

Abstract
Background
NFE2-related factor 2 (Nrf2) is a master regulatory transcription factor for antioxidant genes. Inhibition of its adaptor protein, Kelch-like ECH-associated protein 1 (Keap1), activates Nrf2. Podocyte injury triggers the progressive deterioration of glomerular damage toward glomerulosclerosis. We examined whether modulation of the Keap1-Nrf2 system has an impact on this process.
Methods
Nrf2 null-mutant (KO) and Keap1 hypomorphic knockdown (KD) mice were crossed with NEP25 mice, in which podocyte-specific injury can be induced by an immunotoxin.
Results
Thiobarbituric acid reactive substances, 8-hydroxydeoxyguanosine and phosphorylated JNK were increased in the injured NEP25 kidney. Real-time PCR revealed that Keap1 KD upregulated Nrf2 target genes, including Gclc, Gclm, Gstp1, Gstp2 and Nqo1 in the glomerulus. However, podocyte injury did not upregulate these genes in Keap1 wild-type mice, nor did it further increase the expression of those genes in Keap1 KD mice. Three weeks after the induction of podocyte injury, glomerulosclerosis was considerably more attenuated in Keap1 KD mice than in control mice (median sclerosis index, 0.27 versus 3.03, on a 0–4 scale). Keap1 KD mice also showed considerably preserved nephrin staining (median index, 6.76 versus 0.91, on a 0–8 scale) and decreased glomeruli containing desmin-positive injured podocytes (median percentage, 24.5% versus 85.8%), along with a decrease in mRNAs for Fn1, Tgfb1, Col4a4 and Col1a2.
Conclusions
Thus, podocyte injury cannot effectively activate Nrf2, but Nrf2 activation by Keap1 knockdown attenuates glomerulosclerosis. These results indicate that the Nrf2-Keap1 system is a promising drug target for the treatment of chronic kidney diseases.
Keywords: antioxidant genes, glomerulosclerosis, hypomorphic allele, oxidative stress
INTRODUCTION
The histology of end-stage renal failure is typically characterized by glomerulosclerosis. Many studies have shown that podocyte injury is a key step triggering the progression of glomerulosclerosis [1–5]. A number of factors, including genetic, mechanical, and immunological stresses such as toxins, as well as oxidative stress, can cause podocyte injury [6–11].
Kelch-like ECH-associated protein 1 (Keap1) is a cytoplasmic protein that serves as a substrate adaptor molecule for ubiquitin E3 ligase to regulate proteasomal degradation [12, 13]. The most relevant substrate of Keap1 is the transcriptional factor NFE2-related factor 2 (Nrf2), which serves as a master regulator of phase II detoxification and antioxidative stress enzymes/proteins [14, 15]. Without oxidative stress, Keap1 suppresses Nrf2 activity by promoting ubiquitination of Nrf2 and subsequent degradation by the proteasome. Upon exposure to oxidative stress, reactive cysteine residues in Keap1 become modified, which leads to a decline in the E3 ligase activity, stabilization of Nrf2 and the subsequent robust induction of cytoprotective genes. These include the cystine transporter, enzymes for glutathione production and enzymes metabolizing oxidative intermediates, such as NAD(P)H:quinone oxidoreductase 1 (NQO1) [16–18].
Indeed, Nrf2-mediated transcriptional responses have been shown to have protective effects in various experimental diseases, including lipopolysaccharide-induced sepsis (shock) [19, 20], oxidative lung injury and fibrosis, asthma, smoking-induced emphysema [21–25], and brain ischemia-reperfusion injury [26, 27]. These findings are all derived from studies with Nrf2 knockout mice or pharmacological activation of Nrf2 by small-molecule compounds.
In Keap1-null mice, Nrf2 constitutively accumulated in nuclei, and Nrf2 target genes were constitutively activated [15]. However, the knockout mice were found to die in the neonatal stage, mainly due to hyperkeratosis in the upper digestive tract. The phenotypes seen in the knockout mice were all canceled in the double-mutant mice that were null for the Keap1 and Nrf2 genes, indicating that Keap1-null phenotypes are the result of the constitutive activation of Nrf2 [15]. To generate Keap1 conditional knockout mice, we previously generated mice carrying a mutant allele (Keap1loxP) in which loxP sequences and the EGFP gene were inserted into an intron of the Keap1 gene. Unexpectedly, this allele was found to be hypomorphic. Thus, Keap1loxP/– mice, even in the absence of Cre recombinase, displayed a very low level of Keap1 gene expression, resulting in a constitutive and ubiquitous activation in Nrf2 [28]. As Keap1loxP/− mice (designated as Keap1 knockdown mice) are vital and have no obvious abnormal phenotype, they can provide an opportunity to examine the effect of constitutive Nrf2 activation.
Several papers reported that the Keap1-Nrf2 system is involved in kidney diseases. Thus, Nrf2 knockout mice are more sensitive to experimental diabetic nephropathy [29], and Nrf2-activating compounds (sulforaphane or cinnamic aldehyde) attenuated the progression of renal damage [30]. Ischemia/reperfusion renal injury was exaggerated in Nrf2 knockout mice, and the injury was attenuated by treatment with antioxidants (N-acetyl-cysteine or glutathione) in these mice [31]. However, few studies have been reported to date about whether the Keap1-Nrf2 system plays a role in podocyte injury and the subsequent development of glomerulosclerosis [32], the common pathogenic pathway to chronic renal failure, especially in nondiabetic and nonischemic conditions.
Previously, we established a mouse model of selective and inducible podocyte injury (NEP25) [33]. NEP25 mice express hCD25 selectively on podocytes. After injection of the hCD25-targeted immunotoxin LMB2, only hCD25-expressing podocytes are selectively injured. With a low dose [0.625 ng/g body weight (BW)] of LMB2, NEP25 mice developed moderate proteinuria, which peaked 1–2 weeks after the injection and gradually decreased. After 3–4 weeks, some glomeruli developed global or segmental sclerosis, whereas other glomeruli recovered from the podocyte injury and showed a normal structure [33]. The injury induced by this low dose of LMB2 can be attenuated by angiotensin II inhibition [34]. To clarify whether the Keap1-Nrf2 system is involved in the injury and recovery process of podocytes and subsequent development of glomerulosclerosis, we performed histological and morphological analysis with Keap1loxP/−/Nep25 and Nrf2−/−/Nep25 mice treated with the low dose of LMB2.
MATERIALS AND METHODS
Animal experiments
Keap1loxP/− and Nrf2−/− mice were previously reported [14, 28, 35]. To induce podocyte-specific injury, Nrf2−/− or Keap1loxP/− mice were mated with NEP25 mice on a C57 BL/6 genetic background [33]. Genotyping for Nrf2−, Keap1− and NEP25 was performed on tail DNA by PCR as previously reported [14, 15, 33]. Keap1loxP allele was identified by PCR with primers, 5′-GAAGCAGCACGACTTCTTCAAGTC-3′ and 5′-TGGCGGATCTTGAAGTTCACCTTG-3′.
Ten mice (5 males and 5 females) carrying Keap1+/+/Nep25 and 14 mice (6 males and 8 females) carrying Keap1loxP/−/Nep25 (all 3–5 months of age) were injected with 0.625 ng/g BW of LMB2. Twenty-four-hour urine was collected before and 7, 14 and 21 days after the injection of LMB2. The animals were euthanized 21 days after the injection. Three Keap1loxP/−/Nep25 mice were excluded from the analysis because they were found to have unilateral hydronephrosis on autopsy, which is frequently observed in Keap1loxP/− mice without podocyte injury (unpublished observation). The remaining 11 Keap1loxP/−/Nep25 mice showed no appreciable phenotype that indicated ureteral obstruction—such as enlarged calyx, thinning of medulla and ectopic staining of Tamm–Horsfall protein on the glomerulus (data not shown)—and were therefore subjected to the following analysis. A similar experiment with the same protocol was duplicated using eight Keap1+/+/Nep25 mice (all females) and five Keap1loxP/−/Nep25 mice (two males and three females). In addition, nine Nrf2−/−/Nep25 mice (four males, five females) and six Nrf2+/+/Nep25 mice (three males, three females) were injected with 0.625 ng/g BW of LMB2, and similarly analyzed with the same protocol.
Separately, to evaluate the expression level of Keap1, Nrf2 and Nrf2 target genes, three Keap1+/+/Nep25 and three Keap1loxP/−/Nep25 mice were injected with 0.625 ng/g BW of LMB2, and 5 days later, glomeruli were isolated by perfusing with Dynabeads. Glomeruli were also isolated from six Keap1+/+/Nep25 and six Keap1loxP/−/Nep25 mice without LMB2. RNA was isolated from the glomeruli and subjected to real-time PCR analysis, as described below.
Urinalysis
Concentration of creatinine in urine was determined by enzymatic method in an outside laboratory (SRL, Tokyo, Japan). Since we used both male and female mice in the study and adult male mice excrete a larger and variable amount of small molecular weight proteins, we measured albumin concentration rather than total protein to evaluate proteinuria in this study. Urinary albumin concentration was measured by the nephelometric method as follows. Urine samples were incubated with a rabbit polyclonal anti-mouse albumin antibody (Cappel, Durham, NC, USA) in PBS at room temperature. The samples were then illuminated with an LED light, and the intensity of the scattered light was measured at 840 nm with a Behring Nephelometer (Siemens Health Care Diagnostics, Norwood, MA, USA). The measurements were highly correlated with the total protein concentration in female samples measured by the pyrogallol red method (Supplementary Figure S1), validating the accuracy of the measurements. This supports the reliability of the nephelometric method when used for albumin measurement.
Statistical analysis
Urinary albumin/creatinine ratios showed a logarithmic normal distribution. For these data, geographic means and 95% confidence intervals are reported in the text. To compare between two groups, MANOVA was used after logarithmic transformation. To compare the sclerosis and nephrin staining indices, pJNK and TBARS between the two groups, nonparametric Mann–Whitney U-test was used. For these data, medians, 25 percentiles and 75 percentiles are reported. One-way ANOVA was used to compare the mRNA level among four groups. Values were considered significant at P < 0.05. Other methods are described in the Supplementary data.
RESULTS
Podocyte injury induced oxidative stress in the kidney in NEP25 mice
We induced podocyte injury by injecting LMB2 into NEP25 mice and then quantified the level of TBARS, which is produced by lipid oxidation [36], in the whole kidney. The level of TBARS was significantly greater (1.6-fold) in the NEP25 kidney after podocyte injury than in the control (Figure 1A). Immunostaining revealed that 8-OHdG, a marker of oxidative DNA damage [37], was enhanced in glomeruli and tubules of NEP25 mice after podocyte injury but not in the control (Figure 1B). In addition, western blot analysis showed that pJNK, which is induced by oxidative stress, was increased by 2.4-fold in the NEP25 kidney after podocyte injury when compared with the control (Figure 1C). These data indicate that oxidative stress is induced in response to podocyte injury in kidneys from NEP25 mice.
FIGURE 1:
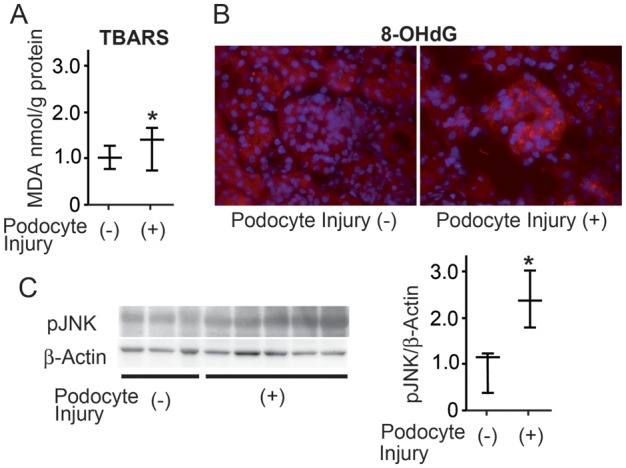
Oxidative stress is induced in NEP 25 kidneys after podocyte injury. NEP25 mice were injected with 2.5 ng/g BW of LMB2, and the kidneys were harvested 7 days later. Kidneys from untreated NEP25 mice were used as controls. (A) The levels of TBARS were increased in NEP 25 kidneys after podocyte injury. (B) Immunostaining for 8-OHdG (red) was enhanced in glomeruli and tubules in the NEP25 kidney after podocyte injury but not in the control kidney. (C) Western blot analysis revealed that pJNK was higher in NEP 25 kidneys after podocyte injury. The horizontal bars represent the median and interquartile ranges. *P < 0.05 compared with untreated NEP25 mice.
Enhancement of Nrf2 signals in the glomeruli of Keap1 knockdown mice
We quantified Keap1 mRNA by real-time RT–PCR in isolated glomeruli. As expected, Keap1 mRNA was markedly lower in Keap1loxP/−/Nep25 than in Keap1+/+/Nep25 glomeruli (Figure 2A). We next analyzed nuclear Nrf2 by western blot analysis in the glomeruli and livers of Keap1loxP/−/Nep25, Keap1+/+/Nep25 and Nrf2−/−/NEP25 mice (Figure 2B). There was clear Nrf2 expression in Keap1+/+/Nep25 liver and even greater expression in Keap1loxP/−/Nep25 liver. Nuclear Nrf2 was not detectable in glomeruli from Keap1+/+/Nep25 mice but was increased in Keap1loxP/−/Nep25 glomeruli. The results indicate that Keap1 knockdown activates Nrf2 in the glomeruli of NEP25 mice.
FIGURE 2:

Keap1 knockdown activated Nrf2 in the glomerulus. (A) Real-time RT–PCR analysis revealed that the level of glomerular Keap1 mRNA was remarkably lower in Keap1loxP/−/Nep25 (F/−) than in Keap1+/+/Nep25 (+/+). The horizontal bars represent the average and standard deviations. *P < 0.05 compared with Keap1+/+. (B) Western blot analysis for nuclear Nrf2 in the glomerulus and the liver. All samples were electrophoresed in the same gel and transferred to the same filter. At the position indicated by the dashed rectangle, a clear band was observed for Nrf2 from livers of Keap1+/+/Nep25 mice, but not from Nrf2−/−/Nep25 mice, whereas from Keap1loxP/−/Nep25 mice, the band was intensified (shown by the arrow). The accompanying larger bands are nonspecific; glomerular nuclei contain larger amounts of the protein causing the nonspecific bands. Nrf2 expression (indicated by the arrow) was only observed in Keap1loxP/−/Nep25 glomeruli. Nrf2 was undetectable in both wild-type and Nrf2−/− glomeruli, using this method.
We next quantified nine mRNAs from Nrf2 target genes by real-time RT–PCR in glomeruli. Without LMB2 injection, the expression of Gclc, Gclm, Gstp1, Gstp2 and Nqo1 was considerably higher in Keap1loxP/−/Nep25 than in Keap1+/+/Nep25 glomeruli, confirming that Nrf2 was indeed activated in Keap1loxP/− glomeruli. The increase was, on average, 2.48-fold for Gclc, 2.33-fold for Gclm, 3.79-fold for Gstp1, 3.08-fold for Gstp2 and 10.37-fold for Nqo1, whereas the expression level of Gpx1, Gsta4, Hox1 and Prdx1 was comparable between the two mouse strains (Figure 3).
FIGURE 3:

Relative mRNA level of Nrf2 target genes in isolated glomeruli. Without LMB2 (-LMB2), Keap1loxP/− (F/−) glomeruli showed significantly greater amount of Gclc (A), Gclm (B), Gstp1 (E), Gstp2 (F) and Nqo1 (G) mRNAs than Keap1+/+ (+/+) glomeruli. Injection of LMB2 did not change the level of these mRNAs in Keap1+/+/Nep25 mice, nor did the level further increase in Keap1F/−/Nep25 mice. The horizontal bars represent the average and standard deviations. *P < 0.05 compared with Keap1+/+ (-LMB2) and #P < 0.05 compared with Keap1+/+ (+LMB2).
Podocyte injury induced by LMB2 did not increase the expression of Nrf2 target genes in Keap1+/+/Nep25 glomeruli. mRNAs for Gclc, Gclm, Gstp1, Gstp2 and Nqo15 did not further increase after podocyte injury in Keap1loxP/−/Nep25 glomeruli, although they are higher than those in Keap1+/+/Nep25 glomeruli with podocyte injury. These results indicate that podocyte injury caused by LMB2 cannot effectively activate the endogenous Nrf2 in glomeruli.
No significant difference was observed in the levels of glomerular Nrf2 mRNA expression between Keap1loxP/−/Nep25 and Keap1+/+/Nep25 mice at baseline, and LMB2 injection did not change the levels of Nrf2 mRNA expression in both groups in this period (Supplementary Figure S2).
Effect of Keap1 knockdown on the progression of glomerulosclerosis
Without LMB2, Keap1loxP/−/Nep25 mice showed no abnormal phenotype in urinary albumin (Figure 4), renal histology, and nephrin and desmin staining (data not shown).
FIGURE 4:

Urinary albumin/creatinine ratio. Both Keap1loxP/−/Nep25 (closed circle) and Keap1+/+/Nep25 (open circle) mice showed moderate albuminuria, which peaked 1 week after the injection. The degree of albuminuria was not statistically significant between the two mouse groups. Data were presented as geographic means and 95% confidence intervals.
After injection of 0.625 ng/g BW of LMB2, both Keap1loxP/−/Nep25 and Keap1+/+/Nep25 mice showed similarly moderate albuminuria, which peaked 1–2 weeks after the injection. The urinary albumin/creatinine ratio was not different between the two groups at any time points (Figure 4). A recent study reported that bardoxolone methyl, an Nrf2 inducer, reduces tubular albumin reabsorption by suppressing megalin in Cynomolgus monkeys [38]. We therefore examined megalin and albumin in a separate set of Nrf2−/−, Keap1loxP/− and wild-type mice without podocyte injury. Notably, there was no appreciable difference in baseline immunoreactivity for megalin among the three genotypes (Supplementary Figure S3). In addition, the megalin mRNA level was also comparable between Keap1loxP/− (relative mRNA level, 2.44 ± 0.46, n = 6) and wild-type mice (2.03 ± 0.78, n = 6). In the Keap1+/+/Nep25 and Keap1loxP/−/Nep25 mice 3 weeks after the LMB2 injection, tubular albumin staining was increased reflecting podocyte damage, and megalin staining was decreased in injured tubules as previously reported [39]. These changes similarly occurred in both types of mice, and there was no difference in megalin and albumin staining between the two genotypes (Supplementary Figure S3).
Histological analysis on renal tissue harvested 3 weeks after the LMB2 injection revealed that the degree of glomerulosclerosis was remarkably attenuated in Keap1loxP/−/Nep25 mice with a median sclerosis index (on a 0–4 scale) of 0.27, compared with Keap1+/+/Nep25 mice with a median sclerosis index of 3.03 (Figures 5 and 6).
FIGURE 5:
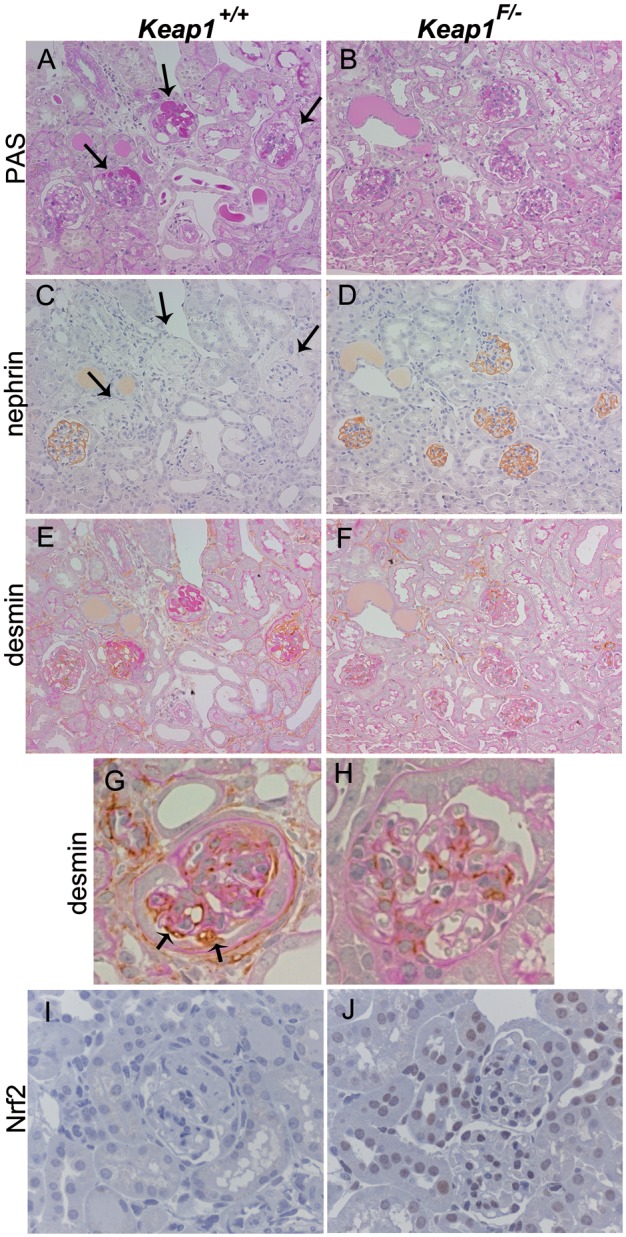
Representative pictures of Keap1+/+/Nep25 and Keap1loxP/−/Nep25 (Keap1F/−/Nep25) after injection of LMB2. Keap1+/+/Nep25 mice showed severe glomerulosclerosis in PAS staining (A, arrows), marked decrease in nephrin staining (C, arrows), and the presence of desmin-positive surface podocytes (E, G, arrows). In contrast, Keap1F/−/Nep25 mice displayed mild glomerulosclerosis (B), almost normal nephrin staining (D), and no desmin-positive surface podocyte (F, H). (I) and (J) show immunostaining for Nrf2. The nuclei in glomerular and tubular cells are positive for Nrf2 staining in Keap1F/−/Nep25 mice (J), but not in Keap1+/+/Nep25 mice. (A), (C) and (E) or (B), (D) and (F) are from adjacent sections. (A–F) (×200), (G–J) (×400).
FIGURE 6:

Knockdown of Keap1 gene attenuated the progression of podocyte injury and glomerulosclerosis. Three weeks after LMB2 injection, Keap1loxP/−/Nep25 (Keap1F/−/Nep25) mice (closed circles) showed significantly attenuated sclerosis index (A), more preserved nephrin staining index (B) and less percentage of glomeruli containing desmin-positive injured podocyte(s) (C), when compared with control Keap1+/+/Nep25 mice (open circles). The horizontal bars represent the medians and interquartile ranges. *P < 0.05 compared with Keap1+/+/Nep25 mice.
Irreversibly injured podocytes intensely express desmin [40]. We quantified the percentage of glomeruli containing desmin-positive surface podocytes for each mouse. This value was significantly lower in Keap1loxP/−/Nep25 mice than in Keap1+/+/Nep25 mice (median 24.5% versus 85.8%) (Figures 5 and 6). Podocyte injury was also assessed by nephrin staining, a more sensitive marker. Nephrin staining was semiquantified using scores of 0 (no staining) to 8 (normal staining). Nephrin staining was also significantly preserved in Keap1loxP/−/Nep25 mice with a median nephrin index of 6.76, compared with Keap1+/+/Nep25 mice with a median nephrin index of 0.91 (Figures 5 and 6).
Moreover, to evaluate the degree of kidney injury more objectively, we performed real-time RT–PCR assay and quantified mRNAs of injury-related genes. The results showed that the levels of Fn1 (the gene for Fibronectin-1; 49.8%), Tgfb1 (the gene for Tgf-β1; 62.9%), Col4a4 (the gene for Collagen, type IV, alpha 4; 61.7%) and Col1a2 (the gene for Collagen, type I, alpha 2; 56.8%) mRNA expression were significantly lower in Keap1loxP/−/Nep25 mice than in Keap1+/+/Nep25 (Figure 7).
FIGURE 7:

Knockdown of Keap1 attenuated the levels of mRNA for injury-related genes. Keap1loxP/− (Keap1F/−) kidneys showed significantly less amounts of mRNA for Fn1 (Fibronectin-1) (A), Tgfb1 (Tgf-β1) (B), Col4a4 (Collagen type IV alpha 4) (D) and Col1a2 (Collagen type I alpha 2) (F) when compared with Keap1+/+ kidneys. The levels of Nphs1 (Nephrin) mRNA (C) and Col1a1(Collagen type I alpha 1) mRNA (E) were not significantly different between the two groups, although Keap1F/−/Nep25 mice show a tendency of protection. *P < 0.05 compared with Keap1+/+/Nep25 mice.
An independent experiment with the same protocol was performed using five Keap1loxP/−/Nep25 mice and eight Keap1+/+/Nep25 mice. Again, Keap1loxP/−/Nep25 mice showed less glomerulosclerosis, less desmin-positive podocytes and more preserved nephrin staining—substantially identical results (Supplementary Figure S4). These results indicate that knockdown of Keap1 gene attenuated the progression of podocyte injury and glomerulosclerosis.
Immunostaining revealed that nuclear Nrf2 staining was present in some glomeruli and tubules in Keap1loxP/−/Nep25 mice, but not in Keap1+/+/Nep25 mice, after LMB2 injection (Figure 5I and J).
Effect of Nrf2 knockout on the progression of glomerulosclerosis
We next examined the effect of Nfr2 knockout on podocyte injury and glomerulosclerosis. Nrf2−/−/Nep25 and Nrf2+/+/Nep25 mice were treated with 0.625 ng/g BW of LMB2 and similarly analyzed. Both types of mice similarly showed moderate albuminuria (Supplementary Figure S5), with no significant difference at any time points. Histological examination at 3 weeks revealed that, although there was a tendency for Nrf2−/−/Nep25 mice to show more severe glomerular injury, the degree of glomerulosclerosis, nephrin staining index and percentage of desmin-positive podocytes were not significantly different between Nrf2−/−/Nep25 and Nrf2+/+/Nep25 mice (Supplementary Figure S6).
DISCUSSION
In the present study, we demonstrated that oxidative stress was induced and systemic knockdown of Keap1 attenuated glomerulosclerosis and podocyte injury in a NEP25 mouse model. Although the beneficial effect was modest, it was reproducibly observed in two independent experiments. This indicates that oxidative stress is involved in the development of glomerulosclerosis.
It had been shown previously that reactive oxygen species (ROS) are increased in the glomerulus of a puromycin aminonucleoside nephritis (PAN) model, and that proteinuria and morphological changes in podocytes in this model are attenuated by decreasing ROS [41–45]. The NEP25 model, which was used in the present study, is basically a focal segmental glomerulosclerosis (FSGS) model, as opposed to the PAN model, which is a minimal change nephropathy model. LMB2, the recombinant immunotoxin used in the present study, is composed of the Fv portion of the anti-hCD25 antibody combined with the Pseudomonas aeruginosa exotoxin PE38. LMB2 itself, unlike adriamycin [32], is not reported to directly generate oxygen radicals. Nonetheless, oxidative stress was modestly increased in the NEP25 kidney, probably by secondary mechanisms. Notably, injected LMB2 is rapidly cleared from the circulation, whereas the podocyte injury progresses over several weeks. Our previous study with chimeric mice indicated that podocyte damage caused by LMB2 secondarily damages other podocytes within the same glomerulus [40]. We therefore speculate that the glomerular phenotype observed 3 weeks after the injection of this low dose (0.625 ng/g BW) of LMB2 reflects both primary and secondary podocyte injury. In that case, the protective effect of Keap1 knockdown may be universally observed in all glomerular diseases with podocyte depletion.
Nrf2-Keap1 is the master regulatory system of antioxidant genes, and, therefore, an attractive drug target. Since high glucose induces ROS production [46, 47] and ROS initiate podocyte apoptosis [48, 49], it is expected that Nrf2 activators have a beneficial effect on diabetic nephropathy. Indeed, pharmacological activation of Nrf2 has been demonstrated to attenuate metabolic disturbance and albuminuria in streptozotocin-induced diabetic nephropathy [30]. Recently, bardoxolone methyl [50–52], a potent Nrf2 inducer, has been shown to elicit significant improvements in the estimated glomerular filtration rate (eGFR) in type 2 diabetes patients with chronic kidney disease in a phase II clinical trial (BEAM study) [53]. In this study, eGFR was elevated within 4 weeks, accompanied with an increase in urinary albumin-creatinine ratio. Renal histology was not evaluated. This raises the possibility that the improvement of renal function is merely attributed to a hemodynamic change, but not to the protection of the underlying glomerular architecture. On the other hand, our study showed that the upregulation of Nrf2 target genes indeed attenuates the development of glomerulosclerosis, indicating that Nrf2 activation can have a therapeutic potential commonly for chronic kidney diseases with various etiology [54, 55].
As expected, western blot and real-time PCR analyses confirmed that nuclear Nrf2 and its target antioxidant genes were upregulated in the glomerulus of Keap1 knockdown mice at baseline. Some Nrf2 target genes were not upregulated, probably reflecting the diversity of the regulatory mechanism among cell types [35]. Notably, the present study showed that the amount of nuclear Nrf2 in normal glomeruli of wild-type mice was lower than that in the liver. In accordance with this observation, podocyte injury per se did not upregulate the antioxidant genes in the glomerulus of wild-type Keap1 mice for at least 5 days. In addition, podocyte injury did not further upregulate the antioxidant genes in Keap1 knockdown mice. In this regard, the glomerulus does not appear to be able to quickly respond to oxidative stress evoked by LMB2-induced podocyte injury.
Keap1 knockdown raised the level of nuclear Nrf2 and enhanced the expression of downstream genes in glomeruli. Immunostaining indicated that Nrf2 was also activated in tubule. Since oxidative stress is induced after podocyte injury, the enhanced antioxidative activities may be the major protective mechanism of Keap1 knockdown, although other mechanisms, e.g. such as the activation of murine double minute (MDM)-2 by Nrf2 [56], cannot be ruled out.
We did not observe that Nrf2 knockout exaggerated podocyte injury and glomerulosclerosis. This negative result may be due to the small number of mice used in the study. Alternatively, this may be explained by low Nrf2 basal activity and ineffective induction of podocyte injury by LMB2 in glomeruli. Absence of the low level of glomerular Nrf2 may not have a great impact on glomerular injury caused by LMB2. However, previous studies reported that Nrf2 knockout exaggerated functional or structural injury in mouse DM nephropathy model [29, 57]. The different results may be because the degree of oxidative stress induced by LMB2 is relatively modest compared with the intense stress resulting from hyperglycemia and because the time for disease development in NEP25 mice is relatively short (3 weeks) when compared with that in the DM model (10–16 weeks).
Despite the reproducible attenuation of glomerulosclerosis in Keap1 knockdown mice, the degree of albuminuria was appreciably indistinguishable between the two types of mice. This is contrasting to the effect of a high dose of losartan, an angiotensin type 1 receptor antagonist, which attenuated both proteinuria and glomerulosclerosis, presumably by reducing glomerular ultrafiltration pressure difference [34]. The Nrf2 activator bardoxolone methyl has been shown to decrease the expression of megalin, a protein involved in the tubular reabsorption of albumin, leading to an increase in albuminuria in monkeys [38]. Therefore, the degree of urinary albumin excretion could be assumed to be overestimated in Keap1 knockdown mice. However, we found that Keap1 knockdown mice similarly express megalin mRNA and protein, and similarly reabsorb albumin to control mice. This indicates that albuminuria similarly reflects glomerular barrier dysfunction in the two mouse groups. The discrepancy in the megalin response between the two studies may be because of the differences between monkeys and mice or because of a possible off-target effect of bardoxolone methyl.
Discrepancy between glomerulosclerosis and proteinuria is often seen in the clinical setting. In this regard, Yu et al. [58] reported that proteinuria is not always in parallel with podocyte loss. Indeed, rats with anti-Thy1.1 nephritis showed transient urinary podocyte loss, whereas proteinuria persisted after urinary podocytes disappeared. We speculate that the kidney of Keap1 knockdown mice is in a similar situation. Thus, Keap1 knockdown may protect podocytes from severe injury that leads to podocyte loss and glomerulosclerosis, but cannot completely preserve their barrier function.
In conclusion, we have demonstrated that Nrf2 activation by genetic Keap1 knockdown attenuated glomerulosclerosis in a mouse model of podocyte-specific injury. This study can provide a foundation for the therapeutic use of Nrf2 activators to prevent the progression of renal failure commonly in chronic kidney diseases with various etiologies.
SUPPLEMENTARY DATA
Supplementary data are available online at http://ndt.oxfordjournals.org.
CONFLICT OF INTEREST STATEMENT
None declared.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Ms Shiho Imai, Ms Chika Sato and Ms Chie Sakurai for excellent technical assistance, Ms Yukiko Tanaka for administrative assistance, Dr Susumu Takekoshi for help with TBARS assay, Dr Takamasa Ishii for help with 8-OHdG staining and pJNK assay. We also thank Dr Reiko Inagi for helpful discussions. Parts of this study were presented in abstract form at the annual meetings of the Japanese Society of Nephrology in 2011. This study was supported by Grant-in-Aid for Scientific Research of Japan Society for the Promotion of Science, MEXT, MEXT-Supported Program for the Strategic Research Foundation at Private Universities (2009–2013), and in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
REFERENCES
- 1.Kwoh C, Shannon MB, Miner JH, et al. Pathogenesis of nonimmune glomerulopathies. Annu Rev Pathol. 2006;1:349–374. doi: 10.1146/annurev.pathol.1.110304.100119. [DOI] [PubMed] [Google Scholar]
- 2.LeHir M, Kriz W. New insights into structural patterns encountered in glomerulosclerosis. Curr Opin Nephrol Hypertens. 2007;16:184–191. doi: 10.1097/MNH.0b013e3280c8eed3. [DOI] [PubMed] [Google Scholar]
- 3.Pollak MR. Focal segmental glomerulosclerosis: recent advances. Curr Opin Nephrol Hypertens. 2008;17:138–142. doi: 10.1097/MNH.0b013e3282f5dbe4. [DOI] [PubMed] [Google Scholar]
- 4.Machuca E, Benoit G, Antignac C. Genetics of nephrotic syndrome: connecting molecular genetics to podocyte physiology. Hum Mol Genet. 2009;18:R185–R194. doi: 10.1093/hmg/ddp328. [DOI] [PubMed] [Google Scholar]
- 5.Patrakka J, Tryggvason K. New insights into the role of podocytes in proteinuria. Nat Rev Nephrol. 2009;5:463–468. doi: 10.1038/nrneph.2009.108. [DOI] [PubMed] [Google Scholar]
- 6.Tryggvason K, Patrakka J, Wartiovaara J. Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med. 2006;354:1387–1401. doi: 10.1056/NEJMra052131. [DOI] [PubMed] [Google Scholar]
- 7.Zenker M, Machuca E, Antignac C. Genetics of nephrotic syndrome: new insights into molecules acting at the glomerular filtration barrier. J Mol Med. 2009;87:849–857. doi: 10.1007/s00109-009-0505-9. [DOI] [PubMed] [Google Scholar]
- 8.Shah SN, He CJ, Klotman P. Update on HIV-associated nephropathy. Curr Opin Nephrol Hypertens. 2006;15:450–455. doi: 10.1097/01.mnh.0000232887.58271.67. [DOI] [PubMed] [Google Scholar]
- 9.Stitt-Cavanagh E, MacLeod L, Kennedy C. The podocyte in diabetic kidney disease. Sci World J. 2009;9:1127–1139. doi: 10.1100/tsw.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Binder CJ, Weiher H, Exner M, et al. Glomerular overproduction of oxygen radicals in Mpv17 gene-inactivated mice causes podocyte foot process flattening and proteinuria: a model of steroid-resistant nephrosis sensitive to radical scavenger therapy. Am J Pathol. 1999;154:1067–1075. doi: 10.1016/S0002-9440(10)65359-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greiber S, Munzel T, Kastner S, et al. NAD(P)H oxidase activity in cultured human podocytes: effects of adenosine triphosphate. Kidney Int. 1998;53:654–663. doi: 10.1046/j.1523-1755.1998.00796.x. [DOI] [PubMed] [Google Scholar]
- 12.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 15.Wakabayashi N, Itoh K, Wakabayashi J, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 16.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31:90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Sykiotis GP, Bohmann D. Stress-activated cap'n'collar transcription factors in aging and human disease. Sci Signal. 2010;3:re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thimmulappa RK, Lee H, Rangasamy T, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thimmulappa RK, Scollick C, Traore K, et al. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. 2006;351:883–889. doi: 10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho HY, Jedlicka AE, Reddy SP, et al. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2002;26:175–182. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 22.Cho HY, Reddy SP, Debiase A, et al. Gene expression profiling of NRF2 mediated protection against oxidative injury. Free Radic Biol Med. 2005;38:325–343. doi: 10.1016/j.freeradbiomed.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 23.Cho HY, Reddy SP, Yamamoto M, et al. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 2004;18:1258–1260. doi: 10.1096/fj.03-1127fje. [DOI] [PubMed] [Google Scholar]
- 24.Rangasamy T, Guo J, Mitzner WA, et al. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rangasamy T, Cho CY, Thimmulappa RK, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao J, Kobori N, Aronowski J, et al. Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci Lett. 2006;393:108–112. doi: 10.1016/j.neulet.2005.09.065. [DOI] [PubMed] [Google Scholar]
- 28.Taguchi K, Maher JM, Suzuki T, et al. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol Cell Biol. 2010;30:3016–3026. doi: 10.1128/MCB.01591-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang T, Huang Z, Lin Y, et al. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:850–860. doi: 10.2337/db09-1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng H, Whitman SA, Wu W, et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011;60:3055–3066. doi: 10.2337/db11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu M, Grigoryev1 DN, Crow MT, et al. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009;76:277–285. doi: 10.1038/ki.2009.157. [DOI] [PubMed] [Google Scholar]
- 32.Tsai P-Y, Ka S-M, Chao T-K, et al. Antroquinonol reduces oxidative stress by enhancing the Nrf2 signaling pathway and inhibits inflammation and sclerosis in focal segmental glomerulosclerosis mice. Free Radic Biol Med. 2011;50:1503–1516. doi: 10.1016/j.freeradbiomed.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 33.Matsusaka T, Xin J, Niwa S, et al. Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol. 2005;16:1013–1023. doi: 10.1681/ASN.2004080720. [DOI] [PubMed] [Google Scholar]
- 34.Matsusaka T, Asano T, Niimura F, et al. Angiotensin receptor blocker protection against podocyte-induced sclerosis is podocyte angiotensin II type 1 receptor-independent. Hypertension. 2010;55:967–973. doi: 10.1161/HYPERTENSIONAHA.109.141994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okawa H, Motohashi H, Kobayashi A, et al. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun. 2006;339:79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- 36.Kanehira T, Takekoshi S, Nagata H, et al. A novel and potent biological antioxidant, Kinobeon A, from cell culture of safflower. Life Sci. 2003;74:87–97. doi: 10.1016/j.lfs.2003.06.033. [DOI] [PubMed] [Google Scholar]
- 37.Onouchi H, Ishii T, Miyazawa M, et al. Mitochondrial superoxide anion overproduction in Tet-mev-1 transgenic mice accelerates age-dependent corneal cell dysfunctions. Invest Ophthalmol Vis Sci. 2012;53:5780–5787. doi: 10.1167/iovs.12-9573. [DOI] [PubMed] [Google Scholar]
- 38.Reisman SA, Chertow GM, Hebbar S, et al. Bardoxolone methyl decreases megalin and activates Nrf2 in the kidney. J Am Soc Nephrol. 2012;23:1663–1673. doi: 10.1681/ASN.2012050457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Motoyoshi Y, Matsusaka T, Saito A, et al. Megalin contributes to the early injury of proximal tubule cells during nonselective proteinuria. Kidney Int. 2008;74:1262–1269. doi: 10.1038/ki.2008.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsusaka T, Sandgren E, Shintani A, et al. Podocyte injury damages other podocytes. J Am Soc Nephrol. 2011;22:1275–1285. doi: 10.1681/ASN.2010090963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diamond JR, Bonventre JV, Karnovsky MJ. A role for oxygen free radicals in aminonucleoside nephrosis. Kidney Int. 1986;29:478–483. doi: 10.1038/ki.1986.24. [DOI] [PubMed] [Google Scholar]
- 42.Thakur V, Walker PD, Shah SV. Evidence suggesting a role for hydroxyl radical in puromycin aminonucleoside-induced proteinuria. Kidney Int. 1988;34:494–499. doi: 10.1038/ki.1988.208. [DOI] [PubMed] [Google Scholar]
- 43.Gwinner W, Landmesser U, Brandes RP, et al. Reactive oxygen species and antioxidant defense in puromycin aminonucleoside glomerulopathy. J Am Soc Nephrol. 1997;8:1722–1731. doi: 10.1681/ASN.V8111722. [DOI] [PubMed] [Google Scholar]
- 44.Marshall CB, Pippin JW, Krofft RD, et al. Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo. Kidney Int. 2006;70:1962–1973. doi: 10.1038/sj.ki.5001965. [DOI] [PubMed] [Google Scholar]
- 45.Kinugasa S, Tojo A, Sakai T, et al. Selective albuminuria via podocyte albumin transport in puromycin nephrotic rats is attenuated by an inhibitor of NADPH oxidase. Kidney Int. 2011;80:1328–1338. doi: 10.1038/ki.2011.282. [DOI] [PubMed] [Google Scholar]
- 46.Hayden MR, Whaley-Connell A, Sowers JR. Renal redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and diabetic nephropathy: paying homage to the podocyte. Am J Nephrol. 2005;25:553–569. doi: 10.1159/000088810. [DOI] [PubMed] [Google Scholar]
- 47.Piwkowska A, Rogacka D, Audzeyenka I, et al. High glucose concentration affects the oxidant-antioxidant balance in cultured mouse podocytes. J Cell Biochem. 2011;112:1661–1672. doi: 10.1002/jcb.23088. [DOI] [PubMed] [Google Scholar]
- 48.Zheng S, Carlson EC, Yang L, et al. Podocyte-specific overexpression of the antioxidant metallothionein reduces diabetic nephropathy. J Am Soc Nephrol. 2008;19:2077–2085. doi: 10.1681/ASN.2007080967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Susztak K, Raff AC, Schiffer M, et al. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 50.Dinkova-Kostova AT, Liby KT, Stephenson KK, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci USA. 2005;102:4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yates MS, Tauchi M, Katsuoka F, et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 2007;6:154–162. doi: 10.1158/1535-7163.MCT-06-0516. [DOI] [PubMed] [Google Scholar]
- 52.Sporn MB, Liby KT, Yore MM, et al. New synthetic triterpenoids: potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J Nat Prod. 2011;74:537–545. doi: 10.1021/np100826q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pergola PE, Raskin P, Toto RD, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365:327–336. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 54.Ruiz S, Pergola PE, Zager RA, et al. Targeting the transcription factor Nrf2 to ameliorate oxidative stress and inflammation in chronic kidney disease. Kidney Int. 2013;83:1029–1041. doi: 10.1038/ki.2012.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zoja C, Benigni A, Remuzzi G. The Nrf2 pathway in the progression of renal disease. Nephrol Dial Transplant. 2014;29(Suppl 1):i19–i24. doi: 10.1093/ndt/gft224. [DOI] [PubMed] [Google Scholar]
- 56.Hagemann JH, Thomasova D, Mulay SR, et al. Nrf2 signalling promotes ex vivo tubular epithelial cell survival and regeneration via murine double minute (MDM)-2. Nephrol Dial Transplant. 2013;28:2028–2037. doi: 10.1093/ndt/gft037. [DOI] [PubMed] [Google Scholar]
- 57.Yoh K, Hirayama A, Ishizaki K, et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells. 2008;13:1159–1170. doi: 10.1111/j.1365-2443.2008.01234.x. [DOI] [PubMed] [Google Scholar]
- 58.Yu D, Petermann A, Kunter U, et al. Urinary podocyte loss is a more specific marker of ongoing glomerular damage than proteinuria. J Am Soc Nephrol. 2005;16:1733–1741. doi: 10.1681/ASN.2005020159. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.