as determined using the following conditions: a Shimadzu HPLC instrument with a Hamilton reversed phase column (HxSil, C18, 3 µm, 2.1 mm × 50 mm (H2)); eluent A consisting of 5% CH3CN in H2O; eluent B consisting of 90% CH3CN in H2O; flow rate of 0.2 mL/ min; UV detection, 254 and 214 nm.
General Procedure for Synthesis of 2-Phenylacetamide Building Blocks
2-Phenylacetamide
Benzyl cyanide (5 g, 42.7 mmol) was added slowly to concentrated sulfuric acid (20 mL) cooled by a water–ice bath. The solution was stirred overnight. The reaction mixture was poured into ice–water and neutralized with 20% NaOH. The aqueous phase was extracted by ethyl acetate (3 × 15 mL). The combined organic layer was washed with water (3 × 10 mL) and brine (3 × 10 mL), dried over anhydrous MgSO4, filtered, concentrated under reduced pressure, and the residue was recrystallized from ethyl acetate and hexane to give the title compound (4.5 g, 78%). 1H NMR (400 MHz, DMSO-d6) δ 7.54 (s, 1H), 7.20–7.32 (m, 5H), 6.87 (s, 1H), 3.38 (s, 2H).
2-(4-Chlorophenyl)acetamide
1H NMR (400 MHz, DMSO-d6) δ 7.49 (s, 1H), 7.34–7.35 (m, 2H), 7.26–7.27 (m, 2H), 6.92 (s, 1H), 3.34–3.37 (m, 2H).
(4-(Trifluoromethyl)phenyl)acetamide
1H NMR (400 MHz, DMSO-d6) δ 7.65 (d, J = 8.0 Hz, 2H), 7.58 (s, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.00 (s, 1H), 3.51 (s, 2H).
General Procedure for the Coupling Reaction between Amide and Aldehyde
General Method 1
N,N′-((4-(Dimethylamino)phenyl)methylene)bis(2-phenylacetamide) (9)
To a suspension of 4-(dimethylamino)benzaldehyde (149 mg, 1 mmol) and 2-phenylacetamide (270 mg, 2 mmol) in anhydrous dichloroethane (2 mL) was added TMSCl (216 mg, 2 mmol).45 The mixture was heated at 70 °C for 12 h, then cooled to room temperature and the crude product precipitated from the solution. The crude product was recrystallized with methanol and hexane to give the final product (140 mg, 35%). 1H NMR (400 MHz, DMSO-d6) δ 8.89 (d, J = 8.0 Hz, 2H), 7.59 (s, 2H), 7.41 (d, J = 8.8 Hz, 2H), 7.21–7.32 (m, 10H), 6.54 (t, J = 8.0 Hz, 1H), 3.52 (dd, J = 14.0, 15.6 Hz, 4H), 3.06 (s, 6H). LC–MS (ESI): m/z 402.1 (M + H)+. HRMS (ESI) for C25H28N3O2 (MH+): calcd, 402.2176; found, 402.2179.
N,N′-(Phenylmethylene)bis(2-phenylacetamide) (11)
Compound 11 was prepared from 2-phenylacetamide and benzaldehyde using method1. Yield: 67%. 1H NMR (400 MHz, DMSO-d6)δ 8.78 (d, J = 7.2 Hz, 2H), 7.21–7.35 (m, 15H), 6.55 (t, J = 7.8 Hz, 1H), 3.50 (dd, J = 13.8, 20.4 Hz, 4H). LC–MS (ESI): m/z 359.3 (M + H)+.
N,N′-((2-Fluorophenyl)methylene)bis(2-phenylacetamide) (12)
Compound 12 was prepared from 2-phenylacetamide and 2-fluorobenzaldehyde using method 1. Yield: 64%. 1H NMR (400 MHz, DMSO-d6) δ 8.87 (d, J = 7.8 Hz, 2H), 7.44 (t, J = 7.8 Hz, 1H), 7.36 (q, J = 6.6 Hz, 1H), 7.17–7.29 (m, 12H), 6.74 (t, J = 7.8 Hz, 1H), 3.48 (dd, J = 14.4, 24.0 Hz, 4H). LC–MS (ESI): m/z 377.2 (M + H)+.
N,N′-((4-Fluorophenyl)methylene)bis(2-phenylacetamide) (14)
Compound 14 was prepared from 2-phenylacetamide and 4-fluorobenzaldehyde using method 1.Yield: 72%. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (d, J = 8.0 Hz, 2H), 7.16–7.36 (m, 14H), 6.54 (t, J = 8.0 Hz, 1H), 3.52 (dd, J = 14.4, 15.6 Hz, 4H). LC–MS (ESI): m/z 377.2 (M + H)+.
N,N′-((4-Chlorophenyl)methylene)bis(2-phenylacetamide) (15)
Compound 15 was prepared from 2-phenylacetamide and 4-chlorobenzaldehyde using method 1. Yield: 71%. 1H NMR (400 MHz, DMSO-d6) δ 8.83 (d, J = 7.8 Hz, 2H), 7.41 (d, J = 7.8 Hz, 2H), 7.21–7.32 (m, 12H), 6.51 (t, J = 7.8 Hz, 1H), 3.50 (dd, J = 14.4, 17.4 Hz, 4H). LC–MS (ESI): m/z 393.2 (M + H)+.
N,N′-(p-Tolylmethylene)bis(2-phenylacetamide) (17)
Compound 17 was prepared from 2-phenylacetamide and 4-methylbenzaldehyde using method 1.Yield: 70%. 1H NMR (400 MHz, DMSO-d6) δ8.71 (d, J = 8.0 Hz, 2H), 7.13–7.32 (m, 14H), 6.52 (t, J = 8.0 Hz, 1H), 3.51 (dd, J = 14.4, 15.6 Hz, 4H), 2.29 (S, 3H). LC–MS (ESI): m/z 373.1 (M +H)+.
N,N′-((4-Methoxyphenyl)methylene)bis(2-phenylacetamide) (19)
Compound 19 was prepared from 2-phenylacetamide and 4-methoxybenzaldehyde using method 1. Yield: 62%. 1H NMR (400 MHz, DMSO-d6) δ 8.70 (d, J = 7.6 Hz, 2H), 7.21–7.31 (m, 12H), 6.89 (d, J = 8.8 Hz, 2H), 6.51 (t, J = 8.0 Hz, 1H), 3.74 (s, 3H), 3.50 (dd, J = 14.0, 17.2 Hz, 4H). LC–MS (ESI): m/z 389.1 (M + H)+.
N,N′-((2-(Trifluoromethyl)phenyl)methylene)bis(2-phenylaceta-mide)(22)
Compound 22 was prepared from 2-phenylacetamide and 2-(trifluoromethyl)benzaldehyde using method 1. Yield: 70%. 1H NMR (400 MHz, DMSO-d6) δ 8.87 (d, J = 7.2 Hz, 2H), 7.78 (d, J = 7.6 Hz, 1H), 7.68–7.73 (m, 2H), 7.54 (t, J = 7.6 Hz, 1H), 7.19–7.30 (m, 10H), 6.83 (t, J = 6.8 Hz, 1H), 3.46 (dd, J = 14.0, 17.6 Hz, 4H). LC–MS (ESI): m/z 427.0 (M + H)+.
N,N′-((4-(Trifluoromethyl)phenyl)methylene)bis(2-phenylaceta-mide)(23)
Compound 23 was prepared from 2-phenylacetamide and 4-(trifluoromethyl)benzaldehyde using method 1. Yield: 75%. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (d, J = 7.6 Hz, 2H), 7.72 (d, J = 7.6 Hz, 2H), 7.51 (d, J = 7.6 Hz, 2H), 7.25–7.30 (m, 10H), 6.58 (t, J = 7.2 Hz, 1H), 3.53 (s, 4H). LC–MS (ESI): m/z 427.2 (M + H)+.
N,N′-((4-Nitrophenyl)methylene)bis(2-phenylacetamide)(24)
Compound 24 was prepared from 2-phenylacetamide and 4-nitro-benzaldehyde using method 1. Yield: 84%. 1H NMR (400 MHz, DMSO-d6) δ 8.98 (d, J = 7.6 Hz, 2H), 8.20–8.23 (m, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.23–7.32 (m, 10H), 6.58 (t, J = 7.6 Hz, 1H), 3.53 (s, 4H). LC–MS (ESI): m/z 404.1 (M + H)+.
N,N′-((4-(Dipropylamino)phenyl)methylene)bis(2-phenylaceta-mide)(27)
Compound 27 was prepared from 2-phenylacetamide and 4-(dipropylamino)benzaldehyde using method 1. Yield: 15%. 1H NMR (400 MHz, CD3OD) δ 7.25–7.30 (m, 10H), 7.10 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 8.8 Hz, 2H), 6.57 (s, 1H), 3.56 (s, 4H), 3.24–3.26 (m, 4H), 1.54–1.64 (m, 4H), 0.93 (t, J = 7.6 Hz, 6H). LC-MS (ESI): m/z 458.2 (M + H)+. HRMS (ESI) for C29H36N3O2 (MH+): calcd, 458.2802; found, 458.2792.
N,N′-((4-(Pyrrolidin-1-yl)phenyl)methylene)bis(2-phenylaceta-mide)(30)
Compound 30 was prepared from 2-phenylacetamide and 4-(pyrrolidin-1-yl)benzaldehyde using method 1. Yield: 12%. 1H NMR (400 MHz, DMSO-d6) δ 8.58 (d, J = 8.0 Hz, 2H), 7.20–7.31 (m, 10H), 7.08 (d, J = 8.4 Hz, 2H), 6.42–6.49 (m, 3H), 3.44–3.45 (m, 4H), 3.18– 3.20 (m, 4H), 1.93–1.96 (m, 4H). LC–MS (ESI): m/z 428.2 (M +H)+. HRMS (ESI) for C27H30N3O2 (MH+): calcd, 428.2333; found, 428.2328.
N,N′-(Phenylmethylene)bis(2-(4-chlorophenyl)acetamide)(32)
Compound 32 was prepared from 2-(4-chlorophenyl)acetamide and benzaldehyde using method 1. Yield: 52%. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (d, J = 7.8 Hz, 2H), 7.25–7.41 (m, 13H), 6.52 (t, J = 7.8 Hz, 1H), 3.50 (s, 4H). LC–MS (ESI): m/z 427.1 (M + H)+.
N,N′-((2-Fluorophenyl)methylene)bis(2-(4-chlorophenyl)-acetamide)(33)
Compound 33 was prepared from 2-(4-chlorophenyl)acetamide and 2-fluorobenzaldehyde using method 1. Yield: 63%. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (d, J = 7.2 Hz, 2H), 7.45 (t, J = 7.2 Hz, 1H), 7.33–7.37 (m, 5H), 7.25–7.26 (m, 4H), 7.18– 7.21 (m, 2H), 6.73 (t, J = 7.2 Hz, 1H), 3.48 (s, 4H). LC–MS (ESI): m/z 445.0 (M + H)+.
N,N′-((4-Fluorophenyl)methylene)bis(2-(4-chlorophenyl)-acetamide)(34)
Compound 34 was prepared from 2-(4-chlorophenyl)acetamide and 4-fluorobenzaldehyde using method 1. Yield: 67%. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (d, J = 7.6 Hz, 2H), 7.34–7.37 (m, 6H), 7.27 (d, J = 8.4 Hz, 4H), 7.19 (t, J = 8.8 Hz, 2H), 6.51 (t, J = 8.0 Hz, 1H), 3.51 (s, 4H). LC–MS (ESI): m/z 444.9 (M + H)+.
N,N′-((4-Chlorophenyl)methylene)bis(2-(4-chlorophenyl)-acetamide)(35)
Compound 35 was prepared from 2-(4-chlorophenyl)acetamide and 4-chlorobenzaldehyde using method 1. Yield: 70%. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (d, J = 7.8 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 7.31–7.35 (m, 6H), 7.26 (d, J = 8.4 Hz, 4H), 6.47 (t, J = 7.8 Hz, 1H), 3.50 (s, 4H). LC–MS (ESI): m/z 460.8 (M + H)+.
N,N′-(p-Tolylmethylene)bis(2-(4-chlorophenyl)acetamide)(36)
Compound 36 was prepared from 2-(4-chlorophenyl)acetamide and 4-methylbenzaldehyde using method 1. Yield: 88%. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.4 Hz, 4H), 7.28 (d, J = 8.4 Hz, 4H), 7.17 (q, J = 8.0 Hz, 4H), 6.50 (t, J = 7.6 Hz, 1H), 3.51 (s, 4H), 2.29 (s, 3H). LC–MS (ESI): m/z 441.3 (M + H)+.
N,N′-((4-Methoxyphenyl)methylene)bis(2-(4-chlorophenyl)-acetamide)(37)
Compound 37 was prepared from 2-(4-chlorophenyl)acetamide and 4-methoxybenzaldehyde using method 1. Yield: 91%. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (d, J = 7.6 Hz, 2H), 7.34 (d, J = 8.8 Hz, 4H), 7.26 (d, J = 8.4 Hz, 4H), 7.21 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 8.8 Hz, 2H), 6.46 (t, J = 8.0 Hz, 1H), 3.74 (s, 3H), 3.49 (s, 4H). LC-MS (ESI): m/z 457.2 (M + H)+.
N,N′-((2-(Trifluoromethyl)phenyl)methylene)bis(2-(4-chlorophenyl)acetamide)(38)
Compound 38 was prepared from 2-(4-chlorophenyl)acetamide and 2-(trifluoromethyl)benzaldehyde using method 1. Yield: 82%.1H NMR (400 MHz, DMSO-d6) δ 8.04 (d,J = 6.0 Hz, 2H), 7.81 (d, J = 7.8 Hz, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.52 (t, J = 7.8 Hz, 1H), 7.27–7.30 (m, 8H), 7.06 (t, J = 7.2 Hz, 1H), 3.52 (s, 4H). LC–MS (ESI): m/z 495.0 (M + H)+.
N,N′-((4-(Trifluoromethyl)phenyl)methylene)bis(2-(4-chlorophenyl)acetamide)(39)
Compound 39 was prepared from 2-(4-chlorophenyl)acetamide and 4-(trifluoromethyl)benzaldehyde using method 1. Yield: 85%.1H NMR (400 MHz, DMSO-d6) δ 8.96 (d,J = 7.6 Hz, 2H), 7.73 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 4H), 7.28 (d, J = 8.4 Hz, 4H), 6.57 (t, J = 7.6 Hz, 1H), 3.54 (s, 4H). LC–MS (ESI): m/z 495.2 (M + H)+.
N,N′-((4-Nitrophenyl)methylene)bis(2-(4-chlorophenyl)-acetamide)(40)
Compound 40 was prepared from 2-(4-chlorophenyl)acetamide and 4-nitrobenzaldehyde using method 1. Yield: 90%. 1H NMR (400 MHz, DMSO-d6) δ 9.02 (d, J = 8.0 Hz, 2H), 8.24–8.25 (m, 2H), 7.56–7.58 (m, 2H), 7.34–7.37 (m, 4H), 7.26–7.29 (m, 4H), 6.55 (t, J = 8.0 Hz, 1H), 3.53 (S, 4H). LC–MS (ESI): m/z 472.0 (M + H)+.
N,N′-(Phenylmethylene)bis(2-(4-(trifluoromethyl)phenyl)-acetamide)(41)
Compound 41 was prepared from 2-(4-(trifluoromethyl)phenyl)acetamide and benzaldehyde using method 1. Yield: 83%. 1H NMR (400 MHz, DMSO-d6) δ 8.94 (d, J = 7.8 Hz, 2H), 7.63 (d, J = 7.8 Hz, 4H), 7.48 (d, J = 8.4 Hz, 4H), 7.29–7.37 (m, 5H), 6.55 (t, J = 7.8 Hz, 1H), 3.63 (s, 4H). LC–MS (ESI): m/z 495.1 (M + H)+.
N,N′-((2-Fluorophenyl)methylene)bis(2-(4-(trifluoromethyl)-phenyl)acetamide)(42)
Compound 42 was prepared from 2-(4-(trifluoromethyl)phenyl)acetamide and 2-fluorobenzaldehyde using method 1. Yield: 86%. 1H NMR (400 MHz, DMSO-d6) δ 8.99 (d, J = 7.2 Hz, 2H), 7.63 (d, J = 7.8 Hz, 4H), 7.46 (d, J = 7.8 Hz, 5H), 7.36–7.40 (m, 1H), 7.21 (t, J = 7.8 Hz, 2H), 6.74 (t, J = 7.8 Hz, 1H), 3.60 (s, 4H). LC–MS (ESI): m/z 513.0 (M + H)+.
N,N′-((4-Fluorophenyl)methylene)bis(2-(4-(trifluoromethyl)-phenyl)acetamide)(43)
Compound 43 was prepared from 2-(4-(trifluoromethyl)phenyl)acetamide and 4-fluorobenzaldehyde using method 1. Yield: 90%. 1H NMR (400 MHz, DMSO-d6) δ 8.95 (d, J = 8.4 Hz, 2H), 7.63 (d, J = 8.4 Hz, 4H), 7.47 d, J = 8.4 Hz, 4H), 7.37 (dd, J = 5.4, 8.4 Hz, 2H), 7.19 (t, J = 8.4 Hz, 2H), 6.51 (t, J = 7.8 Hz, 1H), 3.62 (s, 4H). LC–MS (ESI): m/z 513.2 (M + H)+.
N,N′-((4-Chlorophenyl)methylene)bis(2-(4-(trifluoromethyl)-phenyl)acetamide)(44)
Compound 44 was prepared from 2-(4-(trifluoromethyl)phenyl)acetamide and 4-chlorobenzaldehyde using method 1. Yield: 85%.1H NMR (400 MHz, DMSO-d6) δ 8.96 (d,J = 7.8 Hz, 2H), 7.63 (d, J = 7.2 Hz, 4H), 7.46 (d, J = 7.8 Hz, 4H), 7.43 (dd, J = 1.8, 8.4 Hz, 2H), 7.34–7.35 (m, 2H), 6.50 (t, J = 7.2 Hz, 1H), 3.62 (s, 4H). LC–MS (ESI): m/z 529.0 (M + H)+.
N,N′-(p-Tolylmethylene)bis(2-(4-(trifluoromethyl)phenyl)-acetamide)(45)
Compound 45 was prepared from 2-(4-(trifluoromethyl)phenyl)acetamide and 4-methylbenzaldehyde using method 1. Yield: 81%.1H NMR (400 MHz, DMSO-d6) δ 8.87 (d,J = 8.4 Hz, 2H), 7.63 (d, J = 7.8 Hz, 4H), 7.47 (d, J = 8.4 Hz, 4H), 7.20 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 7.8 Hz, 2H), 6.49 (t, J = 7.8 Hz, 1H), 3.61 (s, 4H), 2.28 (s, 3H). LC–MS (ESI): m/z 509.1 (M + H)+.
N,N′-((4-Methoxyphenyl)methylene)bis(2-(4-(trifluoromethyl)-phenyl)acetamide)(46)
Compound 46 was prepared from 2-(4-(trifluoromethyl)phenyl)acetamide and 4-methoxybenzaldehyde using method 1. Yield: 86%.1H NMR (400 MHz, DMSO-d6) δ 8.85 (d, J = 7.8 Hz, 2H), 7.63 (d, J = 7.8 Hz, 4H), 7.47 (d, J = 7.8 Hz, 4H),7.24 (d, J = 9.0 Hz, 2H), 6.91 (d, J = 8.4 Hz, 2H), 6.48 (t, J = 7.8 Hz, 1H), 3.74 (s, 3H), 3.61(s, 4H). LC–MS (ESI): m/z 525.1 (M + H)+.
N,N′-((4-(Trifluoromethyl)phenyl)methylene)bis(2-(4-(trifluoromethyl)phenyl)acetamide)(47)
Compound 47 was prepared from 2-(4-(trifluoromethyl)phenyl)acetamide and 4-(trifluoromethyl)-benzaldehyde using method 1. Yield: 74%. 1H NMR (400 MHz, DMSO-d6) δ 9.08 (d, J = 7.2 Hz, 2H), 7.73 (d, J = 7.8 Hz, 2H), 7.63 (d, J = 7.8 Hz, 4H), 7.56 (d, J = 7.8 Hz, 2H), 7.48 (d, J = 7.8 Hz, 4H), 6.60 (t, J = 7.8 Hz, 1H), 3.35–3.43 (m, 4H). LC–MS (ESI): m/z 562.9 (M + H)+.
N,N′-((4-(Diethylamino)phenyl)methylene)dibenzamide (51)
Compound 51 was prepared from benzamide and 4-(diethylamino)-benzaldehyde using method 1. Yield: 73%. 1H NMR (400 MHz, CD3OD) δ 9.24 (d, J = 7.6 Hz, 1H), 7.88–7.93 (m, 4H), 7.81 (d, J = 8.8 Hz, 2H), 7.46–7.65 (m, 9H), 7.20 (m, 1H), 3.70–3.81 (m, 4H), 1.17 (t, J = 7.2 Hz, 6H). LC–MS (ESI): m/z 402.2 (M + H)+. HRMS (ESI) for C25H28N3O2 (MH+): calcd, 402.2176; found, 402.2167.
N,N′-((4-(Diethylamino)phenyl)methylene)bis(3-phenylpropana-mide)(52)
Compound 52 was prepared from 3-phenylpropanamide and 4-(diethylamino)benzaldehyde using method 1. Yield: 66%. 1H NMR (400 MHz, CD3OD) δ 8.29–8.30 (m, 2H), 7.17–7.29 (m, 10H), 6.92 (d, J = 8.4 Hz, 2H), 6.56 (d, J = 8.4 Hz, 2H), 6.46 (t, J = 8.0 Hz, 1H), 2.82 (t, J = 7.6 Hz, 4H), 2.42–2.47 (m, 4H), 1.07 (t, J = 6.8 Hz, 6H). LC–MS (ESI): m/z 458.2 (M + H)+. HRMS (ESI) for C29H36N3O2 (MH+): calcd, 458.2802; found, 458.2795.
N,N′-((4-(Diethylamino)phenyl)methylene)bis(3-phenylacryla-mide)(53)
Compound 53 was prepared from cinnamamide and 4-(diethylamino)benzaldehyde using method 1. Yield: 68%. 1H NMR (400 MHz, CD3OD) δ 8.68–8.70 (m, 2H), 7.38–7.58 (m, 12H), 7.19 (d, J = 8.8 Hz, 2H), 6.79 (d, J = 16.0 Hz, 2H), 6.66–6.68 (m, 3H), 3.29–3.35 (m, 4H), 1.08 (t, J = 7.2 Hz, 6H). LC–MS (ESI): m/z 454.2 (M + H)+. HRMS (ESI) for C29H32N3O2 (MH+): calcd, 454.2489; found, 454.2487.
N,N′-((4-(Diethylamino)phenyl)methylene)bis(2-methylpropana-mide)(56)
Compound 56 was prepared from isobutyramide and 4-(diethylamino)benzaldehyde using method 1. Yield: 80%. 1H NMR (400 MHz, CD3OD) δ 7.18 (d, J = 8.4 Hz, 2H), 6.70–6.73 (m, 2H), 6.56 (s, 1H), 3.35–3.50 (m, 2H), 2.47–2.54 (m, 4H), 1.13–1.16 (m, 12H). LC–MS (ESI): m/z 334.2 (M + H)+. HRMS (ESI) for C19H32N3O2 (MH+): calcd, 334.2489; found, 334.2483.
N,N′-((4-(Diethylamino)phenyl)methylene)bis(2,2-dimethylpro-panamide)(57)
Compound 57 was prepared from pivalamide and 4-(diethylamino)benzaldehyde using method 1. Yield: 77%. 1H NMR (400 MHz, DMSO) δ 7.70 (d, J = 8.8 Hz, 2H), 7.03 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 8.8 Hz, 2H), 6.52 (t, J = 8.4 Hz, 1H), 3.30–3.33 (m, 4H), 1.12 (s, 18H), 1.05–1.08 (m, 6H). LC–MS (ESI): m/z 262.2 (M + H)+. HRMS (ESI) for C21H36N3O2 (MH+): calcd, 362.2802; found, 362.2795.
N,N′-((4-(Diethylamino)phenyl)methylene)dipentanamide (58)
Compound 58 was prepared from pentanamide and 4-(diethylamino)-benzaldehyde using method 1. Yield: 69%. 1H NMR (400 MHz, CD3OD) δ 7.18 (d, J = 8.8 Hz, 2H), 6.71 (d, J = 8.8 Hz, 2H), 6.58 (t, J = 8.4 Hz, 1H), 3.33–3.41 (m, 4H), 2.25 (t, J = 7.2 Hz, 4H), 1.58–1.66 (m, 4H), 1.33–1.43 (m, 4H), 1.14 (t, J = 7.2 Hz, 6H), 0.95 (t, J = 2.8 Hz, 6H). LC–MS (ESI): m/z 362.2 (M + H)+. HRMS (ESI) for C21H36N3O2 (MH+): calcd, 362.2802; found, 362.2792.
N,N′-((4-(Diethylamino)phenyl)methylene)dihexanamide (59)
Compound 59 was prepared from hexanamide and 4-(diethylamino)-benzaldehyde using method 1. Yield: 76%. 1H NMR (400 MHz, CD3OD) δ 7.19 (d, J = 8.8 Hz, 2H), 6.71 (d, J = 8.8 Hz, 2H), 6.58 (t, J = 8.4 Hz, 1H), 3.35–3.41 (m, 4H), 2.19–2.26 (m, 4H), 1.61–1.68 (m, 4H), 1.32–1.35 (m, 8H), 1.14 (t, J = 6.8 Hz, 6H), 0.94 (t, J = 2.8 Hz, 6H). LC–MS (ESI): m/z 390.3 (M + H)+. HRMS (ESI) for C23H40N3O2 (MH+): calcd, 390.3115; found, 390.3108.
N,N′-((4-(Diethylamino)phenyl)methylene)dioctanamide (60)
Compound 60 was prepared from octanamide and 4-(diethylamino)-benzaldehyde using method 1. Yield: 68%. 1H NMR (400 MHz, DMSO) δ 8.21 (d, J = 8.0 Hz, 2H), 7.07 (d, J = 8.8 Hz, 2H), 6.61 (d, J = 8.8 Hz, 2H), 6.42 (t, J = 8.0 Hz, 1H), 3.29–3.31 (m, 4H), 2.06–2.14 (m, 4H), 1.47–1.50 (m, 4H), 1.08–1.24 (m, 16H), 1.06 (t, J = 7.2 Hz, 6H), 0.94 (t, J = 7.2 Hz, 6H). LC–MS (ESI): m/z 446.3 (M + H)+. HRMS (ESI) for C27H48N3O2 (MH+): calcd, 446.3741; found, 446.3734.
N,N′-((4-(Diethylamino)phenyl)methylene)bis(decanamide)(61)
Compound 61 was prepared from decanamide and 4-(diethylamino)-benzaldehyde using method 1. Yield: 56%. 1H NMR (400 MHz, DMSO) δ 8.99 (d, J = 9.6 Hz, 1H), 8.38 (d, J = 8.8 Hz, 1H), 6.99 (d, J = 8.8 Hz, 2H), 6.58 (d, J = 8.4 Hz, 2H), 5.84 (d, J = 8.4 Hz, 1H), 3.27–3.30 (m, 4H), 2.33 (t, J = 7.2 Hz, 4H), 2.14 (t, J = 7.2 Hz, 4H), 1.50–1.55 (m, 4H), 1.24–1.28 (m, 24H), 1.06 (t, J = 7.2 Hz, 6H), 0.87 (t, J = 7.2 Hz, 6H). LC–MS (ESI): m/z 502.4 (M + H)+.
General Method 2
N,N′-(2-Phenylethane-1,1-diyl)bis(2-phenyl-acetamide) (48)
To a well stirred suspension of 2-phenylacetamide (540 mg, 4 mmol) in dry dichloromethane (2 mL) was added the 2-phenylacetaldehyde (240 mg, 2 mmol) and trimethylsilyltrifluoro-methane sulfonate (22 mg, 0.1 mmol).46 The mixture was vigorously stirred for 12 h at room temperature, diluted with toluene (4 mL), and filtered. The precipitate was washed several times with toluene which was recrystallized with methanol and hexane to give the final product (560 mg, 76%). 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 7.6 Hz, 2H), 7.14–7.28 (m, 15H), 5.55 (t, J = 7.6 Hz, 1H), 3.39 (s, 4H), 2.93 (d, J = 7.2 Hz, 2H). HPLC–MS (ESI): m/z 373.2 (M + H)+.
N,N′-((3-Fluorophenyl)methylene)bis(2-phenylacetamide) (13)
Compound 13 was prepared from 2-phenylacetamide and 3-fluorobenzaldehyde using method 2. Yield: 77%. 1H NMR (400 MHz, DMSO-d6) δ 8.83 (d, J = 8.0 Hz, 2H), 7.06–7.42 (m, 14H), 6.53 (t, J = 7.6 Hz, 1H), 3.47–3.55 (m, 4H). LC–MS (ESI): m/z 377.2 (M + H)+.
N,N′-((4-Bromophenyl)methylene)bis(2-phenylacetamide) (16)
Compound 16 was prepared from 2-phenylacetamide and 4-bromobenzaldehyde using method 2. Yield: 89%. 1H NMR (400 MHz, DMSO-d6) δ 8.82 (d, J = 7.6 Hz, 2H), 7.53–7.56 (m, 2H), 7.21–7.33 (m, 12H), 6.48 (t, J = 7.6 Hz, 1H), 3.46–3.54 (m, 4H). LC–MS (ESI): m/z 437.0 (M + H)+.
N,N′-((4-Isopropylphenyl)methylene)bis(2-phenylacetamide)(18)
Compound 18 was prepared from 2-phenylacetamide and 4-isopropylbenzaldehyde using method 2. Yield: 65%. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (d, J = 8.0 Hz, 2H), 7.20–7.32 (m, 14H), 6.51 (t, J = 8.0 Hz, 1H), 3.46–3.54 (m, 4H), 2.85–2.89 (m, 1H), 1.19 (d, J = 6.8 Hz, 6H). LC–MS (ESI): m/z 401.2 (M + H)+. HRMS (ESI) for C26H29N2O2 (MH+): calcd, 401.2224; found, 401.2219.
N,N′-((4-Ethoxyphenyl)methylene)bis(2-phenylacetamide) (20)
Compound 20 was prepared from 2-phenylacetamide and 4-ethoxybenzaldehyde using method 2. Yield: 70%. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (d, J = 8.0 Hz, 2H), 7.18–7.31 (m, 10H), 6.88 (d, J = 6.4 Hz, 2H), 6.48 (t, J = 8.0 Hz, 1H), 3.98–4.03 (m, 2H), 3.45–3.52 (m, 4H), 1.31 (t, J = 6.8 Hz, 3H). LC–MS (ESI): m/z 403.1 (M + H)+.
N,N′-((4-Isopropoxyphenyl)methylene)bis(2-phenylacetamide) (21)
Compound 21 was prepared from 2-phenylacetamide and 4-isopropoxybenzaldehyde using method 2. Yield: 81%. 1H NMR (400 MHz, DMSO-d6) δ 8.69 (d, J = 7.6 Hz, 2H), 7.18–7.13 (m, 12H), 6.87 (d, J = 6.8 Hz, 2H), 6.48 (t, J = 8.0 Hz, 1H), 4.58–4.61 (m, 1H), 3.45–3.53 (m, 4H), 1.25 (d, J = 6.0 Hz, 6H). LC–MS (ESI): m/z 417.2 (M + H)+.
N,N′-((4-(Diethylamino)phenyl)methylene)bis(2-phenylaceta-mide) (26)
Compound 26 was prepared from 2-phenylacetamide and 4-(diethylamino)benzaldehyde using method 2. Yield: 78%. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (d, J = 8.0 Hz, 2H), 7.21–7.31 (m, 10H), 7.07 (d, J = 8.4 Hz, 2H), 6.60 (d, J = 8.8 Hz, 2H), 6.43 (t, J = 8.0 Hz, 1H), 3,44–3.52 (m, 4H), 3.29–3.34 (m, 4H), 1.06 (t, J = 7.6 Hz, 6H). LC–MS (ESI): m/z 430.3 (M + H)+. HRMS (ESI) for C27H32N3O2 (MH+): calcd, 430.2489; found, 430.2496.
N,N′-((4-(Dibutylamino)phenyl)methylene)bis(2-phenylaceta-mide) (28)
Compound 28 was prepared from 2-phenylacetamide and 4-(dibutylamino)benzaldehyde using method 2. Yield: 85%. 1H NMR (400 MHz, DMSO-d6) δ 8.58 (d, J = 8.0 Hz, 2H), 7.20–7.31 (m, 10H), 7.06 (d, J = 8.8 Hz, 2H), 6.57 (d, J = 8.8 Hz, 2H), 6.41 (t, J = 8.0 Hz, 1H), 3.47–3.48 (m, 4H), 3.22–3.26 (m, 4H), 1.43–1.50 (m, 4H), 1.26–1.35 (m, 4H), 0.91 (t, J = 7.6 Hz, 6H). LC–MS (ESI): m/z 486.2 (M + H)+.
N,N′-((4-(Piperidin-1-yl)phenyl)methylene)bis(2-phenylaceta-mide)(31)
Compound 31 was prepared from 2-phenylacetamide and 4- (piperidin-1-yl)benzaldehyde using method 2. Yield: 85%. 1H NMR (400 MHz, DMSO-d6) δ 8.80 (d, J = 8.0 Hz, 2H), 7.15–7.32 (m, 14H), 6.51 (t, J = 8.0 Hz, 1H), 3.37–3.52 (m, 8H), 1.61–1.83 (m, 6H). LC–MS (ESI): m/z 442.3 (M + H)+.
N,N′-(3-Phenylpropane-1,1-diyl)bis(2-phenylacetamide) (49)
Compound 49 was prepared from 2-phenylacetamide and 3-phenyl-propanal using method 2.Yield: 92%. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 7.6 Hz, 2H), 7.10–7.31 (m, 15H), 5.26 (t, J = 7.6 Hz, 1H), 3.38–3.46 (m, 4H), 2.47–2.53 (m, 2H), 1.88–1.94 (m, 2H). LC–MS (ESI): m/z 387.3 (M + H)+.
(E)-N,N′-(3-Phenylprop-2-ene-1,1-diyl)bis(2-phenylacetamide) (50)
Compound 50 was prepared from 2-phenylacetamide and cinnamaldehyde using method 2. Yield: 88%. 1H NMR (400 MHz, DMSO-d6) δ 8.61 (d, J = 7.6 Hz, 2H), 7.21–7.37 (m, 15H), 6.41–6.45 (m, 1H), 6.27–6.32 (m, 1H), 6.04–6.09 (m, 1H), 3.44–3.52 (m, 4H). LC–MS (ESI): m/z 385.1 (M + H)+.
N,N′-(Pentane-1,1-diyl)bis(2-phenylacetamide) (54)
Compound 54 was prepared from 2-phenylacetamide and pentanal using method 2. Yield: 83%. 1H NMR (400 MHz, DMSO-d6) δ 8.25 (d, J = 8.0 Hz, 2H), 7.19–7.30 (m, 8H), 5.30 (t, J = 7.6 Hz, 1H), 3.36–3.44 (m, 4H), 1.56–1.62 (m, 2H), 1.14–1.26 (m, 4H), 0.81 (t, J = 7.2 Hz, 3H). LC–MS (ESI): m/z 339.1 (M + H)+.
N,N′-(Hexane-1,1-diyl)bis(2-phenylacetamide) (55)
Compound 55 was prepared from 2-phenylacetamide and hexanal using method 2. Yield: 93%. 1H NMR (400 MHz, DMSO-d6) δ 8.25 (d, J = 8.0 Hz, 2H), 7.19–7.30 (m, 8H), 5.29 (t, J = 7.6 Hz, 1H), 3.39–3.44 (m, 4H), 1.57–1.59 (m, 2H), 1.18–1.23 (m, 6H), 0.82 (t, J = 6.8 Hz, 3H). LC–MS (ESI): m/z 353.3 (M + H)+.
General Method of Reduction
N,N′-((4-Aminophenyl)-methylene)bis(2-phenylacetamide) (25)
To a well stirred suspension of N,N′-((4-nitrophenyl)methylene)bis(2-phenylacetamide) (24) (403 mg, 1 mmol) in ethanol (2 mL) was added the palladium (10%, 3.0 mg) and hydrazine (0.05 mL, 1.5 mmol). The mixture was vigorously stirred for 3 h at 70 °C. After filtration, the filtrate was evaporated to dryness on a rotary evaporator. The crude compound was further purified by recrystallization from ethanol. After the sample was dried in a vacuum at room temperature, 25 was obtained as a yellow solid (370 mg, 99%). 1H NMR (400 MHz, DMSO-d6) δ 8.54 (d, J = 8.0 Hz, 2H), 7.20–7.31 (m, 10H), 6.92 (d, J = 8.4 Hz, 2H), 6.49 (d, J = 8.4 Hz, 2H), 6.39 (t, J = 8.0 Hz, 1H), 5.04 (bs, 2H), 3.43–3.53 (m, 4H). LC–MS (ESI): m/z 374.1 (M + H)+.
General Method of Alkylation
N,N′-((4-(Dibenzylamino)-phenyl)methylene)bis(2-phenylacetamide) (29)
Compound 25 (373 mg, 1 mmol), K2CO3 (0.27 g, 1.95 mmol), and DMF (10 mL) were placed in a flask equipped with a condenser and a magnetic stirrer. Benzyl bromide (376 mg, 2.2 mmol) was added, and the mixture was stirred at room temperature for 12 h. The reaction solution was poured into water and extracted with EA. The combined organic layers were washed with water and brine and then dried over Na2SO4. The mixture was filtered, and the solvent was evaporated in vacuum. The residue was purified by flash chromatography on silica gel to obtain 29 (282 mg, 51%). 1H NMR (400 MHz, DMSO-d6) δ 8.50 (s, 2H), 7.18–7.35 (m, 20H), 7.01 (d, J = 8.8 Hz, 2H), 6.62 (d, J = 8.8 Hz, 2H), 6.39 (t, J = 8.0 Hz, 1H), 4.70 (s, 4H), 3.44–3.45 (m, 4H). LC–MS (ESI): m/z 554.2(M + H)+.
Radioligand Competition Binding Assays
CB ligand competition binding assay was carried out as described previously.31 Briefly, nonradioactive (or cold) ligands (PAM derivatives and reference ligands) were diluted in binding buffer (50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 2.5 mM EGTA, and 0.1% (w/v) fatty acid free BSA), supplemented with 10% dimethyl sulfoxide and 0.4% methylcellulose. Each assay plate well contained a total of 200 µL of reaction mixture comprising 5 µg of CB1 (or CB2) membrane protein, labeled [3H]CP-55,940 ligand at a final concentration of 3 nM, and the unlabeled ligand at its varying dilutions as stated above. Plates were incubated at 30 °C for 1 h with gentle shaking. The reaction was terminated by rapid filtration through Unifilter GF/B filter plates using a UniFilter cell harvester (PerkinElmer). After the plate was allowed to dry overnight, 30 µL MicroScint-0 cocktail (PerkinElmer) was added to each well and the radioactivity was counted by using a PerkinElmer TopCount. All assays were performed in duplicate and data points represented as the mean ± SEM. Bound radioactivity data were analyzed for Ki values using nonlinear regression analysis via GraphPad Prism 5.0 software.
The saturation binding of [3H]CP-55,940 to the membrane proteins was performed as described previously.36 Briefly, the CB1 (or CB2) membrane fractions (5 (µg) were incubated with increasing concentrations of [3H]CP-55,940 (0.05–4 nM) in 96-well plates at 30 °C with slow shaking for 1 h. The incubation buffer was composed of 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 2.5 mM EGTA, and 0.1% (w/v) fatty acid free BSA Ligand was diluted in incubation buffer supplemented with 10% dimethyl sulfoxide and 0.4% methylcellulose. Nonspecific binding was determined in the presence of unlabeled CP-55,940 (5000 nM). The reaction was terminated and the radioactivity was counted as stated above. Nonlinear regression analysis revealed the receptor density (Bmax) and the equilibrium dissociation constant (Kd) of [3H]CP-55,940 for the CB2 receptor.
cAMP Assays
Cellular cAMP levels were measured according to a reported method with modifications using LANCE cAMP 384 kits (PerkinElmer).36 The assay is based on competition between a europium-labeled cAMP trace complex and total cAMP for binding sites on cAMP-specific antibodies labeled with a fluorescent dye. The energy emitted from the Eu chelate is transferred to the dye on the antibodies, which in turn generates a time-resolved fluorescent resonant energy transfer (TR-FRET) signal at 665 nm. The fluorescence intensity (665 nm) decreases in the presence of cAMP from the tested samples, and resulting signals are inversely proportional to the cAMP concentration of a sample. CB2 receptor wild type (WT) transfected CHO cells were seeded in a 384-well white ProxiPlates with a density of 2000 cells per well in 5 µL of RPMI-1640 medium containing 1% dialyzed FBS, 25 mM HEPES, 100 µg/mL pennicilin, 100 U/mL strepmicin, and 200 µg/mL G-418. After culture overnight, 2.5 µL of cAMP antibody and RO20-1724 (final consentration of 50 µM) in stimulation buffer (DPBS 1×, containing 0.1% BSA) was added to each well, followed by addition of either 2.5 µL compound or forskolin (final 5 µM) for agonist-inhibited adenylate cyclase (AC) activity assay. After incubated at room temperature for 45 min, 10 µL of detection reagent was added into each well. The plate was then incubated for 1 h at room temperature and measured in Synergy H1 hybrid reader (BioTek) with excitation at 340 nm and emission at 665 nm. Each cAMP determination was made via at least two independent experiments, each in triplicate. EC50 values were determined by nonlinear regression of dose-response curves (GraphPad Prism 5).
Osteoclast Formation Assay
Human marrow-derived mononuclear cells (2 × 10 cells/well) were seeded in 96-well multiplates at 100 µL/well in a MEM containing 20% horse serum, 10 ng/mL M-CSF, and 25 ng/mL RANKL. The tested compounds at the indicated final concentrations were added to the appropriate wells. Half-medium changes were carried out twice a week using drug-containing medium where appropriate. The culture was incubated for a total of 3 weeks at 37 °C with 5% CO2 and 95% humidity. Differentiation into OCLs was assessed by staining with monoclonal antibody 23c6 using a Vectastatin-ABC-AP kit (Vector Laboratories, Burlingame, CA). The antibody 23c6, which recognizes CD51/61 dimer constituting the OCL vitronectin receptor, was generously provided by Mchael Horton (Rayne Institute, Bone and Mineral Center, London, U.K.). The 23c6-positive multinucleated OCLs containing three or more nuclei per OCL were scored using an inverted microscope.47
Cytotoxicity Assay on Human Mononuclear Cells
Peripheral blood was drawn in a heparinized syringe from healthy fasting volunteers who had been without medication for at least 2 weeks. The peripheral blood mononuclear cell (PBMC) fraction was obtained by gradient centrifugation over Ficoll-Hypaque (Amersham), as described previously.48 PBMC were washed three times with ice-cold PBS, followed by resuspension at 5 × 105/mL in the culture medium supplemented with 10% inactivated FBS, 2 mM glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin (Sigma). The compounds in a stock solution (50 mM in DMSO) were diluted with the culture medium to application conditions and further used for the treatment of PBMC for 3 days. The final DMSO concentrations are always 0.02%. After treatment for 72 h, cell viability was determined using trypan blue exclusion assay. These human cell studies conformed to the guidelines of the Institutional Review Board of the University of Pittsburgh, PA.
Molecular Modeling and CoMFA Studies
Out of the 52 compounds from Tables 1–5, 40 compounds were used in the subsequent 3D QSAR CoMFA studies. Twelve compounds that showed no binding, hence no experimental Ki, were ignored in the analysis. Approximately 75% (29 compounds) and 25% (11 compounds) were randomly selected as a training set and a test set, respectively. Molecular modeling and CoMFA studies were performed using the SYBYL X1.2 from the Tripos molecular modeling package.49 By use of our established protocol, 38–40 molecular dynamic simulations were carried out for the best compound 26. Briefly, dynamic simulations were simulated at 300 K with a time steps of 1 fs for a total duration of 300 ps, and conformation samples were collected at every 1 ps, resulting in 300 conformers of compound 26. All conformers were then minimized and converged into four families. These four representative conformers derived from MD simulations were compared to the docking pose resulting from the molecular docking experiment using our in-house 3D CB2 receptor model. The docking experiment was done using the Surflex-Dock module from the Tripos modeling software. The conformer with maximum agreement between these two experiments was chosen as a preferred conformer for further CoMFA studies. Structural alignments of all molecules in the training and test sets to the preferred conformer of compound 26 were performed using the MultiFit program in Sybyl X1.2. The CoMFA study was then carried out using the SYBYL/CoMFA module. The steric and electrostatic field energies (Gasteiger–Huckel charge) were calculated using the default parameters, namely, the Tripos standard CoMFA field class, distance-dependent dielectric constant, steric and electrostatic field cutoff set at 30 kcal·mol−1 . Leave one-out cross-validation (LOOCV) partial least squares (PLS) analysis was then performed with a minium σ (column filter) value of 5.0 kcal·mol−1 to improve the signal-to-noise ratio by omitting those lattice points whose energy variation was below this threshold. The final model (non-cross-validated analysis) was developed from the LOOCV model with the highest cross-validated r2, using the optimal number of components determined by the LOOCV model.
ACKNOWLEDGMENTS
The authors gratefully acknowledge financial support from NIH Grant R01DA025612 (X.-Q.X.) and the National Natural Science Foundation of China (NSFC81090410, NSFC90913018). K.-Z.M. is a predoctoral trainee supported by NIH T32 Training Grant T32 EB009403 under the Joint CMU-Pitt computational biology Ph.D. program.
ABBREVIATIONS USED
- CB
cannabinoid
- AMTa
anandamide membrane transporter
- FAAH
fatty acid amide hydrolase
- MAGL
monoacylglycerol lipase
- GPCR
G-protein-coupled receptor
- GALAHAD
genetic algorithm-based pharmacophore alignment
- CoMFA
comparative molecular field analysis
- QSAR
quantitative structure-activity relationship
- HB
H-bond
- PAM
phenylacetamide
- OCL
osteoclast
- MD
molecular dynamics
- MM
molecular mechanics
- LOOCV
leave-one-out cross-validation
- MTA
material transfer agreement
- TLC
thin-layer chromatography
- TMSCl
trimethylsilyl chloride
- DMSO
dimethyl sulfoxide
- DMF
dimethylformamide
- EA
ethyl acetate
- BSA
bovine serum albumin
- EGTA
ethylene glycol tetraacetic acid
- TR-FRET
time-resolved fluorescent resonant energy transfer
- WT
wild type
- FBS
fetal bovine serum
- DPBS
Dulbecco’s phosphate buffered saline
- AC
adenylate cyclase
- MEM
minimal essential medium
- M-CSF
macrophage colony-stimulating factor
- RANKL
receptor activator of nuclear factor κB ligand
- PBMC
peripheral blood mononuclear cell
- PLS
partial least squares
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- 2.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 3.Degenhardt BF, Darmani NA, Johnson JC, Towns LC, Rhodes DC, Trinh C, McClanahan B, DiMarzo V. Role of osteopathic manipulative treatment in altering pain biomarkers: a pilot study. Am. Osteopath. Assoc. 2007;107:387–400. [PubMed] [Google Scholar]
- 4.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 5.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of aperipheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 6.Sanchez C, de Ceballos ML, Gomez del Pulgar T, Rueda D, Corbacho C, Velasco G, Galve-Roperh I, Huffman JW, Ramon y, Cajal S, Guzman M. Inhibition of glioma growth in vivo by selective activation of the CB(2) cannabinoid receptor. Cancer Res. 2001;61:5784–5789. [PubMed] [Google Scholar]
- 7.Fernandez-Ruiz J, Romero J, Velasco G, Tolon RM, Ramos JA, Guzman M. Cannabinoid CB2 receptor: a new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007;28:39–45. doi: 10.1016/j.tips.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement involvement in nervous system structures—a short review. Pharmacol., Biochem. Behav. 2008;90:501–511. doi: 10.1016/j.pbb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 9.Hohmann AG. Spinal and peripheral mechanisms of cannabinoid antinociception: behavioral, neurophysiological and neuroanatomical perspectives. Chem. Phys. Lipids. 2002;121:173–190. doi: 10.1016/s0009-3084(02)00154-8. [DOI] [PubMed] [Google Scholar]
- 10.Karsak M, Gaffal E, Date R, Wang-Eckhardt L, Rehnelt J, Petrosino S, Starowicz K, Steuder R, Schlicker E, Cravatt B, Mechoulam R, Buettner R, Werner S, Di Marzo V, Tuting T, Zimmer A. Attenuation of allergic contact dermatitis through the endocannabinoid system. Science. 2007;316:1494–1497. doi: 10.1126/science.1142265. [DOI] [PubMed] [Google Scholar]
- 11.Palazuelos J, Davoust N, Julien B, Hatterer E, Aguado T, Mechoulam R, Benito C, Romero J, Silva A, Guzman M, Nataf S, Galve-Roperh I. The CB2 cannabinoid receptor controls myeloid progenitor trafficking. J. Biol. Chem. 2008;283:13320–13329. doi: 10.1074/jbc.M707960200. [DOI] [PubMed] [Google Scholar]
- 12.Guindon J, Hohmann AG. The endocannabinoid system and cancer: therapeutic implication. Br. J. Pharmacol. 2011;163:1447–1463. doi: 10.1111/j.1476-5381.2011.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oesch S, Gertsch J. Cannabinoid receptor ligands as potential anticancer agents—high hopes for new therapies? J. Pharm. Pharmacol. 2009;61:839–853. doi: 10.1211/jpp/61.07.0002. [DOI] [PubMed] [Google Scholar]
- 14.Idris AI, Ralston SH. Cannabinoids and bone: friend or foe? Calcif. Tissue Int. 2010;87:285–297. doi: 10.1007/s00223-010-9378-8. [DOI] [PubMed] [Google Scholar]
- 15.Izzo AA, Camilleri M. Emerging role of cannabinoids in gastrointestinal and liver diseases: basic and clinical aspects. Gut. 2008;57:1140–1155. doi: 10.1136/gut.2008.148791. [DOI] [PubMed] [Google Scholar]
- 16.Munoz-Luque J, Ros J, Fernandez-Varo G, Tugues S, Morales-Ruiz M, Alvarez CE, Friedman SL, Arroyo V, Jimenez W. Regression of fibrosis after chronic stimulation of cannabinoid CB2 receptor in cirrhotic rats. J. Pharmacol. Exp. Ther. 2008;324:475–483. doi: 10.1124/jpet.107.131896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Compton DR, Rice KC, De Costa BR, Razdan RK, Melvin LS, Johnson MR, Martin B. R Cannabinoid structure- activity relationships: correlation of receptor binding and in vivo activities. J. Pharmacol. Exp. Ther. 1993;265:218–226. [PubMed] [Google Scholar]
- 19.Huffman JW. Cannabimimetic indoles, pyrroles and indenes. Curr. Med. Chem. 1999;6:705–720. [PubMed] [Google Scholar]
- 20.Palmer SL, Thakur GA, Makriyannis A. Cannabinergic ligands. Chem. Phys. Lipids. 2002;121:3–19. doi: 10.1016/s0009-3084(02)00143-3. [DOI] [PubMed] [Google Scholar]
- 21.Raitio KH, Salo OM, Nevalainen T, Poso A, Jarvinen T. Targeting the cannabinoid CB2 receptor: mutations, modeling and development of CB2 selective ligands. Curr. Med. Chem. 2005;12:1217–1237. doi: 10.2174/0929867053764617. [DOI] [PubMed] [Google Scholar]
- 22.Marriott KS, Huffman JW. Recent advances in the development of selective ligands for the cannabinoid CB(2) receptor. Curr. Top. Med. Chem. 2008;8:187–204. doi: 10.2174/156802608783498014. [DOI] [PubMed] [Google Scholar]
- 23.Yang P, Wang L, Xie XQ. Latest advances in novel cannabinoid CB(2) ligands for drug abuse and their therapeutic potential. Future Med. Chem. 2012;4:187–204. doi: 10.4155/fmc.11.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rinaldi-Carmona M, Barth F, Millan J, Derocq JM, Casellas P, Congy C, Oustric D, Sarran M, Bouaboula M, Calandra B, Portier M, Shire D, Breliere JC, Le Fur GL. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J. Pharmacol. Exp. Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- 25.Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, Pertwee RG. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br. J. Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwamura H, Suzuki H, Ueda Y, Kaya T, Inaba T. In vitro and in vivo pharmacological characterization of JTE-907, a novel selective ligand for cannabinoid CB2 receptor. J. Pharmacol. Exp. Ther. 2001;296:420–425. [PubMed] [Google Scholar]
- 27.Lunn CA, Fine JS, Rojas-Triana A, Jackson JV, Fan X, Kung TT, Gonsiorek W, Schwarz MA, Lavey B, Kozlowski JA, Narula SK, Lundell DJ, Hipkin RW, Bober LA. A novel cannabinoid peripheral cannabinoid receptor-selective inverse agonist blocks leukocyte recruitment in vivo. J. Pharmacol. Exp. Ther. 2006;316:780–788. doi: 10.1124/jpet.105.093500. [DOI] [PubMed] [Google Scholar]
- 28.Schuehly W, Paredes JM, Kleyer J, Huefner A, Anavi-Goffer S, Raduner S, Altmann KH, Gertsch J. Mechanisms of osteoclastogenesis inhibition by a novel class of biphenyl-type cannabinoid CB(2) receptor inverse agonists. Chem. Biol. 2011;18:1053–1064. doi: 10.1016/j.chembiol.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 29.Giblin GM, O’Shaughnessy CT, Naylor A, Mitchell WL, Eatherton AJ, Slingsby BP, Rawlings DA, Goldsmith P, Brown AJ, Haslam CP, Clayton NM, Wilson AW, Chessell IP, Wittington AR, Green R. Discovery of 2-[(2,4-dichlorophenyl)-amino]-N-[(tetrahydro-2H-pyran-4-yl)methyl]-4-(trifluoromethyl)-5-pyrimidinecarboxamide, a selective CB2 receptor agonist for the treatment of inflammatory pain. J. Med. Chem. 2007;50:2597–2600. doi: 10.1021/jm061195+. [DOI] [PubMed] [Google Scholar]
- 30.Xi Z-X, Peng X-Q, Li X, Song R, Zhang H-Y, Liu Q-R, Yang H-J, Bi G-H, Li J, Gardner EL. Brain cannabinoid CB2 receptors modulate cocaine’s actions in mice. Nat. Neurosci. 2011;14:1160–1166. doi: 10.1038/nn.2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gertsch J, Leonti M, Raduner S, Racz I, Chen JZ, Xie XQ, Altmann KH, Karsak M, Zimmer A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9099–9104. doi: 10.1073/pnas.0803601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong H, Bowen JP. GALAHAD pharmacophore modeling for drug discovery. J. Am. Chem. Soc. 2007;129:5780–5780. [Google Scholar]
- 33.SYBYL 8.0. St. Louis, MO, U.S: Tripos; 2007. [Google Scholar]
- 34.Xie XQ, Chen J. Data-mining a small molecule drug screening representative subset from NIH PubChem database. J. Chem. Inf. Model. 2008;48:465–475. doi: 10.1021/ci700193u. [DOI] [PubMed] [Google Scholar]
- 35.Francisco ME, Seltzman HH, Gilliam AF, Mitchell RA, Rider SL, Pertwee RG, Stevenson LA, Thomas BF. Synthesis and structure-activity relationships of amide and hydrazide analogues of the cannabinoid CB(1) receptor antagonist N-(piperidinyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carbox-amide (SR141716) J. Med. Chem. 2002;45:2708–2719. doi: 10.1021/jm010498v. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Xie Z, Wang L, Schreiter B, Lazo JS, Gertsch J, Xie XQ. Mutagenesis and computer modeling studies of a GPCR conserved residue W5.43(194) in ligand recognition and signal transduction for CB2 receptor. Int. Immunopharmacol. 2011;11:1303–1310. doi: 10.1016/j.intimp.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anandarajah AP, Schwarz EM, Totterman S, Monu J, Feng CY, Shao T, Haas-Smith SA, Ritchlin CT. The effect of etanercept on osteoclast precursor frequency and enhancing bone marrow oedema in patients with psoriatic arthritis. Ann. Rheum. Dis. 2008;67:296–301. doi: 10.1136/ard.2007.076091. [DOI] [PubMed] [Google Scholar]
- 38.Myint KZ, Xie X-Q. Recent advances in fragment-based QSAR and multi-dimensional QSAR methods. Int. J. Mol. Sci. 2010;11:3846–3866. doi: 10.3390/ijms11103846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J-Z, Han X-W, Liu Q, Makriyannis A, Wang J, Xie X-Q. 3D-QSAR studies of arylpyrazole antagonists of cannabinoid receptor subtypes CB1 and CB2. A combined NMR and CoMFA approach. J. Med. Chem. 2006;49:625–636. doi: 10.1021/jm050655g. [DOI] [PubMed] [Google Scholar]
- 40.Xie XQ, Melvin LS, Makriyannis A. The conformational properties of the highly selective cannabinoid receptor ligand CP-55,940. J. Biol. Chem. 1996;271:10640–10647. doi: 10.1074/jbc.271.18.10640. [DOI] [PubMed] [Google Scholar]
- 41.Xie X-Q, Chen J-Z, Billings EM. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins: Struct., Funct., Bioinf. 2003;53:307–319. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 42.Diaz P, Phatak SS, Xu J, Astruc-Diaz F, Cavasotto CN, Naguib M. 6-Methoxy-N-alkyl isatin acylhydrazone derivatives as a novel series of potent selective cannabinoid receptor 2 inverse agonists: design, synthesis, and binding mode prediction. J. Med. Chem. 2008;52:433–444. doi: 10.1021/jm801353p. [DOI] [PubMed] [Google Scholar]
- 43.Gouldson P, Calandra B, Legoux P, Kerneis A, Rinaldi-Carmona M, Barth F, Le Fur G, Ferrara P, Shire D. Mutational analysis and molecular modeling of the antagonist SR 144528 binding site on the human cannabinoid CB2 receptor. Eur. J. Pharmacol. 2000;401:17–25. doi: 10.1016/s0014-2999(00)00439-8. [DOI] [PubMed] [Google Scholar]
- 44.Poso A, Huffman JW. Targeting the cannabinoid CB2 receptor: modelling and structural determinants of CB2 selective ligands. Br. J. Pharmacol. 2008;153:335–346. doi: 10.1038/sj.bjp.0707567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan JP, Chai YF, Wu JM, Pan YJ. N,N′-(Phenylmethylene)diacetamide analogues as economical and efficient ligands in copper-catalyzed arylation of aromatic nitrogen-containing heterocycles. Synlett. 2008:3068–3072. [Google Scholar]
- 46.Bayer A, Maier ME. Synthesis of enamides from aldehydes and amides. Tetrahedron. 2004;60:6665–6677. [Google Scholar]
- 47.Feng R, Anderson G, Xiao G, Elliott G, Leoni L, Mapara MY, Roodman GD, Lentzsch S. SDX-308, a nonsteroidal anti-inflammatory agent, inhibits NF-kappaB activity, resulting in strong inhibition of osteoclast formation/activity and multiple myeloma cell growth. Blood. 2007;109:2130–2138. doi: 10.1182/blood-2006-07-027458. [DOI] [PubMed] [Google Scholar]
- 48.Feng R, Ma H, Hassig CA, Payne JE, Smith ND, Mapara MY, Hager JH, Lentzsch S. KD5170, a novel mercaptoketone-based histone deacetylase inhibitor, exerts antimyeloma effects by DNA damage and mitochondrial signaling. Mol. Cancer Ther. 2008;7:1494–1505. doi: 10.1158/1535-7163.MCT-08-0183. [DOI] [PubMed] [Google Scholar]
- 49.SYBYL X1.2. 1699 South Hanley Rd, St. Louis, MO, 63144, U.S.: Tripos; software available at www.tripos.com. [Google Scholar]