

Abstract
Human lipoxygenases (LOXs) are a family of iron-containing enzymes which catalyze the oxidation of polyunsaturated fatty acids to provide the corresponding bioactive hydroxyeicosatetraenoic acid (HETE) metabolites. These eicosanoid signaling molecules are involved in a number of physiologic responses such as platelet aggregation, inflammation, and cell proliferation. Our group has taken a particular interest in platelet-type 12-(S)-LOX (12-LOX) because of its demonstrated role in skin diseases, diabetes, platelet hemostasis, thrombosis, and cancer. Herein, we report the identification and medicinal chemistry optimization of a 4-((2-hydroxy-3-methoxybenzyl)amino)benzenesulfonamide-based scaffold. Top compounds, exemplified by 35 and 36, display nM potency against 12-LOX, excellent selectivity over related lipoxygenases and cyclooxygenases, and possess favorable ADME properties. In addition, both compounds inhibit PAR-4 induced aggregation and calcium mobilization in human platelets and reduce 12-HETE in β-cells.
INTRODUCTION
Lipoxygenases are a class of nonheme iron-containing enzymes which regio- and stereospecifically oxidize polyunsaturated fatty acid substrates such as arachidonic acid (AA) and linoleic acid (LA).1 The position at which these cis,cis-1,4-pentadiene substrates are oxidized corresponds to the requisite lipoxygenase, with the three major human lipoxygenases, 5-LOX, 12-LOX, and 15-LOX-1, oxidizing the C-5, C-12, and C-15 positions respectively. Lipoxygenases are involved in the first committed step in a cascade of metabolic pathways, and the products of these enzymes (eicosanoids) are precursors of hormones such as leukotrienes and lipoxins, which mediate a wide array of cellular functions.2 Consequently, the lipoxygenase enzymes and their bioactive metabolites (e.g., hydroxyeicosatetraenoic acid (HETE) and leukotriene A4) have been implicated in a variety of inflammatory diseases and cancers. Specifically, 5-LOX has been implicated in cancer3 and inflammatory diseases such as asthma4 and remains the only lipoxygenase enzyme for which there is an FDA-approved inhibitor (Zileuton) on the market.5 Reticulocyte 15-LOX-1 has received particular attention for its role in atherogenesis,6 neurodegenerative diseases,7 and neuronal damage associated from an acute ischemic stroke event.8 12-LOX exists as three isozymes, platelet-type, leukocyte, and epidermal, however, leukocyte 12-LOX is not found in humans yet is present in rat, mouse, pig, and cow.9 Platelet-type 12-(S)-LOX (12-LOX), which the current study focuses on, has been found to be overexpressed in a variety of tumor tissues including prostate cancer, colorectal cancer, breast cancer, and lung cancer.10 Moreover, 12-HETE levels have been linked to increased cancer cell metastasis by facilitating tumor cell motility and angiogenesis.11 12-LOX is also expressed in human pancreatic islets, which is upregulated and activated by inflammatory cytokines, leading to increased 12-LOX translocation.12 The resulting 12-HETE product leads to reduced insulin secretion, reduced metabolic activity, and pancreatic β-cell death through the amplification of the inflammatory response.13 Toward this end, both non-obese diabetic (NOD) 12-LOX12a and 12-LOX KO12b mice showed significant resistance to the development of diabetes compared to the controls, suggesting 12-LOX is a clear regulator in this disease. 12-LOX and its product 12-HETE have also been implicated in the modulation of hemostasis and thrombosis via their role in regulating platelet function (reactivity, clot formation, calcium mobilization).14 Historically, the precise role of 12-HETE has been controversial, with reports suggesting both an anti- and pro-thrombotic effects.15 One difficulty in being able to clearly define the role of 12-LOX in these systems has been the lack of potent and selective 12-LOX small molecule inhibitors. However, working with one of our previously described inhibitors, N-((8-hydroxy-5-nitroquinolin-7-yl)(thiophen-2-yl)-methyl)propionamide (NCTT-956),16 we were able to show that inhibition of 12-LOX leads to reduced platelet aggregation and calcium mobilization following stimulation by various agonists (PAR1-AP, PAR4-AP, collagen, thrombin).17 These results helped further validate 12-LOX as a potential target for antiplatelet therapy.
Our laboratories18 and others19 have been actively engaged for many years in the discovery of novel potent 12-LOX inhibitors utilizing traditional medicinal chemistry, computational chemistry,20 and natural product isolation.21 However, despite these extensive efforts, no drug-like small molecule that was either chemically tractable or selective had been discovered. Instead, these efforts yielded compounds that were either reductive and/or promiscuous in nature, as exemplified by the inhibitors listed in Figure 1. The known reductive LOX inhibitor, nordihydroguaiaretic acid (NDGA),18a and other natural products including the brominated aryl phenols,18c which are marine derived natural products, all have modest 12-LOX inhibitory activity (micromolar). Importantly, these compounds lack selectivity toward 12-LOX and have undesirable chemical structures. For example, the polyphenolic compound, Baicalein, was once one of the most commonly used 12-LOX inhibitors as it was originally thought to be selective for 12-LOX. However, subsequent studies revealed equipotent in vitro activity toward both 12-LOX and 15-LOX-1.18b
Figure 1.

Representative examples of previously reported 12-LOX inhibitors.
Given the previously stated biological importance of 12-LOX and the lack of high quality probe compounds in the literature, we initiated a high throughput screening campaign as part of the MLPCN program. This effort led to the discovery of an 8-hydroxyquinoline-based compound N-((5-bromo-8-hydroxy-quinolin-7-yl)(thiophen-2-yl)methyl)acetamide (ML127) which exhibited excellent selectivity (>50–100-fold) over related lipoxygenses and cyclooxygenase.22 In contrast to many of the previously reported inhibitors, kinetic experiments revealed that these inhibitors were noncompetitive and nonreductive. However, despite this promising activity, the chemical series was difficult to optimize further, given that subtle structural modifications led to diminished activity.22 Therefore, we sought to reexamine the results from the original HTS to uncover additional compounds that may be amendable to medicinal chemistry optimization. Ultimately, this led to the identification of a 4-((2-hydroxy-3-methoxybenzyl)amino)-benzenesulfonamide-based scaffold which was subjected to medicinal chemistry optimization and biological characterization. The results of these efforts are described herein.
CHEMISTRY
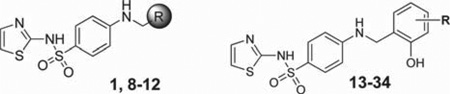
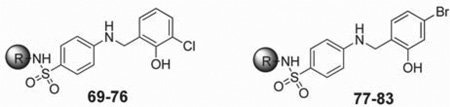
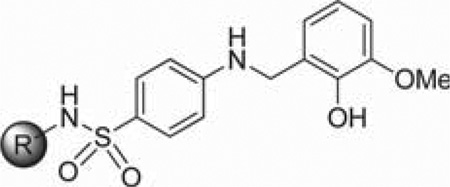
Prior to initiating an extensive SAR campaign around the HTS “hit” molecule (1), we first sought to resynthesize the molecule to confirm its identity and activity. The synthesis of compound 1 involved a reductive amination with 4-aminobenzesulfonamide and 2-hydroxy-3-methoxybenzaldehyde. While this route appeared amenable to the facile preparation of numerous synthetic analogues, the standard mild method for reductive aminations utilizing sodium triacetoxyborohydride and catalytic acetic acid afforded little to no product.23 Instead, a stepwise approach involving preformation of the imine was required with 4-amino-N-(thiazol-2-yl)benzenesulfonamide (2) and the requisite benzaldehyde overnight in ethanol at reflux. Subsequent reduction of the imine with sodium borohydride provided the desired compounds 1, 8–10, and 14–33 (Scheme 1, method A). While this route worked for a majority of the analogues, some analogues (11–13 and 34) required an alternative route in which a Buchwald–Hartwig type coupling was utilized with the commercially available 4-bromo-N-(thiazol-2-yl)benzenesulfonamide (3) and 2-methoxy substituted benzyl amines to provide the desired products 11–13 and 34 (Scheme 1, method B).24 For modifications of the thiazole portion of the molecule, we wanted to introduce diversity in the final step of the synthesis (Scheme 1, method C). Therefore, reaction of commercially available 4-amino-benzenesulfonamide (4) with 2-hydroxy-3-methoxybenzalde-hyde in ethanol at reflux for 6 h, followed by treatment with sodium borohydride, provided compound 5 in 95% yield. The resulting sulfonamide derivative was then subjected to Cu-catalyzed N-arylation conditions using the appropriate hetero-aryl bromides to afford compounds 35, 38, 44–48, 52, 53, 56– 59, 61–69, 71, 74, and 75.25 Despite the general versatility of this method, there was a few isolated cases where the Cucatalyzed reaction failed to produce the desired product (e.g., compounds 77–83) or the heteroaryl bromides were not readily available. For these compounds, a less direct route was developed by heating 4-nitrobenzene-1-sulfonyl chloride (6) and the required heteroaryl amines to 100 °C for 1.5–18 h depending on the reactivity of the amine to give the 4-nitrophenyl-sulfonamide derivatives 7 (Scheme 1, method D). Reduction of the nitro group was achieved using the H-Cube Pro flow reactor with 10% Pd on carbon at 50 °C and a pressure of 50 bar. Alternatively, for less soluble compounds which were not amenable to flow chemistry, a Zn/AcOH reduction was performed at 60 °C in methanol. Once the desired aniline was in hand, a reductive amination was carried out with the corresponding benzyl amine derivative to provide compounds 36, 37, 39–43, 49–51, 54, 55, 60, 70, 72, 73, and 76–83.
Scheme 1.

Synthesis of Analogues 1–83a
aReagents and conditions: (i) RCHO (1.5 equiv), EtOH, 3–18 h, reflux, NaBH4 (2.0 equiv), 0.5–0.6 h at rt; (ii) RCH2NH2 (1.2 equiv), Xantphos (0.06 equiv), Pd2dba3 (0.02 equiv), NaOtBu (2.5 equiv), 1,4-dioxane, MW, 30 min, 100 °C; (iii) 2-hydroxy-3-methoxybenzaldehyde (1.2 equiv), EtOH, 6 h, reflux, NaBH4 (1.5 equiv), 30 min, rt, 95%; (iv) R′Br (1.2 equiv), N,N′-dimethylethylenediamine, CuI (0.05 equiv), K2CO3 (2.5 equiv), 80 °C, 6–8 h; (v) R′NH2, pyridine, 100 °C, 1.5–18 h; (vi) 10% Pd/C, MeOH/EtOAc/THF (1:1:1), 50 bar, 50 °C or zinc (4.0 equiv), AcOH (4.0 equiv), methanol, 60 °C, 30 min−2 h; (vii) 2-hydroxy-R-benzaldehyde (1.2 equiv), EtOH, 18 h, reflux, NaBH4 (3.0 equiv).
RESULTS AND DISCUSSION
After having confirmed the identity and biological activity of the resynthesized lead compound, 1, we began our systematic SAR explorations as shown in Tables 1–3. Importantly, all the IC50 values reported for the prior art compounds were determined in our laboratory using the same assay conditions as the new compounds reported herein. Our initial concern surrounding this chemotype was the presence of the “catechol-like” moiety, as this functionality can often be associated with promiscuous activity and/or metal chelation as seen with NDGA18a and small catechols.26 However, the excellent selectivity of 1 over related lipoxygenase enzymes (5-LOX, 15-LOX-1, and 15-LOX-2) suggested this was not the case. Moreover, the 3-methoxy moiety present on 1 reduces the chelation potential of the molecule compared to a traditional 1,2-dihydroxy benzene (catechol) functionality. Despite this, we chose to focus on modification of the 2-hydroxy-3-methoxybenzyl moiety for our series of analogues. As shown in Table 1, we found that removal of both phenolic groups (8), 3-OMe group (9), or the 2-OH group (10) resulted in a complete loss of activity. Protecting the 2-OH moiety as methyl ether (compounds 11–12) or replacing with an amine (compounds 13 and 15) also negated all 12-LOX inhibitory activity. We tried a bioisosteric replacement of the 2-OH, 3-OMe-phenyl moiety with an indole (17), however this analogue was also inactive. These data seemed to indicate the requirement of the 2-OH group, yet this group alone was not enough for activity. Therefore, we decided to hold the 2-OH group constant and try modifications at the other positions. We found the 3-Cl (19, IC50 = 6.2 µM) to have comparable activity, while the 3-fluoro and 3-bromo derivatives (20, IC50 = 19 µM; 21, IC50 = 13 µM) were ∼4 to ∼2.5-fold fold less active, respectively. An interesting and unexpected result was the 2-fold improvement in activity observed for the 4-bromo derivative (22, IC50 = 2.2 µM) and 4-chloro derivative (27) which had comparable activity (6.3 µM). In contrast, the 4-methoxy analogue (28) had reduced activity compared to compound 1 (IC50 = 22 µM). Other substituents at 3-position such as methyl (23), amino (24), and nitro (25) all resulted in a drastic loss of activity (IC50 > 40 µM) as did all 5-position analogues (29–33). Only one compound with a modification to the 6-position was synthesized, R = OMe (34), and this was also inactive. Thus, we determined the 2-OH to be essential for activity and the 3-position to be most optimal for the methoxy group. However, this preliminary SAR suggested that the 3-OMe could be replaced with a chloro group (19) and a 4-bromo (22) and 4-chloro (27) while maintaining comparable, if not improved, activity to compound 1.
Table 1.
12-LOX Inhibition of Analogues 1, 8–34a
 | ||
|---|---|---|
| compd | R | IC50 [± SD] (µM) |
| 1 | 2-OH, 3-OMe-Ph | 5.1 [0.5] |
| 8 | Ph | >40 |
| 9 | 2-OH-Ph | >40 |
| 10 | 3-OMe-Ph | >40 |
| 11 | 2-OMe-Ph | >40 |
| 12 | 2,3-OMe-Ph | >40 |
| 13 | 2-NH2-Ph | >40 |
| 14 | 3-OH-Ph | >40 |
| 15 | 2-NH2, 3-OMe-Ph | >40 |
| 16 | 3-OH, 4-OMe-Ph | >40 |
| 17 | 7-indole | >40 |
| 18 | 2,3-CI-Ph | >40 |
| 19 | 3-CI | 6.2 [2.0] |
| 20 | 3-F | 19 [4] |
| 21 | 3-Br | 13 [1] |
| 22 | 4-Br | 2.2 [0.5] |
| 23 | 3-Me | >40 |
| 24 | 3-NH2 | >40 |
| 25 | 3-NO2 | >40 |
| 26 | 3-allyl | >40 |
| 27 | 4-Cl | 6.3 [1.0] |
| 28 | 4-OMe | 22 [11] |
| 29 | 5-CI | >40 |
| 30 | 5-OMe | >40 |
| 31 | 5-NH2 | >40 |
| 32 | 5-F | >40 |
| 33 | 5-NO2 | >40 |
| 34 | 6-OMe | >40 |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the UV–vis cuvette-based assay in triplicate.
Table 3.
12-LOX Inhibition of Analogues 69–83a
 | ||||
|---|---|---|---|---|
| 12-LOX |
15-LOX-1 |
|||
| compd | R | IC50 [± SD] (µM) |
IC50 [± SD] (µM) |
% inhb |
| 69 | Ph | 4.5 [0.4] | 49 | |
| 70 | 1-naphthalene | 1.6 [0.2] | 70 | |
| 71 | 2-benzothiazole | 1.3 [0.2] | 1.3 [0.2] | |
| 72 | 2-naphthalene | 2.3 [0.3] | 75 | |
| 73 | 4-bi-Ph | 4.2 [1.0] | 73 | |
| 74 | 8-isoquinoline | 4.5 [0.8] | 63 | |
| 75 | 3-quinoline | 5.3 [0.7] | 58 | |
| 76 | 4-piperidine-Ph | 6.3 [3.0] | 4 | |
| 77 | Ph | 2.9 [0.4] | 82 | |
| 78 | 1-naphthalene | 1.3 [0.3] | 83 | |
| 79 | 2-benzothiazole | 1.7 [0.8] | 100 | |
| 80 | 3-quinoline | 2.3 [0.5] | 74 | |
| 81 | 2-naphthalene | 2.2 [0.3] | 78 | |
| 82 | 4-bi-Ph | 2.5 [0.4] | 80 | |
| 83 | 8-quinoline | 5.6 [2.0] | 55 | |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the UV–vis cuvette-based assay in triplicate.
Represents percent inhibition at 25 µM.
Having established a preliminary SAR profile of this region of the molecule, we next turned our attention to modification of the thiazole group as shown in Table 2. Gratifyingly, unlike our efforts on the phenyl moiety, changes to this region of the molecule led to analogues with improved potency. Replacing the thiazole moiety with a 2-benzothiazole (35, ML355) resulted in an 18-fold improvement in 12-LOX activity while retaining selectivity. The benzoxazole (36) and benzimidazole (37) possessed good potency, and introduction of a methyl group at the 4-position of the benzothiazole ring (39) maintained favorable 12-LOX inhibitory activity (IC50 = 0.24 µM). Potency against 12-LOX was also improved over 10-fold when the thiazole was replaced with a thiophene (38), but the substituted thiazole/isoxazole derivatives (40–43) did not show this increased potency. The phenyl derivative (48), a known thiophene bioisostere, also demonstrated potent inhibition against 12-LOX (IC50 = 0.5 µM). Generally, larger aromatic [1-naphthalene (49) and 2-naphthalene (50)] and heteroaromatic compounds [3-quinoline (46), 8-isoquinoline (47), 2-pyridine (58), and 3-pyridine (59)] were well tolerated and had better potency than the thiazole derivative (1). In an effort to improve solubility by adding solubilizing functionality, we synthesized a few phenyl derivatives substituted with a piperazine moiety at different positions around the aryl ring (53–55). While these changes were tolerated, they had much reduced activity compared to the top actives (e.g., entry 35 and 36).
Table 2.
12-LOX Inhibition of Analogues 35–68a
 | ||||
|---|---|---|---|---|
| 12-LOX |
15-LOX-1 |
|||
| compd | R | IC50 [± SD] (µM) |
IC50 [± SD] (µM) |
% inhb |
| 35 | 2-benzothiazole | 0.34 [0.04] | 9.7 [0.1] | |
| 36 | 2-benzoxazole | 0.79 [0.1] | >100 | |
| 37 | 2-benzimidazole | 0.57 [0.04] | >70 | |
| 38 | 2-thiophene | 0.35 [0.02] | 100 | |
| 39 | 4-Me-2-benzothiazole | 0.24 [0.01] | 0.69 [0.1] | |
| 40 | 4-Me-2-thiazole | 2.7 [0.2] | 14 | |
| 41 | 5-Me-2-thiazole | 3.0 [0.4] | 75 | |
| 42 | 5-Ph-2-thiazole | 91%b | 77 | |
| 43 | 4,5-Me-2-thiazole | 1.4 [0.3] | 37 | |
| 44 | 5-Me-3-isoxazole | 11 [1.2] | ||
| 45 | 3-OMe-Ph | 85%b | 73 | |
| 46 | 3-quinoline | 0.48 [0.1] | 77 | |
| 47 | 8-isoquinoline | 0.70 [0.2] | 70 | |
| 48 | Ph | 0.50 [0.05] | 7.6 [1.0] | |
| 49 | 1-naphthalene | 0.51 [0.06] | 73 | |
| 50 | 2-naphthalene | 0.33 [0.05] | 54 | |
| 51 | 1,4-bi-Ph | 1.3 [0.2] | 60 | |
| 52 | 1,3-bi-Ph | 82%b | 70 | |
| 53 | 3-piperazine-Ph | 3.5 [0.5] | 31 | |
| 54 | 4-piperazine-Ph | 11 [2.7] | ||
| 55 | 4-piperidine-Ph | 3.7 [0.6] | 8 | |
| 56 | 4-piperazine-3-pyr | 4.0 [0.6] | 12 | |
| 57 | 6-methyl-3-pyr | 5.0 [0.5] | 18 | |
| 58 | 2-pyr | 5.0 [0.5] | 3 | |
| 59 | 3-pyr | 7.0 [0.5] | ||
| 60 | 2-pyrimidine | 12 [1.0] | ||
| 61 | 3-tBu-Ph | 0.39 [0.8] | 66 | |
| 62 | 6-OMe-2-benzothiazole | 0.26 [0.3] | 5.1 [0.6] | |
| 63 | 4-Ph-2-thiazole | 0.18 [0.03] | 87 | |
| 64 | 3-morpholine-Ph | 3.8 [0.3] | 50 | |
| 65 | 4N–boc-piperidine-3-Ph | 0.76 [0.05] | 34 | |
| 66 | 3-piperidine-Ph | 1.1 [0.3] | 4.7 | |
| 67 | 3-iPr-Ph | 0.16 [0.02] | >100 | |
| 68 | 6-F-2-benzothiazole | 0.22 [0.05] | 4.5 [0.4] | |
IC50 values represent the half maximal (50%) inhibitory concentration as determined in the UV–vis cuvette-based assay in triplicate.
Represents inhibition at 25 µM.
Given our interest in pursuing multiparameter optimization, we simultaneously tested many of these analogues for activity against 15-LOX-1 to ensure that selectivity was maintained. These compounds were initially tested against 15-LOX-1 at a single concentration (25 µM), and an IC50 was determined only on compounds of particular interest. These studies showed that replacement of the thiazole with a benzothiazole, and its derivatives maintained favorable selectivity with 15-LOX-1/12-LOX ratios of 29- (35), 18- (39), 19- (62), and 20-fold (68). The 15-LOX-1/12-LOX selectivity ratio improved to over 100, with conversion of the benzothiazole to a benzoxazole (36), benzimidazole (37), and m-iPr substituted phenyl (67). Interestingly, the phenyl substitution (48) only had a selectivity ratio of 15. A wide-range of selectivity was observed despite the compounds bearing comparable structures, ranging from almost complete inhibition of 15-LOX-1 (e.g., 38 and 63) to minimal inhibition (e.g., 55, 58, and 66). The dramatic effect on selectivity between 12-LOX and 15-LOX-1 in this portion of the molecule supports the differences between the active sites of the two LOX isozymes, as seen previously by mutational analysis.27
As noted above (Table 1), replacement of the “right-hand” portion of the molecule with a 3-chloro-2-phenol (19), 4-bromo-2-phenol (22), or 4-chloro-2-phenol (27) resulted in comparable potency to 1 with IC50 values of 6.2, 2.2, and 6.3 µM, respectively. Therefore, we sought to explore a matrix library utilizing these groups, with some of the best sulfonamide derivatives discovered as part of the initial SAR efforts (see Table 3, compounds 69–83). Unfortunately, none of the compounds had improved potency. Generally, the 2-benzothiazole moiety gave the best activity with both the 3-chloro-2-phenol (71; IC50 = 1.3 µM) and 4-bromo-2-phenol (79; IC50 = 1.7 µM), although the 1-naphthalene derivative 78 also had comparable potency (IC50 = 1.3 µM).
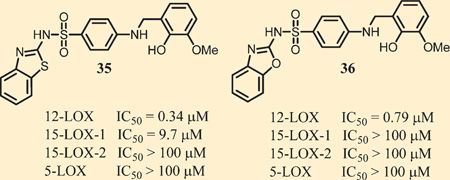
The results of our SAR investigations provided compounds with potency as low as 160 nM against 12-LOX, which prompted us to examine the selectivity of a few of our top analogues (35, 36, and 37) against other human LOX isozymes (5-LOX and 15-LOX-2). In addition, we profiled the compounds against cyclooxygenase−1/−2 (COX−1/−2). As shown in Table 4, we observed no significant inhibition against any of these related enzymes, with the exception of 35, which has modest potency (29-fold less active) toward 15-LOX-1. These results are encouraging because few compounds reported in the literature have achieved both nM potency toward 12-LOX and selectivity against other isozymes.21 It should be noted that while other compounds with more potent 12-LOX inhibition were identified (e.g., 62 (IC50 = 0.26 µM), 63 (IC50 = 0.18 µM), 67 (IC50 = 0.16 µM), and 68 (IC50 = 0.22 µM), each possess unfavorable attributes compared to 35 and 36. Specifically, compound 62 has poor PAMPA permeability (<10 × 10−6 cm/s), fairly high SD values (0.3), and modest potency toward 15-LOX-1 (5.1 µM). Compounds 63 and 67 are markedly less stable to rat liver microsomes, with T1/2 values of 8.7 and 3.3 min, respectively, with 67 also having poor PAMPA permeability (39 × 10−6 cm/s). Finally, compound 68 has comparable activity and selectivity to 35 but worse PAMPA permeability (65 × 10−6 cm/s). Ultimately, it would have been preferred to test a larger number of compounds in the downstream biological assays, but limitations in resources and reagents (e.g., human islet cells) prompted us to limit the number of compounds. As such, we chose compound 35 because it was one of the first compounds tested in a battery of assays and it had generally favorable in vitro ADME properties and potent inhibition, while compounds 36 and 37 were chosen for further investigations because of their excellent selectivity vs 15-LOX-1 and more favorable ADME properties compared to the other analogues described above.
Table 4.
Selectivity and Redox Activity of Representative Analoguesa
| analogue | 12-LOXb | 15-LOX-1b | 15-LOX-2b | 5-LOXb | COX−1/−2c | redox activityd |
|---|---|---|---|---|---|---|
| 1 | 5.1 | >50 | >40 | >200 | NT | NT |
| 35 | 0.34 | 9.7 | >100 | >100 | NI | NI |
| 36 | 0.79 | >100 | >100 | >100 | NI | NI |
| 37 | 0.57 | >100 | >100 | >35 | NI | NT |
Selectivity profiling of 1, 35, 36, and 37.
IC50 values are reported in µM.
Compounds were tested at 15 µM and none of the compound exhibited inhibition above 10%.
UV–vis pseudoperoxidase activity assay was performed on 35 and 36, and no degradation of the hydroperoxide product was observed at 234 nm, indicating a nonreductive inhibitory mechanism; NI = no inhibition and NT = not tested.
LOX inhibitors are known to exhibit a variety of inhibitory mechanisms; therefore the UV pseudoperoxidase assay is often used to investigate if the inhibition is reductive in nature.28 The assay was performed on 35 and 36 with 12-LOX, and no degradation of the hydroperoxide product was observed at 234 nm, indicating a nonreductive inhibitory mechanism (Table 4). To investigate the inhibition mechanism further, steady-state kinetics were performed using both 35 and 36 by monitoring the formation of 12-HPETE as a function of substrate and inhibitor concentration in the presence of 0.01% Triton X-100. Replots of Km/Vmax and 1/Vmax versus inhibitor concentration yielded linear trends for both 35 and 36. The Ki equaled 0.35 ± 0.08 µM for 35 and 0.53 ± 0.2 µM for 36, from the Km/Vmax graphs (Supporting Information, Figures S1 and S3). The Ki′ equaled 0.72 ± 0.1 µM for 35 and 0.62 ± 0.1 µM for 36, from the 1/Vmax graphs (Supporting Information, Figures S2 and S4). The data for both 35 and 36 correlate with their IC50 values (Table 2) and indicate that both molecules are mixed inhibitors, which is a common property of both 12-LOX22 and 15-LOX-129 inhibitors in general.
Upon completion of the SAR campaign, we then sought to explore the activity of 35 and 36 in relevant cell-based systems. As noted previously, 12-LOX has been linked to platelet activation, which plays a central role in the regulation of primary hemostasis and arterial thrombosis. Consequently, failure to attenuate platelet activation results in excessive clot formation, leading to adverse cardiovascular events such as myocardial infarction and stroke. Previous studies have shown that 12-LOX in human platelets is highly activated following stimulation of the protease-activated receptor-4 (PAR4) by the PAR4-activating peptide (PAR4-AP).30 Moreover, the bioactive metabolite of 12-LOX (12-HETE), resulting from the stereospecific oxidation of AA and reduction by peroxidases, demonstrates pro-thrombotic effects in human platelets.31 Therefore, treatment of PAR4-AP-induced human platelets with a small molecule 12-LOX inhibitor should attenuate the platelet aggregation in a dose-dependent manner. The results in Figure 2B show that 35 does in fact significantly reduce PAR4-AP induced platelet aggregation. Further, 12-LOX has been shown to play a role in calcium mobilization in human platelets and inhibition of 12-LOX should lead to a reduced concentration of free calcium in the platelet.32 To study this, we stimulated human platelets with 200 µM PAR4-AP and measured the free calcium in the platelet, using a C6 Accuri flow cytometer with various concentrations of 35 (Figure 2A). These data demonstrate that at concentrations as low as 250 nM of 35 calcium mobilization is reduced significantly (measured as fold change), with complete inhibition of calcium mobilization occurring at ∼5 µM. Comparable results were obtained with compound 36 and are shown in Supporting Information, Figure S5.
Figure 2.

PAR4-AP-induced calcium mobilization (A) and platelet aggregation (B) in human platelets. (A) Washed human platelets (1 × 106 platelets/mL) were stimulated with 200 µM PAR4-AP in the absence or presence of increasing concentrations of 35. Calcium mobilization was decreased as the concentration of 35 was increased. Calcium was measured in real time using a C6 Accuri flow cytometer. The experiments were done in triplicate. (B) Platelet aggregation of human platelets (3 × 108 platelets/mL) was measured in real-time using a Chronolog Lumi-Aggregometer (model 700D) following addition of PAR4-AP. While 10 µM 35 did not inhibit platelet aggregation, 25 µM 35 inhibited 80% platelet aggregation in washed human platelets.
To further validate these compounds, we then assessed the ability of 35 to inhibit 12-LOX in cell-based models relevant for diabetic disease. As discussed above, 12-LOX is expressed in pancreatic β-cells and its metabolic product, 12-HETE, is implicated in cytokine-induced cell death. Specifically, 12(S)-HETE has been shown to reduce metabolic activity, inhibit insulin secretion and ultimately induce cell death in human islets.33 Toward this end, we tested 35 in both a mouse derived β-cell line (BTC3) and human primary donor islets to determine its ability to inhibit AA/calcium ionophore induced stimulation of 12-HETE. Compound 35 was able to potently inhibit 12-HETE in BTC3 cells with an approximate IC50 of 1 µM, as measured by ELISA (Figure 3A). Similar results were obtained for compound 36 (Supporting Information, Figure S7). Given the difficulty in obtaining primary human islets from donated tissues, the activity in human islets was performed at a single concentration. The data presented in Figure 3B demonstrates significant inhibition of AA/IONO-induced 12-HETE production at 10 µM of 35.
Figure 3.

Inhibition of 12-HETE in mouse β-cells (A) and human islets (B). (A) Mouse β cells (BTC3) were treated with arachidonic acid and calcium ionophore (AA/IONO) alone or in the presence of 35. Graphed are the levels of 12-HETE expressed as a percentage of that detected in cells stimulated with AA/IONO alone. (B) The graph represents the increase (above control/unstimulated) in 12-HETE for human primary donor islets stimulated with arachidonic acid and calcium ionophore (AA/IONO) alone or in the presence of 10 µM of 35. 12-HETE was measured by ELISA. The data graphed in (A) is a representative experiment with each plotted data point being performed in triplicate. These plotted data are representative of four separate experimental determinations performed covering a lesser dose range. The graphed data are mean ± SEM, n = 3. Error bars for some points are masked by the symbol. The data was analyzed by nonlinear regression for dose–response curve inhibition, using variable or restricted hill slope, R2 > 0.81. This analysis was facilitated with Prism 5 software. In addition to compound, DMSO (stock solvent) was included with each condition. DMSO is also the solvent for the calcium ionophore (stimulant).
While the above data demonstrates activity in disease relevant cell-based models, in order to validate the potential use these compounds in proof of concept animal models, we needed to determine both its in vitro ADME and in vivo PK properties. These data are summarized in Tables 5 and 6, respectively. Both compounds 35 and 36 demonstrated excellent microsomal stability with both rat (T1/2 >30 min) and mouse (35: T1/2 >300 min) and was found to be stable to mouse plasma over a 2 h period (100% remaining). Compound 35 showed moderate cell permeability in the Caco-2 assay (1.5 × 10−6 cm/s) and does not appear to be a substrate for Pgp given the efflux ratio of <2, and both compounds had good passive permeability (PAMPA assay). Moreover, 35 showed no degradation in various aqueous buffers (pH 2–9) and was stable to 5 mM glutathione suggesting excellent stability (Supporting Information, Figure S7). One remaining liability is its aqueous solubility, which is <5 µM, however improved solubility is observed in the assay buffer (qualitative analysis). Moreover, we recently found that the benzoxazole derivative (36) has much improved solubility in buffer (>10-fold), albeit with slightly weaker potency toward 12-LOX. The drastic difference in the solubility profile was surprising, so the solubilities of these compounds were tested by a different method, chemiluminscent nitrogen detection (CLND), which confirmed the result obtained with LC-UV methodology (as shown in Table 5). Despite this improvement, at this point in the project we had collected more biological data on 35 and it is approximately 2-fold more potent than 36, so we proceeded with preliminary in vivo PK studies with this compound (Table 6).
Table 5.
In Vitro ADME Profile for 35
| microsomal stability |
|||||||
|---|---|---|---|---|---|---|---|
| compd | PBS buffer (pH 7.4) solubility (µg/mL) |
T1/2 (min)a,b | T1/2 (min)a,b | permeability (1 × 10−6 cm/s) |
efflux ratio |
mouse plasma stability remaining at 2 h (%) |
PBS buffer (pH 7.4) stability at 48 h (%) |
| 35 | <5 | >30 (rat) | >300a (mouse) |
1.5 (A→B)a 394c | 1.8 | 100 | 100 |
| 36 | >60 | >30 (rat) | NT | 168c | NT | 100 | 100 |
These experiments were conducted at Pharmaron Inc. All other studies were conducted at NCATS.
Represents the stability in the presence of NADPH. 35 and 36 showed no degradation without NADPH present over a 1 h period.
Represents permeability in the Parallel Artificial Membrane Permeability Assay (PAMPA) at pH 7.4.
Table 6.
In Vivo PK (Mouse) at 3 mpk (IV) and 30 mpk PO for 35a
| compd | routeb | T1/2 (h) | Tmax (h) | Cmax (µM) | AUCinf (µM·h) | Vd (L/kg) | Cl (mL/min/kg) | %F | MRTc |
|---|---|---|---|---|---|---|---|---|---|
| 35 | IV | 3.4 | NA | 112 | 34 | 0.55 | 3.4 | NA | 2.44 h |
| PO | 2.9 | 0.25 | 38 | 67 | NA | NA | 19.8 | NA |
All experiments were conducted at Pharmaron Inc. using male CD1 mice (6–8 weeks of age). Data was collected in triplicate at 8 time points over a 24 h period.
Formulated as a solution (5% DMSO, 10% Solutol, 20% PEG400, 65% water).
Represents the time for elimination of 63.2% of the IV dose.
Exploratory formulation studies led to an appropriate vehicle (DMSO:Solutol:PEG400:water; 5/10/20/65 v/v/v/v) in which 35 was administered as a solution via IV (3 mpk) and PO (30 mpk). These studies demonstrate that 35 is orally bioavailable (%F = ∼20), with a moderate half-life (T1/2 = 2.9 h). The compound has low clearance (3.4 mL/min/kg) and good overall exposure (AUCinf) of 38 µM·h. The volume of distribution (Vd) is 0.55 L/kg, which is low but suggests a reasonable distribution between tissue and blood. Given the favorable microsomal stability (phase I metabolism) yet modest in vivo T1/2, we suspected that the phenolic moiety could be glucuronidated (phase II metabolism), leading to higher clearance than anticipated. Alternatively, the parent compound could be directly eliminated through renal excretion. Subsequently, we incubated compound 35 with UDPGA cofactor instead of NADPH, which led to a T1/2 of ∼8 min (vs >30 min with NADPH). We thought that the sterically hindered environment of the 2-OH would possibly prevent glucuronidation from occurring, yet this data suggests otherwise. Another strategy which has been used to obviate a glucuronidation liability is to introduce electron-withdrawing groups next to the phenolic moiety of interest.34 As such, we employed this strategy with the synthesis of analogue 71 (2-OH, 3-Cl), however, this did not change the rate of glucuronidation. In addition to introducing other electron-withdrawing groups to the ring, another approach is to modify the phenolic hydroxyl using a pro-drug approach. Ideally, the pro-drug would be slowly hydrolyzed to the free phenol after it has bypassed first-pass metabolism. This approach has been used successfully in several marketed drugs, which contain phenolic groups.35 We plan to investigate these various approaches in future studies.
As stated above, previously reported inhibitors of 12-LOX, such as baicalein and nordihydroguairetic acid (NDGA), “bromo-phenols” or “pyrazole derivatives” (see Figure 1), all possess several liabilities. These compounds are not only less potent and less selective but are also not easily amenable to further optimization. Our previously described 12-LOX inhibitors ((8-hydroxy-5-nitroquinolin-7-yl)(thiophen-2-yl)-methyl)propionamide and N-((5-bromo-8-hydroxyquinolin-7-yl)(thiophen-2-yl)methyl)acetamide, Figure 1) do demonstrate potent inhibition (<500 nM) and excellent selectivity, but they were found to exhibit flat SAR, thus providing little opportunity for further modification. In this work, we describe a new chemotype that is structurally distinct from all previously reported inhibitors and possesses a drug-like scaffold. Compound 35 and related top analogues demonstrate potent (<500 nM) inhibition toward 12-LOX and excellent selectivity against related enzymes (15-LOX-1, 5-LOX, 15-LOX-2, COX−1/−2). This series is readily amenable to structural modifications and displays clear and tractable SAR. Compound 35 exhibits a favorable in vitro ADME and in vivo PK profile with activity in disease relevant cell-based systems, such thrombosis (platelet aggregation and calcium mobilization), and diabetes (12-HETE reducing in β-cells). Future studies are aimed at additional biological characterization of other top compounds (e.g., 36, 37) which possess comparable potency to 35 but with improved selectivity and solubility (e.g., 36). We hope these compounds have the potential to further validate 12-LOX as a potential therapeutic target for a variety of diseases, and we make them readily available to the research community.
EXPERIMENTAL SECTION
General Methods for Chemistry
All air or moisture sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Chemical reagents and anhydrous solvents were obtained from commercial sources and used as-is. Preparative purification was performed on a Waters semipreparative HPLC. The column used was a Phenomenex Luna C18 (5 µm, 30 mm × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient of 10–50% acetonitrile over 8 min was used during the purification. Fraction collection was triggered by UV detection (220 nm). Analytical analysis for purity was determined by two different methods denoted as final QC methods 1 and 2. Method 1: Analysis was performed on an Agilent 1290 Infinity series HPLC: UHPLC Long Gradient Equivalent 4–100% acetonitrile (0.05% trifluoroacetic acid) in water over 3.5 min run time of 4 min with a flow rate of 0.8 mL/min. A Phenomenex Kinetex 1.7 µm C18 column (2.1 mm × 100 mm) was used at a temperature of 50 °C. Method 2: Analysis was performed on an Agilent 1260 with a 7 min gradient of 4–100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) over 8 min run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 µm, 3 mm × 75 mm) was used at a temperature of 50 °C. Purity determination was performed using an Agilent diode array detector for both method 1 and method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. All of the analogues for assay have purity greater than 95% based on both analytical methods. 1H and 13C NMR spectra were recorded on Varian 400 (100) MHz or 600 (151) MHz spectrometers. High resolution mass spectrometry was recorded on Agilent 6210 time-of-flight LC/MS system.
Methods
General Synthetic Procedures. Scheme 1, Method A
4-Amino-N-(thiazol-2-yl)benzene sulfonamide (0.39 mmol) (2) and the required benzaldehyde (0.67 mmol) was added to a microwave vial, and ethanol (2 mL) was added. The reaction vessel was sealed and heated to 100 °C for 4–18 h. The reaction mixture was allowed to cool to room temperature, and sodium borohydride (0.80 mmol) was added and stirred for 30 min, during which time the reaction turned clear and then cloudy. The resulting solids were filtered, washed with ethanol, and purified using a prep-HPLC (gradient 10–100% acetonitrile w/0.1% TFA in water w/0.1% TFA) to give the desired product.
Scheme 1, Method B
A solution of 4-bromo-N-(thiazol-2-yl)benzene sulfonamide (0.31 mmol) (3) in dioxane (1 mL) was added to a mixture of sodium tert-butoxide (0.78 mmol), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (Xantphos) (0.02 mmol), and tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3) (6.27 µmol) in dioxane (1 mL). The resulting mixture was degassed with argon for 15 min, and then the requisite benzylamine (0.38 mmol) was added and the vessel was sealed and heated to 100 °C for 30 min in Biotage microwave reactor. The reaction mixture was cooled to room temperature and filtered through Celite. Silicycle silica bound thiol was added and stirred overnight, again filtered through a pad of Celite, concentrated, and purified by prep-HPLC (gradient 10– 100% acetonitrile w/0.1% TFA in water w/0.1% TFA) to give the desired product.
Scheme 1, Method C
4-Aminobenzenesulfonamide (4) (1.00 g, 5.81 mmol) and 2-hydroxy-3-methoxybenzaldehyde (1.00 g, 7.00 mmol) in EtOH (29 mL) was heated to reflux for 6 h until reaction was an orange turbid mixture. The reaction mixture was cooled to room temperature before sodium borohydride (0.33 g, 8.71 mmol) was added and then stirred for an additional 30 min. A white solid formed after 30 min and was collected by filtration and washed with copious amounts of ethanol, dried under vacuum, and used as-is in subsequent reactions. 1H NMR (400 MHz, DMSO-d6) δ 7.60–7.27 (m, 2H), 6.75–6.40 (m, 4H), 6.06 (t, J = 7.63 Hz, 1H), 4.18 (s, 2H), and 3.65 (s, 3H). 13C NMR (101 MHz, DMSO) δ 40.37, 55.32, 108.91, 109.42, 109.55, 111.29, 111.40, 121.05, 125.05, 127.49, 129.92, 150.17, 152.36, and 156.94. LC-MS retention time (method 1): 2.876 min. General procedure Step iv: 4-(2-Hydroxy-3-methoxybenzylamino)benzenesulfonamide (5) (0.58 mmol), arylbromide (0.70 mmol), K2CO3 (1.45 mmol), N,N′-dimethylethylenedi-amine (0.29 mmol), and copper(I)iodide (0.03 mmol) in 1,4-dioxane (1.5 mL) were place under N2 and sealed in a 5 mL sealed tube. The reaction was heated to 80 °C for 6–8 h and monitored by LC/MS analysis. Upon completion, the heterogeneous mixture was cooled to room temperature, filtered, and washed with dioxane. The solution was passed through a thiol cartridge (metal scavenging), diluted with AcOEt, and washed with NH4Cl (2×), water, and brine. The crude material was purified using a prep-HPLC (gradient 10–100% acetonitrile w/0.1% TFA in water w/0.1% TFA) to give the desired product.
Scheme 1, Method D: Representative Example N-(Benzo[d]-thiazol-2-yl)-4-nitrobenzenesulfonamide (7)
Step v: To a stirred solution of benzo [d] thiazol-2-amine (0.50 g, 3.35 mmol) in pyridine (1.60 mL, 20.08 mmol) was added 4-nitrobenzene sulfonyl chloride (6) (0.82 g, 3.68 mmol) in three equal parts. The reaction mixture was heated for 75 min at 100 °C and cooled to room temperature, after which time a yellow precipitate formed. The reaction mixture was allowed to sit at room temperature for 2 h, and then the yellow solid was collected by filtration, washed with ethanol, and dried under reduced pressure overnight to give 1.10 g of the desired product 7. 1H NMR (400 MHz, DMSO-d6) δ 8.90 (ddt, J = 0.75, 1.63, and 5.59 Hz, 1H), 8.54, 8.41–8.28 (m, 1H), 8.13–7.96 (m, 2H), 7.82 (dq, J = 0.80, and 7.96 Hz, 1H), 7.45–7.34 (m, 1H), 7.31–7.19 (m, 1H). LC-MS retention time (method 1): 3.272 min. HRMS: m/z (M + H)+ = calculated for C13H10N3O4S2, 336.0107; found, 336.0107.
4-Amino-N-(benzo[d]thiazol-2-yl)benzenesulfonamide
Step vi: N-(Benzo[d]thiazol-2-yl)-4-nitrobenzenesulfonamide (0.20 g, 0.60 mmol), zinc (0.16 g, 2.39 mmol), and acetic acid (0.14 mL, 2.39 mmol) was dissolved in MeOH (3 mL), and the mixture heated to 60 °C for 2 h. The heterogeneous mixture was filtered through a pad of Celite washed with hot methanol, concentrated, and purified using prep-HPLC (gradient 10–100% acetonitrile w/0.1% NH4OH in water w/0.1% NH4OH) to give the desired product. Alternate nitro reduction: N-(benzo [d] thiazol-2-yl)-4-nitrobenzenesulfonamide was dissolved in MeOH/EtOAc/THF (1:1:1) to give a 0.05 M solution passed through the H-Cube Pro flow reactor using a 10% Pd/C, 70 mm CatCart at 50 bar, and 50 °C for two cycles at 0.9 mL/min. The solution was concentrated to give a pale-yellow solid in a quantitative yield. 1H NMR (400 MHz, DMSO-d6) δ 7.49–7.34 (m, 3H), 7.18 (ddd, J = 0.56, 1.21, and 8.00 Hz, 1H), 7.03 (ddd, J = 1.33, 7.24, and 7.97 Hz, 1H), 6.83 (ddd, J = 1.20, 7.25, and 7.68 Hz, 1H), 6.51–6.38 (m, 2H), 5.42 (s, 2H). LC-MS retention time (method 2): 3.933 min. HRMS: m/z (M + H)+ = calculated for C13H12N3O2S2, 306.0365; found, 306.0360.
N-(Benzo[d]thiazol-2-yl)-4-((2-hydroxy-3-methoxybenzyl)-amino)benzenesulfonamide (Step vii, Representative Example) (35)
4-Amino-N-(benzo[d]thiazol-2-yl)benzenesulfonamide (0.10 g, 0.33 mmol) and 2-hydroxy-3-methoxybenzaldehyde (0.075 g, 0.491 mmol) were heated in EtOH (1.5 mL) at reflux for 18 h. The reaction mixture was allowed to cool to room temperature before the addition of NaBH4 (0.04 g, 0.98 mmol) and stirred for an additional 6 h. After this time, the reaction mixture was quenched with methanol and water then stirred for 20 min, the solids were filtered through Celite, and the filtrate collected and concentrated under reduced pressure to provide a yellow solid. The crude material was purified using a prep-HPLC (gradient 10–100% acetonitrile w/0.1% TFA in water w/0.1% TFA) to give the desired product 35. 1H NMR (400 MHz, DMSO-d6) δ 12.86 (s, 1H), 8.73 (d, J = 0.5 Hz, 1H), 7.75 (ddd, J = 0.6, 1.2, and 7.9 Hz, 1H), 7.54–7.46 (m, 2H), 7.40–7.31 (m, 1H), 7.28–7.16 (m, 2H), 6.93–6.79 (m, 2H), 6.78–6.55 (m, 4H), 4.23 (d, J = 5.8 Hz, 2H) and 3.78 (s, 3H). 13C NMR (DMSO-d6) δ ppm 152.4, 147.7, 144.3, 128.2, 125.7, 122.9, 120.4, 119.0, 111.4, 110.9, 56.2 and 40.8. LC-MS retention time (method 1): 2.260 min. HRMS: m/z (M + H)+ = calculated for C21H19N3O4S2, 441.0817; found, 441.0819.
Biological Reagents
All commercial fatty acids (Sigma-Aldrich Chemical Co.) were repurified using a Higgins HAIsil Semi-Preparative (5 µm, 250 mm × 10 mm) C-18 column. Solution A was 99.9% MeOH and 0.1% acetic acid; solution B was 99.9% H2O and 0.1% acetic acid. An isocratic elution of 85% A:15% B was used to purify all fatty acids, which were stored at −80 °C for a maximum of 6 months.
Human Platelets
Human platelets were obtained from healthy volunteers within the Thomas Jefferson University community and the Philadelphia area. These studies were approved by the Thomas Jefferson University Institutional Review Board, and informed consent was obtained from all donors before blood draw. Blood was centrifuged at 200g for 13 min at room temperature. Platelet-rich plasma was transferred into a conical tube containing a 10% acid citrate dextrose solution (39 mM citric acid, 75 mM sodium citrate, and 135 mM glucose, pH 7.4) and centrifuged at 2000g for 15 min at room temperature. Platelets were resuspended in Tyrode’s buffer (12 mM NaHCO3, 127 mM NaCl, 5 mM KCl, 0.5 mM NaH2PO4, 1 mM MgCl2, 5 mM glucose, and 10 mM HEPES), and the final platelet concentration was adjusted to 3 × 108 platelets/mL after counting with a ZI Coulter particle counter (Beckman Coulter, Fullerton, CA). Reported results are the data obtained using platelets from at least three different subjects. Agonists and inhibitors were used at concentrations indicated in the figures and figure legends.
Overexpression and Purification of Human 12-Lipoxyge-nase, Human 5-Lipoxygenase and the Human 15-Lipoxyge-nases
Human platelet 12-lipoxygenase (12-LOX), human reticulocyte 15-lipoxygenase-1 (15-LOX-1), and human epithelial 15-lipoxygenase-2 (15-LOX-2) were expressed as N-terminally, His6-tagged proteins and purified to greater than 90% purity, as evaluated by SDS-PAGE analysis.36 Human 5-lipoxygenase was expressed as a nontagged protein and used as a crude ammonium sulfate protein fraction, as published previously.37 Iron content of 12-LOX was determined with a Finnigan inductively coupled plasma mass spectrometer (ICP-MS), using cobalt-EDTA as an internal standard. Iron concentrations were compared to standardized iron solutions and used to normalize enzyme concentrations.
High-Throughput Screen Materials
Dimethyl sulfoxide (DMSO) ACS grade was from Fisher, while ferrous ammonium sulfate, Xylenol Orange (XO), sulfuric acid, and Triton X-100 were obtained from Sigma-Aldrich.
12-Lipoxygenase qHTS Assay (AID: 1452)
All screening operations were performed on a fully integrated robotic system (Kalypsys Inc., San Diego, CA) as described elsewhere.38 3 µL of enzyme (approximately 80 nM 12-LOX, final concentration) were dispensed into 1536-well Greiner black clear-bottom assay plates. Compounds and controls (23 nL) were transferred via Kalypsys PinTool equipped with 1536-pin array. The plate was incubated for 15 min at room temperature, and then a 1 µL aliquot of substrate solution (50 µM arachidonic acid final concentration) was added to start the reaction. The reaction was stopped after 6.5 min by the addition of 4 µL of FeXO solution (final concentrations of 200 µM Xylenol Orange (XO) and 300 µM ferrous ammonium sulfate in 50 mM sulfuric acid). After a short spin (1000 rpm, 15 s), the assay plate was incubated at room temperature for 30 min. The absorbances at 405 and 573 nm were recorded using ViewLux high-throughput CCD imager (Perkin-Elmer, Waltham, MA) using standard absorbance protocol settings. During dispensing, enzyme and substrate bottles were kept submerged into a +4 °C recirculating chiller bath to minimize degradation. Plates containing DMSO only (instead of compound solutions) were included approximately every 50 plates throughout the screen to monitor any systematic trend in the assay signal associated with reagent dispenser variation or decrease in enzyme specific activity. Data were normalized to controls, and plate-based data corrections were applied to filter out background noise.
Lipoxygenase UV–vis Assay
The inhibitor compounds were screened initially using one concentration point at 25 µM on a Perkin-Elmer Lambda 40 UV–vis spectrophotometer. The percent inhibition was determined by comparing the enzyme rates of the control (DMSO solvent) and the inhibitor sample by following the formation of the conjugated diene product at 234 nm (ε = 25000 M−1 cm−1 ). The reactions were initiated by adding either of 30 nM 12-LOX, 40 nM 15-LOX-1, 200 nM 15-LOX-2, or 200 nM of 5-LOX crude extract to a cuvette with a 2 mL reaction buffer constantly stirred using a magnetic stir bar at room temperature (22 °C). The concentration of 5-LOX in the crude extract was determined by SDS-PAGE in comparison with purified 5-LOX. It should be noted that LOX isozymes are often expressed in the inactive demetalated form, so it is best to utilize activity to determine the optimal LOX concentration for the assay (optimal rate of approximately 0.001 abs/s at 10 µM AA). Reaction buffers used for various lipoxygenase were as follows: 25 mM HEPES (pH 7.3), 0.3 mM CaCl2, 0.1 mM EDTA, 0.2 mM ATP, 0.01% Triton X-100, 10 µM AA for the crude, ammonium sulfate precipitated 5-LOX, and 25 mM HEPES (pH 7.5), 0.01% Triton X-100, 10 µM AA for 12-LOX, 15-LOX-1, and 15-LOX-2. The substrate concentration was quantitatively determined by allowing the enzymatic reaction to go to completion in the presence of 15-LOX-2. For the inhibitors that showed more than 50% inhibition (25 µM inhibitor) at the one-point screens, IC50 values were obtained by determining the % inhibition, relative to solvent vehicle only, at various inhibitor concentrations (approximate range: 0.1–25 µM inhibitor). The data was then plotted against inhibitor concentration, followed by a hyperbolic saturation curve fit (assuming total enzyme concentration , so ). It should be noted that all of the potent inhibitors displayed greater than 80% maximal inhibition, unless stated in the tables. Inhibitors were stored at −20 °C in DMSO.
Steady-State Inhibition Kinetics
The steady-state kinetics experiments were performed with 35 to determine the mode of inhibition. The inhibitor concentrations of 0, 0.2, 0.5, and 1 µM were used. Reactions were initiated by adding substrate (range 1–5 µM AA) to approximately 30 nM 12-LOX in a constantly stirring 2 mL cuvette containing 25 mM HEPES buffer (pH 7.5) in the presence of 0.01% Triton X-100. Lipoxygenase rates were determined by monitoring the formation of the conjugated product, 12-HPETE, at 234 nm (ε = 25 000 M−1 cm−1 ) with a Perkin-Elmer Lambda 45 UV/vis spectrophotometer. It should be noted that 12-LOX displays higher error in the KM values at low substrate concentrations (<1 µM) due to the limits of the spectrophotometer. The substrate concentration was quantitatively determined by allowing the enzymatic reaction to proceed to completion, using 12-LOX or 15-LOX-2. Kinetic data were obtained by recording initial enzymatic rates, at varied substrate and inhibitor concentrations, and subsequently fitted to the Henri–Michaelis– Menten equation, using KaleidaGraph (Synergy) to determine the microscopic rate constants, Vmax (µmol/min/mg) and Vmax/KM(µmol/min/mg/µM). The kinetic rate constants were subsequently replotted with KM/Vmax and 1/Vmax versus inhibitor concentration, yielding Ki and K i′, respectively.
Pseudoperoxidase Assay
The pseudoperoxidase activity rates were determined with BWb70c as the positive control, 13-(S)-HPODE as the oxidizing product, and 12-LOX or 15-LOX-1 on a Perkin-Elmer Lambda 40 UV–vis spectrophotometer, as described previously.28 Activity was determined by monitoring the decrease at 234 nm (product degradation) in buffer (50 mM sodium phosphate (pH 7.4), 0.3 mM CaCl2, 0.1 mM EDTA, 0.01% Triton X-100, and 20 µM 13-(S)-HPODE). About 60 nM 12-LOX was added to 2 mL of buffer containing 20 µM 13-(S)-HPODE, constantly stirred with a rotating stir bar (22 °C). Reaction was initiated by addition of 20 µM inhibitor (1:1 ratio to product). The percent consumption of 13-(S)-HPODE was recorded, and loss of product less than 20% was not considered as viable redox activity. Individual controls were conducted consisting of enzyme alone with product and 35 alone with enzyme. These negative controls formed the baseline for the assay, reflecting non-pseudoperoxidase dependent hydroperoxide product decomposition. To rule out the autoinactivation of the enzyme from pseudoperoxidase cycling, the 12-LOX residual activity was measured after the assay was complete. Then 20 µM AA was added to the reaction mixture and the residual activity was determined by comparing the initial rates with inhibitor and 13-(S)-HPODE versus inhibitor alone because the inhibitor by itself inherently lowers the rate of the oxygenation. Activity is characterized by direct measurement of the product formation with the increase of absorbance at 234 nm.
Cyclooxygenase Assay
Roughly 2–5 µg of either COX-1 or COX-2 were added to buffer containing 0.1 M Tris-HCl buffer (pH 8.0), 5 mM EDTA, 2 mM phenol, and 1 µM hematin at 37 °C. The selected inhibitors were added to the reaction cell, followed by an incubation of 5 min with either of the COX enzymes. The reaction was then initiated by adding 100 µM AA in the reaction cell. Data was collected using a Hansatech DW1 oxygen electrode, and the consumption of oxygen was recorded. Indomethacin and the solvent DMSO were used as positive and negative controls, respectively, and the percent inhibition of the enzyme was calculated by comparing the rates from samples and the controls
Platelet Aggregation
Washed platelets were adjusted to a final concentration of 3 × 108 platelets/mL. Where indicated, platelets were pretreated with 35 or compound 36 for 10 min at the indicated concentrations for 1 min. The aggregation response to PAR4-AP was measured using an aggregometer with stirring at 1100 rpm at 37 °C.
Calcium Mobilization
Platelets were recalcified to a final concentration of 1 mM followed by preincubation with Fluo-4 AM for 10 min. The platelets were then treated with 35 or compound 36 for 10 min at the indicated concentrations before stimulation with the indicated agonist. Calcium mobilization was measured using the Accuri C6 flow cytometer.
Mouse β-Cells (12-HETE Inhibition) Assay
Cells were gown to 90% confluency in 24-well plates in DMEM (cat. no. 11885092, Life Technologies Grand Island, NY) +10% FBS. Cells were pretreated with 35 and stimulated as for human islets. After four hours, the media was removed and spun at 1000 rpm for 5 min. The cleared supernatant was stored at −80 °C prior to analysis. For analysis, supernatants were extracted on SepPak c18 SPE column (cat. no. WAT054945, Waters Corporation, Milford, MA) and dried under nitrogen gas before reconstitution in 500 µL of 12-HETE ELISA buffer and analysis following manufacturers recommendations (cat. no. 901–050, Enzo Life Sciences, Plymouth Meeting, PA).
Human Islet (12-HETE Inhibition) Assay
Human donor islets obtained from integrated islet distribution program (www.iidp.cohorg. ) were incubated overnight in CMRL media (cat. no. 15–110-CV MediaTech, Inc. Manassas, VA) containing 10% fetal bovine serum, 1 U penicillin, and 1 µg streptomycin (pen/strep). Islets were equilibrated in serum free media (CMRL containing pen/Strep and 1% fatty acid free human serum albumin (cat. no. A1887 Sigma, St. Louis, MO)), for 1 h prior to pretreatment with 10 µM of 35 for 30 min. For 12-HETE induction, islets were treated with 100 µM arachidonic acid (cat. no. BML-FA003–0100, Enzo Life Sciences Plymouth Meeting, PA) and 5 µM A23187 (cat. no. C7522, Sigma, St. Louis, MO) for 4 h at 37 °C. Islets were harvested, centrifuged at 1000 rpm for 5 min with cleared supernatant and islet pellet being stored at −80 °C. For extraction of the supernatants, samples were acidified to pH 3 with 1N HCl for 30 min and spun at 1000 rpm for 5 min. Samples were added to a prepared column (prewashed with 3 mL of EtOH, followed by 3 mL of H2O) and washed with 3 mL of H2O, followed by 3 mL of 15% EtOH and 3 mL of hexane. The samples were eluted with 3 mL of ethyl acetate and dried under nitrogen gas before, reconstitution in 500 mL of 12-HETE ELISA sample buffer (Enzo Life Sciences, Plymouth Meeting, PA). Cell pellets were extracted using CHCl3/MeOH and samples dried under nitrogen gas before reconstitution in 250 µL of ELISA sample buffer. 12-HETE levels in samples were determined using a 12-HETE ELISA kit (cat. no. 901–050, Enzo Life Sciences, Plymouth Meeting, PA).
Supplementary Material
ACKNOWLEDGMENTS
D.K.L., A.Y., E.H.K., L.S., A.J., A.S., and D.J.M. were supported the intramural research program of the National Center for Advancing Translational Sciences and the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research (U54MH084681). Financial support was from the National Institute of Health grants R01 GM56062 (T.R.H.), HL114405 (M.H.), GM105671 (M.H.), and the Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research (R03 MH081283 (T.R.H.)). Additional financial support was from NIH (S10-RR20939 (T.R.H.)), the California Institute for Quantitative Biosciences for the UCSC MS Facility (T.R.H.), and the Cardeza Foundation for Hematologic Research (M.H.). Human islets were provided by the Integrated Islet Distribution Program (IIDP). Additional support was provided by the Juvenile Diabetes Research Foundation to J.L.N., D.T.F., and T.R.H. and NIH RO1 HL112605 to J.L.N. We thank Sam Michael and Richard Jones for automation support, Paul Shinn and Danielle van Leer for the assistance with compound management, William Leister, Heather Baker, Elizabeth Fernandez, and Christopher Leclair for analytical chemistry and purification support, and Kimloan Nguyen for in vitro ADME data.
ABBREVIATIONS USED
- LOX
lipoxygenase
- 15-LOX-1
human reticulocyte 15-lip-oxygenase-1
- 15-LOX-2
human epithelial 15-lipoxygenase-2
- 12-LOX
human platelet 12-lipoxygenase
- 15-rLO
rabbit reticulocyte 15-lipoxygenase
- COX
cyclooxygenase
- NDGA
nordihydroguaiaretic acid
- AA
arachidonic acid
- 15-HPETE
15-(S)-hydroperoxyeicosatetraenoic acid
- 15-HETE
15-(S)-hydroxyeicosatetraenoic acid
- 12-HPETE
12-(S)-hydroperox-yeicosatetraenoic acid
- 12-HETE
12-(S)-hydroxyeicosatetrae-noic acid
- LA
linoleic acid
- Vmax
maximal velocity (mmol/min)
- KM
Henri–Michaelis–Menten constant
- [E]
total active enzyme concentration
- IC50
inhibitor concentration at 50% inhibition
- HTS
high-throughput screening
- MLSMR
Molecular Libraries Small Molecule Repository
- qHTS
quantitative high-throughput screening
- PAR-4
protease-activated receptor-4
- EtOH
ethanol
- MeOH
methanol
- EtOAc
ethyl acetate
- AcOH
acetic acid
- MW
microwave
- Xantphos
4,5-bis-(diphenylphosphino)-9,9-dimethylxanthene
- SD
standard deviation
- MLPCN
Molecular Libraries Probe Production Center Network
Footnotes
Supporting Information
Additional figures, experimental procedures, and spectroscopic data (1H NMR, LC/MS, and HRMS) for representative compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Solomon EI, Zhou J, Neese F, Pavel EG. New insights from spectroscopy into the structure/function relationships of lipoxygenases. Chem. Biol. 1997;4:795–808. doi: 10.1016/s1074-5521(97)90113-7. [DOI] [PubMed] [Google Scholar]; (b) Brash AR. Lipoxygenases: Occurrence, Functions, Catalysis and Acquisition of Substrate. J. Biol. Chem. 1999;274:23679–23682. doi: 10.1074/jbc.274.34.23679. [DOI] [PubMed] [Google Scholar]
- 2.Serhan CN, Petasis NA. Resolvins and Protectins in Inflammation Resolution. Chem. Rev. 2011;111:5922–5943. doi: 10.1021/cr100396c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Ghosh J. Inhibition of Arachidonate 5-Lipoxygenase Triggers Prostate Cancer Cell Death through Rapid Activation of C-Jun N-Terminal Kinase. Biochem. Biophys. Res. Commun. 2003;307:342–349. doi: 10.1016/s0006-291x(03)01201-4. [DOI] [PubMed] [Google Scholar]; (b) Ghosh J, Myers CE. Inhibition of Arachidonate 5-Lipoxygenase Triggers Massive Apoptosis in Human Prostate Cancer Cells. Proc. Natl. Acad. Sci. U. S. A. 1998;95:13182–13187. doi: 10.1073/pnas.95.22.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nakano H, Inoue T, Kawasaki N, Miyataka H, Matsumoto H, Taguchi T, Inagaki N, Nagai H, Satoh T. Synthesis and Biological Activities of Novel Antiallergic Agents with 5-Lipoxygenase Inhibiting Action. Bioorg. Med. Chem. 2000;8:373–280. doi: 10.1016/s0968-0896(99)00291-6. [DOI] [PubMed] [Google Scholar]
- 4.Nakano H, Inoue T, Kawasaki N, Miyataka H, Matsumoto H, Taguchi T, Inagaki N, Nagai H, Satoh T. Synthesis and biological activities of novel antiallergic agents with 5-lipoxygenase inhibiting action. Bioorg. Med. Chem. 2000;8:373–380. doi: 10.1016/s0968-0896(99)00291-6. [DOI] [PubMed] [Google Scholar]
- 5.Berger W, De Chandt MT, Cairns CB. Zileuton: Clinical Implications of 5-Lipoxygenase Inhibition in Severe Airway Disease. Int. J. Clin. Pract. 2007;61:663–676. doi: 10.1111/j.1742-1241.2007.01320.x. [DOI] [PubMed] [Google Scholar]
- 6.(a) Cyrus T, Witztum JL, Rader DJ, Tangirala R, Fazio S, Linton MF, Funk CD. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J. Clin. Invest. 1999;103:1597–1604. doi: 10.1172/JCI5897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Harats D, Shaish A, George J, Mulkins M, Kurihara H, Levkovitz H, Sigal E. Overexpression of 15-lipoxygenase in vascular endothelium accelerates early atherosclerosis in LDL receptor-deficient mice. Arteriosler. Thromb. Vasc. Biol. 2000;20:2100–2105. doi: 10.1161/01.atv.20.9.2100. [DOI] [PubMed] [Google Scholar]
- 7.Pratico D, Zhukareva V, Yao Y, Uryu K, Funk CD, Lawson JA, Trojanowski JQ, Lee VM. 12/15-Lipoxygenase is increased in Alzheimer’s disease: possible involvement in brain oxidative stress. Am. J. Pathol. 2004;164:1655–1662. doi: 10.1016/S0002-9440(10)63724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) van Leyen K, Arai K, Jin G, Kenyon V, Gerstner B, Rosenberg PA, Holman TR, Lo EH. Novel lipoxygenase inhibitors as neuroprotective reagents. J. Neurosci. Res. 2008;86:904–909. doi: 10.1002/jnr.21543. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) van Leyen K, Kim HY, Lee SR, Jin G, Arai K, Lo EH. Baicalein and 12/15-lipoxygenase in the ischemic brain. Stroke. 2006;37:3014–3018. doi: 10.1161/01.STR.0000249004.25444.a5. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto S. Mammalian lipoxygenases: Molecular Structures, functions. Biochim. Biophys. Acta. 1992;1128:117–131. doi: 10.1016/0005-2760(92)90297-9. [DOI] [PubMed] [Google Scholar]; (b) Funk CD, Keeney DS, Oliw EH, Vogel S, Muller-Decker K, Mincheva A, Lichter P, Marks F, Krieg P. Murine epidermal lipoxygenase (Aloxe) encodes a 12-lipoxygenase isoform. FEBS Lett. 1997;402:162–166. doi: 10.1016/s0014-5793(96)01517-7. [DOI] [PubMed] [Google Scholar]
- 10.(a) Catalano A, Procopio A. New aspects on the role of lipoxygenases in cancer progression. Histol. Histopathol. 2005;20:969–975. doi: 10.14670/HH-20.969. [DOI] [PubMed] [Google Scholar]; (b) Nie D, Hillman GG, Geddes T, Tang K, Pierson C, Grignon DJ, Honn KV. Platelet-type 12-lipoxygenase in a human prostate carcinoma stimulates angiogenesis and tumor growth. Cancer Res. 1998;58:4047–4051. [PubMed] [Google Scholar]; (c) Natarajan R, Esworthy R, Bai W, Gu JL, Wilczynski S, Nadler JL. Increase 12-lipoxygenase expression in breast cancer cells and tissues Regulation by epidermal growth factor. J. Clin. Endocrinol. Metab. 1997;82:1790–1798. doi: 10.1210/jcem.82.6.3990. [DOI] [PubMed] [Google Scholar]; (d) Kamitani H, Geller M, Eling T. The possible involvement of 15-lipoxygenase/leukocyte type 12-lipoxygenase in colorectal carcinogenesis. Adv. Exp. Med. Biol. 1999;469:593–598. doi: 10.1007/978-1-4615-4793-8_86. [DOI] [PubMed] [Google Scholar]; (e) Soriano AF, Helfrich B, Chan DC, Heasley LE, Bunn PA, Chou TC. Synergistic effects of new chemopreventive agents and conventional cytotoxic agents against human lung cancer cell lines. Cancer Res. 1999;59:6178–6184. [PubMed] [Google Scholar]
- 11.(a) Nappez C, Liagre B, Beneytout JL. Changes in lipoxygenase activities in human erythroleukemia (HEL) cells during diosgenin-induced differentiation. Cancer Lett. 1995;96:133–140. doi: 10.1016/0304-3835(95)03923-k. [DOI] [PubMed] [Google Scholar]; (b) Timár J, Silletti S, Bazaz R, Raz A, Honn KV. Regulation of melanoma-cell motility by the lipoxygenase metabolite 12-(S)-HETE. Int. J. Cancer. 1993;55:1003–1010. doi: 10.1002/ijc.2910550621. [DOI] [PubMed] [Google Scholar]; (c) Honn KV, Timár J, Rozhin J, Bazaz R, Sameni M, Ziegler G, Sloane B. A lipoxygenase metabolite, 12-(S)-HETE, stimulates protein kinase C-mediated release of cathepsin B from malignant cells. Exp. Cell. Res. 1994;214:120–130. doi: 10.1006/excr.1994.1240. [DOI] [PubMed] [Google Scholar]; (d) Nie D, Tang K, Diglio C, Honn K. Eicosanoid regulation of angiogenesis: role of endothelial arachidonate 12-lipoxygenase. Blood. 2000;95:2304–2311. [PubMed] [Google Scholar]
- 12.(a) McDuffie M, Maybee NA, Keller SR, Stevens BK, Garmey JC, Morris MA, Kropf E, Rival C, Ma K, Carter JD, Tersey SA, Nunemaker CS, Nadler JL. Nonobese diabetic (NOD) mice congenic for a targeted deletion of 12/15-lipoxygenase are protected from autoimmune diabetes. Diabetes. 2008;57:199–209. doi: 10.2337/db07-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bleich D, Chen S, Zipser B, Sun D, Funk CD, Nadler JL. Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J. Clin. Invest. 1999;103:1431–1436. doi: 10.1172/JCI5241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Imai Y, Dobrian AD, Weaver JR, Butcher MJ, Cole BK, Galkina EV, Morris MA, Taylor-Fishwick DA, Nadler JL. Interaction between cytokines and inflammatory cells in islet dysfunction, insulin resistance and vascular disease. Diabetes Obes. Metab. 2013;15(Suppl 3):117–129. doi: 10.1111/dom.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL. 12-Lipoxygenase Products Reduce Insulin Secretion and β-Cell Viability in Human Islets. J. Clin. Endocrinol. Metab. 2010;95:887–893. doi: 10.1210/jc.2009-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, Yang ZD, Smith KM, Carter JD, Nadler JL. Activation of 12-Lipoxygenase in proinflammatory cytokine-mediated β-cell toxicity. Diabetologia. 2005;48:486–495. doi: 10.1007/s00125-005-1673-y. [DOI] [PubMed] [Google Scholar]
- 14.Brash AR. A review of possible roles of the platelet 12-lipoxygenase. Circulation. 1985;72:702–707. doi: 10.1161/01.cir.72.4.702. [DOI] [PubMed] [Google Scholar]
- 15.(a) Aharony D, Smith JB, Silver MJ. Regulation of arachidonate-induced platelet aggregation by the lipoxygenase product, 12-hydroperoxyeicosatetraenoic acid. Biochim. Biophys. Acta. 1982;718:193–200. doi: 10.1016/0304-4165(82)90219-7. [DOI] [PubMed] [Google Scholar]; (b) Chang J, Blazek E, Kreft AF, Lewis AJ. Inhibition of platelet neutrophil phospholipase A2 by hydroxyeicosate-traenoic acids (HETES) A novel pharmacological mechanism for regulating free fatty acid release. Biochem. Pharmacol. 1985;34:1571–1575. doi: 10.1016/0006-2952(85)90701-4. [DOI] [PubMed] [Google Scholar]; (c) Nyby MD, Sasaki M, Ideguchi Y, Wynne HE, Hori MT, Berger ME, Golub MS, Brickman AS, Tuck ML. Platelet lipoxygenase inhibitors attenuate thrombin-and thromboxane mimetic-induced intracellular calcium mobilization and platelet aggregation. J. Pharmacol. Exp. Ther. 1996;278:503–509. [PubMed] [Google Scholar]
- 16.Kenyon V, Rai G, Jadhav A, Schultz L, Armstrong M, Jameson JB, Perry S, Joshi N, Bougie JM, Leister W, Taylor-Fishwick DA, Nadler JL, Holinstat M, Simeonov A, Maloney DJ, Holman TR. Discovery of Potent and Selective Inhibitors of Human Platelet-Type 12-Lipoxygenase. J. Med. Chem. 2011;54:5485–5497. doi: 10.1021/jm2005089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeung J, Apopa PL, Vesci J, Kenyon V, Rai G, Jadhav A, Simeonov A, Holman TR, Maloney DJ, Boutaud O. Holinstat Protein Kinase C Regulation of 12-Lipoxygenase-Mediated Human Platelet Activation. Mol. Pharmacol. 2012;81:420–430. doi: 10.1124/mol.111.075630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Whitman S, Gezginci M, Timmermann BN, Holman TR. Structure-activity relationship studies of nordihydroguaiaretic acid inhibitors toward soybean, 12-human, and 15-human lipoxygenase. J. Med. Chem. 2002;45:2659–2661. doi: 10.1021/jm0201262. [DOI] [PubMed] [Google Scholar]; (b) Deschamps JD, Kenyon VA, Holman TR. Baicalein is a potent in vitro inhibitor against both reticulocyte 15-human and platelet 12-human lipoxygenases. Bioorg. Med. Chem. 2006;14:4295–4301. doi: 10.1016/j.bmc.2006.01.057. [DOI] [PubMed] [Google Scholar]; (c) Segraves EN, Shah RR, Segraves NL, Johnson TA, Whitman S, Sui JK, Kenyon VA, Cichewicz RH, Crews P, Holman TR. Probing the activity differences of simple and complex brominated aryl compounds against 15-soybean, 15-human, and 12-human lipoxygenase. J. Med. Chem. 2004;47:4060–4065. doi: 10.1021/jm049872s. [DOI] [PubMed] [Google Scholar]; (d) Amagata T, Whitman S, Johnson T, Stessmann CC, Carroll J, Loo C, Clardy J, Lobkovsky E, Crews P, Holman TR. Sponge Derived Terpenoids with Selectivity towards Human 15-Lipoxygenase versus Human 12-Lipoxygenase. J. Nat. Prod. 2003;66:230–235. doi: 10.1021/np020462l. [DOI] [PubMed] [Google Scholar]; (e) Cichewicz RH, Kenyon VA, Whitman S, Morales NM, Arguello JF, Holman TR, Crews P. Redox inactivation of human 15-lipoxygenase by marine-derived meroditerpenes and synthetic chromanes: archetypes for a unique class of selective and recyclable inhibitors. J. Am. Chem. Soc. 2004;126:14910–14920. doi: 10.1021/ja046082z. [DOI] [PubMed] [Google Scholar]; (f) Vasquez-Martinez Y, Ohri RV, Kenyon V, Holman TR, Sepulveda-Boza S. Structure-activity relationship studies of flavonoids as potent inhibitors of human platelet 12-hLO, reticulocyte 15-hLO-1, and prostate epithelial 15-hLO-2. Bioorg. Med. Chem. 2007;15:7408–7425. doi: 10.1016/j.bmc.2007.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Sailer ER, Schweizer S, Boden SE, Ammon HPT, Safayhi H. Characterization of an acetyl-11-keto-B-boswellic acid and arachidonate-binding regulatory site of 5-lipoxygenase using photo-affinity labeling. Eur. J. Biochem. 1998;256:364–368. doi: 10.1046/j.1432-1327.1998.2560364.x. [DOI] [PubMed] [Google Scholar]; (b) Malterud KE, Rydland KM. Inhibitors of 15-lipoxygenase from orange peel. J. Agric. Food Chem. 2000;48:5576–5580. doi: 10.1021/jf000613v. [DOI] [PubMed] [Google Scholar]; (c) Moreau RA, Agnew J, Hicks KB, Powell MJ. Modulation of lipoxygenase activity by bacterial hopanoids. J. Nat. Prod. 1997;60:397–398. doi: 10.1021/np960611y. [DOI] [PubMed] [Google Scholar]
- 10.Kenyon V, Chorny I, Carvajal WJ, Holman TR, Jacobson MP. Novel human lipoxygenase inhibitors discovered using virtual screening with homology models. J. Med. Chem. 2006;49:1356–1363. doi: 10.1021/jm050639j. [DOI] [PubMed] [Google Scholar]
- 21.Deschamps JD, Gautschi JT, Whitman S, Johnson TA, Gassner NC, Crews P, Holman TR. Discovery of platelet-type 12-human lipoxygenase selective inhibitors by high-throughput screening of structurally diverse libraries. Bioorg. Med. Chem. 2007;15:6900–6908. doi: 10.1016/j.bmc.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.(a) Kenyon V, Rai G, Jadhav A, Schultz L, Armstrong M, Jameson JB, Perry S, Joshi N, Bougie JM, Leister W, Taylor-Fishwick DA, Nadler JL, Holinstat M, Simeonov A, Maloney DJ, Holman TR. Discovery of Potent and Selective Inhibitors of Human Platelet-Type 12-Lipoxygenase. J. Med. Chem. 2011;54:5485–5497. doi: 10.1021/jm2005089. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rai G, Jadhav A, Schultz L, Kenyon V, Leister W, Simeonov A, Holman TR, vMaloney DJ. Probe Reports from the NIH Molecular Libraries Program [Internet] Bethesda, MD: National Center for Biotechnology Information; 2010. Selective Small Molecule Inhibitors of 12-Human Lipoxygenase (12-hLO) www.ncbi.nlm.nih.gov/books/NBK56235/ [PubMed] [Google Scholar]
- 23.Andreu R, Ronda JC. Synthesis of 3,4-Dihydro-2H-1,3-benzoxazines by Condensation of 2-Hydroxyaldehydes and Primary Amines: Application to the Synthesis of Hydroxy-Substituted and Deuterium-Labeled Compounds. Synth. Commun. 2008;38:2316–2329. [Google Scholar]
- 24.(a) Fulp AB, Johnson MS, Markworth J, Marron BE, Seconi DC, West CW, Wang X, Zhou S. Sodium Channel Blockers. 2008 Jul 14; U.S. Patent WO/2009/012242, [Google Scholar]; (b) Lee YK, Parks DJ, Lu T, Thieu TV, Markotan T, Pan W, McComsey DF, Milkiewicz KL, Crysler CS, Ninan N, Abad MC, Giardino EC, Maryanoff BE, Damiano BP, Player M. R. J. Med. Chem. 2008;51:282–297. doi: 10.1021/jm701217r. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Guram A, Ronk M, Milne JE, Tedrow JS, Faul MM. Copper-catalyzed N-arylation of sulfonamides with aryl bromides. Tetrahedron Lett. 2012;53:7–10. [Google Scholar]
- 26.Nelson MJ. Catecholate Complexes of Ferric Soybean Lipoxygenase 1. Biochemistry. 1988;27:4273–4298. [Google Scholar]
- 27.Gan Q-F, Browner MF, Sloane DL, Sigal E. Defining the arachidonic acid binding site of human 15-lipoxygenase. J. Biol. Chem. 1996;271:25412–25418. doi: 10.1074/jbc.271.41.25412. [DOI] [PubMed] [Google Scholar]
- 28.Hoobler EK, Holz C, Holman TR. Pseudoperoxidase investigations of hydroperoxides and inhibitors with human lipoxygenases. Bioorg. Med. Chem. 2013;21:3894–3899. doi: 10.1016/j.bmc.2013.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rai G, Kenyon V, Jadhav A, Schultz L, Armstrong M, Jameson JB, III, Hoobler E, Leister W, Simeonov A, Holman TR, Maloney DJ. Discovery of Potent and Selective Inhibitors of Human Reticulocyte 15-Lipoxygenase 1. J. Med. Chem. 2010;53:7392–7404. doi: 10.1021/jm1008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikei KN, Yeung J, Apopa PL, Ceja J, Vesci J, Holman TR, Holinstat M. Investigations of human platelet-type 12-Lip-oxygenase: role of lipoxygenase products in platelet activation. J. Lipid Res. 2012;53:2546–2559. doi: 10.1194/jlr.M026385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.(a) Thomas CP, Morgan LT, Maskrey BH, Murphy RC, Kuhn H, Hazen SL, Goodall AH, Hamali HA, Collins PW, O’Donnell VD. Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependant thrombin generation. J. Biol. Chem. 2010;285:6891–6903. doi: 10.1074/jbc.M109.078428. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yeung J, Apopa PL, Vesci J, Stolla M, Rai G, Simeonov A, Jadhav A, Fernandez-Perez P, Maloney DJ, Boutaud O, Holman TR, Holinstat M. 12-Lipoxygenase activity plays an important role in PAR4 and GPVI-mediated platelet reactivity. Thromb. Haemostasis. 2013;110:561–581. doi: 10.1160/TH13-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeung J, Apopa PL, Vesci J, Kenyon V, Rai G, Jadhav A, Simeonov A, Holman TR, Maloney DJ, Boutaud O, Holinstat M. Protein kinase C regulation of 12-lipoxygenase-mediated human platelet activation. Mol. Pharmaceutics. 2012;81:420–430. doi: 10.1124/mol.111.075630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, Nadler JL. 12-Lipoxygenase Products Reduce Insulin Secretion and β-Cell Viability in Human Islets. J. Clin. Endocrinol. Metab. 2010;95:887–893. doi: 10.1210/jc.2009-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madsen P, Ling A, Plewe M, Sams CK, Knudsen LB, Sidelmann UG, Ynddal L, Brand CL, Andersen B, Murphy D, Teng M, Truesdale L, Kiel D, May J, Kuki A, Shi S, Johnson MD, Teston KA, Feng J, Lakis J, Anderes K, Gregor V, Lau J. Optimization of Alkylidene Hydrazide Based Human Glucagon Receptor Antagonists Discovery of the Highly Potent and Orally Available 3-Cyano-4-hydroxybenzoic Acid [1-(2,3,5,6-Tetramethylben-zyl)-1H–indol-4-ylmethylene]hydrazide. J. Med. Chem. 2002;45:5755–5775. doi: 10.1021/jm0208572. [DOI] [PubMed] [Google Scholar]
- 35.Ettmayer P, Amidon GL, Clement B, Testa B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004;47:2393–2404. doi: 10.1021/jm0303812. [DOI] [PubMed] [Google Scholar]
- 36.(a) Amagata T, Whitman S, Johnson TA, Stessman CC, Loo CP, Lobkovsky E, Clardy J, Crews P, Holman TR. Exploring sponge-derived terpenoids for their potency and selectivity against 12-human, 15-human, and 15-soybean lipoxygenases. J. Nat. Prod. 2003;66:230–235. doi: 10.1021/np020462l. [DOI] [PubMed] [Google Scholar]; (b) Ohri RV, Radosevich AT, Hrovat KJ, Musich C, Huang D, Holman TR, Toste FD. A Re(V)-catalyzed C-N bond-forming route to human lipoxygenase inhibitors. Org. Lett. 2005;7:2501–2504. doi: 10.1021/ol050897a. [DOI] [PubMed] [Google Scholar]; (c) Chen X-S, Brash A, Funk C. Purification and characterization of recombinant histidine-tagged human platelet 12-lipoxygenase expressed in a baculovirus/insect cell system. Eur. J. Biochem. 1993;214:845–852. doi: 10.1111/j.1432-1033.1993.tb17988.x. [DOI] [PubMed] [Google Scholar]
- 37.Robinson SJ, Hoobler EK, Riener M, Loveridge ST, Tenney K, Valeriote FA, Holman TR, Crews P. Using enzyme assays to evaluate the structure and bioactivity of sponge-derived meroterpenes. J. Nat. Prod. 2009;72:1857–1863. doi: 10.1021/np900465e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative High-Throughput Screening: A Titration-Based Approach That Efficiently Identifies Biological Activities in Large Chemical Libraries. Proc. Natl. Acad. Sci. U. S. A. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.